

Abstract
This is the first description of the relationship between chronic ethanol self-administration and the brain transcriptome in a non-human primate (rhesus macaque). Thirty-one male animals self-administered ethanol on a daily basis for over 12 months. Gene transcription was quantified with RNA-Seq in the central nucleus of the amygdala (CeA) and cortical Area 32. We constructed coexpression and cosplicing networks, and we identified areas of preservation and areas of differentiation between regions and network types. Correlations between intake and transcription included largely distinct gene sets and annotation categories across brain regions and between expression and splicing; positive and negative correlations were also associated with distinct annotation groups. Membrane, synaptic and splicing annotation categories were over-represented in the modules (gene clusters) enriched in positive correlations (CeA); our cosplicing analysis further identified the genes affected only at the exon inclusion level. In the CeA coexpression network, we identified Rab6b, Cdk18 and Igsf21 among the intake-correlated hubs, while in the Area 32, we identified a distinct hub set that included Ppp3r1 and Myeov2. Overall, the data illustrate that excessive ethanol self-administration is associated with broad expression and splicing mechanisms that involve membrane and synapse genes.
Keywords: addiction, cosplicing, ethanol, network analysis, non-human primate, RNA-Seq
INTRODUCTION
Beginning with Lewohl et al. (2000), there has been substantial progress in characterizing gene expression in human alcoholics (reviewed in Farris & Mayfield 2014). There is now little doubt that chronic ethanol exposure has marked effects on differential gene expression and associated gene networks. Further, there are some examples of aligning gene expression and genome-wide association study data (Zhou et al. 2011b; Farris et al. 2015) that can, to some extent, disentangle predispositions from consequences of excessive ethanol consumption. However, despite this progress, human postmortem analyses are potentially confounded by a number of variables over which the experimenter has no control and frequently little information. These factors include uneven postmortem intervals, marked differences in subject age, environmental insults over the lifetime and at the time of death, nutritional deficiencies, patterns of alcohol consumption, co-morbid psychopathology and the frequency of withdrawals (Kroenke et al. 2014). These uncontrolled variables are likely to obscure alcohol-specific neuroadaptations and highlight the importance of controlled animal studies.
With this perspective in mind, we have examined brain gene expression from a non-human primate model of oral ethanol self-administration that produces individual differences in stable levels of average daily ethanol intake over the course of months to years while controlling organismal, environmental and necropsy variables (Grant et al. 2008a; Baker et al. 2014; Kroenke et al. 2014). The average daily dose of ethanol consumed (0.46–4.03 g/kg/day per monkey) and corresponding average blood ethanol concentrations (BECs) (2.33–141.2 mg/dl per monkey) provide an opportunity to quantify the factors and brain mechanisms associated with developing an alcohol dependence syndrome (Cuzon Carlson et al. 2011; Welsh et al. 2011) and the associated neuropathology (Kroenke et al. 2014; Lovinger & Kash 2015). The sustained level of voluntary daily intake in this model is analogous to that found in human patterns of alcohol use disorders (AUDs) (Baker et al. 2014). Further, the self-administration of ethanol in monkeys classified as very heavy drinkers (>3 g/kg/day or greater than 12 drinks per day) often surpasses elements of binge drinking that are captured in rodent models that allow voluntary drinking e.g. drinking in the dark (Rhodes et al. 2005; Barkley-Levenson & Crabbe 2012, 2014), or two-bottle choice preference/consumption (e.g. Hitzemann et al. 2009; Metten et al. 2014).
RNA-Seq data were obtained from 31 male rhesus macaques (Macaca mulatta) who were first induced to drink 4 percent (w/v) ethanol over 4 months and then allowed to self-administer ethanol for over 12 months under open-access conditions (22 hours/day, 7 days/week) with concurrent meals and continuous access to water (Grant et al. 2008b; Baker et al. 2014). The RNA-Seq data were analyzed with an emphasis on both gene expression and splicing networks as described elsewhere (Iancu et al. 2015). Data were collected from the central nucleus of the amygdala (CeA) and cortical Area 32, elements of the ‘addiction’ circuit (Koob & Volkow 2010), which have key role(s) in modulating ethanol’s acute and chronic effects (Dhaher et al. 2008; Roberto, Gilpin, & Siggins 2012; Gilpin, Herman, & Roberto 2015) and also have key roles in anxiety disorders, which are often co-morbid with AUDs (Gilpin et al. 2015).
Transcriptome-wide changes related to alcohol phenotypes have been detected in both human and mouse models (Iancu et al. 2013; Farris et al. 2015). Gene splicing variability at the individual gene level has also been described, in particular for AMPA receptors in nonhuman primates (Acosta et al. 2011) and in GABA-B receptors in human alcoholics (Lee, Mayfield, & Harris 2014). Iancu et al. (2015) recently developed a cosplicing network construction methodology that facilitates evaluation of transcriptome-wide coordinated splicing. In the present study, we leverage exon-level data from RNA-Seq and evaluate transcriptome-level changes in gene expression as well as changes in splicing. Overall, the results presented here are, to our knowledge, the first transcriptome-level assessment of the correlation between chronic ethanol self-administration and the macaque brain transcriptome.
MATERIALS AND METHODS
Animals
Male rhesus monkeys that were experimentally naive at the onset of alcohol induction (Macaca mulatta, n = 31, 4–11 years, 6–8 kg) were used. The monkeys were from four cohorts, referred to as cohorts 4, 5, 7a and 7b in the Monkey Alcohol Tissue Research Resource (www.MATRR.com; Daunais et al. 2014). As described previously (Baker et al. 2014), the consumption data for these cohorts were well fit by a bounded uniform distribution, facilitating their analysis as a single group. All monkeys were individually housed (quadrant cages, 0.8 m × 0.8 m × 0.9 m) in a room with controlled light cycle (11 hours lights on, 13 hours lights off), temperature (20–22°C) and humidity (65 percent). The monkeys had visual, auditory and olfactory contact with other monkeys. They were weighed weekly. All procedures complied with the National Institutes of Health and the Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at Oregon National Primate Research Center. Ethanol self-administration, circulating steroid hormones and MRI structural brain imaging data from subsets of these monkeys have been published (Helms, Park, & Grant 2014; Kroenke et al. 2014). The self-administration protocol is described elsewhere (Grant et al. 2008b).
Samples
Monkeys were trained in awake blood draws as described (Porcu et al. 2006), and approximately every fifth day, BEC samples were collected 7 hours after the onset of the 22-hour/day session. Preparation of brain tissue was described previously (Davenport et al. 2014), and the tissues were collected as part of the Monkey Alcohol Tissue Research Resource. Necropsy occurred on an open-access drinking day; monkeys were anesthetized with ketamine (10 mg/kg) and maintained on 1 percent isoflurane. The brains were perfused with ice-cold oxygenated artificial cerebral spinal fluid, removed (< 5 minutes postmortem) and sectioned according to each monkey’s individual MRI (Daunais et al. 2010). The CeA was identified by visible landmarks, lateral of the optic tract and ventral of the globus pallidus (Watson, Paxinos, & Tokuno 2010; −4.5 to −9.9 mm). The block of tissue that contained the CeA was dissected and placed into sterile tubes maintained at −80°. The prefrontal cortex was isolated, and Area 32 was dissected and frozen in liquid nitrogen.
RNA-Seq
Library formation (TruSeq Stranded RNA-Seq with RiboZero Gold rRNA depletion) and sequencing on a HiSeq 2000 were all performed according to Illumina’s specifications at the Oregon Health and Science University Massively Parallel Sequencing Shared Resource. Libraries were multiplexed four per lane, yielding approximately 50 million total reads per sample. FastQC was used for quality checks on the raw sequence data. Reads were then aligned to MacaM assembly (Zimin et al. 2014) using STAR version 2.3.0e (Dobin et al. 2013) with default parameters except for the following: outFilterMismatchNmax = 3, outFilterScoreMinOverLread = 0.33, outFilterMismatchNoverLmax = 0.03 and outFilterMultimapNmax = 1. Using the Bedtools suite version 2.17.0 (Quinlan 2014) and the MacaM v7.6.8 GTF annotation file, read counts were summarized at both the gene and exon levels with the following parameters: S and split. For possible ambiguities to be mitigated, only reads that were located on non-overlapping exon portions were retained for further analysis. Data were imported into the R application environment, and the set of samples included in the network and correlation analysis was determined on the basis of manual curation and statistical outlier detection as described previously (Iancu et al. 2012). These procedures identified one sample from each region (10052 and 10048 in CeA and Area 32, respectively). The final transcriptional dataset included 28 CeA and 30 Area 32 samples selected for further analysis. Upper quartile normalization was performed using the edgeR Bioconductor package. Genes with at least 100 average reads per sample and exons with more than 10 average reads were retained for further analysis. For network construction and correlation with phenotypes, we utilized normalized read counts, while for comparing expression and variability across the two regions, we utilized reads per kilobase per million as returned by the edgeR Bioconductor package (Robinson, McCarthy, & Smyth 2010).
Correlations between gene expression, exon usage/splicing and ethanol intake
The average ethanol intake and BEC data are presented in the Supporting Information Table S1. Because the intake and BEC phenotypes display significant correlations (Supporting Information Table S1), we focused on correlating the gene expression data with average daily intake over the 6 months prior to necropsy for further analysis. For gene expression, the Pearson correlation was used; P values were derived from random resamplings (N = 1000). The positive and negative correlations were analyzed separately given marked differences in annotation. For exon usage, a distance measure coupled with Mantel correlations was used (Iancu et al. 2015). This procedure starts by computing the Canberra distance between any two samples, on the basis of exon counts. A similar size distance measure is then computed between all samples, on the basis of absolute value differences in the ethanol phenotype. These similar-size square matrices were next correlated using the Mantel procedure (Mantel 1967). As in Pearson correlation, we derived P values for significance on the basis of N = 10 000 random permutations. Multiple-testing comparison was performed utilizing the sequential goodness-of-fit method for multiple dependent tests (Carvajal-Rodríguez, de Uña-Alvarez, & Rolán-Alvarez 2009). In contrast with other multiple testing adjustment procedures, the sequential goodness of fit has the desirable property of increased power with increasing number of tests.
Coexpression and cosplicing network construction
Gene coexpression and cosplicing networks were constructed utilizing the weighted gene correlation network analysis (WGCNA) approach (Zhang & Horvath 2005). For all networks, the adjacency matrix was constructed by raising the correlations to a power β, which was chosen independently for each network type and brain region in accordance with the scale-free fit criterion (Zhang & Horvath 2005). Gene clustering was performed using the cutTreeDynamic WGCNA function. Gene connectivity was defined as the sum of all network adjacencies; modular connectivity was restricted to the adjacencies within the gene’s module/cluster. Connectivity values are reported on a percentile scale. Module preservation was evaluated utilizing the WGCNA module preservation function. This function takes as input two-network adjacency matrices and the module assignment in only the first network. The output is a set of Z statistics that essentially evaluate whether the module assignment in the first network corresponds to increased coexpression (and other network statistics) in the second network. The Z values are computed by comparing the coexpression values of the (first network) module as compared with random groups of genes selected from the second network. Z values over 2 are considered to signify moderate preservation, while Z values over 10 signify strong preservation. We note that this evaluation does not depend or utilize the module assignment in the second network. For further details of this procedure, we refer the reader to Langfelder et al. (2011).
Functional significance of modules and other gene groups of interest were evaluated using the GOrilla web tool (Eden et al. 2009). The background gene set was the set of genes selected for network construction. To detect modules enriched in intake-correlated genes, we utilized Fisher’s exact test for significant overlap between each set of module genes and the genes significantly associated with drinking at adjusted P value of 0.2. This relatively permissive threshold is justified by our emphasis on network and system level effects; we also report raw and adjusted P values for all individual genes.
RESULTS
Transcriptional network structure across brain regions
We compared the transcriptional structure in the two regions at both the expression and network level. In terms of gene expression levels, we observed very strong preservation between CeA and Area 32 expression levels as quantified by reads per kilobase per million values (Spearman rho = 0.9—see Supporting Information Figure S1). Gene expression coefficient of variability was much less preserved (Spearman rho = 0.47; Supporting Information Figure S2). Next, we constructed coexpression and cosplicing networks, computed whole network gene connectivity values and correlated them across the four networks (Supporting Information Table S2). Network connectivity was preserved to varying extents across networks, with the strongest preservation between the Area 32 coexpression and cosplicing networks (Spearman rho = 0.53, P < 10−15) and the lowest preservation between CeA coexpression and Area 32 cosplicing (Spearman rho = 0.092, significant at P < 10−15).
To further characterize similarities and differences in transcriptional structure, we inquired whether network structure was preserved at the level of modules. We identified 21 modules in the CeA coexpression network, 16 modules in the CeA cosplicing, 32 modules in Area 32 coexpression and 17 modules in the Area 32 cosplicing. Starting with the two CeA networks, we inquired whether modules in these networks also display above-random coexpression in all other CeA and Area 32 networks (see section). As was the case with the overall connectivity values earlier, preservation varied across network types and regions. Of the 21 CeA coexpression modules, 18 were preserved in the CeA cosplicing network, 8 were preserved in the Area 32 coexpression network and 5 were preserved in the Area 32 cosplicing network. Of the 16 CeA cosplicing modules, 8 were preserved on CeA coexpression network, 4 in the Area 32 coexpression network and 2 in the Area 32 cosplicing network. These data indicate that subnetworks/modules vary widely in preservation, while the network as a whole is only mildly preserved as indicated by the correlations in connectivity values (Supporting Information Table S2). We note that module preservation does not necessarily imply that the same modules can be independently detected in the two networks, but rather that those modules in one network display significant coexpression in the second network. Module preservation values are listed in Supporting Information Table S2.
In all networks, we define network hubs as the genes in the top 20 percent of total network connectivity. Across brain regions and network types, the overlap of hubs was relatively modest in CeA and Area 32 (Figure 1—see also Supporting Information Figure S3), signifying that hub identity is highly region and network type specific. Only 135 genes are hubs in all four networks, despite approximately 90 percent of the genes are included in all four networks. Module membership, connectivity values and correlations with intake are listed in Supporting Information Tables S3, S5, S7 and S9.
Figure 1.
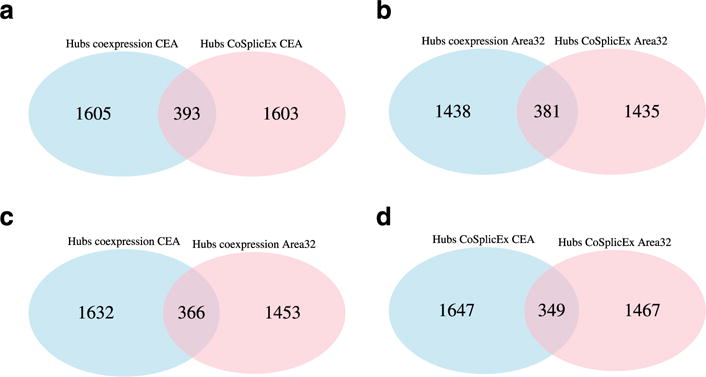
Network structure as quantified by hub identity is distinct across brain regions and network types. (a) CeA coexpression versus cosplicing network hubs. (b) Area 32 coexpression versus cosplicing network hubs. (c) Coexpression, Area 32 versus CeA. (d) Cosplicing, Area 32 versus CeA
Correlation of CeA gene expression with ethanol intake
Pearson correlations were used to determine the relationships between ethanol intake during the 6 months prior to necropsy of self-administration (section) and gene expression. The range of average daily ethanol intake during the 6 months prior to necropsy was 0.52 to 3.79 g/kg. Data for other intake intervals are found in Supporting Information Table S1. The genes correlated with intake (adjusted P < 0.2) (Supporting Information Table S3) were entered into a functional annotation analysis. The positively correlated genes [(+) gene set] (N = 181) were enriched in 15 GO categories with an false discovery rate (FDR) < 0.1 (Supporting Information Table S4). These included membrane annotations (GO:0044425), synapse (GO:0099565), axon (GO:0030516) and process annotations related to the regulation of cell growth (GO:0001558). Genes associated with these annotation categories included Kcnc4 (membrane) and L1cam, Map1b and Limk1 (axon and cell growth). Complete lists of the genes associated with each of the annotation categories are found in Supporting Information Table S4.
For the (+) CeA gene set to be further characterized, the network structure was examined to determine in which modules (sub-networks) the genes were localized and to determine which of the genes were hub nodes (defined as genes with a percentile intramodular connectivity of ≥0.8). Members of the (+) gene set were significantly (P < 2 × 10−4 or better) enriched in four of the 21 CeA coexpression network modules: blue, green, light cyan and turquoise; among these, the light cyan module was the most strongly affected enriched (P < 10−22); it also contains numerous membrane-related and synapse-related genes (Supporting Information Table S4). This module appears specific to CeA coexpression as it has low preservation values in all other networks (Supporting Information Table S2). A number of receptors were associated with the annotation for synaptic signaling, including Chrna7, Chrm3 and Glra2. In contrast to the light cyan module, the green module was significantly less well annotated; however, there was a moderate enrichment of genes associated with clathrin-sculpted GABA vesicular transport.
The negatively correlated genes [(−) gene set] (N = 277) were enriched in 26 GO categories with an FDR of <0.1 (Supporting Information Table S4). These included annotations for nucleic acid metabolic process, nucleic acid binding and transcription factor activity. The network structure was again used to determine which modules were enriched in members of the (−) gene set and to determine which of the genes were hub nodes. Five modules (blue, cyan, green, light cyan and midnight blue) were significantly enriched (Supporting Information Table S3). Note that the blue, green and light cyan modules are also enriched in positively correlated genes. The structure and enrichment of the light cyan module is illustrated in Figure 2. Enrichment with genes from the (+) gene sets in the light cyan module is pronounced (P < 10−22), and additionally, the GO annotations for this module overlap the membrane-related and synapse-related GO categories that characterize the set of positively correlated gene set (Supporting Information Table S4).
Figure 2.
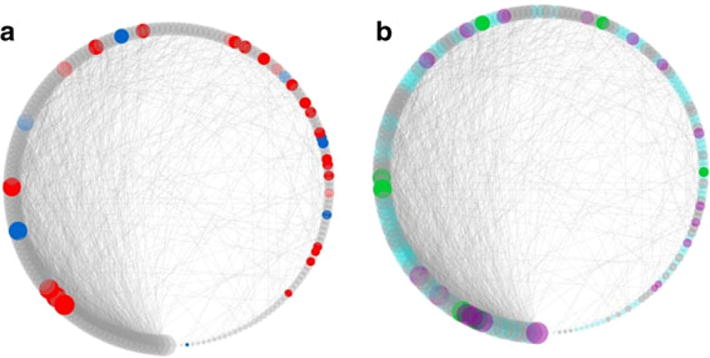
Consumption-correlated genes cluster in a module (light cyan) significantly enriched in synapse and membrane annotations. (a) Positively (red) and negatively (blue) correlated genes. (b) Genes annotated with synapse part are colored in cyan, genes annotated with membrane part are colored in green, and genes with both annotations are colored in purple. Node size is proportional to module connectivity. For visual clarity, only edges with adjacency values over 0.1 are depicted
Among other GO annotations of interest are several splicing-related categories, especially for the blue module, which is enriched in genes from the (−) and (+) gene sets. The genes included in this annotation category include Sfpq (splicing factor proline/glutamine-rich), Srsf11 (serine/arginine-rich splicing factor 11), cleavage stimulation factors such as Cstf2 and Cstf3 and numerous motif-binding and zinc finger proteins.
We additionally correlated the CeA genes with less than 100 average reads per sample with the intake data, finding an additional 227 genes significant at adjusted P value of <0.2 (Supporting Information Table S3). The annotation for these genes was poor, with only a few GO categories significant at FDR 0.1, including plasma membrane-related categories (Supporting Information Table S4).
Correlation of CeA splicing/exon inclusion with ethanol intake
For the splicing/exon inclusion analysis, genes were represented as vectors of exon counts. In this context, a different statistic (the Mantel correlation) was used to detect the relationship between splicing/exon inclusion and intake (Iancu et al. 2015); these data are presented in Supporting Information Table S5. At an adjusted P < 0.2, we detected 480 genes significantly correlated with intake; we note that significant Mantel correlations are positive in nearly all cases as they are generated by pairwise distances. Many of these genes fell into three CeA cosplicing modules: green yellow, salmon and yellow; these modules contained proportionally more significant genes than would be expected given their size (Fisher’s exact test). The salmon module was significantly enriched in mitochondrial, ribosomal and ribosomal protein subunits (Supporting Information Table S6). Overlap of genes correlated to intake in the CeA coexpression network and in the CeA cosplicing network is largely unique (Figure 3). While under ideal conditions the consumption-splicing correlations would also detect all gene level correlations, the two procedures have different levels of power depending on the level of variability in individual exons. The significant GO categories in the cosplicing network were also largely distinct from those detected in the coexpression network, with the exception of the annotation for the salmon module that included several membrane-related categories such as ‘cotranslational protein targeting to membrane’, ‘establishment of protein localization to membrane’ and ‘membrane organization’ (GO:0006613, GO:0090150 and GO:0061024).
Figure 3.
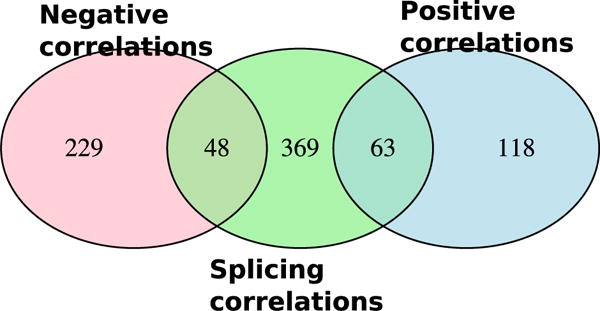
Overlap of three groups of genes correlated to ethanol consumption in the CeA: (1) gene expression negatively correlated to consumption, (2) gene expression positively correlated to consumption, (3) gene splicing correlated to consumption. Correlations at the splicing versus expression level are largely distinct
Correlation of Area 32 gene expression and splicing/exon inclusion with ethanol intake
The results for Area 32 are less pronounced, both in terms of number of genes significantly associated with intake and in terms of GO annotations. In the coexpression network, we detected 122 positively correlated genes and 132 negatively correlated genes (Supporting Information Table S7). Overlap of these genes with genes significant in the CeA is minimal (Supporting Information Figure S4). Annotation for these groups was relatively poor, with only four categories reaching significance for the positive group—these included peptidase and proteasome complexes (Supporting Information Table S8). Although the correlated genes were concentrated in only four modules, only the Area 32 turquoise module had any significant annotation (GO:0044455—mitochondria membrane part). Similarly, splicing effects on Area 32 were minimal, with no genes significant at the threshold of adjusted P values <0.2 (Supporting Information Table S9).
DISCUSSION
Our results identify and annotate a set of correlations between chronic ethanol self-administration and transcriptional structure in rhesus macaques, including changes at the level of exon inclusion rates. By analyzing the transcriptome across two brain regions, we found significant regional specificity in terms of network structure even though gene expression levels are highly preserved. Additionally, we detect some gene hubs that are coordinated only at the exon inclusion/splicing level without being coexpressed at the overall gene expression level; this observation is consistent with our previous analyses in mice (Iancu et al. 2015).
Our analysis of the voluntary intake and transcriptional data proceeded in three steps. The first step was to examine the correlation of gene expression to average daily ethanol intake. The correlation data revealed two important points. One, even within the CeA, the region with more significant correlations, there were only a handful of correlations that exceeded r > 0.6; this observation was taken to be consistent with the idea that chronic ethanol intake is a complex trait, associated with the effects of multiple genes, each with a relatively small influence. However, embedded in the genes associated with intake was a specific annotation structure. For the genes positively correlated to intake and entered into the annotation analysis, 98/181 (54 percent) were annotated as membrane genes (GO:0016020) and 20 clustered within the light cyan module. In turn, this module was richly annotated with synaptic, neuronal and membrane GO categories (GO:0007268, GO:0097458 and GO:0044425, respectively). Additionally, we find among the light cyan genes a number of neurotransmitter receptors (e.g. Chrm3, Chrna4, Chrna7, Glra2, Grm1 and Grm2). A second CeA coexpression module (turquoise) was significantly enriched in positively correlated genes (Fisher’s exact test P < 10−8); this module was also significantly enriched in several membrane-related GO categories (including GO:0044425, GO:0044459 and GO:0031226) as well as neurotransmitter transporter activity (GO:0005326). The present monkey data encompass a larger range of daily intakes (low, binge and heavy drinking) compared with the human alcoholic populations where moderate/binge drinkers are not represented. Nevertheless, the data here are similar to human network data presented elsewhere (e.g. Ponomarev et al. 2012; Farris & Mayfield 2014) and argue that chronic ethanol intake involves large ensembles of genes, with an overrepresentation of genes involved in membrane and/or synaptic function. For example, in Farris et al. (2015), the key hubs in the module (GM15) most closely associated with lifetime alcohol consumption in alcoholics were highly enriched in the GO annotation for synaptic transmission (P < 3 × 10−12).
The genes negatively correlated to intake and entered into the annotation analysis were enriched in GO categories associated with nucleic acid processing and transcription (Supporting Information Table S4). Previously, it was observed that alcohol-preferring inbred mouse strains and animals selectively bred for high alcohol preference have an increased expression of genes associated with transcription, notably key transcription factors (Mulligan et al. 2006). Subsequent studies confirmed the alcohol/transcription interaction (Saba et al. 2006; Hu et al. 2008; Zhou et al. 2011b; Ponomarev et al. 2012). Ponomarev et al. (2012) found, when comparing chronic alcoholics and normal controls, that there was a modest up-regulation of CeA ribosomal genes in the alcoholics. The authors suggested that this may be a consequence of DNA hypomethylation. The data presented here suggest that translation is likely impaired. However, it is unknown if this was a generalized effect or limited to specific protein classes.
The next step in our analysis linked the correlation data with transcriptome network structure. Two pieces of information were extracted. One was to determine which modules (subnetworks) were enriched in the genes correlated with intake. These data provided additional annotation information by identifying the local hubs. For example, the CeA light cyan coexpression module was enriched (P < 10−22) in genes positively correlated to expression. Extending the focus to the six CeA coexpression modules enriched in intake-correlated genes revealed the potential involvement of ion channel complexes, including glutamate and GABA receptor subunits and potassium channels. Beginning with Lewohl, Crane, & Dodd (1997), there is now substantial postmortem evidence that chronic ethanol consumption affects both GABA and glutamate ligand-gated ion channels (Dodd & Lewohl 1998; Mayfield et al. 2002; Buckley et al. 2006; Kuo et al. 2009; Bhandage et al. 2014; Enoch et al. 2014; Farris & Mayfield 2014; Jin et al. 2014). Similar to the data presented here, Farris et al. (2014) have found that network modules strongly associated with lifetime ethanol consumption were enriched in overlapping annotations for potassium channels and transporters (Supporting Information Table S2—Farris & Mayfield 2014).
The network analysis also allowed us to determine which of the genes most strongly associated with intake were highly connected hub nodes. There is compelling evidence, especially from cancer (Bi et al. 2015; Balamurugan 2016) and infectious disease biology (Xu et al. 2014), that targeting hub nodes will significantly affect network structure and, thus, the phenotype of interest. For several of the correlated light cyan hub nodes found in Supporting Information Table S3, we found previous evidences for AUD-related effects. Cdk18 is upregulated in basolateral amygdala in human chronic ethanol drinkers (Ponomarev et al. 2012). Rab6b has been associated with alcohol-related traits in genome-wide association studies (Li et al. 2016). Clstn1 is affected by ethanol exposure in mouse embryo cell cultures (Zhou et al. 2011a). In Area 32, among the hubs significantly correlated with intake, we find Ppp3r1 and Myeov2 in the turquoise coexpression module. Ppp3r1 is located in a genomic region previously associated with ethanol sensitivity (rat alcohol response chromosome 11) (Radcliffe et al. 2004). Myeov2 is located within a mouse alcohol preference quantitative trait locus (QTL) (Alpq5_m) on mouse chromosome 1 (Bice et al. 2009).
The third step of our analysis strategy was to repeat steps one and two but from the perspectives of exon usage and cosplicing networks. For exon usage, a distance measure coupled with Mantel correlations was used (Iancu et al. 2015). This procedure starts by computing the Canberra distance between any two samples, on the basis of exon counts. A similar size distance measure is then computed between all samples, on the basis of absolute value differences in the ethanol phenotype. These similar-size square matrices were next correlated using the Mantel procedure (Mantel 1967). In the CeA, the genes meeting the threshold for inclusion in the annotation analysis were significantly overabundant in three cosplicing modules (green yellow, salmon and yellow—see Supporting Information Table S5). These modules are highly enriched in GO categories associated with protein localization to the endoplasmic reticulum, structural constituent of the ribosome and ribosomal subunit; thus, from the annotation perspective, there was an overlap between the gene and exon level analyses. However, the genes correlated to intake were largely independent between CeA and Area 32 as illustrated in Supporting Information Figure S4. This specificity was also present at the module level: the CeA coexpression light cyan module, which is highly enriched with correlated genes, is not preserved in the other regions and networks. Additionally, the hub identity varied considerably between network types for both CeA and Area 32 (Figure 1). Thus, the exon-level analysis provided a significant amount of additional information on both the system-level transcriptional organization as well as the identity of genes most related to chronic self-administration. At the same time, we note that in the CeA expression data, genes correlated with intake included several genes with role in splicing coordination (Supporting Information Table S4), which suggested that splicing is also associated with intake. Our exon-based cosplicing analysis facilitated the detection of an additional group of genes associated with intake that would be undetectable by only considering overall gene expression.
Overall, the data illustrate that excessive intake is associated with an increased expression of membrane-related genes and decreased expression of translation-related genes, specifically in the CeA, across a range of daily ethanol intakes. A subset of the membrane genes was found to be associated with neuronal function and includes a large number of transmembrane proteins known to regulate synaptic function. We note that most of the genes strongly correlated with excessive intake are among the leaf nodes i.e. genes that are not highly connected. However, understanding the broad transcriptional effects of excessive intake can be facilitated by focusing on hub nodes within key modules. Among these are some hub genes in Area 32 (Ppp3r1 and Myeov2) that reside in previously identified alcohol QTLs and some CeA hub genes that have been previously linked to alcoholism, including Cdk18, Rab6b and Clstn1.
Supplementary Material
Data S1 Supporting info item.
Supplemental Figure 1 Reads per kilobase per million (RPKM) in CeA versus Area32. The expression values in the two regions are remarkably similar (Spearman rho 0.9).
Supplemantal Figure 2 Coefficient of variability of gene expression across the two regions is moderately preserved (Spearman rho 0.47).
Supplemental Figure 3 Hub overlap among the four networks; complements Figure 1 in the text.
Supplemental Figure 4 Overlap between CeA and A32 genes positively and negatively correlated to ethanol consumption. Overlap between gene sets significant in the two regions are largely distinct. Genes included have correlations with adjusted p < 0.2.
Acknowledgments
This study was supported in part by the National Institutes of Health (AA11034, AA13484, AA13510, AA19431, AA109432 and AA10760).
Footnotes
Authors Contribution
JD, KAG and NW were involved in preparing the samples for RNA-Seq. SM, OI, PD, AC (deceased) and RH were involved in data analysis. DO, OI, KAG, NW, CZ, RPS and RH were involved in preparing the manuscript. All authors have critically reviewed content and approved the final version submitted for publication.
SUPPORTING INFORMATION
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site:
References
- Acosta G, Freidman DP, Grant KA, Hemby SE. Alternative splicing of AMPA subunits in prefrontal cortical fields of cynomolgus monkeys following chronic ethanol self-administration. Front Psychiatry. 2011;2:72. doi: 10.3389/fpsyt.2011.00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker EJ, Farro J, Gonzales S, Helms C, Grant KA. Chronic alcohol self-administration in monkeys shows long-term quantity/frequency categorical stability. Alcohol Clin Exp Res. 2014;38:2835–2843. doi: 10.1111/acer.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balamurugan K. HIF-1 at the crossroads of hypoxia, inflammation, and cancer. Int J Cancer. 2016;138:1058–1066. doi: 10.1002/ijc.29519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkley-Levenson AM, Crabbe JC. Ethanol drinking microstructure of a high drinking in the dark selected mouse line. Alcohol Clin Exp Res. 2012;36:1330–1339. doi: 10.1111/j.1530-0277.2012.01749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkley-Levenson AM, Crabbe JC. High drinking in the dark mice: a genetic model of drinking to intoxication. Alcohol Fayettev N. 2014;48:217–223. doi: 10.1016/j.alcohol.2013.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandage AK, Jin Z, Bazov I, Kononenko O, Bakalkin G, Korpi ER, Birnir B. GABA-A and NMDA receptor subunit mRNA expression is altered in the caudate but not the putamen of the postmortem brains of alcoholics. Front Cell Neurosci. 2014;8:415. doi: 10.3389/fncel.2014.00415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi D, Ning H, Liu S, Que X, Ding K. Gene expression patterns combined with network analysis identify hub genes associated with bladder cancer. Comput Biol Chem. 2015;56:71–83. doi: 10.1016/j.compbiolchem.2015.04.001. [DOI] [PubMed] [Google Scholar]
- Bice P, Valdar W, Zhang L, Liu L, Lai D, Grahame N, Flint J, Li T-K, Lumeng L, Fraud T. Genomewide SNP screen to detect quantitative trait loci for alcohol preference in the high alcohol preferring and low alcohol preferring mice. Alcohol Clin Exp Res. 2009;33:531–537. doi: 10.1111/j.1530-0277.2008.00866.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley ST, Foley PF, Innes DJ, Loh E-W, Shen Y, Williams SM, Harper CG, Tannenberg AEG, Dodd PR. GABA(A) receptor beta isoform protein expression in human alcoholic brain: interaction with genotype. Neurochem Int. 2006;49:557–567. doi: 10.1016/j.neuint.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Carvajal-Rodríguez A, de Uña-Alvarez J, Rolán-Alvarez E. A new multitest correction (SGoF) that increases its statistical power when increasing the number of tests. BMC Bioinformatics. 2009;10:209. doi: 10.1186/1471-2105-10-209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuzon Carlson VC, Seabold GK, Helms CM, Garg N, Odagiri M, Rau AR, Daunais J, Alvarez VA, Lovinger DM, Grant KA. Synaptic and morphological neuroadaptations in the putamen associated with long-term, relapsing alcohol drinking in primates. Neuropsychopharmacology. 2011;36:2513–2528. doi: 10.1038/npp.2011.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daunais JB, Davenport AT, Helms CM, Gonzales SW, Hemby SE, Friedman DP, Farro JP, Baker EJ, Grant KA. Monkey Alcohol Tissue Research Resource: banking tissues for alcohol research. Alcohol Clin Exp Res. 2014;38:1973–1981. doi: 10.1111/acer.12467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daunais JB, Kraft RA, Davenport AT, Burnett EJ, Maxey VM, Szeliga KT, Rau AR, Flory GS, Hemby SE, Kroenke CD, et al. MRI-guided dissection of the nonhuman primate brain: a case study. Methods San Diego Calif. 2010;50:199–204. doi: 10.1016/j.ymeth.2009.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport AT, Grant KA, Szeliga KT, Friedman DP, Daunais JB. Standardized method for the harvest of nonhuman primate tissue optimized for multiple modes of analyses. Cell Tissue Bank. 2014;15:99–110. doi: 10.1007/s10561-013-9380-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhaher R, Finn D, Snelling C, Hitzemann R. Lesions of the extended amygdala in C57BL/6J mice do not block the intermittent ethanol vapor-induced increase in ethanol consumption. Alcohol Clin Exp Res. 2008;32:197–208. doi: 10.1111/j.1530-0277.2007.00566.x. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-Seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodd PR, Lewohl JM. Cell death mediated by amino acid transmitter receptors in human alcoholic brain damage: conflicts in the evidence. Ann N Y Acad Sci. 1998;844:50–58. [PubMed] [Google Scholar]
- Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enoch M-A, Rosser AA, Zhou Z, Mash DC, Yuan Q, Goldman D. Expression of glutamatergic genes in healthy humans across 16 brain regions; altered expression in the hippocampus after chronic exposure to alcohol or cocaine. Genes Brain Behav. 2014;13:758–768. doi: 10.1111/gbb.12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farris SP, Mayfield RD. RNA-Seq reveals novel transcriptional reorganization in human alcoholic brain. Int Rev Neurobiol. 2014;116:275–300. doi: 10.1016/B978-0-12-801105-8.00011-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farris SP, Arasappan D, Hunicke-Smith S, Harris RA, Mayfield RD. Transcriptome organization for chronic alcohol abuse in human brain. Mol Psychiatry. 2015;20:1438–1447. doi: 10.1038/mp.2014.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilpin NW, Herman MA, Roberto M. The central amygdala as an integrative hub for anxiety and alcohol use disorders. Biol Psychiatry. 2015;77:859–869. doi: 10.1016/j.biopsych.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant KA, Helms CM, Rogers LSM, Purdy RH. Neuroactive steroid stereospecificity of ethanol-like discriminative stimulus effects in monkeys. J Pharmacol Exp Ther. 2008b;326:354–361. doi: 10.1124/jpet.108.137315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant KA, Leng X, Green HL, Szeliga KT, Rogers LSM, Gonzales SW. Drinking typography established by scheduled induction predicts chronic heavy drinking in a monkey model of ethanol self-administration. Alcohol Clin Exp Res. 2008a;32:1824–1838. doi: 10.1111/j.1530-0277.2008.00765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helms CM, Park B, Grant KA. Adrenal steroid hormones and ethanol self-administration in male rhesus macaques. Psychopharmacology (Berl) 2014;231:3425–3436. doi: 10.1007/s00213-014-3590-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitzemann R, Edmunds S, Wu W, Malmanger B, Walter N, Belknap J, Darakjian P, McWeeney S. Detection of reciprocal quantitative trait loci for acute ethanol withdrawal and ethanol consumption in heterogeneous stock mice. Psychopharmacology (Berl) 2009;203:713–722. doi: 10.1007/s00213-008-1418-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Saba L, Kechris K, Bhave SV, Hoffman PL, Tabakoff B. Genomic insights into acute alcohol tolerance. J Pharmacol Exp Ther. 2008;326:792–800. doi: 10.1124/jpet.108.137521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iancu OD, Colville A, Oberbeck D, Darakjian P, McWeeney SK, Hitzemann R. Cosplicing network analysis of mammalian brain RNA-Seq data utilizing WGCNA and Mantel correlations. Front Genet. 2015;6:174. doi: 10.3389/fgene.2015.00174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iancu OD, Darakjian P, Kawane S, Bottomly D, Hitzemann R, McWeeney SK. Detection of expression quantitative trait loci in complex mouse crosses: impact and alleviation of data quality and complex population substructure. Stat Genet Methodol. 2012;3:157. doi: 10.3389/fgene.2012.00157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iancu OD, Oberbeck D, Darakjian P, Metten P, McWeeney S, Crabbe JC, Hitzemann R. Selection for drinking in the dark alters brain gene coexpression networks. Alcohol Clin Exp Res. 2013;37:1295–1303. doi: 10.1111/acer.12100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z, Bhandage AK, Bazov I, Kononenko O, Bakalkin G, Korpi ER, Birnir B. Expression of specific ionotropic glutamate and GABA-A receptor subunits is decreased in central amygdala of alcoholics. Front Cell Neurosci. 2014;8:288. doi: 10.3389/fncel.2014.00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacol. Off Publ Am Coll Neuropsychopharmacol. 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroenke CD, Rohlfing T, Park B, Sullivan EV, Pfefferbaum A, Grant KA. Monkeys that voluntarily and chronically drink alcohol damage their brains: a longitudinal MRI study. Neuropsychopharmacol Off Publ Am Coll Neuropsychopharmacol. 2014;39:823–830. doi: 10.1038/npp.2013.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo P-H, Kalsi G, Prescott CA, Hodgkinson CA, Goldman D, Alexander J, van den Oord EJ, Chen X, Sullivan PF, Patterson DG, et al. Associations of glutamate decarboxylase genes with initial sensitivity and age-at-onset of alcohol dependence in the Irish affected sib pair study of alcohol dependence. Drug Alcohol Depend. 2009;101:80–87. doi: 10.1016/j.drugalcdep.2008.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P, Luo R, Oldham MC, Horvath S. Is my network module preserved and reproducible? PLoS Comput Biol. 2011;7:e1001057. doi: 10.1371/journal.pcbi.1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Mayfield RD, Harris RA. Altered gamma-aminobutyric acid type B receptor subunit 1 splicing in alcoholics. Biol Psychiatry. 2014;75:765–773. doi: 10.1016/j.biopsych.2013.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewohl JM, Crane DI, Dodd PR. Expression of the alpha 1, alpha 2 and alpha 3 isoforms of the GABAA receptor in human alcoholic brain. Brain Res. 1997;751:102–112. doi: 10.1016/s0006-8993(96)01396-0. [DOI] [PubMed] [Google Scholar]
- Lewohl JM, Wang L, Miles MF, Zhang L, Dodd PR, Harris RA. Gene expression in human alcoholism: microarray analysis of frontal cortex. Alcohol Clin Exp Res. 2000;24:1873–1882. [PubMed] [Google Scholar]
- Li MJ, Liu Z, Wang P, Wong MP, Nelson MR, Kocher J-PA, Yeager M, Sham PC, Chanock SJ, Xia Z, et al. GWASdb v2: an update database for human genetic variants identified by genome-wide association studies. Nucleic Acids Res. 2016;44:D869–D876. doi: 10.1093/nar/gkv1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, Kash TL. Mechanisms of neuroplasticity and ethanol’s effects on plasticity in the striatum and bed nucleus of the stria terminalis. Alcohol Res Curr Rev. 2015;37:109–124. doi: 10.35946/arcr.v37.1.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantel N. The detection of disease clustering and a generalized regression approach. Cancer Res. 1967;27:209–220. [PubMed] [Google Scholar]
- Mayfield RD, Lewohl JM, Dodd PR, Herlihy A, Liu J, Harris RA. Patterns of gene expression are altered in the frontal and motor cortices of human alcoholics. J Neurochem. 2002;81:802–813. doi: 10.1046/j.1471-4159.2002.00860.x. [DOI] [PubMed] [Google Scholar]
- Metten P, Iancu OD, Spence SE, Walter NAR, Oberbeck D, Harrington CA, Colville A, McWeeney S, Phillips TJ, Buck KJ, et al. Dual-trait selection for ethanol consumption and withdrawal: genetic and transcriptional network effects. Alcohol Clin Exp Res. 2014;38:2915–2924. doi: 10.1111/acer.12574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan MK, Ponomarev I, Hitzemann RJ, Belknap JK, Tabakoff B, Harris RA, Crabbe JC, Blednov YA, Grahame NJ, Phillips TJ, et al. Toward understanding the genetics of alcohol drinking through transcriptome meta-analysis. Proc Natl Acad Sci U S A. 2006;103:6368–6373. doi: 10.1073/pnas.0510188103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomarev I, Wang S, Zhang L, Harris RA, Mayfield RD. Gene coexpression networks in human brain identify epigenetic modifications in alcohol dependence. J Neurosci Off J Soc Neurosci. 2012;32:1884–1897. doi: 10.1523/JNEUROSCI.3136-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcu P, Rogers LSM, Morrow AL, Grant KA. Plasma pregnenolone levels in cynomolgus monkeys following pharmacological challenges of the hypothalamic–pituitary–adrenal axis. Pharmacol Biochem Behav. 2006;84:618–627. doi: 10.1016/j.pbb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Quinlan AR. BEDTools: the Swiss-Army tool for genome feature analysis. Curr Protoc Bioinformatics. 2014;47:1–34. doi: 10.1002/0471250953.bi1112s47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radcliffe RA, Erwin VG, Draski L, Hoffmann S, Edwards J, Deng X-S, Bludeau P, Fay T, Lundquist K, Asperi W, et al. Quantitative trait loci mapping for ethanol sensitivity and neurotensin receptor density in an F2 intercross derived from inbred high and low alcohol sensitivity selectively bred rat lines. Alcohol Clin Exp Res. 2004;28:1796–1804. doi: 10.1097/01.alc.0000148106.71801.d7. [DOI] [PubMed] [Google Scholar]
- Rhodes JS, Best K, Belknap JK, Finn DA, Crabbe JC. Evaluation of a simple model of ethanol drinking to intoxication in C57BL/6J mice. Physiol Behav. 2005;84:53–63. doi: 10.1016/j.physbeh.2004.10.007. [DOI] [PubMed] [Google Scholar]
- Roberto M, Gilpin NW, Siggins GR. The central amygdala and alcohol: role of γ-aminobutyric acid, glutamate, and neuropeptides. Cold Spring Harb Perspect Med. 2012;2:a012195. doi: 10.1101/cshperspect.a012195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinforma Oxf Engl. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saba L, Bhave SV, Grahame N, Bice P, Lapadat R, Belknap J, Hoffman PL, Tabakoff B. Candidate genes and their regulatory elements: alcohol preference and tolerance. Mamm Genome Off J Int Mamm Genome Soc. 2006;17:669–688. doi: 10.1007/s00335-005-0190-0. [DOI] [PubMed] [Google Scholar]
- Watson CR, Paxinos G, Tokuno H. Using a panel of immunomarkers to define homologies in mammalian brains. Front Hum Neurosci. 2010;4:13. doi: 10.3389/neuro.09.013.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsh JP, Han VZ, Rossi DJ, Mohr C, Odagiri M, Daunais JB, Grant KA. Bidirectional plasticity in the primate inferior olive induced by chronic ethanol intoxication and sustained abstinence. Proc Natl Acad Sci U S A. 2011;108:10314–10319. doi: 10.1073/pnas.1017079108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Ye B, Han Z, Huang M, Zhu Y. Comparison of transcriptional profiles between CD4+ and CD8+ T cells in HIV type 1-infected patients. AIDS Res Hum Retroviruses. 2014;30:134–141. doi: 10.1089/aid.2013.0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4 doi: 10.2202/1544-6115.1128. Article17. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Yuan Q, Mash DC, Goldman D. Substance-specific and shared transcription and epigenetic changes in the human hippocampus chronically exposed to cocaine and alcohol. Proc Natl Acad Sci U S A. 2011b;108:6626–6631. doi: 10.1073/pnas.1018514108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FC, Zhao Q, Liu Y, Goodlett CR, Liang T, McClintick JN, Edenberg HJ, Li L. Alteration of gene expression by alcohol exposure at early neurulation. BMC Genomics. 2011a;12:124. doi: 10.1186/1471-2164-12-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimin AV, Cornish AS, Maudhoo MD, Gibbs RM, Zhang X, Pandey S, Meehan DT, Wipfler K, Bosinger SE, Johnson ZP, Tharp GK, Marçais G, Roberts M, Ferguson B, Fox HS, Treangen T, Salzberg SL, Yorke JA, Norgren RB., Jr A new rhesus macaque assembly and annotation for next-generation sequencing analyses. Biol Direct. 2014;9:1–15. doi: 10.1186/1745-6150-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1 Supporting info item.
Supplemental Figure 1 Reads per kilobase per million (RPKM) in CeA versus Area32. The expression values in the two regions are remarkably similar (Spearman rho 0.9).
Supplemantal Figure 2 Coefficient of variability of gene expression across the two regions is moderately preserved (Spearman rho 0.47).
Supplemental Figure 3 Hub overlap among the four networks; complements Figure 1 in the text.
Supplemental Figure 4 Overlap between CeA and A32 genes positively and negatively correlated to ethanol consumption. Overlap between gene sets significant in the two regions are largely distinct. Genes included have correlations with adjusted p < 0.2.