

Abstract
Bisegmented dsRNA viruses that infect most or all isolates of apicomplexan parasite Cryptosporidium parvum are currently assigned to a single species, Cryptosporidium parvum virus 1, in genus Cryspovirus, family Partitiviridae. An analysis of existing sequence data suggested that the complete sequences of both cryspovirus genome segments, dsRNA1 and dsRNA2, had yet to be determined. We therefore set out to accomplish this for the virus strain that infects C. parvum isolate Iowa. The results suggest that several previous cryspovirus sequences are indeed truncated at one or both segment termini and also identify sequences at or near the termini that are conserved in both segments. Complete sequences of other cryspovirus strains, including ones from other Cryptosporidium species, are needed for refining their classification into one or more virus species.
Introduction
Cryptosporidia, members of the diverse phylum Apicomplexa, are protozoan parasites of humans and other vertebrates. A number of Cryptosporidium species, but primarily C. parvum (formerly C. parvum genotype 2) and C. hominis (formerly C. parvum genotype 1), are causes of human disease worldwide. The most common symptom of cryptosporidiosis is watery diarrhea, which is self-limited in healthy adults but more persistent and severe in immunocompromised individuals, including untreated HIV/AIDs patients and undernourished children [2].
One notable feature of many C. parvum and hominis isolates is their cryptic infection by a bisegmented dsRNA virus [5–16, 18–20, 21, 22]. In addition to numerous partial sequences [5–7, 11–15, 18, 20, 21, 22], putatively complete sequences of both genome segments have been reported from only two bovine isolates of C. parvum: KSU1 [10] and Changchun [16]. The plus-strand sequence of each segment encompasses a single long ORF, encoding the viral RdRp in dsRNA1 and the viral CP in dsRNA2 [7–10]. The virus from C. parvum KSU1 is the exemplar strain of type species Cryptosporidium parvum virus 1, in genus Cryspovirus, family Partitiviridae [19]. The virus from C. parvum Changchun is closely related to CSpV1-KSU1, sharing >97% identity across their deduced protein sequences (see Fig. 1B).
Fig. 1.
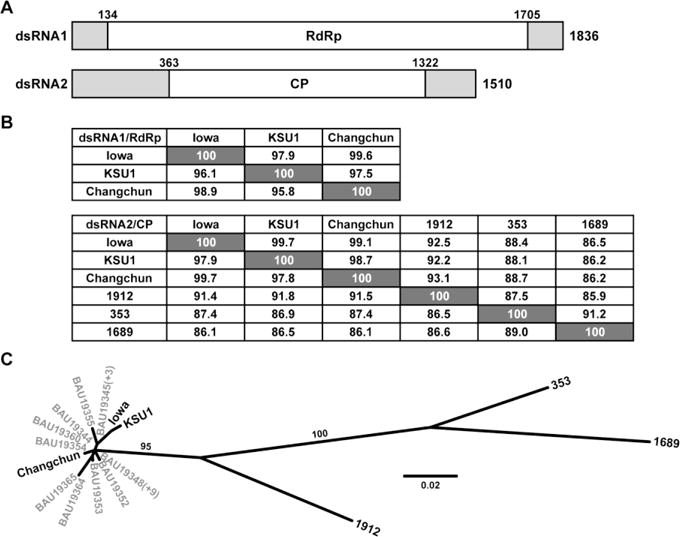
(A) Diagram of the genomic RNA plus strands of CSpV1-Iowa. The beginning and end of the long ORF encoding RdRp or CP are indicated by nt positions in each RNA. Terminal NTRs are shaded. (B) Pairwise alignment scores (%). Values at the lower left of each panel are for the protein-encoding RNA sequences of each genome segment; values at the upper right are for the deduced protein sequences. (C) Maximum-likelihood phylogenetic tree (substitution model, LG; gamma shape parameter, 0.751; proportion of invariant, 0.184) for the CP sequences of the indicated cryspovirus strains (see main text). Branch support values (%) are shown for only the two long branches. Some of the C. parvum-derived strains from Murakoshi et al. [18] share the same CP sequence, indicated by (+N) in the figure.
In addition to the putatively complete sequences from two C. parvum isolates, Leoni et al. [13] have reported the putatively complete sequences of dsRNA2 from three other species: C. hominis (isolate 1912), felis (isolate 353), and meleagridis (isolate 1689). These results indicate that viruses from four different cryptosporidia share >85% identity in pairwise comparisons of their deduced CP sequences (see Fig. 1B), suggesting either slow evolutionary divergence of cryspoviruses in these different host species or recent/ongoing exchange of the viruses among them. It is also noteworthy that the dsRNA2 lengths reported from C. hominis, felis, and meleagridis are similar to one another (1481–1502 bp) [13], but longer than those reported from C. parvum (1374–1375 bp) [10, 16]. This discrepancy suggested to us that the sequences from C. parvum might be truncated at one or both termini of each genome segment. Consistent with this suggestion is that Leoni et al. [13] used methods for rapid amplification of cDNA ends (RACE) to sequence the segment termini, whereas the others did not. We therefore set out to determine the complete sequences of both dsRNA1 and dsRNA2 from the same C. parvum isolate, which seemed not to have yet been done.
Provenance of the virus material
Previous authors [6, 7, 10, 21] have shown evidence that C. parvum isolate Iowa, originally dating from the 1980s and obtained from at least three different suppliers for the cited studies, is infected with CSpV1. For the current study, we purchased oocysts from Bunch Grass Farm (Idaho) and after allowing them to excyst in vitro, used the sporozoites to infect Caco-2 cells [17]. At 48 h postinfection, total RNA was extracted. Primers designed from reported dsRNA1 and dsRNA2 sequences [10, 16] were then used for RT-PCR, and the resulting amplicons were subjected to direct Sanger sequencing. Following cellulose enrichment of dsRNA from the total RNA [1, 4], RNA-ligase-mediated rapid amplification of 3′ cDNA ends (3′RLM-RACE) was also performed [3, 4], allowing the terminal sequences of both segments to be determined. Complete sequences for dsRNA1 and dsRNA2 of CSpV1-Iowa were thereby obtained and deposited in GenBank.
ORFs were identified using EMBOSS getorf, and pairwise sequence alignments were performed using EMBOSS Water or Needleall (http://www.bioinformatics.nl/emboss-explorer/). Multiple sequence alignments were performed using Clustal Omega (http://www.ebi.ac.uk/Tools/msa/). Phylogenetic analyses were performed using PhyML 3.0 (https://www.hiv.lanl.gov/content/sequence/PHYML/interface.html) with parameters Sequence type/model, Amino acids/JTT, LG, rtREV, or WAG (each yielded very similar results); Proportion of invariable sites, estimated from data; Gamma shape parameter, estimated from data; Starting tree(s) optimization, Tree topology and branch length; Tree improvement, Best of NNI and SPR; Branch support, Approximate Likelihood Ratio Test (aLRT), SH-like supports.
Sequence properties
The dsRNA1 sequence of CSpV1-Iowa (GenBank KY884720) spans 1836 bp, longer than reported for CSpV1-KSU1 (1786 bp) and Changchun (1783 bp) [10, 16] (Table 1). The plus-strand sequence encompasses one long ORF spanning 1572 nt from the first in-frame AUG codon at nt positions 134–136 through the first in-frame stop codon at nt positions 1703–1705 (Fig. 1A). The ORF encodes a deduced protein 523 aa long (62 kDa; pI, 9.45) and exhibiting strong sequence similarities to viral RdRps, as expected from previous work [10]. This RdRp length matches that reported for CSpV1-Changchun but is a single residue shorter than for CSpV1-KSU1, due to a 1-codon deletion corresponding to nt positions 1305–1307 in KSU1. Sixteen partial dsRNA1 sequences that cross this region (all from C. parvum isolates) [18] are also missing this codon.
Table 1.
Cryspovirus sequence features
| Genome segment | Host species and isolate | Lengths: RNA (nt) | protein (aa) | NTRs (nt): 5′ | 3′a | GenBank acc. no. |
|---|---|---|---|---|---|---|
| dsRNA1 | C. parvum KSU1 | 1786 | 524 | 133 | 78 | U95995 |
| C. parvum Changchun | 1783 | 523 | 133 | 78 | EU183403 | |
| C. parvum Iowa | 1836 | 523 | 133 | 131 | KY884720 | |
| dsRNA2 | C. parvum KSU1 | 1374 | 319 | 247 | 167 | U95996 |
| C. parvum Changchun | 1375 | 319 | 247 | 168 | EU183404 | |
| C. hominis 1912 | 1492 | 319 | 346 | 186 | DQ193518 | |
| C. felis 353 | 1502 | 335/319 b | 307/355 b | 187 | DQ193520 | |
| C. meleagridis 1689 | 1481 | 319 | 345 | 176 | DQ193519 | |
| C. parvum Iowa | 1510 | 319 | 362 | 188 | KY884721 |
Not including the ORF stop codon.
The first in-frame AUG codon in C. felis 353 dsRNA2 (nt positions 308–310) yields a deduced CP that is 335 aa long. However, this AUG codon is not conserved in the other cryspoviruses. The first in-frame AUG codon that is conserved in all strains yields a deduced CP that is 319 aa long. Thus, in C. felis 353 dsRNA2, it might be the second in-frame AUG codon (nt positions 356–358) that is used for translation initiation [13].
The dsRNA2 sequence of CSpV1-Iowa (GenBank KY884721) spans 1510 bp, longer than reported for CSpV1-KSU1 (1374 bp) and Changchun (1375 bp) [10, 16] but similar to those reported from C. hominis 1912, felis 353, and meleagridis 1689 (1481–1502 bp) [13] (Table 1). The plus-strand sequence encompasses one long ORF spanning 960 nt from the first in-frame AUG codon at nt positions 363–365 through the first in-frame stop codon at nt positions 1320–1322 (Fig. 1A). The ORF encodes a deduced protein 319 aa long (37 kDa; pI, 8.22), presumably the viral CP [8, 9]. This CP length matches those of both CSpV1-KSU1 and Changchun, and also those of the viruses from C. hominis 1912, meleagridis 1689, and possibly C. felis 353 (Table 1).
The complete or putatively complete sequences now available for nine different cryspovirus genome segments (3 dsRNA1, 6 dsRNA2) were compared via pairwise alignments. The results indicate that it is proper to identify CSpV1-Iowa as another strain of species Cryptosporidium parvum virus 1, along with strains CSpV1-KSU1 and Changchun, given the high degree of conservation in both the RNA sequences (95.8–99.7% identity) and the protein sequences (97.5–99.7% identity) of these three viruses from C. parvum (Fig. 1B). The viruses from C. hominis 1912, felis 353, and meleagridis 1689, on the other hand, are somewhat more divergent in their dsRNA2 sequences (86.1–91.8% identity) and CP sequences (85.9–93.1% identity), though the degree of conservation is still high. The CP sequences were also compared by phylogenetic analysis, including the nearly complete CP sequences reported from 22 additional C. parvum isolates [18]. The results again show greater divergence of the viruses from C. hominis 1912, felis 353, and meleagridis 1689 vs. the lesser divergence among all 25 viruses derived from C. parvum (Fig. 1C). Complete genome sequences for other cryspovirus strains are needed before robust conclusions can be drawn, but this host-specific pattern of divergence suggests that exchange of viruses among some of these different host species may be infrequent [7, 12, 13]. Partial CSpV1-Iowa sequences reported previously [6, 21] and the complete ones reported here are highly similar across their regions of overlap (≥96.9%), and the observed divergence is likely explained by the different passage histories of the C. parvum Iowa samples obtained from different suppliers up to 20 years apart.
Based on the start and stop codons for dsRNA1 and dsRNA2 suggested above, the terminal nontranslated regions (NTRs) in the CSpV1-Iowa plus-strand sequences span 133 and 131 nt at the 5′ and 3′ ends of dsRNA1, and 362 and 188 nt at the 5′ and 3′ ends of dsRNA2 (Table 1). The 5′-NTR of dsRNA1 has the same length as reported for CSpV1-KSU1 and Changchun [10, 16], suggesting that all three of these sequences are complete at that end. The 3′-NTR of dsRNA1, on the other hand, is longer than reported for CSpV1-KSU1 and Changchun, suggesting that the latter are 3′-truncated. The 5′- and 3′-NTRs of dsRNA2 are each longer than reported for CSpV1-KSU1 and Changchun, suggesting that the latter sequences are truncated at both ends. The 5′- and 3′-NTRs of dsRNA2 are nearer the sizes reported from C. hominis 1912, felis 353, and meleagridis 1689 [13], but even the latter appear to be truncated by a few nucleotides based on their sequence alignments with CSpV1-Iowa (Fig. S1B).
Notably, dsRNA1 and dsRNA2 of CSpV1-Iowa share several stretches of conserved plus-strand sequences (Fig. S1), including 5 nt at their 5′ ends and 17 nt at their 3′ ends. An even longer conserved sequence (39 nt) is set back from the 3′ ends, but wholly within the 3′ NTRs of both segments, at nt positions 1767–1805 in dsRNA1 and 1431–1469 in dsRNA2. Lastly, dsRNA1 and dsRNA2 share a shorter conserved region (13 nt) within their central protein-coding regions: UAAGAAAGUACCU, at nt positions 691–703 in dsRNA1 and 581–593 in dsRNA2. These motifs conserved between dsRNA1 and dsRNA2 in CSpV1-Iowa are also wholly or partly conserved in CSpV1-KSU1 and Changchun, as well as in the viruses from C. hominis 1912, felis 353, and meleagridis 1689 (Fig. S1). The central motif is also conserved in both segments of numerous strains from the Murakoshi et al. study [18]. These sequences conserved between dsRNA1 and dsRNA2 in multiple cryspovirus strains seem likely to be involved in specific functions, such as plus-strand RNA packaging into nascent particles, RdRp binding for minus- and/or plus-strand RNA synthesis, and possibly even translation. Since partitiviruses package their two segments in separate particles, both segments might be expected to contain similar packaging signals.
In conclusion, we report the first complete sequences of both genome segments from a single strain of type species Cryptosporidium parvum virus 1, in genus Cryspovirus, family Partitiviridae. We propose that this strain, CSpV1-Iowa, should be substituted for CSpV1-KSU1 as the exemplar strain of the species and genus, since the reported sequences of the latter appear to be terminally truncated. Complete genome sequences of other cryspovirus strains, from different Cryptosporidium species, are needed for ascertaining whether genus Cryspovirus should continue to contain only the single current species.
Supplementary Material
Fig. S1 Multiple sequence alignments of complete or putatively complete cryspovirus genome segments. Only sequences from the terminal regions of dsRNA1 (A) and dsRNA2 (B) are shown. The extents of sequences shown were chosen to encompass the putative start codon (shaded) in the 5′-terminal region or the stop codon (shaded) in the 3′-terminal region of each segment. The upstream AUG uniquely in frame in dsRNA2 of the C. felis 353 cryspovirus is also shaded. Primary sequences conserved between dsRNA1 and dsRNA2, as discussed in the main text, are overlined. *, wholly conserved residue at each alignment position.
Acknowledgments
Funding M.V. was supported in part by NIH grant T32 GM007598 to the doctoral program in Molecules, Cells & Organisms. J.G.L. was supported in part by NIH grants T32 AI07077 to the Sackler School of Graduate Biomedical Sciences Program in Immunology and T32 GM008448 to the School of Medicine Medical Scientist Training Program at Tufts University. M.L.N. was supported in part by a subcontract from NIH grant R01 GM033050.
Footnotes
Compliance with ethical standards
Conflict of interest All four authors declare that they have no conflict of interest.
Ethical approval This article contains no studies with human participants or animals performed by any of the authors.
Contributor Information
Minh Vong, Department of Microbiology & Immunobiology, Harvard Medical School, Boston, MA 02115, USA.
Jacob G. Ludington, Division of Geographic Medicine & Infectious Disease, Tufts Medical Center, Boston, MA 02116, USA
Honorine D. Ward, Division of Geographic Medicine & Infectious Disease, Tufts Medical Center, Boston, MA 02116, USA
Max L. Nibert, Department of Microbiology & Immunobiology, Harvard Medical School, Boston, MA 02115, USA
References
- 1.Castillo A, Cottet L, Castro M, Sepúlveda F. Rapid isolation of mycoviral double-stranded RNA from Botrytis cinerea and Saccharomyces cerevisiae. Virol J. 2011;8:38. doi: 10.1186/1743-422X-8-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Checkley W, White AC, Jr, Jaganath D, et al. A review of the global burden, novel diagnostics, therapeutics, and vaccine targets for cryptosporidium. Lancet Infect Dis. 2015;15:85–94. doi: 10.1016/S1473-3099(14)70772-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coutts RHA, Livieratos IC. A rapid method for sequencing the 50- and 30-termini of dsRNA viral templates using RLM-RACE. J Phytopathol. 2003;151:525–527. [Google Scholar]
- 4.Depierreux D, Vong M, Nibert ML. Nucleotide sequence of Zygosaccharomyces bailii virus Z: Evidence for +1 programmed ribosomal frameshifting and for assignment to family Amalgaviridae. Virus Res. 2016;217:115–124. doi: 10.1016/j.virusres.2016.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Green J, Gallimore CI, Clewley JP, Brown DW. Genomic characterisation of the large segment of a rabbit picobirnavirus and comparison with the atypical picobirnavirus of Cryptosporidium parvum. Arch Virol. 1999;144:2457–2465. doi: 10.1007/s007050050658. [DOI] [PubMed] [Google Scholar]
- 6.Jenkins MC, Higgins J, Abrahante JE, et al. Fecundity of Cryptosporidium parvum is correlated with intracellular levels of the viral symbiont CPV. Int J Parasitol. 2008;38:1051–1055. doi: 10.1016/j.ijpara.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 7.Khramtsov NV, Chung PA, Dykstra CC, et al. Presence of double-stranded RNAs in human and calf isolates of Cryptosporidium parvum. J Parasitol. 2000;86:275–282. doi: 10.1645/0022-3395(2000)086[0275:PODSRI]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 8.Khramtsov NV, Upton SJ. Association of RNA polymerase complexes of the parasitic protozoan Cryptosporidium parvum with virus-like particles: heterogeneous system. J Virol. 2000;74:5788–5795. doi: 10.1128/jvi.74.13.5788-5795.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khramtsov NV, Upton SJ. dsRNAs of Cryptosporidium parvum. J Parasitol. 2003;89:S165–S168. [PubMed] [Google Scholar]
- 10.Khramtsov NV, Woods KM, Nesterenko MV, Dykstra CC, Upton SJ. Virus-like, double-stranded RNAs in the parasitic protozoan Cryptosporidium parvum. Mol Microbiol. 1997;26:289–300. doi: 10.1046/j.1365-2958.1997.5721933.x. [DOI] [PubMed] [Google Scholar]
- 11.Leoni F, Gallimore CI, Green J, McLauchlin J. A rapid method for identifying diversity within PCR amplicons using a heteroduplex mobility assay and synthetic polynucleotides: application to characterisation of dsRNA elements associated with Cryptosporidium. J Microbiol Methods. 2003;54:95–103. doi: 10.1016/s0167-7012(03)00014-9. [DOI] [PubMed] [Google Scholar]
- 12.Leoni F, Gallimore CI, Green J, McLauchlin J. Molecular epidemiological analysis of Cryptosporidium isolates from humans and animals by using a heteroduplex mobility assay and nucleic acid sequencing based on a small double-stranded RNA element. J Clin Microbiol. 2003;41:981–992. doi: 10.1128/JCM.41.3.981-992.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leoni F, Gallimore CI, Green J, McLauchlin J. Characterisation of small double stranded RNA molecule in Cryptosporidium hominis, Cryptosporidium felis and Cryptosporidium meleagridis. Parasitol Int. 2006;55:299–306. doi: 10.1016/j.parint.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 14.Leoni F, Gómez-Couso H, Ares-Mazás ME, McLauchlin J. Multilocus genetic analysis of Cryptosporidium in naturally contaminated bivalve molluscs. J Appl Microbiol. 2007;103:2430–2437. doi: 10.1111/j.1365-2672.2007.03508.x. [DOI] [PubMed] [Google Scholar]
- 15.Leoni F, Mallon ME, Smith HV, Tait A, McLauchlin J. Multilocus analysis of Cryptosporidium hominis and Cryptosporidium parvum isolates from sporadic and outbreak-related human cases and C. parvum isolates from sporadic livestock cases in the United Kingdom. J Clin Microbiol. 2007;45:3286–3294. doi: 10.1128/JCM.02536-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li W, Zhang N, Liang X, et al. Transient transfection of Cryptosporidium parvum using green fluorescent protein (GFP) as a marker. Mol Biochem Parasitol. 2009;168:143–148. doi: 10.1016/j.molbiopara.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 17.Ludington JG, Ward HD. The Cryptosporidium parvum C-Type lectin CpClec mediates infection of intestinal epithelial cells via interactions with sulfated proteoglycans. Infect Immun. 2016;84:1593–1602. doi: 10.1128/IAI.01410-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murakoshi F, Ichikawa-Seki M, Aita J, et al. Molecular epidemiological analyses of Cryptosporidium parvum virus 1 (CSpV1), a symbiotic virus of Cryptosporidium parvum, in Japan. Virus Res. 2016;211:69–72. doi: 10.1016/j.virusres.2015.09.021. [DOI] [PubMed] [Google Scholar]
- 19.Nibert ML, Woods KM, Upton SJ, Ghabrial SA. Cryspovirus: a new genus of protozoan viruses in the family Partitiviridae. Arch Virol. 2009;154:1959–1965. doi: 10.1007/s00705-009-0513-7. [DOI] [PubMed] [Google Scholar]
- 20.Sharma P, Khurana S, Sharma A, Sehgal R, Malla N. Presence of intracellular viruses in human Cryptosporidium isolates. Ann Parasitol. 2016;62:139–147. doi: 10.17420/ap6202.46. [DOI] [PubMed] [Google Scholar]
- 21.Strong WB, Nelson RG. Preliminary profile of the Cryptosporidium parvum genome: an expressed sequence tag and genome survey sequence analysis. Mol Biochem Parasitol. 2000;107:132. doi: 10.1016/s0166-6851(99)00225-x. [DOI] [PubMed] [Google Scholar]
- 22.Xiao L, Limor J, Bern C, Lal AA, Epidemic Working Group Tracking Cryptosporidium parvum by sequence analysis of small double-stranded RNA. Emerg Infect Dis. 2001;7:141–145. doi: 10.3201/eid0701.010121. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Multiple sequence alignments of complete or putatively complete cryspovirus genome segments. Only sequences from the terminal regions of dsRNA1 (A) and dsRNA2 (B) are shown. The extents of sequences shown were chosen to encompass the putative start codon (shaded) in the 5′-terminal region or the stop codon (shaded) in the 3′-terminal region of each segment. The upstream AUG uniquely in frame in dsRNA2 of the C. felis 353 cryspovirus is also shaded. Primary sequences conserved between dsRNA1 and dsRNA2, as discussed in the main text, are overlined. *, wholly conserved residue at each alignment position.