

Abstract
Introduction
Genetic mutations in KCNQ2 which encodes hERG, the alpha subunit of the potassium channel responsible for the IKr current, cause Long QT Syndrome, an inherited cardiac arrhythmia disorder. Electrophysiology techniques are used to correlate genotype with molecular phenotype to determine which mutations identified in patients diagnosed Long QT Syndrome are disease causing, and which are benign. These investigations are usually done using heterologous expression in cell lines, and often, epitope fusion tags are used to enable isolation and identification of the protein of interest.
Methods and Results
Here, we demonstrate through electrophysiology techniques and immunochemistry, that both N-terminal and C-terminal myc fusion tags may perturb hERG protein channel expression and kinetics of the IKr current. We also characterize the impact of two previously reported inadvertent cDNA variants on hERG channel expression and half-life.
Conclusion
Our results underscore the importance of careful characterization of the impact of epitope fusion tags and conformational sequencing prior to genotype-phenotype studies for ion channel proteins such as hERG.
Keywords: hERG, KCNH2, epitope tag, Long QT Syndrome, electrophysiology
Introduction
Ion channels are essential for maintaining normal physiology. Disruption of usual channel function by genetic mutation may lead to a wide variety of conditions known as channelopathies. Examples of channelopathies can be found in every organ system. Ion channelopathies are well suited for the correlation between genotype and molecular phenotype due to the techniques available to probe the electrophysiology profile of the variant ion channel proteins. Examples of channelopathies include Cystic Fibrosis 1, susceptibility to Malignant Hyperthermia2, Bartter’s Syndrome 3, and hyperinsulinemic hypoglycemia4. In the heart, hereditary cardiac arrhythmias are often due to hereditary ion channelopathies. Conditions such as Brugada Syndrome, Long QT Syndrome, Short QT Syndrome, and Catecholaminergic Polymorphic Ventricular Tachycardia5, are all caused by ion channel dysfunction due to mutations in channel-coding genes or proteins that interact with cardiac channels. Molecular functional phenotyping and correlation with genotype provide invaluable insight into mechanisms of disease. This investigation will focus on the studies of molecular phenotype for Long QT Syndrome.
Congenital Long QT Syndrome (LQTS) is a genetic disorder that disrupts the timing of the cardiac myocyte membrane repolarizing phase of the action potential. There are 15 genes assigned to genetic loci for Long QT Syndrome. Most genotype-positive patients (>90%) with LQTS have mutations in one of three channel genes, KCNQ1 (LQT1), KCNH2 (LQT2) or SCN5A (LQT3). Although the determination of genetic susceptibility to LQTS and the diagnosis of LQTS are in the clinical realm, investigation of these proteins and the resultant mutations provide additional information on arrhythmia mechanisms. Most of the molecular investigation of LQTS has utilized heterologous cell culture expression systems (such as HEK293T, CHO, COS-7 and Xenopus oocytes). Through this method of inquiry, our understanding of normal and abnormal physiology of the heart continues to be enriched.
Functional studies of ion channel proteins using mutant cDNA expression plasmids requires careful assessment of the plasmid being used. Expression studies frequently employ epitope fusions for protein detection (immuno-epitope tags), visual confirmation of cellular transduction (fluorescent protein fusions), or affinity purification (epitopes such as FLAG-tags or His-Tags). Moreover, cloning, sub-cloning, and amplification of site-directed mutant cDNAs can be subject to inadvertent and spurious nucleotide variants that are not always detected. Protein fusion moieties and inadvertent variants may affect channel protein properties in unpredictable ways. Altered channel behavior may perturb functional properties of ion conductance and gating. Furthermore, channel protein translation, folding, assembly, and stability can be affected by tags and variations. Fully understanding the consequences of such experimentally induced changes is paramount for accurate analysis of expression studies of proteins.
In this investigation, we describe three examples of vector constructs for expression of hERG, the protein product of KCNH2 that exhibit plasmid-specific expression variation. In one case, an N-terminal myc-tag, caused significant gating changes in the IKr. In the second case, a PCR-introduced, double missense mutation inserted when hERG was first described (V198E (c. T593A), P202L (c. C605T))6, 7 did not significantly affect IKr, but perturbed channel protein expression and membrane trafficking. In the third case, a C-terminal myc-tag affects deactivation in the IKr current. This study highlights the impact that plasmid construct features may have on hERG functional expression, and how they may interfere with accurate analysis of intended molecular manipulations.
Materials and Methods
Approval from the Institutional Review Board was not needed to conduct this investigation.
Plasmid Preparation
The hERG cDNA, with and without the mutations V198E/P202L were inserted into the pcDNA 3.1+ vector (Invitrogen, ThermoFisher Scientific, Waltham, MA) using the BamHI and XbaI restriction enzymes (New England Bio Labs, Ipswich, MA). The hERG cDNA from the start to stop codon was inserted into the vector, and included a GCCACC Kozak sequence prior to the start codon. The wild type and V198E/P202L mutant inserts for the pCDNA 3.1+ were generated from other plasmids using the Q5 PCR enzyme (New England Biolabs) using the following primers: NT Forward: 5′-TAA GCA GGA TCC GCC ACC ATG CCG GTG CGG AG-3′, Reverse: 5′-TAA GCA TCT AGA CTA ACT GCC CGG GTC-3′.The amino-terminal myc-tagged wild type hERG construct was created by cloning the hERG insert into the pCMV tag 3a vector (Stratagene, La Jolla, CA) at the BAMH1 and HINDIII sites using the Q5 enzyme and the following forward and reverse primers: 5′-TAA TGG ATC CAA TGC CGG TGC GGA GG-3′ and 5′-TCC TAA GCT TCT AAC TGC CCG GGT CCG AG-3′, respectively. A 5-amino acid linker (5′-Ala-Arg-Ala-Asp-Pro-3′) conjoins the myc epitope to the start codon of hERG. The N-terminal myc-tagged hERG construct with V198E/P202L mutations were previously obtained through a gift from Dr. Gail Robertson6, 7. This construct has since been removed from circulation and replaced by the authors of the characterization paper with the correct hERG cDNA7. The C-terminal myc-tagged construct used is the pCI-Neo construct first described in McDonald, et. al 19978. This construct includes a C-terminal myc tag, along with a change of the last 3 amino acids in the hERG channel. The D219V pcDNA 3.1+ construct was generated through site-directed mutagenesis of the WT-hERG pcDNA3.1+ plasmid using the Q5 enzyme and the following primers: forward 5′-GAC AGC CAT GGT CAA CCA CGT G-3′ and reverse 5′-CAC GTG GTT GAC CAT GGC TGT C-3′ (IDT, Coralville, IA, USA) 9. The mutated gene was inserted into the pcDNA 3.1+ vector through the BamHI and XbaI restriction sites. Sequences for all plasmids were confirmed on a nucleotide level using GeneWiz Sanger sequencing (Genewiz, New Jersey).
Throughout this manuscript, WT hERG refers to the untagged hERG pcDNA 3.1+ plasmid, V198E/P202L hERG refers to the hERG pcDNA3.1+ construct with the V198E/P202L mutations. N-myc hERG refers to the wild-type hERG with the N-terminal myc tag. N-myc V198E/P202L hERG refers to the V198E/P202L hERG with the N-terminal myc tag. C-myc hERG refers to the pCI-Neo construct with the C-terminal myc tag. D219V hERG indicates the hERG gene carrying the D219V (c.656A>T) mutation in the pcDNA 3.1+. Figure 1 demonstrates the features of each of the 4 plasmids being compared in this investigation.
Figure 1. Impact of hERG channel protein additions on protein expression and trafficking.
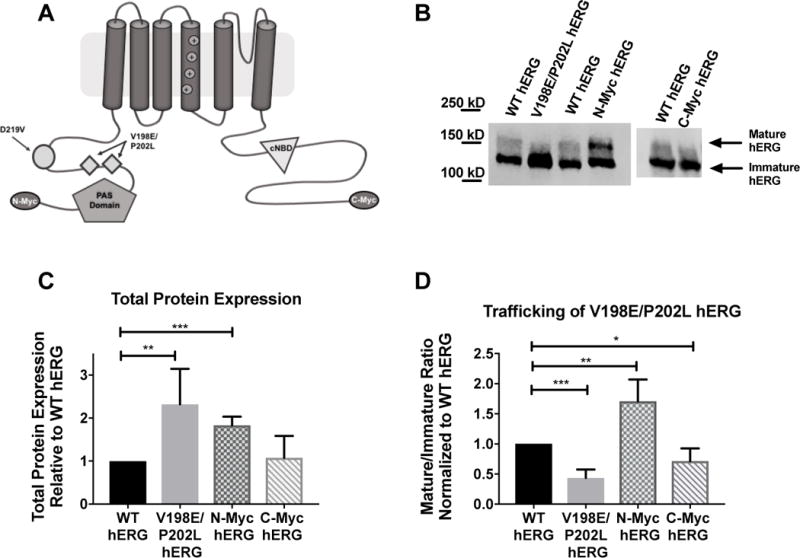
A) Cartoon graphic of a hERG channel subunit demonstrating the 4 variations discussed in this manuscript. B) Immunoblot of WT hERG, along with the inadvertent variation V198E/P202L, the N-terminal myc-tag and C-terminal myc-tag. C) Summary data for total hERG protein expression for each of the 3 variations shown in B. V198E/P202L has increased protein expression relative to WT hERG (n=4, p= 0.0182), the N-terminal myc fusion tag increased protein expression relative to WT hERG (n=4, p=0.0002), and the C-terminal myc fusion tag does not impact protein expression relative to WT hERG (n=5). D) Summary data for mature/immature ratio (155kDa/135kDa), or trafficking, for the 3 variants relative to WT hERG. V198E/P202L hERG has decreased trafficking efficiency (n=4, p=0.0002), N-Myc has increased trafficking efficiency relative to WT hERG (n=4, p= 0.0076) and C-Myc has decreased mature/immature ratio relative to WT hERG (n=5, p=0.0165)
Cell Maintenance and Transfection
Every experiment performed was done using transient transfections in Human Embryonic Kidney (HEK) 293T cells. Cells were cultured in RPMI media (Hyclone, GE Health Care Life Sciences, Pittsburgh, PA) supplemented with 10% Fetal Bovine Serum (Hyclone) and 10,000 IU Penicillin/Streptomycin (Hyclone). Cells were maintained at 37°C in a 5% CO2 environment. All transfections were done using Fugene 6 (Promega, Madison WI) in a 8:1 Fugene (uL) to DNA (ug) ratio. Experiments were performed at 48-hours post-transfection unless otherwise noted.
Immunoblot
HEK293T cells were maintained and transiently transfected in 6-well plates (Fisher Scientific International Inc., Hampton, NH) that were seeded with 3.5 ×105 cells 24 hours prior to transfection. Cells were lysed approximately 48 hours post-transfection in NDET buffer (1% IGEPAL (CA-630), 0.4% Deoxycholic acid, 5 mM EDTA, 25 mM Tris, 150 mM NaCl, pH 7.5) supplemented with complete protease inhibitor (Roche, Basel, Switzerland). The Bradford Assay was performed on the cell lysate using the Bradford Reagent (Bio-Rad Laboratories, Hercules, CA) and Bradford Standard BSA (Bio-Rad Laboratories, Hercules, CA). SDS-PAGE loading buffer was added to 50 μg total protein from the cell lysate and kept at 37°C for 30 min. The proteins were then separated on a 7.5% SDS-PAGE gel. The proteins were transferred onto nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA) using a semi-dry blotting cassette. The membrane was subjected to REVERT total protein stain (Li-Cor, Lincoln, NE), and imaged using the 700 channel of the Odyssey FC (Li-Cor). The membranes were then blocked in 5% nonfat milk (Tris-Buffered-Saline, supplemented with 0.5% Tween-20, and 5% nonfat dry milk for 30 minutes (Santa Cruz, Dallas, TX)). The membrane was washed 3 times in TBS-T and stained over night at 4°C in antibody H175 Rabbit anti-hERG (Santa Cruz) diluted 1:500 in 5% milk. Secondary stain was done with IRDye 800CW anti-Rabbit secondary antibody (Li-Cor) diluted 1:10,000 in 5% milk, stained 30 minutes at room temperature and imaged using the 800 channel of the Odyssey-FC. hERG protein signal was normalized to the total protein signal10 obtained from the REVERT reading. Statistical analysis, including t-tests, was performed using GraphPad Prism. Statistical significance was determined at p<0.05.
Protein Half-Life Determination
Approximately 1.75 ×105 HEK293T cells were seeded in a 12-well dish (Falcon, Lincoln Park, NJ). The following day, cells were transfected with 250 ng of plasmid using Fugene6 in a manner as described above. ~17 hours post transfection cells were starved of methionine by replacing the media with methionine-free RPMI (Gibco, Grand Island, NY) containing 10% dialyzed FBS (Sigma, St. Louis, MO) with Penicillin/Streptomycin. After one hour L-Azidohomoalanine (AHA) (Kerafast, Boston, MA) was added to the cells (200 uM concentration in solution) for 45 minutes. The ‘pulse’ media was replaced with standard growth media supplemented with 1 mM l-methionine (Acros, Bridgewater, NJ). Cells lysates were collected in 500 ul NDET buffer supplemented with protease inhibitor and transferred to the −20°C freezer at the time points indicated on the figure legend. The lysates were thawed then centrifuged at 21,000 XG. Supernatants were transferred to new tubes and 1 ug of hERGC20 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) antibody was added to each sample. After rocking for a minimum of two hours at room temperature 50 ul of resuspended Protein G Magnetic Beads (Bio-Rad, Hercules, CA) were added to each tube and the samples incubated at 4°C overnight with constant movement. The following day the beads were washed with 950 ul of prechilled PBS (Gibco, Grand Island, NY). 35 ul of PBS was added to each tube and a ‘click-it’ reaction was performed in each tube using a click-it buffer reagent kit (Thermo Scientific, Waltham, MA) to conjugate IRDye 800CW alkyne (~10 uM in solution, LI-COR, Lincoln, NE) to the azido moiety-containing analog of methionine. Following a one hour incubation the beads were washed with 950 ul of prechilled PBS, resuspended in SDS-PAGE loading buffer with DTT. Heated at 37°C for 30 minutes and the samples loaded into and run on 7.5% SDS-PAGE gels. The gels were directly scanned using the 800 channel of the Odyssey FC and the resulting images were acquired with the Image Studio Lite Version 5.2 program (Licor, Lincoln, NE) to determine densitometry values.
Prediction of Protein Damage due to Mutation
Three different predictive software programs were used to assess the impact of the V198E/P202L mutations on the hERG channel protein. These software programs cannot asses two mutations in a single prediction, so the impact of the mutations was assessed on an individual basis. SIFT11 (http://sift.jcvi.org/) PolyPHEN212 (http://genetics.bwh.harvard.edu/pph2/) and KvSNP 13 (http://www.bioinformatics.leeds.ac.uk/KvDB/KvSNP.html) were all used. To generate predictions from SIFT, the SIFT Human Protein program was selected. Then the Ensembl protein identifier (ENSP00000262186) was entered, along with the mutation of interest (either V198E or P202L), and a prediction and SIFT score were obtained. For PolyPHEN2, the Ensembl gene identifier for KCNH2 (ENSG00000055118) along with the amino acid substitution (V198E and P202L) was entered, and the software program predicted how damaging the mutation would be to the protein. For KvSNP, the P0386; KCNH2 protein was selected. Then, the wild-type residue V(Valine) or P (Proline) was selected, residue number 198 or 202 was entered and the changed amino acid E (Glutamate) or L (Leucine) was selected. KvSNP then generated a prediction for the disease-impact of the mutation (both predicted to be disease-causing (y/n) and probability of causing disease (from 0 to 1)).
Electrophysiology
For all electrophysiology experiments, HEK 293T cells were transiently transfected with one of the following constructs: WT hERG, V198E/P202L hERG, N-myc hERG, C-myc hERG, or D219V hERG using the transfection protocol described above. Cells were plated on sterile glass cover slips 24 hours after transfection and subsequently placed into an acrylic/polystyrene perfusion chamber (Warner Instruments, Hamden, CT, USA). Chambers were mounted on an inverted microscope equipped with fluorescence optics and patch pipette micromanipulators. All recordings were done at room temperature (∼22°C) and 48–72 hours post transfection. Cells were bathed in extracellular solution (150 mM NaCl, 1.8 mM CaCl2, 4 mM KCl, 1 mM MgCl2, 5 mM glucose, and 10 mM HEPES buffer pH 7.4). Intracellular solution was used in the micropipette (126 mM KCl, 4 mM Mg-ATP, 2 mM MgSO4, 5 mM EGTA, 0.5 mM CaCl2, and 25 mM HEPES buffer pH 7.2). Patch clamp pipettes with a tip resistance of approximately 2–3 mega-ohms were used to obtain the whole-cell configuration14. A MultiClamp 700B amplifier (Molecular Devices, LLC, Sunnyvale, CA, USA) was used and patch-clamp protocols were performed through the pCLAMP10 acquisition and analysis software (Molecular Devices). The pipette offset potential in these solutions was zeroed just prior to seal formation. Whole-cell capacitance (generally 10–30pF) was compensated electronically. Whole-cell series resistance was compensated to 85–90% using amplifier circuitry so that the voltage errors for currents of 2nA were always less than 6 mV. A holding potential of −80 mV was used for patching and whole-cell configurations, and figure insets illustrate the voltage command protocol. The data filtering (8-pole Bessel) at 1.4 kHz and sampling at 5 kHz was done for both the activation and deactivation protocols. For inactivation protocols, data were filtered at 2 kHz with sampled at 10 kHz. All data were analyzed with CLAMPFIT software (Molecular Devices) and figures were created and statistics were performed using GraphPad Prism Software. ANOVA, t-tests and Multiple t-tests were used to test significance, with a p-value <0.05 determined as significant.
Results
In Silico Analysis
Ensembl (ensembl.org), dbSNP (https://www.ncbi.nlm.nih.gov/snp), and ExAC (http://exac.broadinstitute.org/) databases documenting human genetic variations and disease causing mutations were queried. It was determined that neither the V198E mutation nor the P202L mutation exist as a known human variant. These mutations resulted solely from PCR-introduced error. The predicted impact of the individual mutations varies. PolyPhen212 predicted that the V198E mutation is possibly damaging (0.650/1) and the P202L mutation is benign (0.016/1). SIFT11 predicted both mutations to be tolerated (V198E = 0.38/1 and P202L = 0.13/1). KvSNP13 predicted that neither mutation is a disease-causing mutation, with the V198E probability of causing disease at 0.124 and the P202L mutation probability of causing disease at 0.433. Although these mutations were analyzed individually, and the impact of both mutations cannot be predicted, these results suggest that mutations are tolerated in the hERG protein, with the V198E mutation having a higher likelihood of being deleterious to protein function.
N-terminal Myc Tag and V198E/P202L independently impact hERG protein channel expression
SDS-PAGE-immunoblot techniques were used to analyze the hERG protein expression. On an immunoblot, this protein appears as two distinct bands, one at 135 kD and one at 155 kD. The 135 kD band represents the immature protein in the endoplasmic reticulum and cis-Golgi. This protein has only been core glycosylated and has not trafficked through the cell. The band at 155 kD represents the mature protein. This protein has been complexly glycosylated as it passes through the trans-Golgi on the way to the surface of the cell. Taken together, the ratio of 155/135 kD, or the ratio of mature/immature hERG, is an accepted surrogate measure of surface expression of the hERG channel protein15.
When investigating the impact of either the N-terminal myc-tag or the V198E/P202L double mutation, the total protein expressed as well as the mature/immature protein ratio was compared to WT-hERG. (Figure 1C and 1D). The immunoblots demonstrate that both the N-terminal myc-tag as well as the double mutation resulted in significantly increased protein expression. (p= 0.0002 for the N-terminal mac-tag and p = 0.0187 for V198E/P202L-hERG). The N-myc hERG trafficked to the surface of the cell more efficiently than the WT hERG, as demonstrated by the mature/immature protein ratio. (p= 0.0076). An inverse effect however, was observed for V198E/P202L hERG trafficking. WT hERG displays significantly more surface expression than V198E/P202L hERG. (p= 0.0002).
C-Terminal Myc Tag does not affect total protein expression, but modestly decreases hERG channel trafficking
Immunoblot analysis was used as described above to compare WT hERG to C-terminal-myc hERG. Total protein expression and the mature/immature hERG protein ratio for each of the two samples were determined (Figure 1C and 1D, respectively). There was no significant difference between the total protein expression for the two channels. The C-terminal-myc hERG construct demonstrated decreased trafficking, or decreased mature/immature ratio when compared to WT hERG (p=0.0165). However, the C-terminal-myc fusion seems to have less impact on the hERG protein expression overall compared to the N-terminal-myc or inadvertent V192E/P202L variants.
Protein Half Life
To investigate the potential effect of either the amino terminal myc epitope or the double mutations (V198E/P202L) on the stability of the hERG channel, half-life assays were performed. HEK293T transiently expressing either untagged or N-terminally myc tagged hERG both with and without the V198E/P202L mutations were metabolically pulse labeled with L-Azidohomoalanine. Analysis of the total remaining protein at various time points into the chase period indicated the presence of the double mutations increased the half-life of both the untagged (figure 2) (2.8 vs 4.3 hours) and myc-tagged (4.1 vs 7.2 hours) hERG constructs when transiently expressed in HEK-293T cells (Figure 2). Interestingly the N-terminal-myc fusion on wild type hERG augmented the half-life of the channel (2.8 hours vs 4.1 hours.
Figure 2. The amino terminal myc-tag and the V198E/P202L double mutation increase hERG channel stability.
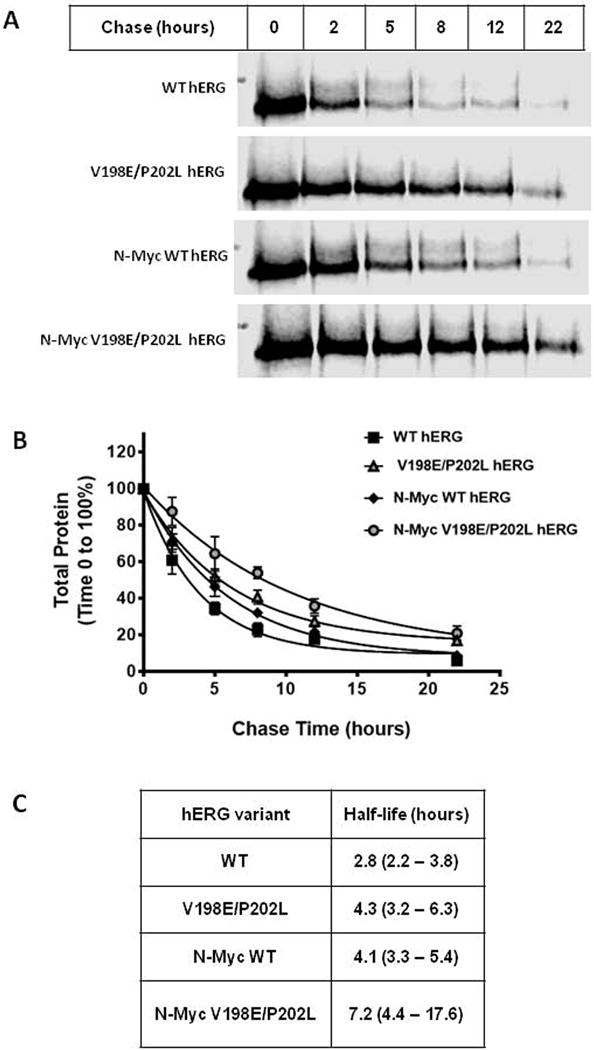
A) Pulse chase in-gel fluorescence scan of the WT hERG, V198E/P202L hERG, N-terminal-myc WT hERG, and N-terminal-myc V198E/P202L hERG expressed in HEK293T cells. Assay is representative of data determined from 4 independent experiments. B) Analysis of pulse chase data from 4 experiments. For each assay the 100% total protein value (immature and mature hERG protein) was defined by the densitometry at chase time 0 hours. The data points represent the mean of the experiments with the standard error of the mean (S.E.M.) displayed as error bars. C) the half-life values for hERG variants from panel B. The 95% confidence intervals are shown in parenthesis.
N-terminal Myc-Tag modifies hERG channel gating
Whole cell configuration of the patch clamp was used to measure potassium currents carried by the hERG channel. To determine channel activation, the membrane was stepped to various depolarizing levels (−60 to 60mV) for 1.5 sec, then repolarized to −40mV for 0.5 sec and finally hyperpolarized to −120 mV for 0.5 sec. A voltage-dependent activation curve (Figure 3C) was generated by measuring the peak outward current at the −40mV repolarizing step and plotted against the preceding activation voltage. The voltage-dependent activation (VDA) data were fit to a Boltzmann function: I = 1/(1+exp[(V1⁄2-V)/k]), where I is the relative tail current amplitude, V is the applied membrane voltage, V1⁄2 is the voltage at half-maximal activation, and k is the slope factor (Figure 3D). The V1⁄2 is for WT hERG is −7.182 ± 0.1371 and the V1⁄2 for N-myc hERG is −17.72 ± 0.9231. This indicates that the N-terminal myc tag produced a hyperpolarizing shift in the voltage dependence of activation. Current density was determined by obtaining the peak current at the depolarization step, and was normalized to cell capacitance. There was a trend for increased current density when the N-terminal myc-tag was included in the hERG protein channel (mean current density (pA/pF) for N-Myc hERG was 214.7 ± 33.4, while WT hERG mean was 153.7 ±29.4), which is consistent with the immunoblot data demonstrating an increased amount of N-myc hERG protein, as well as increased trafficking of N-myc hERG protein to the surface of the cell. (Figure 3B).
Figure 3. Effects of N-terminal-myc hERG on channel activation and deactivation.
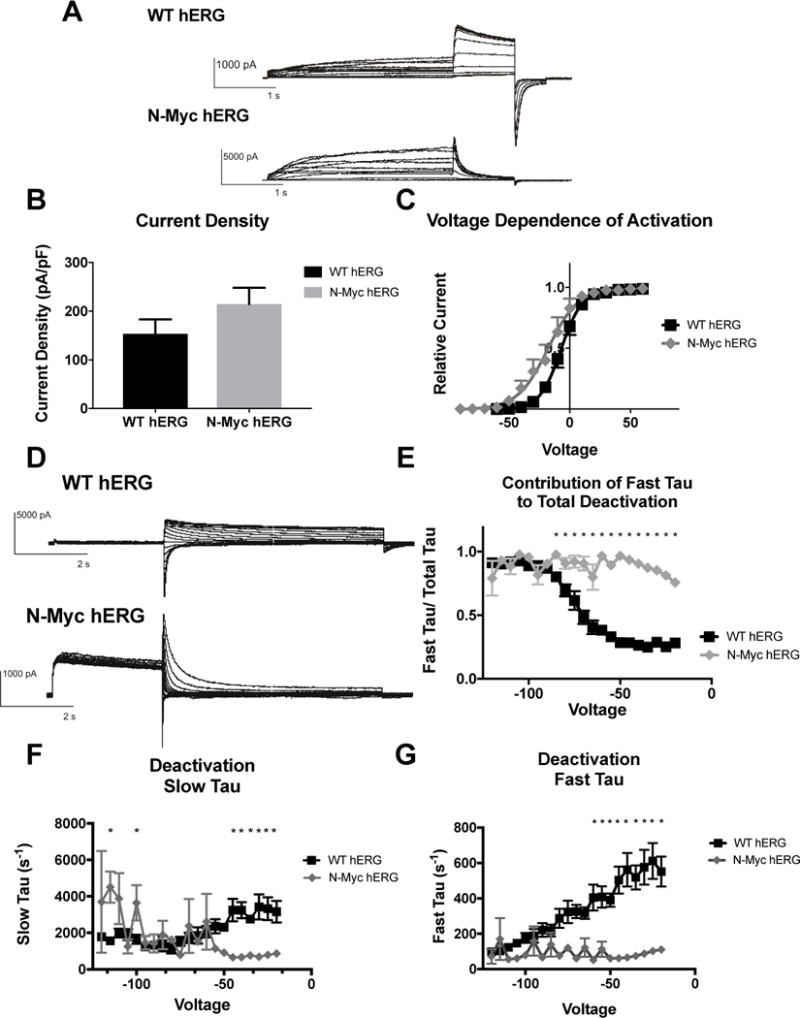
A) Whole cell patch clamp traces of WT hERG and N-terminal-myc hERG activation. Voltage clamp protocol shown below trace. B) Histogram representation of the current density for WT hERG and N-terminal-myc hERG, normalized to cell capacitance. There are no significant differences in current density (n= 12 (WT) and 6 (N-terminal-myc)). C) Representation of Voltage Dependent Activation (VDA) of WT hERG and N-terminal-myc hERG. VDA of N-terminal-myc hERG is hyperpolarized significantly compared to WT hERG. V1/2 of WT hERG is −7.182 ± 0.1371 and the V1/2 of N-terminal-myc hERG is −17.72 ± 0.9231. D) Whole cell patch clamp traces of WT hERG and N-terminal-myc hERG deactivation. Voltage clamp protocol shown below trace. N= 12 (WT hERG) and 8 (N-terminal-myc). E) Contribution of Fast Tau to total time constant of deactivation. There are significant differences from −20 to −85 mV (p<0.05), indicating that N-Myc Fast Tau contributes significantly more to total deactivation. F) Graphical depiction of WT and N-Myc hERG Slow Tau deactivation. There are significant differences at voltages from −20 to −40 and at −100 and −115 (p<0.05), although these may be less relevant differences. G) Representation of Fast Tau deactivation. There are significant differences between WT hERG and N-terminal-myc hERG at voltages from −20 – −60 (p<0.05). All results demonstrate that N-terminal-myc hERG deactivates significantly faster than WT hERG.
The impact of the N-terminal myc tag on hERG channel deactivation was investigated. To determine deactivation, a pulse depolarizing the membrane to +20mV for 1.7 sec was applied, followed by voltage steps from −20 to −120mV for 4.9 sec. Deactivation of the current during the final voltage steps was fitted with a double-exponential function (Itail = A0 + AMPF*expt/τF + AMPS*expt/τS, where A0 is the initial amplitude, AMPF and AMPS are the relative amplitudes of the fast and slow components and τF (Fast Tau) and τS (Slow Tau) are the time constants. (Sanguinetti, 1995). There were significant differences in both Fast and Slow Tau between N-terminal-myc hERG and WT hERG. N-myc hERG demonstrated faster deactivation in both the Slow and Fast Tau from −20mV to −45mV and at −60mV (Fast) (Figure 3F and 3G). The contribution of Fast Tau to the overall rate of deactivation in N-myc is significantly greater than WT from −20mV to −85mV (Figure 3E). A myc-tag on the N-terminus of hERG changes the channel kinetics from a slow deactivation characteristic of the IKr to a rapid deactivation.
V198E/P202L hERG does not significantly affect hERG channel function
The impact of the two mutations, V198E and P202L, on hERG function were investigated in the same fashion as described for N-terminal myc-tagged hERG. Activation and deactivation protocols were performed as described above. There were no significant differences between WT hERG and V198E/P202L hERG in current density, and the Boltzmann curves generated for the VDA were identical. (Figure 4B and 4C). The activation of both channels demonstrates WT IKr. There was not a significant difference in current density, although there was a trend towards decreased current density in the V198E/P202L hERG mutation (mean current density (pA/pf) for V198E/P202L hERG was 58.58 ± 13.87, and WT hERG was 76.3 ±13.93), confirming the description of the original mutation (Trudeau, Science 1996). There were also not significant differences in deactivation between WT hERG and V198E/P202L hERG. Both channels demonstrated comparable fast and slow time constants for deactivation (Figure 4E). V198E/P202L hERG produces WT IKr.
Figure 4. Effects of V198E/P202L hERG on channel activation and deactivation.
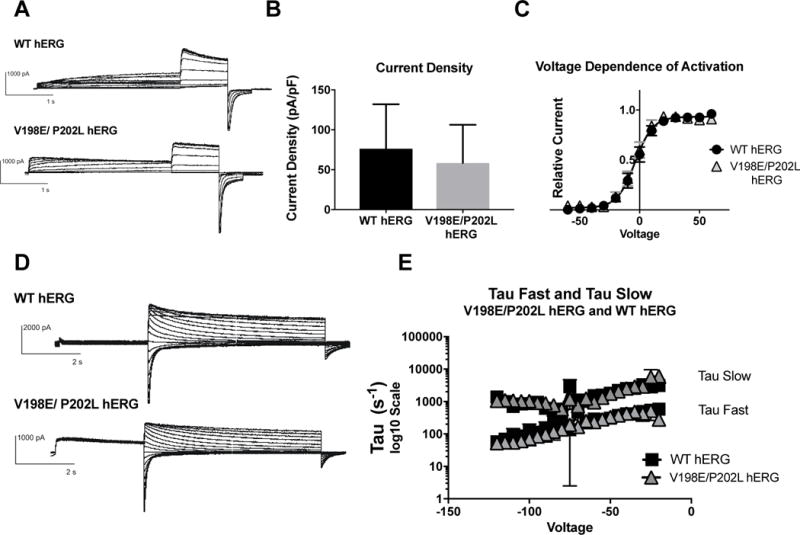
A) Whole cell patch clamp traces of WT hERG and V198E/P202L hERG activation. Voltage clamp protocol shown below trace. B) Current density for WT hERG and V198E/P202L hERG shown in graph form, normalized to cell capacitance. There are no significant differences in current density (n= 8 (WT) and 9 (V198E/P202L hERG)), although there is a trend towards decreased current density in the V198E/P202L hERG. C) Voltage Dependent Activation (VDA) for WT hERG and V198E/P202L hERG shows an identical curve. D) Whole cell patch clamp traces of WT hERG and V198E/P202L hERG deactivation. Voltage clamp protocol shown below trace. N= 2 (WT hERG) and 3 (V198E/P202L hERG). E) Graph of WT and V198E/P202L hERG Slow Tau deactivation and Fast Tau deactivation on a Log10 scale y-axis. There are no differences between WT hERG and V198E/P202L hERG. The V198E/P202L variants do not affect hERG channel deactivation kinetics.
The C-terminal Myc Fusion tag impacts hERG channel kinetics
The impact of the C-terminal myc fusion tag on hERG function was investigated with the same methods used for N-terminal myc-tagged hERG. Differences between WT hERG and C-myc hERG were noted in both current density (Figure 5B) and VDA (Figure 5C). Current density was not significantly different, but demonstrated a trend of decreased current (WT hERG: pA/pf = 153.7 ± 29.4, and C-Myc hERG pA/pF = 82.77 ±11.94), consistent with the decreased hERG trafficking seen in Figure 1D. The V1⁄2 is for WT hERG is −7.182 ± 0.1371 and the V1⁄2 is for C-myc fusion tagged hERG is −3.421 ± 0.1778, and the Boltzmann curve fit demonstrated two different curves to fit the WT hERG and C-terminal-myc hERG data. This indicates that a C-terminal myc fusion tag causes a slight depolarizing shift in voltage-dependent activation.
Figure 5. Effects of C-Terminal myc Fusion on hERG channel activation and deactivation.
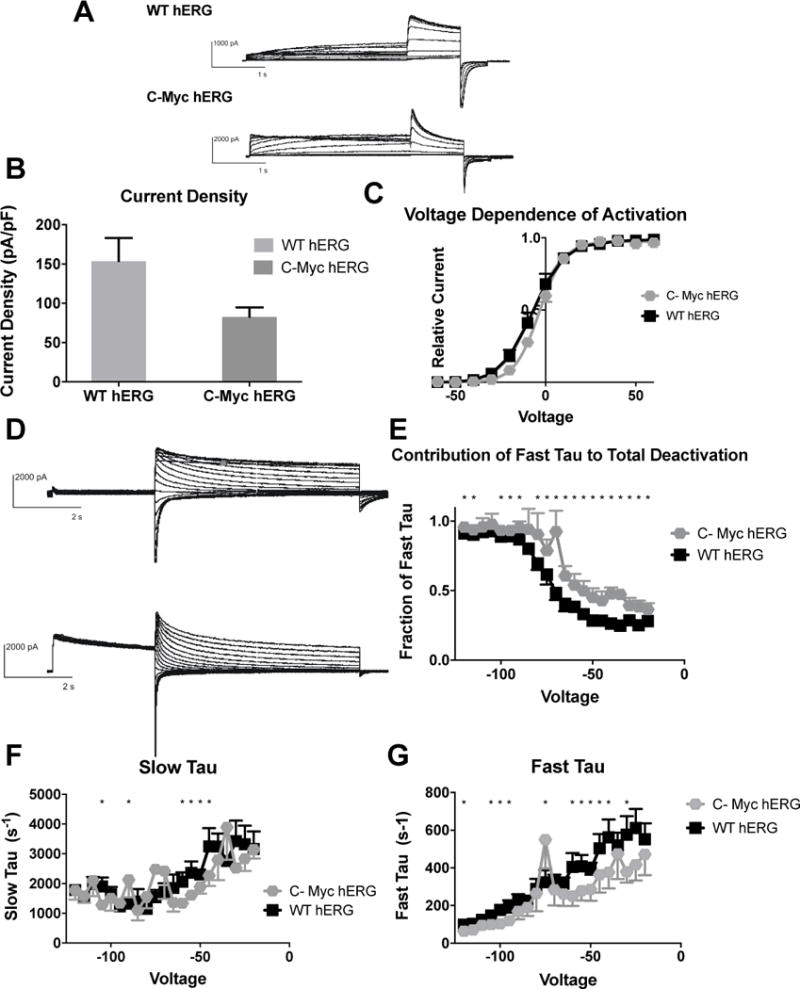
A) Whole cell patch clamp traces of WT hERG and C-terminal-myc hERG activation. Voltage clamp protocol shown below trace. B) Current density for WT hERG and C-terminal-myc hERG shown in graph form, normalized to cell capacitance. There are no significant differences in current density (n= 6 (WT) and 9 (C-terminal-myc c hERG)), although there is a trend towards decreased current density in the C-terminal-myc hERG (p=0.0943). C) Voltage Dependent Activation (VDA) shown for WT hERG and C-terminal-myc hERG. The C-terminal-myc fusion tag is associated with a small depolarizing shift in the VDA. V1/2 for WT hERG is −7.182 ± .3884 and V1/2 for C-terminal-myc hERG is −3.421 ± .1778 D) Whole cell patch clamp traces of WT hERG and C-terminal-myc hERG activation. N= 12 (WT hERG) and 7 (C-Myc hERG). E) Contribution of Fast Tau to total time constant of deactivation. C-terminal-myc hERG has significantly more contribution of Fast deactivation to total time of deactivation at every voltage (p<0.05) except −85, −105 and −110, and here there is a trend towards faster deactivation (p<0.1) F) Graph demonstrates WT and C-terminal-myc hERG Slow Tau deactivation. There are significant differences between the two hERG channels at −45–−60 mV, −75, −90 and −105 mV (p<0.05). G) Graphical representation of Fast Tau deactivation. There are differences between WT hERG and C-terminal-myc hERG at −30 mV, −40 – −60 mV, −75, −95–−105, −120 mV. The C-terminal-myc variants do affect hERG channel deactivation kinetics, primarily in the amount Fast Tau contributes to the overall deactivation rate of the channel.
The major impact that the C-terminal-myc fusion tag has on the hERG channel function is in the deactivation of the channel. There are significant differences in both the slow and fast time components of deactivation (Figures 5F and G). There are also significant differences in the amount that Fast Tau contributes to total deactivation, showing that Fast Tau in C-terminal-myc hERG plays a much larger role in deactivation when compared to WT hERG (Figure 5E). However, it is important to note that the changes in the deactivation characteristics between WT hERG and C-terminal-myc hERG are much less severe than the changes between WT hERG and N-myc hERG.
N-terminal Myc-tag confounds analysis of a clinical LQT2 mutation in hERG
We previously reported a clinical LQT2 variant, D219V, with rapid deactivation kinetics as the primary defect9. In this case, the N-terminal myc-tag construct was used and two inadvertent PCR variants V198E and P202L were included in the sequence. To determine if the epitope tag and variants significantly influenced our functional analysis we compared the D219V hERG variant without the N-terminal fusion myc tag and the noted V198E/P202L variant to WT untagged hERG. Protein expression and channel function were assayed as described above. Although there was no difference in mature/immature ratio between WT hERG and D219V hERG, there was a significant decrease in total protein expression of D219V hERG when compared to WT hERG (Figure 6), and a trend towards decreased protein expression for the 50/50 D219V/WT hERG (WT hERG normalized to 1, 50/50 D219V/WT hERG normalized to WT was 0.845 ± 0.22).
Figure 6. Analysis of LQT2 mutant D219V hERG channel protein abundance.
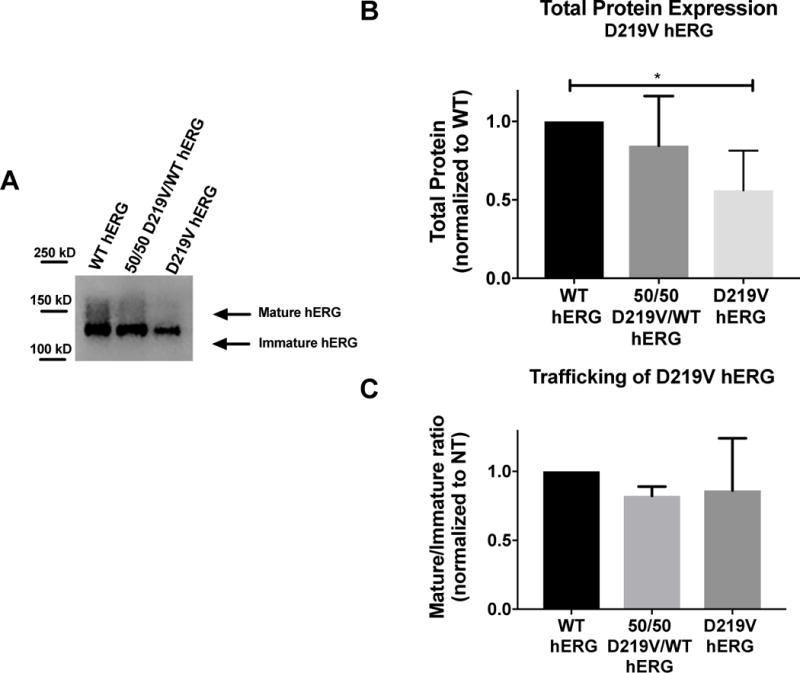
A) Representative immunoblot of the D219V hERG and WT hERG. B) Summary data comparing total protein expression between WT hERG and D219V hERG, normalized to total protein loaded into the immunoblot, and then normalized to WT hERG. N=4. D219V hERG expresses significantly less protein than WT hERG (p=0.0132) C) Summary data for mature/immature ratio (155kDa/135kDa), or trafficking of hERG for WT hERG and D219V hERG. There are no significant differences in trafficking between these two protein channels
When comparison of D219V hERG channel function to WT hERG was done with electrophysiology, only differences in current density were noted. When voltage-dependent activation was examined, D219V hERG and WT hERG data was fit with the same Boltzmann curve, indicating no differences in the voltage-dependence of D219V and WT hERG activation. (Figure 7B). Current density for D219V hERG was significantly lower than WT hERG, consistent with the channel expression reduction (Figure 7C) (p= 0.0326). Furthermore, D219V hERG deactivation time constants were comparable to WT hERG channels (Figure 7E and 7F).
Figure 7. Impact of D219V mutation on hERG channel activation.
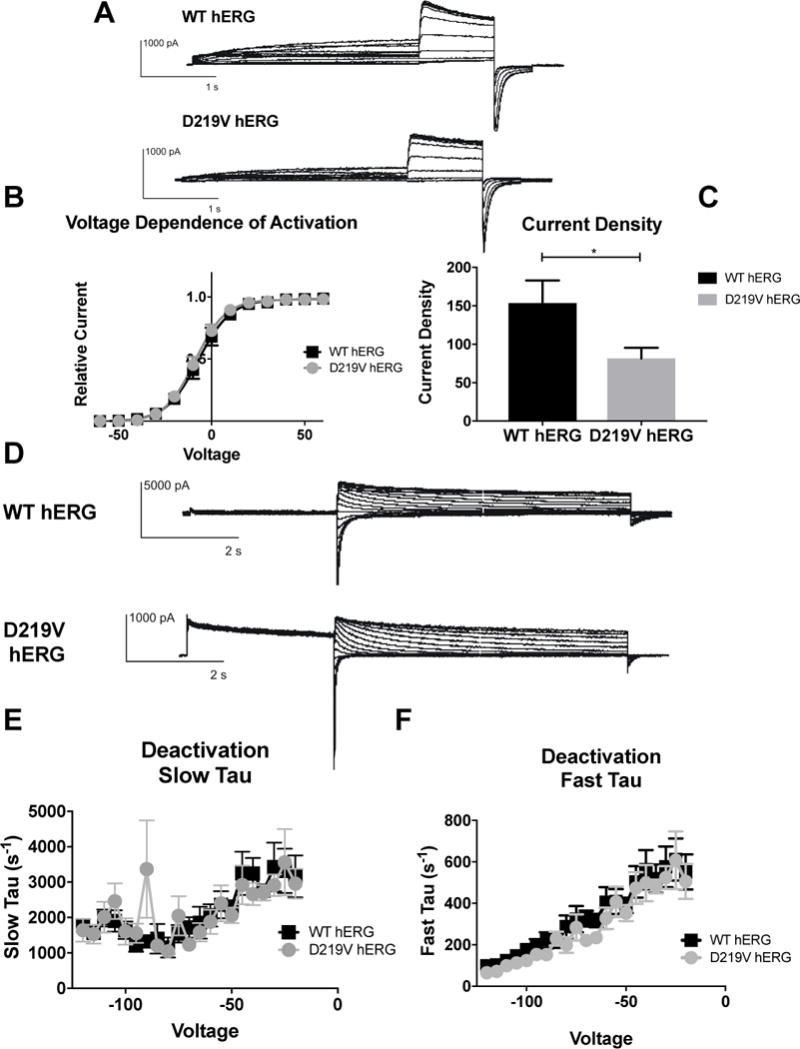
A) Whole cell patch clamp traces of WT hERG and C-terminal myc hERG activation. Voltage clamp protocol shown below trace. B) Current density for WT hERG and D219V hERG shown in graph form, normalized to cell capacitance. There are significant differences in current density. D219V hERG has a decreased current density (n= 12 (WT) and 13 (D219V hERG), p=0.0326). C) Voltage Dependent Activation (VDA) shown for WT hERG and D219V hERG. The VDA curve is similar for both D219V hERG and WT hERG, although D219V hERG is slightly left shifted (WT-hERG V1/2= −7.182 ± 0.1371 and D219V hERG V1/2 = −8.886 ± 0.143). D) Whole cell patch clamp traces of WT hERG and D219V hERG activation. N= 12 (WT hERG) and 9 (D219V hERG). E) Graph shows WT and D219V hERG Slow Tau deactivation. There are no significant differences between WT and D219V hERG. F) Graphical representation of Fast Tau deactivation. There are no differences between WT hERG and D219V hERG. The D219V mutation does not affect hERG channel deactivation kinetics.
Discussion
Here we describe the impact of an N-terminal myc fusion tag, a C-terminal myc fusion tag and a PCR-induced double mutation not found in population sequence databases on the WT hERG channel protein. We also characterize the consequences of the D219V mutation without the double mutation or the N-terminal myc-tag.
Myc tags are one of the first and most commonly used protein fusion tags for immuno-detection16. Immuno-epitope tags may be inserted in numerous locations in various vectors, and there are well characterized antibodies available for easy detection of any protein of interest. Although immuno-epitope tags are quite useful, they have the potential to cause unexpected changes in the protein they are fused with. In this study, we investigated the expression of hERG in three constructs with different features, including an N-terminal myc fusion tag, a C-terminal myc fusion tag, and two inadvertent nucleotide changes. hERG, the alpha subunit of the potassium channel responsible for the rapidly-activating delayed-rectifier potassium current (IKr). hERG plays a critical role in repolarization of the cardiomyocytes and the IKr has distinct patterns of activation, inactivation and deactivation that allow for its unique role in repolarization. Here, we have shown that a myc-tag fused to the N-terminal of WT hERG can cause an increase in expression, an increase in trafficking to the surface of the cell, an increase in protein half-life and can cause significant changes in the hERG kinetics. This agrees with the previously published study17, 18 which determined that the electrophysiological properties of hERG were more sensitive to the presence of a green fluorescent protein (GFP) tag on the amino as opposed to the carboxy-terminus of the hERG subunit. The N-terminal myc-tagged WT hERG demonstrated a hyperpolarizing shift in voltage dependence of activation. Deactivation was the characteristic that was most affected by the N-terminal-myc-tag, with N-myc hERG showing rapid deactivation uncharacteristic for the IKr. Interestingly, these kinetics changes are similar to the kinetic changes on hERG when the N-Cap (the first 23 amino acids of the protein) is deleted. Evidence from multiple groups shows that the N-cap domain has a role in regulating WT hERG kinetics, and that the EAG domain (comprised of the N-Cap and the PAS domain (amino acids 23–135)) interacts with various other regions of the channel to facilitate slow deactivation19,20,21,22, 23,24. Some of these important contacts have been demonstrated to be in the N-Cap, and interaction of polar regions, as well as N-cap flexibility, are crucial. The first 9 amino acids of the cap are unstructured25 and when this was deleted, rapid deactivation occurred. It was demonstrated that this unstructured region played an important role in correct, slow deactivation for the IKr. The N-terminal fusion of the myc-tag may have prevented necessary interactions that the unstructured N-cap makes or may have created an added structure to the N-cap, preventing necessary movement for deactivation. This N-cap fused tag may also be responsible for the left shift in voltage-dependent activation. Deletion of the N-cap causes a right shift in VDA, so addition of excess protein may cause a decreased voltage necessary for activation26. This study demonstrates the importance of not using N-terminal myc-tags on the WT hERG protein due to changes in protein expression and trafficking. The evidence that a fusion protein on the N-terminal end of this protein causes issues should also lead future investigators to carefully test the impact of any N-terminal tag on protein expression and trafficking prior to usage.
As demonstrated in this study, the C-terminal myc fusion tag also induced significant changes in the kinetics of hERG activation and deactivation, as well as changes in protein trafficking. It has been previously reported that deletion of small portions of the C-terminus from the C-terminal end of hERG (from ∆163 or ∆215 amino acids) will result in decreased current amplitude27. This finding is similar to the one described here for C-myc fusion tag causing a trend towards decreased current. The deactivation of other shortened C-terminal hERG proteins (∆236, ∆278) had faster deactivation rates but there was no difference in the deactivation rates of the shortest truncations27. This is less consistent with the findings of the C-terminal myc fusion hERG, but these data, taken together, indicate that the C-terminal myc fusion tag is likely blocking important contacts made by the distal C-terminal region of hERG, and impacting both protein trafficking, current density and channel deactivation.
Although there were significant changes caused by the C-terminal-myc fusion tag, the changes were less severe than the changes in hERG function and protein expression caused by the N-terminal myc fusion tag. If a tag must be added to hERG to facilitate investigation, the addition of the tag to the C-terminal end of hERG might be preferable to an N-terminal addition.
In addition to monitoring carefully fusion protein tags that are added on to the WT hERG protein, it is also important to ensure that the hERG sequence is not altered from the accepted consensus sequence prior to experimentation. It has been documented that the two mutations (V198E and P202L) resulting from PCR error at the earliest characterization of hERG do not affect IKr7, a finding that we confirm here. Although these two mutations do not have impact on the hERG channel function, and are predicted to be tolerated mutations, they do increase total protein expression and decrease protein trafficking. The most common mechanism for disease causing mutations in LQT2 are trafficking mutations, and if the constructs used to investigate newly documented mutations carry these other “benign” mutations, incorrect designation of “trafficking mutant” might be given to the new mutations.
The increase in half-life caused by either the myc tag or the inadvertent variant V198E/P202L likely attributes to the increased steady state expression levels observed for both constructs compared to the wild type channel. However, it also suggests that changes to the half-life/stability of a channel cannot be readily predicted by alterations in channel maturation caused by mutations or other environmental factors as the myc tag, which enhanced trafficking, and the double mutation which decreased trafficking, both resulted in channels with elongated half-lives. Future investigators need to ensure that they are not using hERG constructs obtained prior to 1996, and are aware of the impact that any fusion tag is having on their protein of interest.
As an example of the impact these changes can have on mutation analysis, D219V hERG was characterized again. D219V hERG was originally a mutation found from a genetic screen done in an 11-year-old male with clinically documented Long QT Syndrome. He had a mild presentation of Long QT Syndrome, with only 2 episodes of syncope throughout his life. The original analysis of the mutation indicated that it did not affect protein expression or trafficking compared to WT hERG9. In the present study, when both the N-terminal myc-tag was removed and the double mutants corrected, D219V hERG demonstrated a clear decrease in total protein expression. Both the N-terminal myc tag and the double mutations cause an increase in the hERG protein expression that may have corrected for the decrease in total protein expression caused by the D219V mutation. The D219V hERG protein was originally also believed to have significantly faster deactivation rates in both the slow tau and the fast tau. This was believed to be the main mechanism of disease. However, this change can now be attributed to the n-terminal myc-tag. This study demonstrated a significant decrease in current density corresponding to the decreased total expression of D219V hERG is responsible for the LQT2 phenotype in the proband. This mutation, rather than being expressed normally and having kinetic changes that lead to disease, causes decreased protein expression, likely due to a class 1 loss of function mechanism such as increased protein degradation or reduced translation efficiency. This mutation characterization discrepancy is evidence to the importance of characterizing all unknown aspects of any plasmid being used in an experiment.
Conclusion
This paper demonstrates the importance of testing every aspect of the plasmid and confirming correct sequencing prior to experimentation with the hERG protein. Changes in the gene or tags in the plasmid can impact protein expression and can alter protein function. It was shown that an N-terminal myc-tag caused significant increases in the hERG channel expression and trafficking, and caused changes in function, specifically changing the deactivation kinetics of the channel. A C-terminal myc fusion tag caused decreased trafficking and a trending decrease in current density, along with a faster deactivation rate in the hERG IKr current. It was also shown that two inadvertent cDNA variations in the hERG construct attributable to the mutations in the original construct issued before 19966, 7 caused an increase in protein expression, a decrease in trafficking and an increase in the half-life of hERG. These features of the hERG construct led to incorrect characterization of a new mutation, D219V, found in an isolated patient with LQT2. The Long QT Syndrome and hERG protein research community should take care to no longer use either an N-terminal myc-tag or the hERG construct issued before 1996 that contain the V198E/P202L mutations. In addition, this investigation demonstrates the importance of validating plasmids through sequencing prior to experimentation, and the importance of ensuring tags used to make protein identification simpler do not affect protein expression or function.
Acknowledgments
This work was supported in part by grants from the U.S. National Institutes of Health/National Heart, Lung, and Blood Institute (F30 HL126283 to M.L.O B. and 1R01HL118437 to T.V.M.)
Footnotes
Disclosures: None
References
- 1.Cutting GR. Cystic fibrosis genetics: from molecular understanding to clinical application. Nature reviews Genetics. 2015;16(1):45–56. doi: 10.1038/nrg3849. Epub 2014/11/19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Correia AC, Silva PC, da Silva BA. Malignant hyperthermia: clinical and molecular aspects. Revista brasileira de anestesiologia. 2012;62(6):820–37. doi: 10.1016/s0034-7094(12)70182-4. Epub 2012/11/28. [DOI] [PubMed] [Google Scholar]
- 3.Koulouridis E, Koulouridis I. Molecular pathophysiology of Bartter’s and Gitelman’s syndromes. World journal of pediatrics : WJP. 2015;11(2):113–25. doi: 10.1007/s12519-015-0016-4. Epub 2015/03/11. [DOI] [PubMed] [Google Scholar]
- 4.Senniappan S, Shanti B, James C, Hussain K. Hyperinsulinaemic hypoglycaemia: genetic mechanisms, diagnosis and management. Journal of inherited metabolic disease. 2012;35(4):589–601. doi: 10.1007/s10545-011-9441-2. Epub 2012/01/11. [DOI] [PubMed] [Google Scholar]
- 5.Abriel H, Zaklyazminskaya EV. Cardiac channelopathies: genetic and molecular mechanisms. Gene. 2013;517(1):1–11. doi: 10.1016/j.gene.2012.12.061. Epub 2012/12/26. [DOI] [PubMed] [Google Scholar]
- 6.Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science (New York, NY) 1995;269(5220):92–5. doi: 10.1126/science.7604285. Epub 1995/07/07. [DOI] [PubMed] [Google Scholar]
- 7.Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. HERG sequence correction. Science (New York, NY) 1996;272(5265):1087c. doi: 10.1126/science.272.5265.1087c. Epub 1996/05/24. [DOI] [PubMed] [Google Scholar]
- 8.McDonald TV, Yu Z, Ming Z, Palma E, Meyers MB, Wang KW, et al. A minK-HERG complex regulates the cardiac potassium current I(Kr) Nature. 1997;388(6639):289–92. doi: 10.1038/40882. Epub 1997/07/17. [DOI] [PubMed] [Google Scholar]
- 9.Osterbur ML, Zheng R, Marion R, Walsh C, McDonald TV. An Interdomain KCNH2 Mutation Produces an Intermediate Long QT Syndrome. Human mutation. 2015;36(8):764–73. doi: 10.1002/humu.22805. Epub 2015/04/29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eaton SL, Roche SL, Llavero Hurtado M, Oldknow KJ, Farquharson C, Gillingwater TH, et al. Total protein analysis as a reliable loading control for quantitative fluorescent Western blotting. PloS one. 2013;8(8):e72457. doi: 10.1371/journal.pone.0072457. Epub 2013/09/12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature protocols. 2009;4(7):1073–81. doi: 10.1038/nprot.2009.86. Epub 2009/06/30. [DOI] [PubMed] [Google Scholar]
- 12.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nature methods. 2010;7(4):248–9. doi: 10.1038/nmeth0410-248. Epub 2010/04/01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stead LF, Wood IC, Westhead DR. KvDB; mining and mapping sequence variants in voltage-gated potassium channels. Human mutation. 2010;31(8):908–17. doi: 10.1002/humu.21295. Epub 2010/06/05. [DOI] [PubMed] [Google Scholar]
- 14.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Archiv : European journal of physiology. 1981;391(2):85–100. doi: 10.1007/BF00656997. Epub 1981/08/01. [DOI] [PubMed] [Google Scholar]
- 15.Zhou Z, Gong Q, Epstein ML, January CT. HERG channel dysfunction in human long QT syndrome. Intracellular transport and functional defects. The Journal of biological chemistry. 1998;273(33):21061–6. doi: 10.1074/jbc.273.33.21061. Epub 1998/08/08. [DOI] [PubMed] [Google Scholar]
- 16.Gloor S, Pongs O, Schmalzing G. A vector for the synthesis of cRNAs encoding Myc epitope-tagged proteins in Xenopus laevis oocytes. Gene. 1995;160(2):213–7. doi: 10.1016/0378-1119(95)00226-v. Epub 1995/07/28. [DOI] [PubMed] [Google Scholar]
- 17.Claassen S, Schwarzer S, Ludwig J, Zunkler BJ. Electrophysiological and fluorescence microscopy studies with HERG channel/EGFP fusion proteins. The Journal of membrane biology. 2008;222(1):31–41. doi: 10.1007/s00232-008-9101-0. Epub 2008/04/17. [DOI] [PubMed] [Google Scholar]
- 18.Huang N, Lian JF, Huo JH, Liu LY, Ni L, Yang X, et al. The EGFP/hERG fusion protein alter the electrophysiological properties of hERG channels in HEK293 cells. Cell biology international. 2011;35(3):193–9. doi: 10.1042/cbi20100022. Epub 2010/12/15. [DOI] [PubMed] [Google Scholar]
- 19.Gustina AS, Trudeau MC. HERG potassium channel regulation by the N-terminal eag domain. Cellular signalling. 2012;24(8):1592–8. doi: 10.1016/j.cellsig.2012.04.004. Epub 2012/04/24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J, Trudeau MC, Zappia AM, Robertson GA. Regulation of deactivation by an amino terminal domain in human ether-a-go-go-related gene potassium channels. The Journal of general physiology. 1998;112(5):637–47. doi: 10.1085/jgp.112.5.637. Epub 1998/11/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ke Y, Hunter MJ, Ng CA, Perry MD, Vandenberg JI. Role of the cytoplasmic N-terminal Cap and Per-Arnt-Sim (PAS) domain in trafficking and stabilization of Kv11.1 channels. The Journal of biological chemistry. 2014;289(20):13782–91. doi: 10.1074/jbc.M113.531277. Epub 2014/04/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen J, Zou A, Splawski I, Keating MT, Sanguinetti MC. Long QT syndrome-associated mutations in the Per-Arnt-Sim (PAS) domain of HERG potassium channels accelerate channel deactivation. The Journal of biological chemistry. 1999;274(15):10113–8. doi: 10.1074/jbc.274.15.10113. Epub 1999/04/03. [DOI] [PubMed] [Google Scholar]
- 23.de la Pena P, Machin A, Fernandez-Trillo J, Dominguez P, Barros F. Mapping of interactions between the N- and C-termini and the channel core in HERG K+ channels. The Biochemical journal. 2013;451(3):463–74. doi: 10.1042/bj20121717. Epub 2013/02/20. [DOI] [PubMed] [Google Scholar]
- 24.Gianulis EC, Trudeau MC. Rescue of aberrant gating by a genetically encoded PAS (Per-Arnt-Sim) domain in several long QT syndrome mutant human ether-a-go-go-related gene potassium channels. The Journal of biological chemistry. 2011;286(25):22160–9. doi: 10.1074/jbc.M110.205948. Epub 2011/05/04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ng CA, Hunter MJ, Perry MD, Mobli M, Ke Y, Kuchel PW, et al. The N-terminal tail of hERG contains an amphipathic alpha-helix that regulates channel deactivation. PloS one. 2011;6(1):e16191. doi: 10.1371/journal.pone.0016191. Epub 2011/01/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tan PS, Perry MD, Ng CA, Vandenberg JI, Hill AP. Voltage-sensing domain mode shift is coupled to the activation gate by the N-terminal tail of hERG channels. The Journal of general physiology. 2012;140(3):293–306. doi: 10.1085/jgp.201110761. Epub 2012/08/15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aydar E, Palmer C. Functional characterization of the C-terminus of the human ether-a-go-go-related gene K(+) channel (HERG) The Journal of physiology. 2001;534(Pt 1):1–14. doi: 10.1111/j.1469-7793.2001.t01-3-00001.x. Epub 2001/07/04. [DOI] [PMC free article] [PubMed] [Google Scholar]