

Abstract
Quantitative proteomics using high-resolution and accuracy mass spectrometry promises to transform our understanding of biological systems and disease. Recent development of parallel reaction monitoring (PRM) using hybrid instruments substantially improved the specificity of targeted mass spectrometry. Combined with high-efficiency ion trapping, this approach also provided significant improvements in sensitivity. Here, we investigated the effects of ion isolation and accumulation on the sensitivity and quantitative accuracy of targeted proteomics using the recently developed hybrid quadrupole-Orbitrap-linear ion trap mass spectrometer. We leveraged ultrahigh efficiency nano-electrospray ionization under optimized conditions to achieve yoctomolar sensitivity with more than seven orders of linear quantitative accuracy. To enable sensitive and specific targeted mass spectrometry, we implemented an automated, two-dimensional (2D) ion exchange-reversed phase nanoscale chromatography system. We found that automated 2D chromatography improved the sensitivity and accuracy of both PRM and an intact precursor scanning mass spectrometry method, termed accumulated ion monitoring (AIM), by more than 100-fold. Combined with automated 2D nano-scale chromatography, AIM achieved subattomolar limits of detection of endogenous proteins in complex biological proteomes. This allowed quantitation of absolute abundance of the human transcription factor MEF2C at ∼100 molecules/cell, and determination of its phosphorylation stoichiometry from as little as 1 μg of extracts isolated from 10,000 human cells. The combination of automated multidimensional nano-scale chromatography and targeted mass spectrometry should enable ultrasensitive high-accuracy quantitative proteomics of complex biological systems and diseases.
The emerging ability to measure cellular and physiological states accurately and quantitatively promises to transform our understanding of biology and disease (1). For example, time-resolved and multiparametric quantitative analyses of cellular signaling are enabling the elucidation of fundamental paradigms of cell development and homeostasis (2, 3). Likewise, accurate measurements of human disease states are enabling improved diagnostic markers and refined mechanisms of disease pathophysiology (4, 5).
In large part, these advances were made possible by the development of increasingly accurate and sensitive methods for quantitative analysis of proteins and their post-translational modifications in complex biological proteomes. For example, selected reaction monitoring (SRM)1 uses quadrupole mass analyzers to filter specific precursor and fragment ions produced by collision-induced dissociation (CID) (6–9). This method benefits from high-efficiency continuous ion beams, and relatively high sensitivity of direct dynode detection, but is subject to interference effects because of the relatively low unit-mass resolution of quadrupole mass analyzers. As a result, SRM methods require specialized approaches to control for variable specificity, hindering their widespread use (7, 10–12).
To overcome these limitations, parallel reaction monitoring (PRM) has been developed by leveraging high-resolution Orbitrap mass analyzers to improve assay specificity because of monitoring fragment ions with parts per million (ppm) mass accuracy (13, 14). Likewise, the incorporation of high-resolution time-of-flight (TOF) mass analyzers has been used to improve the accuracy of reaction monitoring methods, including their use in data-independent approaches such as SWATH (15, 16). Thus far, targeted mass spectrometry methods exhibit at least 10-fold better sensitivity than data-independent approaches (15). Consequently, recent efforts have focused on improving the ion transfer efficiencies of these methods, such as the recently introduced parallel accumulation-serial fragmentation technique (17).
The requirement for high ion transfer efficiencies for accurate quantitative proteomics led to the incorporation of ion trapping devices in modern mass spectrometry instruments. For example, the use of external ion storage improved the accumulation efficiency of Fourier transform-ion cyclotron resonance (FT-ICR) mass spectrometers (18, 19). Likewise, implementation of Orbitrap mass analyzers necessitated the incorporation of external ion storage devices with improved electrodynamic concentration properties (20), as originally required for coupling bright continuous ion sources to ICR mass analyzers (21). Recent implementations of ion storage on hybrid instruments, such as the Q Exactive quadrupole-Orbitrap (Q-OT), and the Fusion quadrupole-Orbitrap-linear ion trap (Q-OT-IT) mass spectrometers, use high-capacity multipole ion traps, which permit accumulation and routing of ions before analysis (22, 23).
Here, we investigated the effects of ion selection and accumulation on the sensitivity and quantitative accuracy of targeted proteomics using the recently developed hybrid Fusion quadrupole-Orbitrap-linear ion trap mass spectrometer. We leveraged high efficiency low μm-scale electrospray ionization to analyze synthetic peptides in neat solvent and thus determine the absolute limits of sensitivity, achieving yoctomolar absolute limits of quantitation with more than seven orders of linear quantitative accuracy. We observed that ion coisolation led to significant reduction in sensitivity in analyses of complex human cellular proteomes. To partially overcome this limitation, we implemented an automated, scalable two-dimensional ion exchange-reversed phase nano-scale chromatography system, suitable for robust, high-resolution, high-capacity separations necessary for quantitative targeted mass spectrometry. By quantifying the endogenous transcription factor MEF2C and its phosphorylation stoichiometry in 1 μg of extracts from as few as 10,000 human cells, we achieved significant improvements in the sensitivity and quantitative accuracy of both PRM and accumulation monitoring (AIM) methods, permitting the detection and quantitation of ∼100 molecules/cell.
EXPERIMENTAL PROCEDURES
Reagents
Mass spectrometry grade (Optima LC/MS) water, acetonitrile (ACN), and methanol were from Fisher Scientific (Fair Lawn, NJ). Formic acid of 99%+ purity (FA) was obtained from Thermo Scientific. Ammonium formate and all other reagents at MS-grade purities were obtained from Sigma-Aldrich (St. Louis, MO).
Synthetic Peptides and Proteome Preparation
MRFA peptide was obtained from Sigma-Aldrich. Based on consensus protein sequences in the UniProt database (as of January 30th, 2015), N-terminally isotopically labeled (13C615N2 lysine and 13C615N4 arginine) MEF2C and MARK4 peptides (Table I) were synthesized using solid phase chemistry by New England Peptides (Gardner, MA), and purified by reversed phase chromatography. Extinction coefficients were calculated as described by Kuipers and Gruppen (24), and listed in supplemental Table S1. Peptides were quantified using UV absorbance spectroscopy at 214 nm using 3 mm QS quartz cuvettes (Hellma, Plainview, NY) and the SpectraMax M5 analytical spectrophotometer (Molecular Devices, Sunnyvale, CA). A standard peptide mixture was created by mixing individual peptides at final concentration 10 pmol/μl in a clean glass vial. For direct infusion experiments, the peptide mixture was serially diluted 1:10 in 30% ACN, 0.1% FA containing 1 μg/ml MRFA peptide (Sigma-Aldrich).
Table I. Peptide properties.
| ID | Sequence | m/z |
|---|---|---|
| M1 | SEPVSPPR | 439.7339 |
| M2 | SEPV(pS)PPR | 479.7171 |
| M3 | NSPGLLVSPGNLNK | 709.3981 |
| M4 | N(pS)PGLLVSPGNLNK | 749.3813 |
| M5 | YTEYNEPHESR | 717.8116 |
| M6 | LFEVIETEK | 558.3073 |
| M9 | VLIPPGSK | 409.7649 |
Human OCI-AML2 cells were obtained from the German Collection of Microorganisms and Cell Cultures (Brunswick, Germany). Cells were cultured as described (25), collected while in exponential growth phase, washed twice in ice-cold PBS, snap frozen and stored at −80°C. Protein extraction and proteolysis was performed as previously described (26). Briefly, frozen pellets of 5 million cells were thawed on ice, resuspended in 100 μl of 6 m guanidinium hydrochloride, 100 mm ammonium bicarbonate at pH 7.6 (ABC) containing PhoStop phosphatase inhibitors (Roche Diagnostics GbmH, Mannheim, Germany), and lysed using the E210 adaptive focused sonicator (Covaris, Woburn, CA). The protein content in cell lysate was determined using the BCA assay, according to the manufacturer's instructions (Pierce, Rockford, IL). On reduction and alkylation, proteomes were digested using 1:100 w/w (protease/proteome) LysC endopeptidase (Wako Chemical, Richmond, VA) and 1:50 w/w MS sequencing-grade modified trypsin (Promega, Madison WI). Digestion was stopped by acidifying the reactions to pH 3 using formic acid (Thermo Scientific), and peptides were subsequently desalted using solid phase extraction using C18 Macro Spin columns (Nest Group, Southborough, MA). Peptides were eluted in 60% acetonitrile, 1% formate in water, lyophilized using vacuum centrifugation, and stored at −20°C. Tryptic peptides were reconstituted in 0.1% formate, 3% acetonitrile to a final concentration of 0.5 μg/μl. For experiments in cellular proteomes, the synthetic peptide mixture was initially diluted to 5 pmol/μl in a tryptic digest of whole OCI-AML2 cell proteome at 0.5 μg/μl, and subsequently serially diluted 1:10 in the same solution.
Nanoscale Liquid Chromatography
Detailed description of the instrumental and operational parameters, as well as step-by-step protocol for system construction and operation are provided in supplementary Materials. Both direct infusion sample delivery and liquid chromatography experiments were performed using the Ekspert NanoLC 425 chromatograph (Eksigent, Redwood City, CA), equipped with an autosampler module, two 10-port and one 6-port rotary valves, and one isocratic and two binary pumps. Polyimide-coated fused silica capillaries (365 μm outer diameter, variable inner diameters) were obtained from Polymicro Technologies (Phoenix, AZ). Unions and fittings were obtained from Valco (Houston, TX). For direct infusion, samples were initially aspirated into a 10 μl PEEK sample loop. On valve switching, the content of the loop was ejected using a gradient pump (30% ACN, 0.1% FA at 100 nl/min) into an empty silica capillary (20 μm inner diameter) in-line with the DPV-566 PicoView nano-electrospray ion source (New Objective, Woburn, MA).
Chromatographic columns were fabricated by pressure filling the stationary phase into silica capillaries fritted with K-silicate, as previously described (27). Strong-cation exchange columns were fabricated by packing Polysulfoethyl A 5 μm silica particles (PolyLC, Columbia, MD) into 150 μm × 10 cm fritted capillary. Reversed phase columns were fabricated by packing Reprosil 1.9 μm silica C18 particles (Dr. Meisch, Ammerbauch-Entrigen, Germany) into 75 μm x 40 cm fritted capillaries. Trap columns were fabricated by packing Poros R2 10 μm C18 particles (Life Technologies, Norwalk, CT) into 150 μm x 4 cm fritted capillaries.
Vented trap-elute architecture was used for chromatography (28). One of the 10-port valves was set to include in the flow path either the SCX column or an empty capillary of equal inner volume. Samples were initially loaded into a 10 μl PEEK sample loop, and subsequently delivered at 1 μl/min by the isocratic pump using 0.1% FA in water into either the empty capillary (for one-dimensional chromatography) or the SCX column (for two-dimensional chromatography). A step gradient of 50, 100, 150, 300, and 1000 mm ammonium formate (AF) in water, pH 3 was delivered in 3.5 μl (0.5 column volume) increments from auto-sampler vials to elute peptides into the trap column, where peptides were desalted. Finally, peptides were resolved by reversed phase chromatography hyphenated to the nano-electrospray ion source. On valve switch to connect the trap column in line with the analytical reversed phase column and ion emitter, the pressure was equilibrated at a flow of 250 nl/min for 5 min in 5% buffer B (ACN, 0.1% FA) in buffer A (water, 0.1% FA). Subsequently, a 60-min linear gradient of 5–38% of buffer B was used to resolve peptides, followed by a 5 min 38–80% gradient before column wash at 80% buffer B for 30 min.
Electrospray Ionization and Mass Spectrometry
Electrospray emitters with terminal opening diameter of 2–3 μm were fabricated from silica capillaries as previously described (29). The emitter was connected to the outlet of the reversed phase column using a metal union that also served as the electrospray current electrode. Electrospray ionization was achieved using variable voltage, programmed from 1750 to 1450 V with 50 V steps over 60 min of the gradient elution. During column loading, the electrospray emitter was washed with 50% aqueous methanol using the DPV-565 PicoView ion source (New Objective).
For all measurements, we used the Orbitrap Fusion mass spectrometer (Thermo Scientific, San Jose, CA). During AIM measurements, the mass spectrometer was programmed to iteratively perform precursor ion scans with 8 Th isolation windows targeting both endogenous light and synthetic heavy peptides using Q1 isolation and S-lens voltage of 60 V (22). Unless otherwise specified, ions were accumulated for a maximum of 200 ms with automatic gain control of 105 ions, and scanned at 240,000 resolution. For PRM scans, precursor ions were isolated using 2 Th isolation windows, and fragmented by HCD with normalized collision energy set at 32% before analysis of the fragment ions in the Orbitrap at 30,000 resolution. Optimal fragmentation conditions were preliminarily established for each target peptide by manual inspection of MS2 spectra collected within the same analysis on fragmentation with HCD energy 30, 32, 36, and 38% (30).
Data Analysis
Ion signal intensities and total ion currents were analyzed using Xcalibur Qual Browser 3.0.63 (Thermo Fisher Scientific). Automated chromatographic peak area integration was performed using Skyline 3.5.0 (31), with mass tolerance set at 0.0075 Da corresponding to 10 ppm for m/z of 750 Da, and integration boundaries were manually verified for all peaks. Numerical and statistical analyses were performed using Origin Pro 9.0 (OriginLab Corporation, Northampton, MA). All raw and processed mass spectrometry data as well as Skyline chromatogram documents are available via ProteomeXchange with the identifier PXD006236 (32). Quantitative data from serially diluted synthetic peptides were linearly fitted to obtain a signal-response function for each peptide. These functions were subsequently used to calculate amounts of endogenous peptides. Phosphorylation stoichiometry was defined as the fraction of each peptide being chemically modified, as described (33). The protein content of OCI-AML2 cells was established by BCA assay quantification of total protein extracted from cells, which were manually counted using a Neubauer hemocytometer.
Experimental Design and Statistical Rationale
The study evaluated the sensitivity and specificity of targeted detection using peptides delivered either by direct infusion or by chromatography with variable peak capacities. Technical variability was established under direct infusion regime by collecting seven measurements for each data point. For chromatographically resolved peptides, triplicate measurements of the intensity of synthetic peptides were performed at three experimental conditions. Endogenous peptide measurements were performed in parallel with isotopolog targeting, for a total of 18 replicate measurements. To compare AIM and PRM, assays were performed within the same experiment to control for possible variation in chromatography and ionization performance.
RESULTS
Accumulated Ion Monitoring for Targeted Proteomics using Hybrid Quadrupole-Orbitrap-Linear Ion Trap (Q-OT-IT) Instrument
Sensitivity of mass spectrometric detection is in principle determined by two factors: the minimum number of ions necessary to produce a measureable electronic signal, and the baseline instrumental noise. In the case of high-resolution mass analyzers coupled to external ion storage devices, the number of ions delivered to the mass analyzer can thus be increased by prolonged ion accumulation before detection. The incorporation of high-capacity ion routing multipoles (IRM) on the recently developed Q Exactive (Q-OT) and Fusion (Q-OT-IT) mass spectrometers permits ion accumulation and storage for as long as 5 s.
An acquisition strategy was thus designed, combining quadrupole precursor ion filtering, with precursor quantification in the Orbitrap and parallel identification of fragmentation spectra in the linear ion trap. This method, termed accumulated ion monitoring (AIM), combines high sensitivity and specificity for targeted mass spectrometry. Although conceptually related to selected ion monitoring (SIM) by the use of quadrupole filtering (34), AIM is specifically defined by the use of multipolar ion storage and precursor accumulation before high-resolution, high-mass accuracy precursor ion quantification. AIM is also related to parallel reaction monitoring (PRM) (13, 14), insofar as both methods achieve high-specificity quantification by high-resolution detection of trapped ions, but is distinguished by the identification of fragment ions using parallel ion scanning in the linear ion trap, as specifically enabled by the so-called tribrid architecture of the Fusion Q-OT-IT instrument (22, 23).
Establishing the Limits of Quantitation and Quantitative Accuracy of AIM
In principle, extended ion accumulation of AIM and PRM could enhance the detectability of peptides and extend the quantitative accuracy and overall sensitivity of targeted proteomics. To test this hypothesis, we determined the absolute sensitivity of AIM detection and its dependence on ion accumulation and storage, using synthetic peptides delivered by continuous infusion. We initially chose tryptic peptides derived from human transcription factor MEF2C and kinase MARK4 given their physicochemical and ionization properties that are generally representative of human tryptic peptides (Table I, supplemental Table S1). To control for possible adsorptive losses, peptides were serially diluted in neat solvent containing the MRFA peptide as a carrier. To maximize nano-electrospray ionization efficiencies while maintaining robust emitter performance (35–38), we fabricated fused silica emitter tips with tip openings of 2–3 μm, as determined using scanning electron and optical microscopy (supplemental Fig. S1A and S1B) (29). This enabled ionization efficiencies of 3–23% for the monitored peptides, as estimated by the time required to achieve the automatic gain control (AGC) threshold values (supplemental Fig. S2).
Using these parameters, we analyzed the absolute sensitivity of AIM by measuring the signal response of synthetic peptides serially diluted in MRFA-containing neat solvent. Remarkably, this achieved specific detection of less than 1 ymol/ms of peptides, corresponding to less than 100 ions per scan, and nearly 7 orders of magnitude of linear signal response (Fig. 1A, supplemental Figs. S3, Supplemental Table S2). We observed no significant differences in the limits of quantitation by increasing the ion accumulation time from 25 to 2500 ms (Fig. 1A).
Fig. 1.

A, AIM acquisition enables detection of 2–5 yoctomoles/ms of target peptides serially diluted in neat solvent, and seven order of magnitude of linear dynamic range. The MS intensity within 10 ppm from the theoretical m/z was recorded for peptide NSPGLLVSPGNLNK (m/z 709.3981) with maximum injection time (max IT) 25 (red), 250 (black), and 2500 (gray) ms (n = 7, error bars represent standard deviation of independent experiments). Noise levels recorded at baseline level (i.e. with no target peptide infused) is displayed for each maximum injection times as continuous line. Overlapping data points were horizontally offset when needed for clarity. B, Ion accumulation times obtained for each target peptide with maximum injection time set at 5s. Ion injection time in the baseline samples (i.e. with no target present) is denoted as “B.” Comparison of spectra recorded with target flow 10e7 (C) and 100 ymol/ms (D) reveals that target accumulation is practically limited by coisolated contaminant ions (ion intensity on absolute linear scale).
Analysis of ion accumulation times under non-limiting maximum injection times revealed two distinct regimes of operation (Fig. 1B). As long the target peptide was the dominant ion in the isolation window, i.e. delivered at flow rates greater than ∼10 zmol/ms, ion accumulation times scaled linearly with peptide concentration (Fig. 1B). However, for peptide targets delivered at less than 10 zmol/ms, contaminant ions from solvent, silica and ambient air limited accumulation times to ∼2 s (Fig. 1B), as confirmed by spectral analysis (Fig. 1C and 1D). Thus, AIM can achieve sensitivities on the order of 100 ions/scan, but target coisolation substantially limits target ion accumulation even under optimized analytical conditions.
AIM Quantitative Proteomics using Automated Two-Dimensional Nanoscale Chromatography
Because target purity affects ions accumulation, which would be further exacerbated in the analysis of complex biological proteomes, we sought to enhance the sensitivity of ion trap-targeted method by reducing contaminant coisolation. Coisolation is normally controlled by gas phase fractionation using quadrupolar mass filters (39, 40). However, this strategy is potentially affected by variable transmittance, particularly for narrow selected m/z ranges, and may introduce biases from independent selection of isotopologs when the isolation window is smaller than their m/z difference. In addition, we reasoned that high-resolution chromatography could be used to improve the isolation of specific peptides and consequently their sensitive quantitation by both AIM and other targeted proteomics methods such as PRM (41). Based on prior implementations of multidimensional separations (28, 42–46), we designed an automated two-dimensional (2D) strong cation exchange (SCX) and reversed phase (RP) nano-scale chromatography architecture (Fig. 2). This implementation benefits from scalability, allowing for integration of additional separation dimensions, and direct interoperability between two- and one-dimensional (1D) modes of operation, allowing for rigorous quantitative analysis of chromatographic performance (Fig. 3). Reduction of SCX chromatographic resolution was previously described because of isocratic mass elution, particularly during step gradient elution (47). To address this issue, we devised an optimized method that minimizes the volumes of mobile phase flowing through the SCX column (supplementary Methods). Detailed description of the instrumental features, as well as step-by-step construction and operation instructions are provided in the supplementary Materials.
Fig. 2.
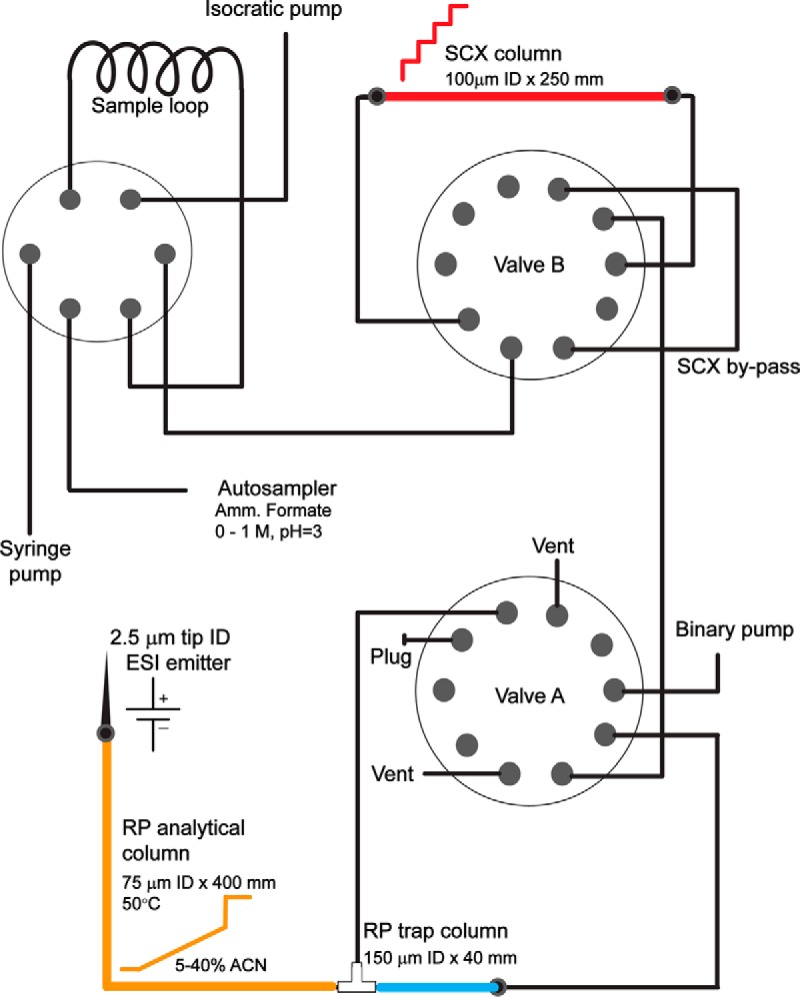
Overview of the automated 2D chromatography system.
Fig. 3.

A, Automated 2D chromatography system enables efficient resolution of target peptides. Average recovery and SCX fractionation efficiency of peptides are plotted relative to 1D (n = 4, error bars represent standard deviation of fraction of target in best SCX fraction. Quantification by PRM). B, 2D chromatography reduces total ion current from coisolated contaminant ions, in absence of synthetic targets (n = 3, error bars = standard deviation). C, 2D chromatography improves ion accumulation, in absence of synthetic targets (n = 3, error bars = standard deviation). Maximum ion injection time (200 ms, dashed line) is achieved for phosphorylated peptides. D, 2D chromatography improves the ratio of MS signal specific for target ions (specific ion current), over total ion current within the scan (10 fmol target peptide, currents at apex of chromatographic peak). E, Improvement of SIC/TIC by SCX-RP chromatography is statistically significant (p = 0.0054, paired t test, n = 6).
To establish the chromatographic performance of this system, we analyzed the separation efficiency of synthetic isotopically-labeled targets diluted in 1 μg of tryptic peptides isolated from human cells, and analyzed under identical conditions and operational parameters in 2D as compared with 1D separations. First, we assessed the recovery of peptides by quantifying each of them in SCX fractions (obtained by stepwise elution with increasing concentrations of ammonium formate at pH 3) and comparing with the complete column elution from 1D chromatography (Fig. 3A). Using this approach, we observed essentially complete recovery of target peptides in 2D versus 1D separations, with minimal losses because of incomplete retention, as assessed by analysis of the flow-through fractions (Fig. 3A). Notably, the two most hydrophilic peptides exhibited significantly increased (2.6–3-fold) signal intensities in 2D as compared with 1D separations, consistent with their improved retention by the final reverse phase column on SCX fractionation (Fig. 3B). We found that retention times in the final reverse phase separations were highly reproducible for 2D-separated peptides, with a constant delay of 30–90 s, as compared with 1D-separated analytes (supplemental Fig. S4). Peptide LFEVIETEK was not recovered from the reverse phase column regardless of pre-fractionation because of poor retention properties, and was therefore omitted in further analyses. Importantly, 2D separation enabled nearly complete (93% average) resolution of target peptides in individual SCX fractions, as assessed by the analysis of signal intensities detected across fractions (Fig. 3A). Using recent formalizations of multidimensional chromatographic separations (48), we estimated that our current 2D implementation produced ∼6-fold increase in the chromatographic efficiency, as compared with conventional 1D chromatography alone.
To test the hypothesis that automated 2D nano-scale chromatography can reduce coelution of background ion and thus their coisolation in AIM and PRM, we compared the total ion currents (TIC) observed at the expected retention times of each target peptides in 1 μg of tryptic human cell extract, resolved by either 2D or 1D under otherwise identical conditions. Consistent with the expected reduction of contaminant coisolation, we observed significant reduction of TIC levels for most target peptides (Fig. 3B). We found that target ion accumulation, as assessed by the effective injection time, significantly increased for most target peptides under 2D as compared with 1D separation (Fig. 3C). Lastly, we estimated the effective purification of specific target peptides by analyzing their fractional specific ion current (SIC) as compared with the total ion current (TIC). This analysis showed that the SIC/TIC ratios for most target peptides were significantly increased by 2D as compared with 1D separations (average > 3-fold, p = 5.4e-3, t test, Figs. 3D and 3E).
Automated Two-Dimensional Nano-scale Chromatography Improves Sensitivity and Quantitative Accuracy of AIM and PRM Targeted Proteomics
Having confirmed improved target accumulation by the automated 2D chromatography, we next sought to determine its effect on the sensitivity of targeted detection in complex biological proteomes. To test this, we used AIM and PRM to quantify synthetic isotopically-labeled peptides (Table I) serially diluted in 1 μg of tryptic human cell proteome (Fig. 4A and 4B, supplemental Fig. S5). For AIM, we used a quadrupole isolation window of 8 Th to enable simultaneous and unbiased coisolation of both endogenous (light) and synthetic (heavy) peptides, and compared the performance of automated 2D versus 1D separations under otherwise identical conditions. Targeted fragment spectra under optimized HCD condition were recorded for PRM along with AIM scanning within the same experiment, allowing for unbiased comparison of the two methods.
Fig. 4.

The improvement in sensitivity from 2D depends on efficiency of fractionation. A, Phosphorylated peptide N(pS)PGLLVSPGNLNK is efficiently resolved from isobaric contaminants, resulting in a 2 amol AIM sensitivity after SCX compared with 1 fmol in 1D. B, PRM analysis achieves 100 amol LOD both in 1D and 2D. Peptide NSPGLLVSPGNLNK is poorly resolved by SCX from the bulk of doubly charged tryptic peptides, resulting in similar sensitivity in 1D (black) and 2D (red) by both AIM (C) and PRM (D). Horizontal lines represent average noise levels (n = 3) for 1D (black) and 2D (red) in AIM. Error bars represent technical variability as standard deviation (n = 3) measured at noise level, 10 amol and 100 fmol. Overlapping data points are horizontally offset when needed for clarity. Vertical arrows mark the LOD in 1D (black) and 2D (red) respectively.
We observed that for some peptides, and particularly phosphorylated peptides which are efficiently resolved by SCX chromatography, AIM 2D chromatography achieved subattomolar limits of detection, with an improvement of nearly 3 orders of magnitude as compared with 1D (Fig. 4A, supplemental Fig. S5C). However, this improvement was not universal, as doubly charged peptides that tend to coelute with most human tryptic peptides, exhibited similar limits of detection (Fig. 4B, supplemental Fig. S5), in agreement with their similar fractional SIC/TIC values in 1D and 2D (Fig. 3D). Nonetheless, even for these peptides, AIM achieved limits of detection in the 1–10 attomolar range (Fig. 4B, supplemental Fig. S5). In agreement with prior studies (14), PRM achieved limits of detection in the 10–100 attomolar range (Fig. 4C and 4D), consistent with its dependence on ion fragmentation for quantitation (defined as at least 3 fragment ions in 5 consecutive scans), in contrast to AIM which only uses fragment ions for identification. Similarly, we found that automated 2D chromatography improved the sensitivity of PRM detection of some peptides to the 10 attomolar range (supplemental Fig. S5). In all, automated 2D chromatography improved the sensitivity and quantitative accuracy of AIM and PRM by nearly 100-fold with subattomolar sensitivity in complex biological proteomes.
Quantitative Proteomics with Ultrahigh Accuracy and Precision using Automated 2D AIM and PRM
To establish the analytical performance of automated 2D chromatography for quantitative proteomics, we determined the absolute abundance of endogenous peptides derived from the master transcription factor MEF2C (49), including its two functional phosphorylation sites, in human leukemia cells OCI-AML2. We analyzed 1 μg of tryptic peptides extracted from 10,000 cells, having diluted isotopically-labeled synthetic peptides as reference standards for absolute quantitation. We found that both automated 2D AIM and PRM accurately quantified the abundance of nonmodified endogenous MEF2C peptides, at the mean level of 180 amol/μg total cell lysate, corresponding to on average 11,000 molecules/cell (Fig. 5). Consistent with the improved sensitivity of 2D versus 1D targeted proteomics (Figs. 3 and 4), more targeted peptides were accurately quantified by 2D AIM and PRM, as compared with their 1D versions (Fig. 5A and 5B). Likewise, 2D AIM exhibited superior sensitivity as compared with 2D PRM, particularly for phospho-peptides, one of which (MEF2C SPEV(pS396)PPR) was not detectable by 2D PRM, because of its low attomolar abundance, corresponding to ∼100 molecules/cell (Fig. 5A and 5B). Notably, 2D AIM enabled both quantitation of absolute abundance and phosphorylation stoichiometry for endogenous MEF2C S222, achieving accurate mean measurements of 42 amol of phosphorylated peptide per μg total cell lysate, with phosphorylation stoichiometry of 26% (Figs. 5A and 5B) (33). Finally, whereas AIM exhibited improved sensitivity, PRM had comparatively lower variability, partly because of reduced mass accuracy for highly concentrated and pure target peptides, which decreased the accuracy of peak profiling (supplemental Fig. S6–S7). Thus, automated two-dimensional nano-scale chromatography enables ultrasensitive high-accuracy quantitative mass spectrometry of complex biological proteomes.
Fig. 5.

Cellular amount of endogenous peptides from protein MEF2C in 1 μg sample, from ∼10,000 cells, as determined by 1D (black) and 2D (red) using A, AIM and B, PRM acquisition. Quantification of endogenous phosphorylated peptides was exclusively achievable using SCX. Each dot represents an independent measurements (full horizontal lines indicate average). LOD for each assay, established from serially diluted isotopologs, is denoted with dashed lines in correspondence.
DISCUSSION
Continuous improvements in chromatography and mass spectrometry instrumentation have now established targeted mass spectrometry as a sensitive and reliable tool to elucidate cellular functions and their regulatory mechanisms (50). Nonetheless, limits of detection of current implementations of targeted mass spectrometry, combined with the low microgram loading capacity of nano-scale chromatographic columns, imply that quantitative analysis of human proteomes is mostly limited to proteins with medium and high cellular copy number (51). Even for such targets, the sensitivity of current methods is often insufficient to detect peptides bearing low stoichiometry post-translational modifications (PTMs), which may also deteriorate ionization and fragmentation efficiencies of peptides (52, 53).
Our current work systematically examined factors affecting the sensitivity and quantitative accuracy of targeted mass spectrometry using the recently developed hybrid Fusion quadrupole-Orbitrap-linear ion trap mass spectrometer. First, we found that under optimized electrospray ionization conditions, current mass analyzer sensitivity and ion transmission enable detection of less than one thousand ions with up to seven orders of magnitude of linear quantitative accuracy. Considering the low-μg loading capacity of current nano-scale chromatographic systems, this sensitivity per se would be sufficient to detect molecules present at a few copies per cells or isolated from single cells. However, we also found that coisolation of background ions nearly isobaric to the target peptides practically impedes extended accumulation before detection. In consideration of the excellent transmission efficiency and detector sensitivity of current state-of-the-art mass spectrometers, coaccumulation of contaminants is thus the principal factor limiting sensitivity of targeted methods leveraging ion traps, such as AIM and PRM.
Current targeted mass spectrometry methods generally rely on fragment ion quantitation, as this enables the specificity necessary for the analysis of complex proteomes. Here, we demonstrate that targeted analysis of intact precursor ions, termed accumulated ion monitoring (AIM), enables sensitivity equal or superior to that of optimized parallel reaction monitoring methods, with specificity of detection controlled by isotopically encoded reference standards and parallel fragment ion identification. AIM acquisition enables simultaneous and unbiased isolation of both endogenous targets and reference isotopologs, which can be leveraged to reduce duty cycle requirements as compared with current PRM implementations for rare analytes that may require extended ion accumulation. The dependence on efficient fragmentation may hinder PRM or SRM quantification of certain peptides. For example, even under ideal fragmentation conditions, the amount of specific fragment ions is inherently a fraction of their respective precursor ions, in turn limiting the sensitivity of PRM for the quantification of relatively low abundance peptides. AIM partly circumvents this limitation, as its quantitation is based on precursor ion currents, whereas its specificity can be maintained even under conditions of reduced fragmentation efficiency.
To reduce contaminant coisolation and thus facilitate quantitative proteomics of complex biological proteomes, we implemented an automated two-dimensional nano-scale chromatography architecture, based on prior developments in multidimensional separations (28, 38–44). Unlike previous implementation of 2D chromatography that were deployed in combination with data-dependent methods, operation of our system was specifically configured to achieve reproducible and high-resolution sample pre-fractionation suitable for quantitative mass spectrometry. This automated 2D chromatography system, accompanied by detailed construction and operation instructions described in the supplementary Materials, also permits scalable and seamless chromatography mode switching. Automated 2D chromatography improved the detection limits of targeted analysis of both precursor and fragment ions by effectively reducing coelution of isobaric contaminants in the final reverse phase chromatography. Such improvement depends on the specific retention properties of each target, and strong cation exchange chromatography was particularly effective in improving, as expected, the analytical exposure of phosphorylated peptides. This enabled detection of endogenously phosphorylated peptides from 1 μg of extracts from as little as 10,000 cells, a scale that is mostly unsuitable for conventional phospho-proteomics methods (54). This proof-of-concept suggests that targeted quantification of specific classes of peptides could be achieved by leveraging high-resolution, high-capacity chromatographic separations orthogonal to reverse phase under acidic conditions, without dedicated affinity enrichment (47, 55, 56).
In addition, robust reproducibility and high efficiency of fractionation using automated 2D chromatography permit multidimensional retention time scheduling of targeted assays, with consequent increase in the number of measurable targets per experiment at otherwise constant mass spectrometer duty cycle. Such increased assay scheduling capacity further expands the scope of recently described strategies such as isotopolog-triggered scanning (57), and can be coupled to ultrahigh capacity chromatography systems (41), as recently described (58). As a result, the number of targeted assays that can be deployed per sample injection should increase from the hundreds to the thousands. Furthermore, nano-scale 2D chromatography enables efficient fractionation of low-μg samples, such as those obtained from primary samples and disease specimens, enabling targeted analysis of rare cell populations. Multidimensional fractionation can reduce throughput as compared with singly dimensioned analyses. However, the increased sensitivity and breadth of targeting afforded by automated multidimensional separation should permit comprehensive panels of targeted mass spectrometry assays to elucidate rare regulatory events that control functional biological processes.
DATA AVAILABILITY
All raw and processed mass spectrometry data as well as Skyline chromatogram documents are available via ProteomeXchange [30] with identifier PXD006236.
Supplementary Material
Acknowledgments
We thank Scott Ficarro and Jarrod Marto for technical advice, Michael Senko for critical discussion, Avantika Dhabaria for technical assistance, Christopher Lima and Laurent Cappadocia for spectrophotometric analyses, and Nina Lampen for electron microscopy.
Footnotes
Author Contributions: P.C. and A.K. conceived and designed the study, P.C. performed the experiments, P.C. and A.K. performed analyses, P.C. and A.K. wrote the manuscript.
* This work was supported by the NIH R21 CA188881, R01 CA204396, P30 CA008748, Burroughs Wellcome Fund, Josie Robertson Investigator Program, Rita Allen Foundation, Alex's Lemonade Stand Foundation, American Society of Hematology, and Gabrielle's Angel Foundation (A.K.), and American Italian Cancer Foundation (P.C.). A.K. is the Damon Runyon-Richard Lumsden Foundation Clinical Investigator.
 This article contains supplemental material.
This article contains supplemental material.
The authors have no competing financial interests.
1 The abbreviations used are:
- SRM
- selected reaction monitoring
- ABC
- ammonium bicarbonate
- ACN
- acetonitrile
- AIM
- accumulated ion monitoring
- AGC
- automatic gain control
- CID
- collision induced dissociation
- FA
- formic acid
- HCD
- higher-energy collisional dissociation
- HPLC
- high pressure liquid chromatography
- IRM
- ion routing multipole
- LOD
- limit of detection
- PRM
- parallel reaction monitoring
- PTM
- post-translational modification
- Q1
- (first) quadrupolar mass filter
- SCX
- strong cation exchange
- SIC
- specific ion current
- SIM
- selected ion monitoring
- TSQ
- triple stage quadrupole (also QQQ)
- XIC
- extracted ion chromatogram.
REFERENCES
- 1. Steen H., and Pandey A. (2002) Proteomics goes quantitative: measuring protein abundance. Trends Biotechnol. 20, 361–364 [DOI] [PubMed] [Google Scholar]
- 2. Olsen J. V., Vermeulen M., Santamaria A., Kumar C., Miller M. L., Jensen L. J., Gnad F., Cox J., Jensen T. S., Nigg E. A., Brunak S., and Mann M. (2010) Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 3, ra3. [DOI] [PubMed] [Google Scholar]
- 3. Dephoure N., and Gygi S. P. (2012) Hyperplexing: a method for higher-order multiplexed quantitative proteomics provides a map of the dynamic response to rapamycin in yeast. Sci. Signal. 217, rs2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kentsis A., Shulman A., Ahmed S., Brennan E., Monuteaux M. C., Lee Y. H., Lipsett S., Paulo J. A., Dedeoglu F., Fuhlbrigge R., Bachur R., Bradwin G., Arditi M., Sundel R. P., Newburger J. W., Steen H., and Kim S. (2013) Urine proteomics for discovery of improved diagnostic markers of Kawasaki disease. EMBO Mol. Med. 5, 210–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cima I., Schiess R., Wild P., Kaelin M., Schüffler P., Lange V., Picotti P., Ossola R., Templeton A., Schubert O., Fuchs T., Leippold T., Wyler S., Zehetner J., Jochum W., Buhmann J., Cerny T., Moch H., Gillessen S., Aebersold R., and Krek W. (2011) Cancer genetics-guided discovery of serum biomarker signatures for diagnosis and prognosis of prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 108, 3342–3347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lange V., Picotti P., Domon B., and Aebersold R. (2008) Selected reaction monitoring for quantitative proteomics: a tutorial. Mol. Syst. Biol. 4, 222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carr S. A. Abbatiello S. E. Ackermann B. L., Borchers C. Domon B. Deutsch E. W. Grant R. P. Hoofnagle A. N. Hüttenhain R. Koomen J. M. Liebler D. C. Liu T. MacLean B. Mani D. R. Mansfield E. Neubert H. Paulovich A. G. Reiter L. Vitek O. Aebersold R. Anderson L. Bethem R. Blonder J. Boja E. Botelho J. Boyne M. Bradshaw R. A. Burlingame A. L. Chan D. Keshishian H. Kuhn E. Kinsinger C. Lee J. S. H. Lee S.-W. Moritz R. Oses-Prieto J. Rifai N. Ritchie J. Rodriguez H. Srinivas P. R. Townsend R. R. Van Eyk J. Whiteley G. Wiita A. and Weintraub S. (2014) Targeted peptide measurements in biology and medicine: best practices for mass spectrometry-based assay development using a fit-for-purpose approach. Mol. Cell. Proteomics 13, 907–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Picotti P., and Aebersold R. (2012) Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nat. Methods 9, 555–566 [DOI] [PubMed] [Google Scholar]
- 9. Picotti P., Bodenmiller B., Mueller L. N., Domon B., and Aebersold R. (2009) Full dynamic range proteome analysis of S. cerevisiae by targeted proteomics. Cell 138, 795–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Abbatiello S. E., Mani D. R., Keshishian H., and Carr S. A. (2010) Automated detection of inaccurate and imprecise transitions in peptide quantification by multiple reaction monitoring mass spectrometry. Clin. Chem. 56, 291–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sherman J. McKay M. J. Ashman K. and Molloy M. P. (2009) How specific is my SRM?: The issue of precursor and product ion redundancy. Proteomics 9, 1120–1123 [DOI] [PubMed] [Google Scholar]
- 12. Bao Y. Waldemarson S. Zhang G. Wahlander A. Ueberheide B. Myung S. Reed B. Molloy K. Padovan J. C. Eriksson J. Neubert T. A. Chait B. T. and Fenyö D. (2013) Detection and correction of interference in SRM analysis. Methods 61, 299–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Peterson A. C., Russell J. D., Bailey D. J., Westphall M. S., and Coon J. J. (2012) Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol. Cell. Proteomics 11, 1475–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gallien S., Duriez E., Crone C., Kellmann M., Moehring T., and Domon B. (2012) Targeted proteomic quantification on quadrupole-orbitrap mass spectrometer. Mol. Cell. Proteomics 11, 1709–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gillet L. C., Navarro P., Tate S., Röst H., Selevsek N., Reiter L., Bonner R., and Aebersold R. (2012) Targeted data extraction of the MS/MS spectra generated by data-independent acquisition: a new concept for consistent and accurate proteome analysis. Mol. Cell. Proteomics 11, O111.016717, Jun. 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu Y., Hüttenhain R., Surinova S., Gillet L. C. J., Mouritsen J., Brunner R., Navarro P., and Aebersold R. (2013) Quantitative measurements of N-linked glycoproteins in human plasma by SWATH-MS. Proteomics 13, 1247–1256 [DOI] [PubMed] [Google Scholar]
- 17. Meier F., Beck S., Grassl N., Lubeck M., Park M. A., Raether O., and Mann M. (2015) Parallel Accumulation-Serial Fragmentation (PASEF): Multiplying Sequencing Speed and Sensitivity by Synchronized Scans in a Trapped Ion Mobility Device. J. Proteome Res. 14, 5378–5387 [DOI] [PubMed] [Google Scholar]
- 18. Martin S. E., Shabanowitz J., Hunt D. F., and Marto J. A. (2000) Subfemtomole MS and MS/MS peptide sequence analysis using nano-HPLC micro-ESI fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 72, 4266–4274 [DOI] [PubMed] [Google Scholar]
- 19. Weisbrod C. R., Hoopmann M. R., Senko M. W., and Bruce J. E., (2013) Performance evaluation of a dual linear ion trap-Fourier transform ion cyclotron resonance mass spectrometer for proteomics research. J. Proteomics 88, 109–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hardman M., and Makarov A. A. (2003) Interfacing the orbitrap mass analyzer to an electrospray ion source. Anal. Chem. 75, 1699–1705 [DOI] [PubMed] [Google Scholar]
- 21. Senko M. W., Hendrickson C. L., Emmett M. R., Shi S. D. H., and Marshall A. G. (1997) External accumulation of ions for enhanced electrospray ionization fourier transform ion cyclotron resonance mass spectrometry. J. Am. Soc. Mass Spectrom. 8, 970–976 [Google Scholar]
- 22. Senko M. W., Remes P. M., Canterbury J. D., Mathur R., Song Q., Eliuk S. M., Mullen C., Earley L., Hardman M., Blethrow J. D., Bui H., Specht A., Lange O., Denisov E., Makarov A., Horning S., and Zabrouskov V. (2013) Novel parallelized quadrupole/linear ion trap/Orbitrap tribrid mass spectrometer improving proteome coverage and peptide identification rates. Anal. Chem. 85, 11710–11714 [DOI] [PubMed] [Google Scholar]
- 23. Eliuk S., and Makarov A. (2015) Evolution of orbitrap mass spectrometry instrumentation. Annu. Rev. Anal. Chem. 8, 61–80 [DOI] [PubMed] [Google Scholar]
- 24. Kuipers B. J. H., and Gruppen H. (2007) Prediction of molar extinction coefficients of proteins and peptides using UV absorption of the constituent amino acids at 214 nm to enable quantitative reverse phase high-performance liquid chromatography-mass spectrometry analysis. J. Agric. Food Chem. 55, 5445–5451 [DOI] [PubMed] [Google Scholar]
- 25. Kentsis A., Reed C., Rice K. L., Sanda T., Rodig S. J., Tholouli E., Christie A., Valk P. J. M., Delwel R., Ngo V., Kutok J. L., Dahlberg S. E., Moreau L. A., Byers R. J., Christensen J. G., Vande Woude G., Licht J. D., Kung A. L., Staudt L. M., and Look A. T. (2012) Autocrine activation of the MET receptor tyrosine kinase in acute myeloid leukemia. Nat. Med. 18, 1118–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dhabaria A., Cifani P., Reed C., Steen H., and Kentsis A. (2015) A high-efficiency cellular extraction system for biological proteomics. J. Proteome Res. 14, 3403–3408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dhabaria A., Cifani P., and Kentsis A. (2015) Fabrication of capillary columns with integrated frits for mass spectrometry. Protocol Exchange [Google Scholar]
- 28. Ficarro S. B., Zhang Y., Carrasco-Alfonso M. J., Garg B., Adelmant G., Webber J. T., Luckey C. J., and Marto J. A. (2011) Online nanoflow multidimensional fractionation for high efficiency phosphopeptide analysis. Mol. Cell. Proteomics 10, O111.011064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cifani P., Dhabaria A., and Kentsis A. (2015) Fabrication of Nanoelectrospray Emitters for LC-MS. Protocol Exchange [Google Scholar]
- 30. Zhang Y., Ficarro S. B., Li S., and Marto J. A. (2009) Optimized Orbitrap HCD for quantitative analysis of phosphopeptides. J. Am. Soc. Mass Spectrom. 20, 1425–1434 [DOI] [PubMed] [Google Scholar]
- 31. MacLean B., Tomazela D. M., Shulman N., Chambers M., Finney G. L., Frewen B., Kern R., Tabb D. L., Liebler D. C., and MacCoss M. J. (2010) Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vizcaíno J. A., Côté R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J., O'Kelly G., Schoenegger A., Ovelleiro D., Pérez-Riverol Y., Reisinger F., Ríos D., Wang R., and Hermjakob H. (2013) The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–D1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cifani P., Shakiba M., Chhangawala S., and Kentsis A. (2017) ProteoModlR for functional proteomic analysis. BMC Bioinformatics 18, 153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stellaard F., and Paumgartner G. (1985) Measurement of isotope ratios in organic compounds at picomole quantities by capillary gas chromatography/quadrupole electron impact mass spectrometry. Biomed. Mass Spectrom. 12, 560–564 [DOI] [PubMed] [Google Scholar]
- 35. Wilm M., and Mann M. (1996) Analytical properties of the nanoelectrospray ion source. Anal. Chem. 68, 1–8 [DOI] [PubMed] [Google Scholar]
- 36. Marginean I., Kelly R. T., Prior D. C., LaMarche B. L., Tang K., and Smith R. D. (2008) Analytical characterization of the electrospray ion source in the nanoflow regime. Anal. Chem. 80, 6573–6579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marginean I. Tang K. Smith R. D. and Kelly R. T. (2014) Picoelectrospray ionization mass spectrometry using narrow-bore chemically etched emitters. J. Am. Soc. Mass Spectrom. 25, 30–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ficarro S. B., Zhang Y., Lu Y., Moghimi A. R., Askenazi M., Hyatt E., Smith E. D., Boyer L., Schlaeger T. M., Luckey C. J., and Marto J. A. (2009) Improved electrospray ionization efficiency compensates for diminished chromatographic resolution and enables proteomics analysis of tyrosine signaling in embryonic stem cells. Anal. Chem. 81, 3440–3447 [DOI] [PubMed] [Google Scholar]
- 39. Yi E. C., Marelli M., Lee H., Purvine S. O., Aebersold R., Aitchison J. D., and Goodlett D. R. (2002) Approaching complete peroxisome characterization by gas-phase fractionation. Electrophoresis 23, 3205–3216 [DOI] [PubMed] [Google Scholar]
- 40. Scherl A., Shaffer S. A., Taylor G. K., Kulasekara H. D., Miller S. I., and Goodlett D. R. (2008) Genome-specific gas-phase fractionation strategy for improved shotgun proteomic profiling of proteotypic peptides. Anal. Chem. 80, 1182–1191 [DOI] [PubMed] [Google Scholar]
- 41. Burgess M. W., Keshishian H., Mani D. R., Gillette M. A., and Carr S. A. (2014) Simplified and efficient quantification of low-abundance proteins at very high multiplex via targeted mass spectrometry. Mol. Cell. Proteomics 13, 1137–1149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhou F., Lu Y., Ficarro S. B., Adelmant G., Jiang W., Luckey C. J., and Marto J. A. (2013) Genome-scale proteome quantification by DEEP SEQ mass spectrometry. Nat. Commun. 4, 2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mohammed S., and Heck A. (2011) Strong cation exchange (SCX) based analytical methods for the targeted analysis of protein post-translational modifications. Curr. Opin. Biotechnol. 22, 9–16 [DOI] [PubMed] [Google Scholar]
- 44. Wolters D. A., Washburn M. P., and Yates J. R. (2001) An automated multidimensional protein identification technology for shotgun proteomics. Anal. Chem. 73, 5683–5690 [DOI] [PubMed] [Google Scholar]
- 45. Taouatas N., Altelaar A. F. M., Drugan M. M., Helbig A. O., Mohammed S., and Heck A. J. R. (2009) Strong cation exchange-based fractionation of Lys-N-generated peptides facilitates the targeted analysis of post-translational modifications. Mol. Cell. Proteomics 8, 190–200 [DOI] [PubMed] [Google Scholar]
- 46. Gilar M., Olivova P., Daly A. E., and Gebler J. C. (2005) Orthogonality of separation in two-dimensional liquid chromatography. Anal. Chem. 77, 6426–6434 [DOI] [PubMed] [Google Scholar]
- 47. Le Bihan T., Duewel H. S., and Figeys D. (2003) On-line strong cation exchange micro-HPLC-ESI-MS/MS for protein identification and process optimization. J. Am. Soc. Mass Spectrom. 14, 719–727 [DOI] [PubMed] [Google Scholar]
- 48. Li X., Stoll D. R., and Carr P. W. (2009) Equation for peak capacity estimation in two-dimensional liquid chromatography. Anal. Chem. 81, 845–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Canté-Barrett K., Pieters R., and Meijerink J. P. P. (2014) Myocyte enhancer factor 2C in hematopoiesis and leukemia. Oncogene 33, 403–410 [DOI] [PubMed] [Google Scholar]
- 50. Aebersold R., and Mann M. (2016) Mass-spectrometric exploration of proteome structure and function. Nature 537, 347–355 [DOI] [PubMed] [Google Scholar]
- 51. Wiśniewski J. R., Hein M. Y., Cox J., and Mann M. (2014) A “proteomic ruler” for protein copy number and concentration estimation without spike-in standards. Mol. Cell. Proteomics 13, 3497–3506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Steen H., Jebanathirajah J. A., Rush J., Morrice N., and Kirschner M. W. (2006) Phosphorylation analysis by mass spectrometry: myths, facts, and the consequences for qualitative and quantitative measurements. Mol. Cell. Proteomics 5, 172–181 [DOI] [PubMed] [Google Scholar]
- 53. Boersema P. J., Mohammed S., and Heck A. J. R. (2009) Phosphopeptide fragmentation and analysis by mass spectrometry. J. Mass Spectrom. 44, 861–878 [DOI] [PubMed] [Google Scholar]
- 54. Villén J., and Gygi S. P. (2008) The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat. Protoc. 3, 1630–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zarei M., Sprenger A., Metzger F., Gretzmeier C., and Dengjel J. (2011) Comparison of ERLIC-TiO2, HILIC-TiO2, and SCX-TiO2 for global phosphoproteomics approaches. J. Proteome Res. 10, 3474–3483 [DOI] [PubMed] [Google Scholar]
- 56. Ritorto M. S., Cook K., Tyagi K., Pedrioli P. G. A., and Trost M. (2013) Hydrophilic strong anion exchange (hSAX) chromatography for highly orthogonal peptide separation of complex proteomes. J. Proteome Res. 12, 2449–2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gallien S., Kim S. Y., and Domon B. (2015) Large-scale targeted proteomics using internal standard triggered-parallel reaction monitoring (IS-PRM). Mol. Cell. Proteomics 14, 1630–1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Urisman A., Levin R. S., Gordan J. D., Webber J. T., Hernandez H., Ishihama Y., Shokat K. M., and Burlingame A. L. (2017) An optimized chromatographic strategy for multiplexing in parallel reaction monitoring mass spectrometry: insights from quantitation of activated kinases. Mol. Cell. Proteomics 16, 265–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw and processed mass spectrometry data as well as Skyline chromatogram documents are available via ProteomeXchange [30] with identifier PXD006236.