

Abstract
Glycoprotein A repetitions predominant (GARP) (encoded by the Lrrc32 gene) plays important roles in cell-surface docking and activation of TGFβ. However, GARP's role in organ development in mammalian systems is unclear. To determine the function of GARP in vivo, we generated a GARP KO mouse model. Unexpectedly, the GARP KO mice died within 24 h after birth and exhibited defective palatogenesis without apparent abnormalities in other major organs. Furthermore, we observed decreased apoptosis and SMAD2 phosphorylation in the medial edge epithelial cells of the palatal shelf of GARP KO embryos at embryonic day 14.5 (E14.5), indicating a defect in the TGFβ signaling pathway in the GARP-null developing palates. Of note, the failure to develop the secondary palate and concurrent reduction of SMAD phosphorylation without other defects in GARP KO mice phenocopied TGFβ3 KO mice, although GARP has not been suggested previously to interact with TGFβ3. We found that GARP and TGFβ3 co-localize in medial edge epithelial cells at E14.5. In vitro studies confirmed that GARP and TGFβ3 directly interact and that GARP is indispensable for the surface expression of membrane-associated latent TGFβ3. Our findings indicate that GARP is essential for normal morphogenesis of the palate and demonstrate that GARP plays a crucial role in regulating TGFβ3 signaling during embryogenesis. In conclusion, we have uncovered a novel function of GARP in positively regulating TGFβ3 activation and function.
Keywords: apoptosis; cell signaling; development; embryo; TGFβ; GARP, palatogenesis
Introduction
GARP2 is a type 1 transmembrane protein with 20 leucine-rich repeats in the extracellular domain, a single transmembrane domain, and a short 14-amino acid cytoplasmic tail (1–5). GARP is highly expressed by platelets, activated regulatory T cells (Tregs) (6–9), mesenchymal stromal cells (10), hepatic stellate cells (11), and transformed tumor cells (12, 13). Multiple studies have established that GARP plays a role in activating TGFβ by mediating the surface expression and integrin-mediated activation of latent TGFβ (1, 2, 8, 14–18). GARP associates with latency-associated peptide to form an alternative cell-surface platform for latent TGFβ presentation (1, 2). GARP participates in the regulatory function of activated Tregs, demonstrated by impaired suppressive activity upon GARP silencing (4, 6) and inefficient support by GARP KO T cells for the generation of inducible Tregs (19). Aberrant overexpression of GARP in the tumor microenvironment also promotes Treg generation and immune escape (12, 13). Recently, we further identified that gp96 plays an essential role in surface GARP–TGFβ complex expression by chaperoning GARP in the endoplasmic reticulum (5). Further, GARP has been shown to function in promoting immune tolerance and tumor progression in cancer (5, 12, 15, 17). Thus, GARP is a key posttranslational regulator of TGFβ biogenesis and immune tolerance.
The TGFβ superfamily is a crucial cytokine family for both development and immunity, performing various functions in cell proliferation, differentiation, and cancer (20–22). There are three TGFβ isoforms. GARP reportedly binds only to TGFβ1 and TGFβ2 but not TGFβ3 (1). Genetically modified mice lacking Tgfb1 are born with autoimmune, endocrine, reproductive, vascular, and developmental abnormalities and die at 3–4 weeks of age (23). Tgfb2-null mice show perinatal mortality and multiple developmental defects, including cardiac, lung, craniofacial, limb, spinal column, eye, inner ear, and urogenital defects (24). Tgfb3 KO mice have a cleft palate caused by defects in medial edge epithelial (MEE) seam degeneration and palate fusion, leading to death within the first day after birth (25–27). Although all three TGFβs are expressed in the palate during mouse palate development, only inhibition of TGFβ3, but not TGFβ1 or TGFβ2, results in the palate fusion defect (28, 29), demonstrating the isoform-specific role of TGFβ family members in vivo. However, the regulation of TGFβ3 biogenesis and signaling remains largely unknown.
To determine the GARP function in vivo, we have developed a reversible GARP KO mouse model using the flexible accelerated STOP TetO (FAST) knockin system (30), the GARP FAST model. The homozygous GARP KO mice died within 24 h after birth. Unexpectedly, the only developmental defect observed in KO mice was a cleft palate, which is identical to Tgfb3 KO mice (26, 31). We further performed studies to establish the critical roles of GARP in binding and activating TGFβ3 and identified a novel role of GARP in TGFβ3 biogenesis and function.
Results
Knockout of Lrrc32 causes postnatal lethality and a cleft palate
Successful disruption of GARP was accomplished by inserting a stop cassette and TetO element into the endogenous promoter region of the Lrrc32 locus. Genomic PCR and flow cytometry analysis on platelets of day 0 live pups confirmed the absence of GARP expression in the homozygous GARP FAST neonates (supplemental Fig. S1).
Immediately postpartum, the ratios of wild-type, heterozygous, and homozygous offspring (total n = 94) from intercrossing between heterozygotes followed the expected Mendelian pattern (24.5% +/+, 23.4% −/−, 52.1% +/−; supplemental Table S1), demonstrating that the loss of GARP does not cause prenatal mortality. However, no offspring with homozygous mutated alleles survived 24 h after birth. The dead GARP-null offspring showed no visible milk in their stomachs. Further gross anatomical analysis and IHC of the KO mice showed no apparent abnormalities in the major organs, including the heart, lung, spleen, and liver (data not shown).
Importantly, cleft palates were observed in all homozygous mutated pups (Fig. 1). No developmental defects were identified in heterozygous mice. Thus, we subsequently focused on analyzing palate development in GARP mutant embryos and wild-type littermate controls to specify the cause of the pathology. At embryonic day 13.5 (E13.5), when the palatal shelves form from the maxillary processes, proliferate, and undergo elevation to reach the horizontal position, the palates in mutants and controls were indistinguishable (data not shown). The differences between GARP KO and wild-type embryos became evident at E14.5, when the palatal shelves begin to fuse (Fig. 1). At E14.5, the formation and elevation of the palatal shelf was comparable between wild-type and GARP mutant mice at the rostral region (Fig. 1, A and E). However, at the caudal region, where palatal fusion has begun, a midline epithelial seam is seen in WT palate (Fig. 1B, arrowhead) but is absent from GARP mutants (Fig. 1F, asterisk). From E15.5 to E16.5, palatal fusion is completed throughout the entire palate in the wild type (Fig. 1, C and D) but not observed in the mutants (Fig. 1, G and H). Instead, a large gap can be seen in GARP mutants (Fig. 1, G and H, asterisk), demonstrating a cleft palate phenotype. Together, these observations suggest that GARP is required for epithelial fusion during palatogenesis.
Figure 1.

Ablation of GARP/Lrrc32 causes cleft palate in mice. A–H, H&E histological analysis (A–C and E–G) and whole-mount (D and H) of palatogenesis of wild-type (A–D) and GARP KO mice (E–H). Formation and elevation of the palatal shelf (P) are shown for both the wild-type (A) and KO (E) at the rostral region of E14.5 embryos. A midline epithelial seam is seen in the WT palate (B, arrowhead) but absent from GARP KO mice (F, asterisk). From E15.5 to E16.5 embryos, palatal fusion is completed throughout the entire palate in the wild type (C and D) but not observed in the mutant (G and H). Instead, a large gap can be seen in GARP mutants (asterisk), demonstrating a cleft palate phenotype. T, tongue.
Palatal edge epithelial cell proliferation, apoptosis, and TGFβ signaling in the absence of GARP
We next analyzed the possible mechanism underlying the defective palatogenesis caused by GARP deletion. First, by staining for and quantifying phospho-histone H3, we found no difference in palatal edge epithelial cell proliferation between mutant and WT embryos (supplemental Fig. S2), suggesting that the proliferation abnormality is not the cause for the cleft palate.
MEE cell apoptosis has been shown to be important for palate fusion (32). We therefore used TUNEL assays to determine whether apoptotic signaling is compromised in KO palates. Indeed, we observed a significant decrease of apoptosis at the MEE seam in the mutant palate (Fig. 2). Because GARP is known to enhance TGFβ activation, and the TGFβ3-SMAD2/3 signaling pathway plays an essential role in palatogenesis (32–34), we next focused on discerning the possible TGFβ signaling defect in the developing palates of KO mice. Indeed, decreased pSMAD2 was observed in E14.5 mutant palates, revealing clear defective canonical TGFβ signaling in the absence of GARP (Fig. 3).
Figure 2.
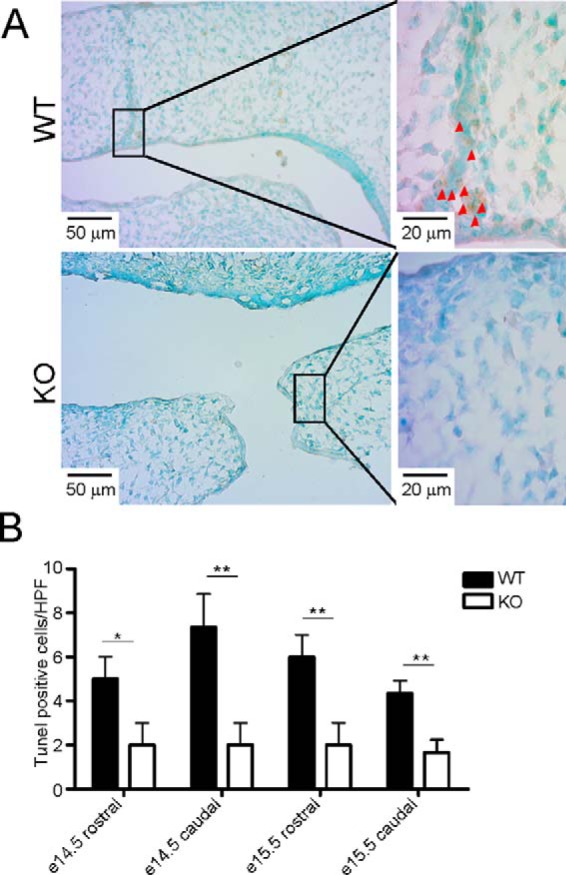
Decreased apoptosis in GARP KO palates. A, a TUNEL assay was used to detect apoptotic cells at the MEE region in E14.5 WT and corresponding KO palates. Arrowheads indicate TUNEL-positive apoptotic cells. B, quantification of apoptotic cells in WT and KO mice at the indicated time points. HPF, high-power field. The values are expressed as mean ± S.D. (n = 3). *, p < 0.05; **, p < 0.01. The data are representative of two independent experiments.
Figure 3.

Ablation of GARP results in reduction of SMAD2/3 phosphorylation in palatal edge epithelial cells. IHC was performed to determine the expression of pSMAD2/3 at the MEE region in E14.5 WT and the corresponding KO palates. The immunohistochemical staining was performed with antibody against pSMAD2/3. Slides were counterstained with hematoxylin. Representative images are shown. Arrowheads points to pSMAD2/3-positive cells. The staining intensity scores for pSMAD2/3 are expressed as mean ± S.D. (n = 4). *, p < 0.05.
Expression and co-localization of GARP and TGFβ3 in palatal edge epithelial cells
The isolated phenotype observed in GARP KO mice is conspicuously similar to Tgfb3 KO mice (25, 26). This suggests the possibility that GARP and TGFβ3 function together in vivo to regulate palatogenesis. If so, then GARP and TGFβ3 are expected to co-express at the MEE region. To address this prediction, we performed GARP IHC using a sheep anti-mouse GARP antibody and discovered that GARP was expressed at the MEE region in WT E14.5 palates but not in the corresponding KO palates (Fig. 4A). Consistently, TGFβ3 was also detected in WT MEE cells (Fig. 4B). However, as IHC cannot differentiate various forms of TGFβ3, we did not observe a clear difference in TGFβ3 levels between WT and KO palates, proving that GARP does not affect total TGFβ3 expression. Altogether, these results suggest that strategically expressed GARP in MME cells is required for TGFβ3 activation and palatogenesis.
Figure 4.

Expression and localization of GARP and TGFβ3 in palatal edge epithelial cells. A, IHC was performed to detect GARP expression at the MEE region in E14.5 WT and the corresponding KO palates. B, TGFβ3 was detected in E14.5 WT and KO palate MMEs. The staining intensity scores are expressed as mean ± S.D. (n = 4). **, p < 0.01.
GARP interacts with proTGFβ3 and is required for its cell-surface expression
Up to this point, the literature has suggested that GARP regulates both TGFβ1 and TGFβ2 but not TGFβ3 (1). We next revisited this question by performing extensive in vitro biochemical studies. We co-overexpressed proTGFβ3 and HA-tagged mouse GARP in HEK293 cells, followed by immunoprecipitation analysis. As expected, TGFβ3 was co-pulled down with GARP (Fig. 5, A and B). In addition, we overexpressed proTGFβ3 in a mouse mammary carcinoma cell line, 67NR, which was made to express either mouse full-length GARP or the soluble extracellular domain of GARP fused with the Fc domain of IgG (GARP-Fc), which can interact with protein A/G beads (supplemental Fig. S3). Thus, using protein A/G beads, we were able to immunoprecipitate GARP-Fc with TGFβ3 from the cell lysates and the supernatants of cells that express both GARP-Fc and TGFβ3. These results demonstrate a direct interaction between the two proteins.
Figure 5.
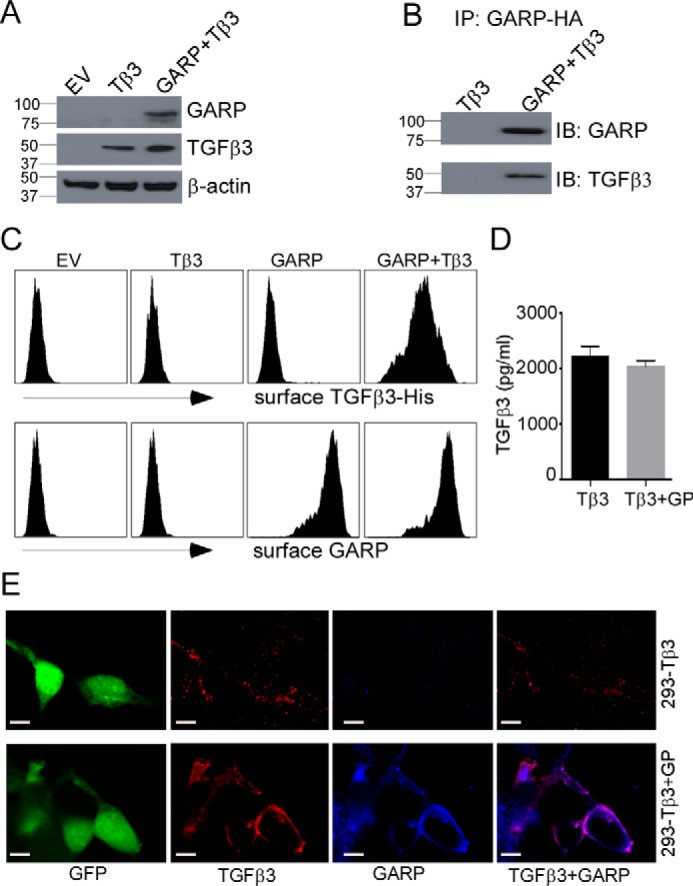
GARP interacts with TGFβ3 and is required for TGFβ3 surface expression. A, human TGFβ3 was transiently overexpressed in HEK293 cells or HEK293 cells stably expressing HA-tagged mouse GARP. Two days after transfection, the cell pellets were collected. The whole-cell lysates were subjected to immunoblot analysis. EV, empty vector. B, pulldown of GARP-HA was performed using an anti-HA antibody. The immunoprecipitated (IP) proteins were analyzed by immunoblot (IB) analysis. C–E, cell surface expression of His-tagged TGFβ3 depends on GARP. C, His-TGFβ3 was expressed in HEK293 or 293-GARP cells, followed by surface staining with anti-His antibody and flow cytometry analysis. D, total TGFβ3 levels were quantified by ELISA from conditioned medium of the indicated cells. The values are expressed as mean ± S.D. (n = 3). Statistical significance was calculated with respect to the control (*p < 0.05). E, confocal microscopy analysis was performed to demonstrate the co-localization of GARP (blue) and TGFβ3 (red) in 293 cells. Scale bars = 20 μm. The data are representative of two independent experiments. GP, GARP; Tβ3, TGFβ3.
Because GARP is a cell-surface TGFβ docking receptor (1, 2, 4–6), we next investigated whether GARP is important for the surface expression of TGFβ3. Similar to what was previously done with TGFβ1 (35), we tagged human TGFβ3 with a His tag right after the furin cleavage site. The tagged TGFβ3 (His-TGFβ3) was expressed in HEK293 cells with or without GARP. We demonstrated that surface His-TGFβ3 is only detected in cells that co-express GARP (Fig. 5, C and E), despite similar levels of secreted TGFβ3 from GARP+ and GARP− cells (Fig. 5D). Finally, by confocal microscopic analysis, co-localization of GARP and His-TGFβ3 on the cell surface was demonstrated (Fig. 5E). Altogether, we identified TGFβ3 as a novel GARP ligand and that GARP is required for cell surface expression of TGFβ3.
Discussion
TGFβ is a pleotropic cytokine whose activity is subject to extensive regulation to control its expression level, activation status, spatial and temporal availability, and downstream signaling. GARP is a non-signaling receptor that is primarily responsible for cell-surface docking of TGFβ onto Tregs and platelets. Not surprisingly, the research focus on GARP has been in the area of immune regulation. However, there is evidence for the notion that GARP also functions in cell differentiation and embryonic development. As early as in 1996, Roubin et al. (36) reported that GARP transcripts are expressed in various tissues in mouse embryos. Importantly, the highest levels were detected in E13.5 and E15.5 embryos (36), with unclear significance. This study is the first to establish a role for GARP in mouse embryonic development.
Although 100% lethality was observed with GARP KO neonates, analysis of embryos and day 1 neonates did not reveal other malformations in any other organs except for a cleft palate. The isolated defect in palatogenesis in GARP KO mice is identical to the one observed in TGFβ3 mice. Thus, although GARP is broadly expressed in embryos, its most important function during embryogenesis appears to be selectively facilitating the function of TGFβ3. Indeed, we found that both GARP and TGFβ3 are co-expressed in MEE cells at E14.5. More importantly, we showed that GARP directly interacts with TGFβ3 and is responsible for cell-surface expression of TGFβ3, challenging a previous study claiming that GARP binds to the latent forms of TGFβ1 and TGFβ2 but not TGFβ3 (1). Sequencing alignment of all three TGFβs (supplemental Fig. S4) showed that the Cys-4 of proTGFβ1, which is important for interaction with GARP (2), is conserved among proTGFβ1, proTGFβ2, and proTGFβ3. In addition, the RGD motif is present in proTGFβ1 and proTGFβ3 but not in proTGFβ2. This suggests that TGFβ1 and TGFβ3 share similar mechanisms of activation by GARP and αV integrins (αVβ6 or αVβ8).
It should be noted that GARP-null embryos show no clear phenotype in TGFβ1- or TGFβ2-null embryos. Does this mean that GARP is more important for the function of TGFβ3 than that of TGFβ1 and TGFβ2 in vivo? The answer is not immediately clear, but it is plausible that the loss of GARP in controlling TGFβ1 and TGFβ2 activity is compensated by alternative or GARP-independent mechanisms of TGFβ regulation. In addition to GARP, the large TGFβ-binding protein can also be involved in binding, transporting, and activating proTGFβs. In addition, as the concentrations of TGFβ1 and TGFβ2 in mouse blood are much higher than that of TGFβ3 (below the detection limit by conventional assays), it is possible that active TGFβ1 and TGFβ2 can be delivered into the embryo through the placenta from the heterozygote mother, compensating for the loss of GARP-dependent TGFβ1 and TGFβ2 activation. Our discovery that GARP is expressed at a high level in MEE cells also further underscores the unique function of TGFβ3 in palatogenesis (28, 29). In TGFβ3 KO mice, the expression of GARP in MEEs most likely ensures abundant TGFβ1 and TGFβ2 in this location. However, for reasons that are unclear, neither of these two molecules is able to compensate for the loss of TGFβ3 (28, 29, 37, 38).
Interestingly, a recent study observed no pups for CMV-cre/GARPfl/fl mice and suggested an “embryonic lethal phenotype” (39). As neither embryos nor dead pups have been analyzed for defects of CMV-cre/GARPfl/fl mice/embryos, whether the two kinds of GARP KO mice have the same lethal defects need to be further determined by analyzing CMV-cre/GARPfl/fl mice. Nevertheless, the study further confirms the essential role of GARP in embryonic development.
Cleft palate in humans occurs in about one in 700 live births worldwide. Although mutations in a few genes have been suggested to contribute to cleft lip and cleft palate (40), our knowledge of genetic factors that contribute to the more common isolated cases of cleft palate is still incomplete. In addition to family genetics, a cleft palate is present in many different chromosome disorders, such as Patau syndrome. A study has reported the de novo deletion of chromosome 11q13.4-q14.3 in a boy with microcephaly, including a cleft palate, ptosis, and developmental delay (41). A 2014 review further summarized that seven cases among 32 patients with deletion of 11q13-q23.2 have a cleft palate (42). Based on this study and the knowledge that Lrrc32 locates in chromosome 11q13.5, we suggest that GARP mutation in patients may constitute a novel mechanism of cleft palate clinically.
In summary, we have uncovered, genetically, an important developmental function of GARP in regulating palatogenesis. Mechanistically, it is the first time that GARP is linked to regulate TGFβ3 function in vivo. Contrary to a previous report, we demonstrated that GARP binds directly to TGFβ3 and that it is important for TGFβ3 expression on the cell surface. Our study points to another mechanism of human craniofacial deformities. The GARP-targeted strategy will also be useful for probing other unique aspects of TGFβ3 function, such as in immune regulation, in the future.
Experimental procedures
The GARP-FAST mouse model
All animal procedures in this study were approved by the Medical University of South Carolina Institutional Animal Care and Use Committee. We developed a reversible GARP KO mouse model using the flexible accelerated STOP TetO knockin system (inGenious Targeting Laboratory) (30). The GARP-FAST mouse model was generated by inserting the promoter region of Lrrc32 with a stop cassette and a TetO-responsive element, which collectively deactivate the endogenous Lrrc32 promoter and enable the use of the tetracycline transcription system to induce/silence GARP expression.
Male and female GARP-FAST+/− mice were intercrossed to generate GARP-FAST−/− (KO) mice, whose genotypes were confirmed by genomic PCR (primers: FAST-SQ1, 5′-ACA CCT CCC CCT GAA CCT GAA AC-3′; SC1, 5′-GCG ACA AAT ACC GAG GCA AAG CTC-3′; SQ1, 5′-AGC CTC TTG AGT TCC AGA ATA CCA C-3′). Noon of the day of plug appearance was counted as day 0.5. Embryos at different gestation stages were collected surgically and investigated.
Sample preparation and H&E staining
All samples for histological analysis were fixed in 4% formaldehyde and processed into optimal cutting temperature compound–embedded sections. Sections (6 μm) were mounted on poly-l-lysine–coated slides. For general morphology, sections were stained with hematoxylin and eosin using standard procedures.
Apoptosis assay
Following treatment with proteinase K for 30 min at room temperature, apoptotic cells on histological slides were assayed by the TUNEL procedure using a terminal deoxynucleotidyltransferase in situ apoptosis detection kit (R&D Systems) according to the manufacturer's protocol.
Immunohistochemistry
Immunohistochemical staining was performed with antibodies against GARP (1:100 dilution, Roche), pSMAD2/3 (Abcam, 1:100), and TGFβ3 (R&D Systems, 1:100). Slides were counterstained with hematoxylin and visualized using a standard bright-field microscope. The staining intensity was graded with the sample identity blinded as described previously (12): 0, negative; 1, faint; 2, moderate; 3, strong but less intense than 4; and 4, intense.
Immunoblot analysis and immunoprecipitation
A human TGFβ3 expression vector (pLVE-hTGFB3-IRES-Red) was purchased from Addgene and transiently overexpressed in HEK293 cells or HEK293 cells stably expressing HA-tagged mouse GARP. Two days after transfection, the cell pellets were collected. The cell pellets were lysed in radioimmune precipitation assay buffer and subjected to immunoblot analysis using antibodies against mouse GARP (Roche) and TGFβ3 (R&D Systems). Pulldown of GARP-HA was performed using an anti-HA antibody (Proteintech) and protein A/G beads (Bio-Rad) following the the manufacturer's protocol. Briefly, the cell lysates were incubated with the beads and antibody overnight at 4 °C. The immunoprecipitated protein was released by treating the beads with 2× SDS Laemmli buffer for immunoblot analysis.
Surface detection of TGFβ3 expression
A nine-histidine (His) repeat was inserted between Arg-300 and Ala-301 in human TGFβ3. The tagged TGFβ3 (His-TGFβ3) was cloned into a MigR1 retroviral vector with GFP as a reporter. His-TGFβ3 was expressed in HEK293 cells or 293-GARP cells, followed by surface staining with an anti-His antibody (1:100, Proteintech) and a secondary anti-mouse-allophycocyanin antibody. TGFβ3 surface expression was then detected by a flow cytometer (BD FACSVerse). For microscopy analysis, the above cells were fixed with 4% formaldehyde for 10 min and permeabilized with cold methanol for 5 min, followed by anti-His and GARP antibody staining. The images were taken by an Olympus FV10i confocal microscope.
ELISA
The TGFβ3 ELISA was performed using an ELISA kit (R&D Systems, DY243) according to the manufacturer's instruction.
Statistical analysis
Results are expressed as mean ± S.D. Comparisons among groups were made by Student's t test. A p value of 0.05 or less was considered statistically significant.
Author contributions
B. X. W., A. L., L. L., C. W., S. K., and S. L. performed the experiments and data analyses. B. X. W., X. L., and Z. L. designed the study and wrote the manuscript.
Supplementary Material
This work was supported by NHLBI, National Institutes of Health Grant R01HL136921 and National Institutes of Health Grant R01DK110477 (to X. L.) and NCI, National Institutes of Health Grant P01CA186866 02 and NIAID, National Institutes of Health Grant R01AI070603 (to Z. L.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S4.
- GARP
- glycoprotein A repetitions predominant
- Treg
- regulatory T cell
- MEE
- medial edge epithelial
- FAST
- flexible accelerated STOP TetO
- E
- embryonic day
- IHC
- immunohistochemistry.
References
- 1. Tran D. Q., Andersson J., Wang R., Ramsey H., Unutmaz D., and Shevach E. M. (2009) GARP (LRRC32) is essential for the surface expression of latent TGF-β on platelets and activated FOXP3+ regulatory T cells. Proc. Natl. Acad. Sci. U.S.A. 106, 13445–13450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang R., Zhu J., Dong X., Shi M., Lu C., and Springer T. A. (2012) GARP regulates the bioavailability and activation of TGFβ. Mol. Biol. Cell 23, 1129–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang H., Peters T., Sindrilaru A., Kess D., Oreshkova T., Yu X. Z., Seier A. M., Schreiber H., Wlaschek M., Blakytny R., Röhrbein J., Schulz G., Weiss J. M., and Scharffetter-Kochanek K. (2008) TGF-β-dependent suppressive function of Tregs requires wild-type levels of CD18 in a mouse model of psoriasis. J. Clin. Invest. 118, 2629–2639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wang R., Kozhaya L., Mercer F., Khaitan A., Fujii H., and Unutmaz D. (2009) Expression of GARP selectively identifies activated human FOXP3+ regulatory T cells. Proc. Natl. Acad. Sci. U.S.A. 106, 13439–13444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhang Y., Wu B. X., Metelli A., Thaxton J. E., Hong F., Rachidi S., Ansa-Addo E., Sun S., Vasu C., Yang Y., Liu B., and Li Z. (2015) GP96 is a GARP chaperone and controls regulatory T cell functions. J. Clin. Invest. 125, 859–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang R., Wan Q., Kozhaya L., Fujii H., and Unutmaz D. (2008) Identification of a regulatory T cell specific cell surface molecule that mediates suppressive signals and induces Foxp3 expression. PLoS ONE 3, e2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Macaulay I. C., Tijssen M. R., Thijssen-Timmer D. C., Gusnanto A., Steward M., Burns P., Langford C. F., Ellis P. D., Dudbridge F., Zwaginga J. J., Watkins N. A., van der Schoot C. E., and Ouwehand W. H. (2007) Comparative gene expression profiling of in vitro differentiated megakaryocytes and erythroblasts identifies novel activatory and inhibitory platelet membrane proteins. Blood 109, 3260–3269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cuende J., Lienart S., Dedobbeleer O., van der Woning B., De Boeck G., Stockis J., Huygens C., Colau D., Somja J., Delvenne P., Hannon M., Baron F., Dumoutier L., Renauld J. C., De Haard H., et al. (2015) Monoclonal antibodies against GARP/TGF-β1 complexes inhibit the immunosuppressive activity of human regulatory T cells in vivo. Sci. Transl. Med. 7, 284ra256. [DOI] [PubMed] [Google Scholar]
- 9. Edwards J. P., Fujii H., Zhou A. X., Creemers J., Unutmaz D., and Shevach E. M. (2013) Regulation of the expression of GARP/latent TGF-β1 complexes on mouse T cells and their role in regulatory T cell and Th17 differentiation. J. Immunol. 190, 5506–5515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carrillo-Galvez A. B., Cobo M., Cuevas-Ocaña S., Gutiérrez-Guerrero A., Sánchez-Gilabert A., Bongarzone P., García-Pérez A., Muñoz P., Benabdellah K., Toscano M. G., Martín F., and Anderson P. (2015) Mesenchymal stromal cells express GARP/LRRC32 on their surface: effects on their biology and immunomodulatory capacity. Stem Cells 33, 183–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li Y., Kim B. G., Qian S., Letterio J. J., Fung J. J., Lu L., and Lin F. (2015) Hepatic stellate cells inhibit T cells through active TGF-β1 from a cell surface-bound latent TGF-β1/GARP complex. J. Immunol. 195, 2648–2656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Metelli A., Wu B. X., Fugle C. W., Rachidi S., Sun S., Zhang Y., Wu J., Tomlinson S., Howe P. H., Yang Y., Garrett-Mayer E., Liu B., and Li Z. (2016) Surface expression of TGFβ docking receptor GARP promotes oncogenesis and immune tolerance in breast cancer. Cancer Res. 76, 7106–7117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hahn S. A., Neuhoff A., Landsberg J., Schupp J., Eberts D., Leukel P., Bros M., Weilbaecher M., Schuppan D., Grabbe S., Tueting T., Lennerz V., Sommer C., Jonuleit H., and Tuettenberg A. (2016) A key role of GARP in the immune suppressive tumor microenvironment. Oncotarget 7, 42996–43009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dedobbeleer O., Stockis J., van der Woning B., Coulie P. G., and Lucas S. (2017) Cutting edge: active TGF-β1 released from GARP/TGF-β1 complexes on the surface of stimulated human B lymphocytes increases class-switch recombination and production of IgA. J. Immunol. 199, 391–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rachidi S., Metelli A., Riesenberg B., Wu B. X., Nelson M. H., Wallace C., Paulos C. M., Rubinstein M. P., Garrett-Mayer E., Hennig M., Bearden D. W., Yang Y., Liu B., and Li Z. (2017) Platelets subvert T cell immunity against cancer via GARP-TGFβ axis. Sci Immunol 2, eaai7911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rachidi S., Sun S., Wu B. X., Jones E., Drake R. R., Ogretmen B., Cowart L. A., Clarke C. J., Hannun Y. A., Chiosis G., Liu B., and Li Z. (2015) Endoplasmic reticulum heat shock protein gp96 maintains liver homeostasis and promotes hepatocellular carcinogenesis. J. Hepatol. 62, 879–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Shevach E. M. (2017) Garp as a therapeutic target for modulation of T regulatory cell function. Expert Opin. Ther. Targets 21, 191–200 [DOI] [PubMed] [Google Scholar]
- 18. Stockis J., Dedobbeleer O., and Lucas S. (2017) Role of GARP in the activation of latent TGF-β1. Mol. Biosyst. 13, 1925–1935 [DOI] [PubMed] [Google Scholar]
- 19. Edwards J. P., Hand T. W., Morais da Fonseca D., Glass D. D., Belkaid Y., and Shevach E. M. (2016) The GARP/Latent TGF-β1 complex on Treg cells modulates the induction of peripherally derived Treg cells during oral tolerance. Eur. J. Immunol. 46, 1480–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu M. Y., and Hill C. S. (2009) Tgf-β superfamily signaling in embryonic development and homeostasis. Dev. Cell 16, 329–343 [DOI] [PubMed] [Google Scholar]
- 21. Massagué J. (2008) TGFβ in cancer. Cell 134, 215–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li M. O., and Flavell R. A. (2008) TGF-β: a master of all T cell trades. Cell 134, 392–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bonyadi M., Rusholme S. A., Cousins F. M., Su H. C., Biron C. A., Farrall M., and Akhurst R. J. (1997) Mapping of a major genetic modifier of embryonic lethality in TGF β 1 knockout mice. Nat. Genet. 15, 207–211 [DOI] [PubMed] [Google Scholar]
- 24. Sanford L. P., Ormsby I., Gittenberger-de Groot A. C., Sariola H., Friedman R., Boivin G. P., Cardell E. L., and Doetschman T. (1997) TGFβ2 knockout mice have multiple developmental defects that are non-overlapping with other TGFβ knockout phenotypes. Development 124, 2659–2670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kaartinen V., Voncken J. W., Shuler C., Warburton D., Bu D., Heisterkamp N., and Groffen J. (1995) Abnormal lung development and cleft palate in mice lacking TGF-β 3 indicates defects of epithelial-mesenchymal interaction. Nat. Genet. 11, 415–421 [DOI] [PubMed] [Google Scholar]
- 26. Proetzel G., Pawlowski S. A., Wiles M. V., Yin M., Boivin G. P., Howles P. N., Ding J., Ferguson M. W., and Doetschman T. (1995) Transforming growth factor-β 3 is required for secondary palate fusion. Nat. Genet. 11, 409–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Taya Y., O'Kane S., and Ferguson M. W. (1999) Pathogenesis of cleft palate in TGF-β3 knockout mice. Development 126, 3869–3879 [DOI] [PubMed] [Google Scholar]
- 28. Brunet C. L., Sharpe P. M., and Ferguson M. W. (1995) Inhibition of TGF-β 3 (but not TGF-β 1 or TGF-β 2) activity prevents normal mouse embryonic palate fusion. Int. J. Dev. Biol. 39, 345–355 [PubMed] [Google Scholar]
- 29. Yang L. T., and Kaartinen V. (2007) Tgfb1 expressed in the Tgfb3 locus partially rescues the cleft palate phenotype of Tgfb3 null mutants. Dev. Biol. 312, 384–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tanaka K. F., Ahmari S. E., Leonardo E. D., Richardson-Jones J. W., Budreck E. C., Scheiffele P., Sugio S., Inamura N., Ikenaka K., and Hen R. (2010) Flexible accelerated STOP tetracycline operator-knockin (FAST): a versatile and efficient new gene modulating system. Biol. Psychiatry 67, 770–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kaartinen V., Cui X. M., Heisterkamp N., Groffen J., and Shuler C. F. (1997) Transforming growth factor-β3 regulates transdifferentiation of medial edge epithelium during palatal fusion and associated degradation of the basement membrane. Dev. Dyn. 209, 255–260 [DOI] [PubMed] [Google Scholar]
- 32. Huang X., Yokota T., Iwata J., and Chai Y. (2011) Tgf-β-mediated FasL-Fas-caspase pathway is crucial during palatogenesis. J. Dent. Res. 90, 981–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cui X. M., Chai Y., Chen J., Yamamoto T., Ito Y., Bringas P., and Shuler C. F. (2003) TGF-β3-dependent SMAD2 phosphorylation and inhibition of MEE proliferation during palatal fusion. Dev. Dyn. 227, 387–394 [DOI] [PubMed] [Google Scholar]
- 34. Cui X. M., Shiomi N., Chen J., Saito T., Yamamoto T., Ito Y., Bringas P., Chai Y., and Shuler C. F. (2005) Overexpression of Smad2 in Tgf-β3-null mutant mice rescues cleft palate. Dev. Biol. 278, 193–202 [DOI] [PubMed] [Google Scholar]
- 35. Wolfraim L. A., Alkemade G. M., Alex B., Sharpe S., Parks W. T., and Letterio J. J. (2002) Development and application of fully functional epitope-tagged forms of transforming growth factor-β. J. Immunol. Methods 266, 7–18 [DOI] [PubMed] [Google Scholar]
- 36. Roubin R., Pizette S., Ollendorff V., Planche J., Birnbaum D., and Delapeyriere O. (1996) Structure and developmental expression of mouse Garp, a gene encoding a new leucine-rich repeat-containing protein. Int. J. Dev. Biol. 40, 545–555 [PubMed] [Google Scholar]
- 37. Murillo J., Maldonado E., Barrio M. C., Del Río A., López Y., Martínez-Sanz E., González I., Martín C., Casado I., and Martínez-Alvarez C. (2009) Interactions between TGF-β1 and TGF-β3 and their role in medial edge epithelium cell death and palatal fusion in vitro. Differentiation 77, 209–220 [DOI] [PubMed] [Google Scholar]
- 38. Barrio M. C., Del Río A., Murillo J., Maldonado E., López-Gordillo Y., Paradas-Lara I., Hernandes L., Catón J., and Martínez-Álvarez C. (2014) Epidermal growth factor impairs palatal shelf adhesion and fusion in the Tgf-β 3 null mutant. Cells Tissues Organs 199, 201–211 [DOI] [PubMed] [Google Scholar]
- 39. Vermeersch E., Denorme F., Maes W., De Meyer S. F., Vanhoorelbeke K., Edwards J., Shevach E. M., Unutmaz D., Fujii H., Deckmyn H., and Tersteeg C. (2017) The role of platelet and endothelial GARP in thrombosis and hemostasis. PLoS ONE 12, e0173329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cox T. C. (2004) Taking it to the max: the genetic and developmental mechanisms coordinating midfacial morphogenesis and dysmorphology. Clin. Genet. 65, 163–176 [DOI] [PubMed] [Google Scholar]
- 41. Wincent J., Schoumans J., and Anderlid B. M. (2010) De novo deletion of chromosome 11q13.4-q14.3 in a boy with microcephaly, ptosis and developmental delay. Eur. J. Med. Genet. 53, 50–53 [DOI] [PubMed] [Google Scholar]
- 42. Nacinovich R., Villa N., Redaelli S., Broggi F., Bomba M., Stoppa P., Scatigno A., Selicorni A., Dalprà L., and Neri F. (2014) Interstitial 11q deletion: genomic characterization and neuropsychiatric follow up from early infancy to adolescence and literature review. BMC Res. Notes 7, 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.