

Abstract
Scorpion toxins can kill other animals by inducing paralysis and arrhythmia, which limits the potential applications of these agents in the clinical management of diseases. Antitumor-analgesic peptide (AGAP), purified from Buthus martensii Karsch, has been proved to possess analgesic and antitumor activities. Trp38, a conserved aromatic residue of AGAP, might play an important role in mediating AGAP activities according to the sequence and homology-modeling analyses. Therefore, an AGAP mutant, W38G, was generated, and effects of both AGAP and the mutant W38G were examined by whole-cell patch clamp techniques on the sodium channels hNav1.4 and hNav1.5, which were closely associated with the biotoxicity of skeletal and cardiac muscles, respectively. The data showed that both W38G and AGAP inhibited the peak currents of hNav1.4 and hNav1.5; however, W38G induced a much weaker inhibition of both channels than AGAP. Accordingly, W38G exhibited much less toxic effect on both skeletal and cardiac muscles than AGAP in vivo. The analgesic activity of W38G and AGAP were verified in vivo as well, and W38G retained analgesic activity similar to AGAP. Inhibition to both Nav1.7 and Nav1.8 was involved in the analgesic mechanism of AGAP and W38G. These findings indicated that Trp38 was a key amino acid involved in the biotoxicity of AGAP, and the AGAP mutant W38G might be a safer alternative for clinical application because it retains the analgesic efficacy with less toxicity to skeletal and cardiac muscles.
Keywords: electrophysiology, pain, peptides, sodium channel, toxicity, AGAP, Trp38, hNav1.4, hNav1.5
Introduction
Scorpion venom contains a large variety of biologically active components, including enzymes, peptides, nucleotides, lipids, mucoproteins, biogenic amines, and other unknown substances (1, 2), which might potentially act as new compounds in clinical medications, including epilepsy, convulsion, facial paralysis, hemiplegia, Parkinson's disease, tumor, and pain (3–11). In recent years, a range of scorpion analgesic peptides has been obtained and proved to target voltage-gated sodium channels (VGSCs),3 such as Buthus martensii Karsch AngM1, B. martensii Karsch AGP-SYPU1, B. martensii Karsch AS, B. martensii Karsch BTx, and BmNaL-3SS2 (12–15), although the exact interaction of toxins with VGSCs has remained unclear. Moreover, some severe adverse reactions have been caused, and the toxicological mechanism has been less explored despite the satisfactory analgesic effect of venoms (16).
VGSCs are considered to be involved in the generation and propagation action potential. Nine genes have been identified to encode functional sodium channel isoforms, namely Nav1.1–Nav1.9. Voltage-gated sodium channel 1.4 (Nav1.4), encoded by SCN4A, is mainly expressed in skeletal muscle and essential for the generation and propagation of the action potential crucial to skeletal muscle contraction. Loss-of-function mutations in SCN4A lead to a decrease in peak sodium current (INap), resulting in severe fetal hypokinesia or some classical congenital myopathy (17). The cardiac voltage-gated sodium channel (Nav1.5) is encoded by SCN5A and mainly expressed in cardiac muscle. In the intercellular communication via gap junctions, Nav1.5 is a major determinant of impulse conduction velocity in cardiac tissues (18). Loss-of-function mutations in SCN5A lead to defects in the generation and conduction of the cardiac electrical impulse and are associated with various arrhythmia phenotypes, including brugada syndrome, sick sinus syndrome, cardiac conduction diseases, and possibly dilated cardiomyopathy (19–22). Therefore, compounds blocking Nav1.4 and Nav1.5 might induce the dysfunction of muscular and cardiac system, leading to muscular and cardiac diseases and even death. Nav1.4 and Nav1.5 could also be viewed as one of the targets for the treatment of diseases associated with muscular and cardiac dysfunction. Some sodium channel isoforms have been reported to be potentially involved in the production of pain, of which Nav1.7 and Nav1.8 have drawn more attention because much evidence has been obtained of their close association with the triggering and propagation of pain.
Our group has been engaged in exploring new peptides from the Chinese scorpion B. martensii Karsch for many years. In previous studies (5, 23), antitumor-analgesic peptide (AGAP), one of the long-chain scorpion toxins purified from B. martensii Karsch, was proved to exhibit both analgesic and anti-tumor effects using in vivo models. Several site-directed mutations of AGAP were carried out based on the homology modeling and 3D structure analysis so as to improve its activity or decrease its toxicity (24) (supplemental Fig. S1). Among them, Trp38, a conserved aromatic residue located in the core domain as well as a link between Face A and Face B, is assumed to be an important residue in maintaining AGAP function.
In this paper, the effects of an AGAP mutant, W38G, on both hNav1.4 and hNav1.5 were examined and compared with those of AGAP by using whole-cell patch clamp to evaluate its potential effects on the function of skeletal and cardiac muscles. The toxicity of W38G and AGAP on skeletal and cardiac muscles was examined in vivo as well, and the analgesic activity of W38G and AGAP was further verified by a thermal pain test and a formalin-induced paw-licking test. The results indicated that the mutant W38G displayed lower toxicities to skeletal and cardiac muscles than AGAP, whereas it retained an analgesic activity similar to that of AGAP. The decreased toxicity of W38G might result from its much lower inhibition of activities of both hNav1.4 (skeletal muscle) and hNav1.5 (cardiac muscle). Inhibition of the activities of both hNav1.7 and hNav1.8 might be involved in the analgesic mechanism of AGAP and W38G.
Results
Effects of AGAP and W38G on hNav1.4 and hNav1.5
Effects of AGAP and W38G on the amplitude of sodium currents evoked from hNav1.4 and hNav1.5
The effects of AGAP and W38G on the amplitude of sodium currents evoked from hNav1.4 and hNav1.5 expressed in CHO cells were determined and compared. Currents were elicited by test pulses ranging from −80 to +80 mV for 50 ms with increments of 10 mV. AGAP inhibited the amplitude of sodium currents in a concentration-dependent manner with a calculated IC50 value of 10 nm for hNav1.4 and 12 nm for hNav1.5, respectively. The inhibition to hNav1.4 and hNav1.5 was significantly weakened by W38G with a calculated IC50 value of 600 and 5 μm, respectively. Even at a concentration of 10 μm, the peak of the sodium currents was only inhibited by W38G to 75% for hNav1.4 and 56% for hNav1.5, respectively (Fig. 1, A and D).
Figure 1.

Effects of AGAP and W38G on the Na+ peak current of hNav1.4 and hNav1.5. The Na+ currents were obtained by plotting current peak amplitudes with a function of test potentials ranging from −80 to +80 mV for 50 ms from the holding potential of −80 mV in increments of 10 mV. Normalized Na+ peak currents were depolarized at −20 mV after a 5-min treatment in the absence and presence of 10 pm, 100 pm, 1 nm, 10 nm, 100 nm, 1 μm, and 10 μm AGAP and W38G (A and D). The current traces were evoked by −20 mV for 50 ms from a holding potential of −80 mV in the absence and presence of 100 nm AGAP and W38G. Averaged current traces were obtained from hNav1.4-CHO (B) and hNav1.5-CHO (E) cells in each group. Current-voltage relationships of hNav1.4 (C) and hNav1.5 (F) were evoked by 100 nm AGAP and W38G. *, p < 0.05; **, p < 0.01; significant difference between AGAP and W38G values; paired one-way ANOVA. Each data point represents mean ± S.E. (error bars) (n = 6).
At a concentration of 100 nm, AGAP potently inhibited the activity of both hNav1.4 and hNav1.5, with the amplitude of INaP significantly reduced to 32 and 29%, respectively. As for W38G, the INaP was only reduced to 90% on hNav1.4 and 75% on hNav1.5, which indicated a much weaker inhibition of both channels (Fig. 1) induced by W38G.
Effects of AGAP and W38G on dynamic properties of hNav1.4 and hNav1.5
The effects of AGAP and W38G were determined and compared on channel properties of hNav1.4 and hNav1.5. After exposure to 100 nm AGAP, the I-V curve was significantly shifted to the hyperpolarized direction with V½ values of activation decreased by 4.1 mV on hNav1.4 and 5.1 mV on hNav1.5. The slope factor was also changed by 1.9 and 1.5 against the control, respectively. 100 nm W38G shifted the V½ of activation by −6.6 mV on hNav1.4 and −8.4 mV on hNav1.5 with few changes of the slope factors compared with the control (Fig. 2 (A and D) and Table 1). With regard to the inactivation phase, a 100 nm concentration of either AGAP or W38G had no significant effect on V½ values of inactivation on both hNav1.4 and hNav1.5. AGAP evoked a change of slope factor by 3.4 for hNav1.5 compared with control, whereas W38G evoked few changes of the slope factor for both hNav1.4 and hNav1.5. Both hNav1.4 and hNav1.5 were somewhat difficult to recover from inactivation to resting state after exposure to 100 nm AGAP and W38G, although the duration of the recovery process changed little (Fig. 2, C and F).
Figure 2.

Effects of AGAP and W38G on voltage dependence of activation and inactivation of hNav1.4 and hNav1.5 and effects of AGAP and W38G on recovery from inactivation of Na+ current. Na+ current was generated by applying pulses from −80 to +40 mV at 10-mV steps for 20 ms from the holding potential of −80 mV in increments of 10 mV. Peak currents were converted into conductance, and normalized conductance of hNav1.4 (A) and hNav1.5 (D) was plotted against the voltages of conditioning pulses. Each point represents mean ± S.E. (n = 6). The currents were elicited with a test pulse at −20 mV for 20 ms following 30-ms prepulses ranging from −100 to 0 mV at 5-mV steps. Peak currents were normalized, and the inactivation curves of hNav1.4 (B) and hNav1.5 (E) were plotted against the command potentials. Each point represents mean ± S.E. (error bars) (n = 6). The currents were obtained at two pulses, consisting of a 200-ms prepulse to −20 mV followed by resting at −80 mV for a time varying from 2 to 56 ms in increments of 3 ms and a test pulse to −20 mV for 10 ms. The I/Imax ratio represented the recovery of Na+ current from inactivation for hNav1.4 (C) and hNav1.5 (F). Each point represents mean ± S.E. (n = 6).
Table 1.
Effects of AGAP and W38G on the kinetic and recovery properties of hNav1.4 and hNav1.5
*, p < 0.05; significant difference between control and toxins on hNav1.4 and hNav1.5; one-way ANOVA. #, p < 0.05; significant difference between AGAP and W38G on hNav1.4 and hNav1.5; one-way ANOVA. **, p < 0.01; significant difference between control and toxins on hNav1.4 and hNav1.5; one-way ANOVA. ##, p < 0.01; significant difference between AGAP and W38G on hNav1.4 and hNav1.5; one-way ANOVA.
| Group | hNav1.4 |
hNav1.5 |
||||
|---|---|---|---|---|---|---|
| Control | AGAP (100 nm) | W38G (100 nm) | Control | AGAP (100 nm) | W38G (100 nm) | |
| Activation | ||||||
| V½ (mV) | −34.18 ± 0.65 | −38.31 ± 0.36* | −40.81 ± 0.76* | −34.29 ± 0.26 | −39.35 ± 0.26* | −42.66 ± 0.31*# |
| k | 3.75 ± 0.24 | 5.66 ± 0.73* | 4.81 ± 0.24* | 3.90 ± 0.28 | 5.42 ± 0.25* | 4.47 ± 0.26 |
| Inactivation | ||||||
| V½ (mV) | −36.48 ± 0.67 | −37.31 ± 0.32 | −39.40 ± 0.72 | −42.09 ± 0.34 | −43.67 ± 0.71 | −45.73 ± 0.69 |
| k | 8.78 ± 0.62 | 8.90 ± 0.33 | 7.64 ± 0.36 | 6.25 ± 0.28 | 9.68 ± 0.69* | 7.06 ± 0.55 |
| Recovery | ||||||
| τ(ms) | 7.51 ± 0.53 | 8.21 ± 0.46 | 7.56 ± 0.51 | 4.32 ± 0.66 | 6.26 ± 0.45 | 7.09 ± 0.49* |
Compared with AGAP, W38G exhibited a similar effect on V½ values of hNav1.4 and hNav1.5 in both activation and inactivation phases with no significant differences. W38G exhibited the same effect in the recovery phase from the inactivated state to the resting state. The activation, inactivation, and recovery parameters are summarized in Table 1.
Toxicity of AGAP and W38G to skeletal and cardiac muscle in vivo
Acute toxicity of AGAP and W38G to skeletal and cardiac muscle in vivo
Due to the much less inhibitory effects on hNav1.4 and hNav1.5 in vitro, W38G is assumed to be a potential toxin analog with lower side effects on the cardiac and muscular system. In a previous study, AGAP showed distinct analgesic activity at a concentration of 0.5 mg/kg. Therefore, doses up to 5- and 10-fold higher were used to examine the potential muscular and cardiac toxicity of AGAP and W38G in mice. As shown in Fig. 3, 2.5 mg/kg AGAP and 2.5 and 5.0 mg/kg W38G exhibited no significant effects on heart rate compared with saline control. By contrast, 5.0 mg/kg AGAP decreased the heart rate at 15 min after intravenous injection. 2.5 and 5.0 mg/kg W38G had no effects on activity levels of creatine kinase (CK) and lactate dehydrogenase (LDH) as well, whereas both 2.5 and 5.0 mg/kg of AGAP significantly increased the activity levels of CK and LDH, indicating that potential damage to skeletal or cardiac muscle was induced by AGAP.
Figure 3.

The effects of AGAP and W38G on heart rate (A and D) and enzyme activities (B, C, E, and F) of CK and LDH at doses up to 2.5 and 5.0 mg/kg. In each experiment, data points represent mean ± S.E. (error bars) (n = 10 for saline and 2.5 mg/kg W38G, n = 8 for 5.0 mg/kg W38G, n = 6 for 2.5 mg/kg AGAP, n = 4 for 5.0 mg/kg AGAP). Statistically significant differences compared with the saline control group (calculated using one-way ANOVA) are indicated by p < 0.05 (*) or p < 0.01 (**).
Subacute toxicity of AGAP and W38G to skeletal and cardiac muscle in vivo
Survival analysis was carried out, and survival curves of 20 mice in each group are shown in Fig. 4. At doses of 2.5 mg/kg, all mice died by the day 6 in the AGAP-treated group, whereas 16 mice survived in the W38G-treated group (Fig. 4A). At doses of 5.0 mg/kg, all mice died by day 4 in the AGAP-treated group, whereas four mice survived in the W38G-treated group (Fig. 4B). A log-rank test of the survival analysis showed a rather lower toxicity of W38G than of AGAP, although 5.0 mg/kg W38G still retained considerable toxicity when continuously administered to mice.
Figure 4.

One-week survival of mice treated with AGAP and W38G. W38G improves survival time of AGAP at doses of 2.5 and 5.0 mg/kg with 20 mice in each group. Mice treated with normal saline are shown as a comparison. Comparison of the survival curves of the AGAP and W38G groups was calculated using log-rank test analysis. *, p < 0.05; **, p < 0.01; statistically significant differences.
Mice surviving a 7-day treatment of 2.5 mg/kg W38G or the same amount of saline were randomly and equally divided into two groups, respectively. To investigate the effect of W38G on motor functions in mice, the forced swimming test and rotarod test were administered immediately 20 min after the last injection on day 7. A corresponding amount of saline was injected in the exact same way as the control. The data showed that the accumulation of W38G did not affect the motor functions of mice in both the forced swimming test and rotarod test compared with control (Fig. 5, A and B). No significant differences were found between the W38G-treated group and the control group with reference to the activity levels of serum CK and LDH (Fig. 5, C and D). These results indicated that 2.5 mg/kg W38G caused much lower toxicities to skeletal and cardiac muscles even after 7-day treatment.
Figure 5.
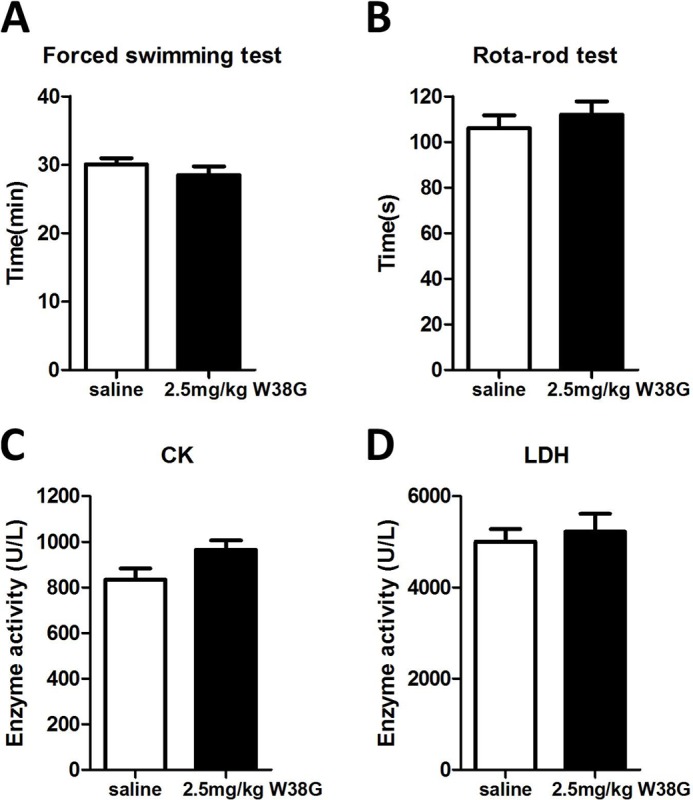
The effects of W38G on motor function (A and B) and enzyme activities (C and D) of CK and LDH after intravenous injection for 7 days. In each experiment, data points represent mean ± S.E. (error bars) (n = 10 for saline, n = 8 for 2.5 mg/kg W38G). Statistically significant differences compared with the saline control group (calculated using one-way ANOVA) are indicated by p < 0.05 (*) and p < 0.01 (**).
Analgesic activities of W38G compared with AGAP in vivo
Analgesic effects of AGAP and W38G were further verified in rodent pain models induced by heat or noxious chemical.
W38G similarly reduced the pain induced by a hot plate when compared with AGAP. The licking time increased significantly in a concentration-dependent method in both AGAP-treated groups and W38G-treated groups. By 15 min, the licking time of mice had increased from 9.5 to 17.6 s (0.25 mg/kg), 24.5 (0.5 mg/kg), and 28.3 s (1.0 mg/kg) in the AGAP-treated group, whereas those times had increased from 10.6 to 19.7 s (0.25 mg/kg), 28.7 s (0.5 mg/kg), and 31.5 s (1.0 mg/kg) in the W38G-treated group. By 45 min, the licking time of mice had increased from 9.5 to 17.3 s (0.25 mg/kg), 26.7 s (0.5 mg/kg), and 27.4 s (1.0 mg/kg) in the AGAP-treated group, whereas those times had increased from 10.6 to 19.3 s (0.25 mg/kg), 29.2 s (0.5 mg/kg), and 31.2 s (1.0 mg/kg) in the W38G-treated group (Fig. 6A). The analgesic effect of AGAP and W38G initiated at 15 min and was sustained until 60 min or longer in the hot-plate model (Fig. 6B).
Figure 6.

Analgesic effects of AGAP and W38G in mice. Both AGAP and W38G have analgesic effects in rodent pain models. W38G was similarly effective as AGAP at reducing thermal-induced pain (A and B). W38G was similarly effective as AGAP in attenuating nociceptive behavior (duration of paw licking and flinching time) during phase I (0–5 min postinjection) (C) and phase II (20–40 min postinjection) (D) following intraplantar injection of formalin. Data points are shown as mean ± S.E. (error bars) (n = 10). Statistically significant differences compared with the saline control group (calculated using one-way ANOVA) are indicated by p < 0.05 (*) and p < 0.01 (**).
Intraplantar injection of formalin can produce two phases of nociceptive behaviors in mice. The nociceptive behaviors in the first phase (0–5 min postinjection), was caused by direct stimulation of TRPA1 in a subpopulation of C-fiber nociceptors and followed by a quiescent period. The nociceptive behaviors in the second phase (20–40 min postinjection) were caused by peripheral inflammation together with central nervous sensitization. Nociceptive responses were relieved significantly and dose-dependently in both phase I and phase II by intravenous injection of AGAP and W38G (Fig. 6, C and D). In phase I, the average licking time was significantly attenuated to 76.2, 63.9, and 54.8% of that in control group by 0.25, 0.5, and 1.0 mg/kg AGAP and 69.7, 58.7, and 53.7% of that in control group by 0.25, 0.5, and 1.0 mg/kg W38G (Fig. 6C). This time was also attenuated for AGAP and W38G in phase II, with a rate of 95.1, 73.6, and 60.5% and 92.9, 69.6, and 54.7% at concentrations of 0.25, 0.5, and 1.0 mg/kg, respectively (Fig. 6D).
Effects of AGAP and W38G on hNav1.7 and hNav1.8
To explore the possible mechanisms for analgesic activities, effects of AGAP and W38G were examined on the activity of hNav1.7 and hNav1.8. At a concentration of 100 nm, AGAP potently inhibited the activity of both hNav1.7 and hNav1.8, with the amplitude of INaP significantly reduced to 32 and 75%, respectively. As for W38G, the amplitude of INaP was reduced to 57% on hNav1.7 and 80% on hNav1.8. It seemed that W38G induced a weaker inhibition to hNav1.7 than AGAP (Fig. 7), whereas it induced a similar inhibition to hNav1.8 with AGAP (Fig. 8).
Figure 7.

Effects of AGAP and W38G on hNav1.7. The Na+ currents were obtained by plotting current peak amplitudes with a function of test potentials ranging from −80 to +80 mV for 50 ms from the holding potential of −80 mV in increments of 10 mV. The current traces were evoked by −20 mV for 50 ms from a holding potential of −80 mV in the absence and presence of 100 nm AGAP and W38G. Averaged current traces were obtained from hNav1.7 (A) cells in each group. The current-voltage relationship of hNav1.7 (B) was evoked by 100 nm AGAP and W38G. Peak currents were converted into conductance, and normalized conductance of hNav1.7 (C) was plotted against the voltages of conditioning pulses. The currents were elicited with a test pulse at −20 mV for 20 ms following 30-ms prepulses ranging from −100 to 0 mV at 5-mV steps. Peak currents were normalized, and the inactivation curves of hNav1.7 (D) were plotted against the command potentials. Each data point represents mean ± S.E. (error bars) (n = 6).
Figure 8.

Effects of AGAP and W38G on hNav1.8. The Na+ currents were obtained by plotting current peak amplitudes with a function of test potentials ranging from −80 to +80 mV for 50 ms from the holding potential of −80 mV in increments of 10 mV. The current traces were evoked by +10 mV for 50 ms from a holding potential of −80 mV in the absence and presence of 100 nm AGAP and W38G. Averaged current traces were obtained from hNav1.8 (A) cells in each group. The current-voltage relationship of hNav1.8 (B) was evoked by 100 nm AGAP and W38G. Peak currents were converted into conductance, and normalized conductance of hNav1.8 (C) was plotted against the voltages of conditioning pulses. The currents were elicited with a test pulse at +10 mV for 20 ms following 30-ms prepulses ranging from −100 to 0 mV at 5-mV steps. Peak currents were normalized, and the inactivation curves of hNav1.8 (D) were plotted against the command potentials. Each data point represents mean ± S.E. (error bars) (n = 5).
After exposure to 100 nm AGAP, the I-V curve was significantly shifted to the hyperpolarized direction with V½ values of activation decreased by 6.0 mV on hNav1.7 and 3.9 mV on hNav1.8. Few changes of the slope factors were evoked by AGAP on both hNav1.7 and hNav1.8. 100 nm W38G shifted the V½ values of activation by −8.0 mV on hNav1.7 and −5.7 mV on hNav1.8 with few changes of the slope factors compared with the control (Figs. 7C and 8C) (Table 2). With regard to the inactivation phase, a 100 nm concentration of either AGAP or W38G had no significant effect on V½ values and slope factors on both hNav1.7 and hNav1.8 (Figs. 7D and 8D) (Table 2).
Table 2.
Effects of AGAP and W38G on the kinetic of hNav1.7 and hNav1.8
*, p < 0.05; significant difference between control and toxins on hNav1.7 and hNav1.8; one-way ANOVA. #, p < 0.05; significant difference between AGAP and W38G on hNav1.7 and hNav1.8; one-way ANOVA. **, p < 0.01; significant difference between control and toxins on hNav1.7 and hNav1.8; one-way ANOVA. ##, p < 0.01; significant difference between AGAP and W38G on hNav1.7 and hNav1.8; one-way ANOVA.
| Group | hNav1.7 |
hNav1.8 |
||||
|---|---|---|---|---|---|---|
| Control | AGAP (100 nm) | W38G (100 nm) | Control | AGAP (100 nm) | W38G (100 nm) | |
| Activation | ||||||
| V½ (mV) | −35.38 ± 0.33 | −41.35 ± 0.78* | −43.35 ± 0.95* | 0.79 ± 0.55 | −3.13 ± 0.75* | −4.88 ± 0.9* |
| k | 5.11 ± 0.27 | 6.15 ± 0.69 | 5.05 ± 0.78 | 10.77 ± 0.5 | 11.39 ± 0.68 | 11.35 ± 0.8 |
| Inactivation | ||||||
| V½ (mV) | −54.81 ± 0.98 | −52.60 ± 0.82 | −55.24 ± 0.70 | −67.84 ± 0.63 | −70.16 ± 0.56 | −67.8 ± 0.48 |
| k | 9.34 ± 0.92 | 9.65 ± 0.77 | 9.33 ± 0.65 | 10.93 ± 0.57 | 10.84 ± 0.52 | 9.15 ± 0.44 |
Discussion
Scorpion toxins can prolong the action potentials of excitable cells in vivo and kill other animals by inducing paralysis and arrhythmia, which contributes greatly to their major toxicity. According to the size of the peptide chain, scorpion toxins can be characterized as short-chain toxins, usually targeting potassium channels, and long-chain toxins, usually targeting sodium channels (25, 26). B. martensii Karsch AGAP is composed of 66 amino acids and viewed as a long-chain toxin, although the association of its function with sodium channels has not yet been elucidated clearly.
In this study, Nav1.4 and Nav1.5 were considered as the targets to screen the possible toxicity to skeletal and cardiac muscles due to their specific distributions and roles in maintaining the skeletal and cardiac electrophysiology. Effects of AGAP and its several mutants were first examined on the amplitude of sodium channel currents evoked from hNav1.4 and hNav1.5 expressed in CHO cells. W38G was selected to be further studied due to its distinctly reduced inhibition to both hNav1.4 and hNav1.5 at a concentration of 100 nm (supplemental Fig. S2). The data in this study showed that AGAP displayed a significantly inhibitory effect on both hNav1.4 and hNav1.5, which might contribute to its toxicity to skeletal and cardiac muscles. The W38G mutation significantly decreased the inhibition of INap with the calculated IC50 value increased 60,000-fold for hNav1.4 and 420-fold for hNav1.5 (Fig. 1). However, the kinetic characteristics of both channels evoked by W38G exhibited a variation similar to that evoked by AGAP, including activation, inactivation, and recovery process. Therefore, the reduced inhibition of W38G to hNav1.4 and hNav1.5 seemed more likely to be due to its decreased blocking action on both channels than its changes on kinetic processes. These results indicated that Trp38 might be the key amino acid to maintain the crucial structure of AGAP interacting with hNav1.4 and hNav1.5. Also, the structural nuances of AGAP and W38G might explain their different performance on channels, because W38G had been proved to change the CD spectra of AGAP in a previous study (24). A detailed comparison of the surface formed by the interaction of toxins with VGSCs might be necessary to validate this hypothesis; however, this seemed difficult to carry out in the current work.
Due to its weakened inhibition to Nav1.4 and Nav1.5, W38G is assumed to possess lower adverse effects on the muscular and cardiac system. Acute and subacute experiments were carried out to verify this assumption. Heart rate is the most direct measure for arrhythmia. CK and LDH exist in higher amounts in the cytoplasm and mitochondria of cardiac and skeletal muscle cells. Increased activity levels of CK and LDH in serum are commonly considered as a highly specific and sensitive measure of muscle cell death (27, 28), such as in the presence of acute myocardial infarction and skeletal muscle damage (29–32). In this study, W38G exhibited no significant effects on heart rate, activity levels of CK and LDH, and motor function in both acute and subacute toxicity tests, whereas 5.0 mg/kg AGAP decreased the heart rate at 15 min after intravenous injection and significantly increased the activity levels of CK and LDH in the serum of mice. In survival analysis, mice in W38G-treated groups survived much longer than those in AGAP-treated groups. Although the half-life of most peptides is generally short, the toxicity induced by AGAP indicated an accumulation of AGAP by continuous administration. The results confirmed that W38G exhibited much lower toxicity to skeletal and cardiac muscles compared with AGAP. Although multiple complicated mechanisms might be involved in the reduced toxicity of W38G in vivo, the decreased inhibition of hNav1.4 and hNav1.5 was assumed to be one major reason.
In this study, W38G and AGAP exhibited similar analgesic effects in rodent pain models induced by heat and noxious chemicals; however, the analgesic mechanism remains uncertain. Nav1.7 and Nav1.8 are the most prominent potential targets associated closely with both acute and chronic pain. Nav1.7, one of the tetrodotoxin-sensitive voltage-gated sodium channel isoforms, is abundantly expressed in dorsal root ganglion and sympathetic ganglion pain-sensing neurons (33–35). In recent years, Nav1.7 has been thought to be a major contributor to pain signaling in humans, according to genetic and functional studies (36–39), and to be associated with erythromelalgia, paroxysmal extreme pain disorder, and congenital insensitivity to pain (39, 40). Nav1.8, one of the tetrodotoxin-resistant voltage-gated sodium channel isoforms, is prominently expressed in primary sensory neurons, including dorsal root ganglion neurons (41, 42). A previous study (43) showed that knockdown of Nav1.8 can attenuate neuropathic and inflammatory pain in rodents. AGAP potently inhibited the activity of both hNav1.7 (Fig. 7) and hNav1.8 (Fig. 8), indicating that both channel isoforms were involved in the analgesic mechanism of AGAP. W38G evoked a similar inhibitory effect on hNav1.8 but a much weaker inhibition of hNav1.7 compared with AGAP. Considering the similar analgesic effects, it seemed that the inhibition of hNav1.8 might be more responsible for the analgesic effects of AGAP exhibited in rodent pain models induced by heat and noxious chemicals.
Another notable point is that AGAP had been defined as an α-type scorpion toxin by the bioinformational analysis of the structure in a previous study (5). However, one of the characteristics of α-type scorpion toxins is that they are well-known to affect the inactivation process of VGSCs in excitable membranes by binding to receptor site 3. In this study, AGAP induced little changes in the inactivation processes of hNav1.4, hNav1.5, hNav1.7, and hNav1.8. On the contrary, the activation processes of these four channels were changed with I-V curves left-shifted to the hyperpolarized direction after exposure to 100 nm AGAP (Figs. 2, 7, and 8). A prepulse protocol was used to determine whether AGAP is a β-type scorpion toxin (44). The current-voltage relationships for AGAP-modified sodium currents were analyzed and revealed a negative shift in the voltage dependence of activation after priming depolarizations in the presence of AGAP (supplemental Fig. S3). Therefore, it is more reasonable to define AGAP as a β-type scorpion toxin rather than an α-type toxin according to its electrophysiological characteristics upon the interaction with hNav1.4, hNav1.5, hNav1.7, and hNav1.8. More evidence should be obtained in further studies to verify this assumption.
In conclusion, the mutant W38G exhibited much lower inhibitory effects on hNav1.4 and hNav1.5 compared with AGAP. Correspondingly, W38G displayed much lower toxicities to skeletal and cardiac muscles than AGAP, whereas it retained analgesic activity similar to that of AGAP in vivo. Nav1.7 and Nav1.8 might be involved in the analgesic mechanism of AGAP and W38G, indicating that the conserved Trp38 might be a key amino acid involved in the biotoxicity and bioactivity of AGAP, and mutant W38G might be a safer alternative for clinical use because it retains pharmaceutical efficacy with few adverse effects. These findings might facilitate the modification of other scorpion toxins with high preference for their bioactivity target with lower side effects as well.
Experimental procedures
Animals
Healthy female and male Kunming mice were obtained from the Experimental Animal Center of Shenyang Pharmaceutical University (Shenyang, China). They were maintained in standard laboratory conditions of temperature 25 ± 1 °C and a 12-h light/12-h dark cycle with free access to food and water for the duration of the study. The mice were acclimatized for 2 weeks before all tests. Animal care was in accordance with the Guidelines for Animal Experimentation of Shenyang Pharmaceutical University, and the protocol was approved by the Animal Ethics Committee of the institution. All of the cells and tissues of the mice were authorized to be used for scientific purposes.
Cell cultures
IMDM, DMEM, 0.25% trypsin-EDTA, FBS, penicillin/streptomycin, HT supplement, GlutaMAX, and hygromycin were obtained from Life Technologies, Inc., and G418 was purchased from Calbiochem. CHO cells were cultured in IMDM with 2 mm GlutaMAX and supplemented with 100 μm hypoxanthine, 16 μm thymidine, and 10% FBS. ND7/23 cells were cultured in DMDM with 2 mm GlutaMAX and 10% FBS. hNav1.4-CHO, hNav1.5-CHO, and hNav1.7-CHO cells were cultured in IMDM with 2 mm GlutaMAX and supplemented with 100 μm hypoxanthine, 16 μm thymidine, 10% FBS, and 200 μg/ml G418. hNav1.8-ND7/23 cells were cultured in DMDM with 2 mm GlutaMAX, 10% FBS, and 200 μg/ml hygromycin. All cells were cultured routinely as monolayers on a poly-d-lysine–coated dish in an atmosphere of 5% CO2 and 37 °C.
Purification of recombinant scorpion toxins
Escherichia coli BL21 (DE3) cells harboring expression vectors of pSYPU/AGAP(W38G) were grown and harvested as described previously (5), following purification by nickel metal chelating ion affinity chromatography. All purified toxins were analyzed by SDS-PAGE and diluted in external solution/normal saline for the whole-cell patch clamp/animal tests.
Automated patch clamp electrophysiology recording
Whole-cell patch clamp recording was used to examine functional properties of hNav1.4-CHO, hNav1.5-CHO, hNav1.7-CHO, and hNav1.8-ND7/23 cell lines using an automated electrophysiology platform (NPC-1 and Port-a-patch system, Nanion Technologies GmbH, Munich, Germany) and an EPC10 Amplifier (HEKA Elektronik, Lambrecht, Germany). Cells were detached with 0.25% trypsin-EDTA, terminated in fresh culture medium, centrifuged (100 × g, 3 min), and resuspended in external solution to reach a density of 1 × 106 cells/ml. Patchcontrol HT software (Nanion Technologies) was used to control the application of solutions and pressures necessary to establish the whole-cell configuration. Sodium currents were recorded at room temperature (20–25 °C) using external (140 mm NaCl, 4 mm KCl, 1 mm MgCl2, 2 mm CaCl2, 5 mm d-glucose monohydrate, 10 mm HEPES/NaOH, pH 7.4) and internal solutions (50 mm CsCl, 10 mm NaCl, 60 mm CsF, 20 mm EGTA, 10 mm HEPES/CsOH, pH 7.2) identical to those used for conventional patch clamp experiments. Following cell contact with the 2–3.5–megaohm planar electrode, seal enhancer solution (80 mm NaCl, 3 mm KCl, 35 mm CaCl2, 10 mm MgCl2, 10 mm HEPES/NaOH, pH 7.4) was added to the external solution to promote gigaohm seal formation. After establishing the whole-cell configuration, the seal enhancer solution was replaced with two washes of fresh external solution, and the series resistance was compensated automatically. Patchmaster software (HEKA Elektronik, Lambrecht, Germany) was used to automatically compensate for whole-cell capacitance and series resistance and perform voltage clamp protocols. Whole-cell currents were low-pass–filtered at 5 kHz and digitized at 50 kHz (45).
The holding potentials were −80 mV. The Na+ currents (I) were elicited by test pulses ranging from −80 to +80 mV for 50 ms with increments of 10 mV. The amplitudes of transient sodium currents (Ic and I) were recorded before and after the applications of different concentrations of AGAP and W38G. For determining the inhibition of peak current induced by AGAP and W38G, normalized current = I/Ic was used. For determining the voltage dependence of activation, the sodium conductance was calculated using G = I/(Vm − Vr), where Vr is reversal potential. The G value for each test potential was normalized to Gmax and plotted against the test potential to produce a voltage dependence of activation curves, which were fitted using Boltzmann functions, G/Gmax = 1/(1 + exp((Vm − V½)/k)), where V½ is the voltage where half-maximal activation occurs, and k is a slope factor. Inactivation curves were fitted with the Boltzmann equation, I/Imax = 1/(1 + exp((Vm − V½)/k)), where I/Imax is normalized current, V½ is the potential for half-maximal inactivation, and k is the slope factor. The time course of recovery of the sodium currents from inactivation was fitted with a tertiary exponential function, I/Imax = A(1 − exp(−Δt/τ)), where Imax is the maximal current amplitude, I is the current after a recovery period of Δt, τ is the time constant for recovery from inactivation, and A is the amplitude coefficient.
Skeletal and cardiac toxicity of AGAP and W38G in vivo
Heart rate
The mice were randomly divided into five groups of 10 individuals each: 1) normal saline control group; 2) AGAP, 2.5 mg/kg; 3) AGAP, 5.0 mg/kg; 4) W38G, 2.5 mg/kg; 5) W38G, 5.0 mg/kg. The heart rate was detected using an RM6240 multichannel physiological recorder. The mice were intravenously injected through the caudal vein within 10 s under anesthetic using pentobarbital sodium. The heart rate was counted from the start to 60 min of each group.
Blood assays
The mice were randomly divided into five groups of 10 individuals each: 1) normal saline control group; 2) AGAP, 2.5 mg/kg; 3) AGAP, 5.0 mg/kg; 4) W38G, 2.5 mg/kg; 5) W38G, 5.0 mg/kg. One hour after pretreatment, blood samples were collected from mice inner canthus in tubes and centrifuged at 6000 × g for 10 min, after which the serum fraction was transferred to another clean tube. Cardiac and skeletal muscle injury was assessed by determining the activity levels of serum CK and LDH using a CK detection kit (Beijing Solarbio Science and Technology) and LDH detection kit (Beijing Solarbio Science and Technology). Results are expressed in international units/liter.
Forced swimming test
The forced swimming exercise test was used as described previously with some modifications (46). Mice were pretreated with normal saline, AGAP (2.5 or 5.0 mg/kg), and W38G (2.5 or 5.0 mg/kg) once a day at the same time by intravenous injection within 10 s through the caudal vein for 7 consecutive days. 20 min after the last administration, an exhaustive swimming test was given. The surviving mice were placed individually in a columnar swimming pool (length 65 cm and radius 20 cm) with 40-cm water depth maintained at 28 ± 1 °C. A weight equivalent to 5% of body weight was attached to the root of the mouse tail, and the endurance for each mouse was measured as swimming times recorded from the beginning to exhaustion, which was determined by observing loss of coordinated movements and failure to return to the surface.
Rotarod test
The rotarod test was used as described previously with some modifications (47). One day before the test, the animals were trained twice. On the day of the test, only the mice that were able to stay balanced on the rotating rod between 80 and 120 s were selected for testing. The performance time was measured before and at 20 min after treatment. Mice were pretreated with saline, AGAP (2.5 or 5.0 mg/kg), and W38G (2.5 or 5.0 mg/kg) once a day at the same time by intravenous injection within 10 s through the caudal vein for 7 consecutive days. 20 min after the last administration, a rotarod test followed with a rod-rotating speed of 16 rpm. The integrity of motor coordination was assessed on the basis of endurance time of the animals on the rotating rod.
Analgesic effects of AGAP and W38G in vivo
Thermal pain test
Female mice of 18–22 g were put on the hot plate, which was at a temperature of 55 ± 0.5 °C to induce pain. In response, the mice would lick or kick their feet, so that the time from putting the mice onto the hot plate to the licking or kicking action could be counted to reflect the intensity of pain. To perform the bioassay, samples were injected intravenously through the caudal vein in advance. Then the time was measured from putting it on the hot plate to its licking or kicking action at the 15, 30, 45, and 60 min. If this time was >60 s, the mice were taken away from the hot plate at once to prevent damage, and the time was counted as 60 s. The pain killer morphine was used as a positive control.
Formalin-induced paw licking
Pain was induced into mice by intraplantar injection of formalin, and pain attenuation was compared in mice injected intravenously through the caudal vein with either morphine, AGAP, W38G dissolved in 100 μl of saline. Control and normal saline mice received the same volume of saline. After a 15-min pretreatment, animals were injected with 10 μl of 2% (v/v) formalin at the plantar surface of the right hind paw, whereas normal saline mice were injected with 10 μl of saline. Mice were then placed individually into open polyvinyl cages. The time spent licking the injected paw was counted during phase I (0–5 min postinjection) and phase II (20–40 min postinjection). The painkiller morphine was used as a positive control.
Data analysis
Statistical analysis of the data is provided as mean ± S.E. Group statistical significance was assessed using one-way ANOVA. Group statistical significance was assessed using log-rank test analysis in the survival experiment. p < 0.05 (*) and p < 0.01 (**) were considered statistically significant.
Author contributions
Y. X., X. M., X. H., and J. S. carried out electrophysiology studies. Y. X., J. S., Y. S., and Y. N. carried out toxicity experiments. Y. X., X. K., Y. S., and Z. L. carried out analgesic experiments. Y. M., Y. S., and Y. C. carried out peptide purification. Y. X., X. M., M. Z., and J. Z. conceived and designed the experiments. Y. X. and M. Z. wrote the manuscript, to which all authors contributed.
Supplementary Material
This work was supported by National Key Scientific Project for New Drug Discovery and Development of China Grant 2014ZX09101044-001, National Natural Science Foundation of China Grant 81202855, Natural Science Foundation of Liaoning Province Grants 2015020726 and 2015020743, and Career Development Support Program of Shenyang Pharmaceutical University for Young and Middle-aged Teachers Grant 13. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S3.
- VGSC
- voltage-gated sodium channel
- CK
- creatine kinase
- INap
- peak sodium current
- LDH
- lactate dehydrogenase
- AGAP
- antitumor-analgesic peptide
- IMDM
- Iscove's modified Dulbecco's medium
- ANOVA
- analysis of variance.
References
- 1. Zlotkin E., Miranda F., and Rochat H. (1978) Venoms of Buthinae: chemistry and pharmacology of Buthinae scorpion venoms. In Arthropod Venoms (Bettini S., ed) pp. 317–369, Springer, New York [Google Scholar]
- 2. Possani L. D. (1984) Structure of scorpion toxins. In Handbook of Natural Toxins (Tu A. T., ed) pp. 513–550, CRC Press, New York [Google Scholar]
- 3. Zhou X. H. (1984) The biochemical research on scorpion venoms and their application in therapy. Progr. Biochem. Biophys. 56, 20–29 [Google Scholar]
- 4. He X., Peng F., Zhang J., Li W., Zeng X., and Liu H. (2003) Inhibitory effects of recombinant neurotoxin BmK IM on seizures induced by pentylenetetrazol in rats. Chin. Med. J. 116, 1898–1903 [PubMed] [Google Scholar]
- 5. Liu Y. F., Ma R. L., Wang S. L., Duan Z. Y., Zhang J. H., Wu L. J., and Wu C. F. (2003) Expression of an antitumor-analgesic peptide from the venom of Chinese scorpion Buthus martensii karsch in Escherichia coli. Protein Expr. Purif. 27, 253–258 [DOI] [PubMed] [Google Scholar]
- 6. Chai Z. F., Zhu M. M., Bai Z. T., Liu T., Tan M., Pang X. Y., and Ji Y. H. (2006) Chinese-scorpion (Buthus martensi Karsch) toxin BmK αIV, a novel modulator of sodium channels: from genomic organization to functional analysis. Biochem. J. 399, 445–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhao R., Zhang X. Y., Yang J., Weng C. C., Jiang L. L., Zhang J. W., Shu X. Q., and Ji Y. H. (2008) Anticonvulsant effect of BmK IT2, a sodium channel-specific neurotoxin, in rat models of epilepsy. Br. J. Pharmacol. 154, 1116–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cao P., Yu J., Lu W., Cai X., Wang Z., Gu Z., Zhang J., Ye T., and Wang M. (2010) Expression and purification of an antitumor-analgesic peptide from the venom of Mesobuthus martensii Karsch by small ubiquitin-related modifier fusion in Escherichia coli. Biotechnol. Prog. 26, 1240–1244 [DOI] [PubMed] [Google Scholar]
- 9. Du J., Fu Y., Wang J., and Liang A. (2013) Adenovirus-mediated expression of BmK CT suppresses growth and invasion of rat C6 glioma cells. Biotechnol. Lett. 35, 861–870 [DOI] [PubMed] [Google Scholar]
- 10. Zhang X. G., Wang X., Zhou T. T., Wu X. F., Peng Y., Zhang W. Q., Li S., and Zhao J. (2016) Scorpion venom heat-resistant peptide protects transgenic Caenorhabditis elegans from β-amyloid toxicity. Front. Pharmacol. 7, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhu H., Wang Z., Jin J., Pei X., Zhao Y., Wu H., Lin W., Tao J., and Ji Y. (2016) Parkinson's disease-like forelimb akinesia induced by BmK I, a sodium channel modulator. Behav. Brain Res. 308, 166–176 [DOI] [PubMed] [Google Scholar]
- 12. Cao Z. Y., Mi Z. M., Cheng G. F., Shen W. Q., Xiao X., Liu X. M., Liang X. T., and Yu D. Q. (2004) Purification and characterization of a new peptide with analgesic effect from the scorpion Buthus martensi Karch. J. Pept. Res. 64, 33–41 [DOI] [PubMed] [Google Scholar]
- 13. Meng X., Xu Y., Wang F., Zhao M., Hou X., Ma Y., Jin Y., Liu Y., Song Y., and Zhang J. (2017) The roles of conserved aromatic residues (Tyr5 and Tyr42) in interaction of scorpion toxin BmK AGP-SYPU1 with human Nav1.7. Int. J. Biol. Macromol. 99, 105–111 [DOI] [PubMed] [Google Scholar]
- 14. Chen J., Feng X. H., Shi J., Tan Z. Y., Bai Z. T., Liu T., and Ji Y. H. (2006) The anti-nociceptive effect of BmK AS, a scorpion active polypeptide, and the possible mechanism on specifically modulating voltage-gated Na+ currents in primary afferent neurons. Peptides 27, 2182–2192 [DOI] [PubMed] [Google Scholar]
- 15. Lin S., Wang X., Hu X., Zhao Y., Zhao M., Zhang J., and Cui Y. (2017) Recombinant expression, functional characterization of two scorpion venom toxins with three disulfide bridges from the Chinese scorpion Buthus martensii Karsch. Protein Pept. Lett. 24, 235–240 [DOI] [PubMed] [Google Scholar]
- 16. Deng Y. P., Xu G. Z., Wang W., Shen X. H., Chen Q. T., Gao H. Z., Pan Y. Z., Zhu T. Y., Zhu D. M., Zhou X. M., Liu Y. L., and Cai Z. J. (2002) Clinical evaluation of analgesic effect and safety of scorpion venom injection. Chin. Pharm. J. 37, 459–462 [Google Scholar]
- 17. Zaharieva I. T., Thor M. G., Oates E. C., van Karnebeek C., Hendson G., Blom E., Witting N., Rasmussen M., Gabbett M. T., Ravenscroft G., Sframeli M., Suetterlin K., Sarkozy A., D'Argenzio L., Hartley L., et al. (2016) Loss-of-function mutations in SCN4A cause severe foetal hypokinesia or “classical” congenital myopathy. Brain 139, 674–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sheets P. L., Heers C., Stoehr T., and Cummins T. R. (2008) Differential block of sensory neuronal voltage-gated sodium channels by lacosamide [(2R)-2-(acetylamino)-N-benzyl-3–3-methoxypropana mide], lidocaine, and carbamazepine. J. Pharmacol. Exp. Ther. 326, 89–99 [DOI] [PubMed] [Google Scholar]
- 19. Anger T., Madge D. J., Mulla M., and Riddall D. (2001) Medicinal chemistry of neuronal voltage-gated sodium channel blockers. J. Med. Chem. 44, 115–137 [DOI] [PubMed] [Google Scholar]
- 20. Royer A., van Veen T. A., Le Bouter S., Marionneau C., Griol-Charhbili V., Léoni A. L., Steenman M., van Rijen H. V., Demolombe S., Goddard C. A., Richer C., Escoubet B., Jarry-Guichard T., Colledge W. H., Gros D., et al. (2005) Mouse model of SCN5A-linked hereditary Lenegre's disease: age-related conduction slowing and myocardial fibrosis. Circulation 111, 1738–1746 [DOI] [PubMed] [Google Scholar]
- 21. Márquez M. F., Bonny A., Hernández-Castillo E., De Sisti A., Gómez-Flores J., Nava S., Hidden-Lucet F., Iturralde P., Cárdenas M., and Tonet J. (2012) Long-term efficacy of low doses of quinidine on malignant arrhythmias in Brugada syndrome with an implantable cardioverter-defibrillator: a case series and literature review. Heart Rhythm 9, 1995–2000 [DOI] [PubMed] [Google Scholar]
- 22. King J. H., Huang C. L., and Fraser J. A. (2013) Determinants of myocardial conduction velocity: implications for arrhythmogenesis. Front. Physiol. 4, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mao Q., Ruan J., Cai X., Lu W., Ye J., Yang J., Yang Y., Sun X., Cao J., and Cao P. (2013) Antinociceptive effects of analgesic-antitumor peptide (AGAP), a neurotoxin from the scorpion Buthus martensii Karsch, on formalin-induced inflammatory pain through a mitogen-activated protein kinases-dependent mechanism in mice. PLoS One 8, e78239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cui Y., Guo G. L., Ma L., Hu N., Song Y. B., Liu Y. F., Wu C. F., and Zhang J. H. (2010) Structure and function relationship of toxin from Chinese scorpion Buthus martensii Karsch (BmKAGAP): gaining insight into related sites of analgesic activity. Peptides 31, 995–1000 [DOI] [PubMed] [Google Scholar]
- 25. Quintero-Hernández V., Ramírez-Carreto S., Romero-Gutiérrez M. T., Valdez-Velázquez L. L., Becerril B., Possani L. D., and Ortiz E. (2015) Transcriptome analysis of scorpion species belonging to the Vaejovis genus. PLoS One 10, e0117188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Santibáñez-López C. E., and Possani L. D. (2015) Overview of the Knottin scorpion toxin-like peptides in scorpion venoms: insights on their classification and evolution. Toxicon 107, 317–326 [DOI] [PubMed] [Google Scholar]
- 27. Galen R. S. (1975) The enzyme diagnosis of myocardial infarction. Hum. Pathol. 6, 141–155 [DOI] [PubMed] [Google Scholar]
- 28. Roark S. F., Wagner G. S., Izlar H. L. Jr., and Roe C. R. (1976) Diagnosis of acute myocardial infarction in a community hospital. Circulation 53, 965–969 [DOI] [PubMed] [Google Scholar]
- 29. Henderson A. R., and Moss D. W. (1986) Enzymes. In Tietz Fundamentals of Clinical Chemistry, 5th Ed (Burtis C. A., and Ashwood E. R., eds) pp. 356–366, Elsevier, Philadelphia [Google Scholar]
- 30. Alpert J. S., Thygesen K., Antman E., and Bassand P. J. (2000) Myocardial infarction redefined a consensus document of the Joint European Society of Cardiology Committee for the redefinition of myocardial infarction. J. Am. Coll. Cardiol. 36, 959–969 [DOI] [PubMed] [Google Scholar]
- 31. Lewandrowski K., Chen A., and Januzzi J. (2002) Cardiac markers for myocardial infarction: a brief review. Am. J. Clin. Pathol. 118, S93–S99 [DOI] [PubMed] [Google Scholar]
- 32. Nathwani R. A., Pais S., Reynolds T. B., and Kaplowitz N. (2005) Serum alanine aminotransferase in skeletal muscle diseases. Hepatology 41, 380–382 [DOI] [PubMed] [Google Scholar]
- 33. Black J. A., Dib-Hajj S., McNabola K., Jeste S., Rizzo M. A., Kocsis J. D., and Waxman S. G. (1996) Spinal sensory neurons express multiple sodium channel α-subunit mRNAs. Brain Res. Mol. Brain Res. 43, 117–131 [DOI] [PubMed] [Google Scholar]
- 34. Sangameswaran L., Fish L. M., Koch B. D., Rabert D. K., Delgado S. G., Ilnicka M., Jakeman L. B., Novakovic S., Wong K., Sze P., Tzoumaka E., Stewart G. R., Herman R. C., Chan H., Eglen R. M., and Hunter J. C. (1997) A novel tetrodotoxin-sensitive, voltage-gated sodium channel expressed in rat and human dorsal root ganglia. J. Biol. Chem. 272, 14805–14809 [DOI] [PubMed] [Google Scholar]
- 35. Toledo-Aral J. J., Moss B. L., He Z. J., Koszowski A. G., Whisenand T., Levinson S. R., Wolf J. J., Silos-Santiago I., Halegoua S., and Mandel G. (1997) Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc. Natl. Acad. Sci. U.S.A. 94, 1527–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang S., Xiao Y., Kang D., Liu J., Li Y., Undheim E. A., Klint J. K., Rong M., Lai R., and King G. F. (2013) Discovery of a selective NaV1.7 inhibitor from centipede venom with analgesic efficacy exceeding morphine in rodent pain models. Proc. Natl. Acad. Sci. U.S.A. 110, 17534–17539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thomas-Tran R., and Du Bois J. (2016) Mutant cycle analysis with modified saxitoxins reveals specific interactions critical to attaining high-affinity inhibition of hNaV1.7. Proc. Natl. Acad. Sci. U.S.A. 113, 5856–5861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vetter I., Deuis J. R., Mueller A., Israel M. R., Starobova H., Zhang A., Rash L. D., and Mobli M. (2017) NaV1.7 as a pain target: from gene to pharmacology. Pharmacol. Ther. 172, 73–100 [DOI] [PubMed] [Google Scholar]
- 39. Dib-Hajj S. D., Yang Y., Black J. A., and Waxman S. G. (2013) The Na(v)1.7 sodium channel: from molecule to man. Nat. Rev. Neurosci. 14, 49–62 [DOI] [PubMed] [Google Scholar]
- 40. Cox J. J., Reimann F., Nicholas A. K., Thornton G., Roberts E., Springell K., Karbani G., Jafri H., Mannan J., Raashid Y., Al-Gazali L., Hamamy H., Valente E. M., Gorman S., Williams R., McHale D. P., Wood J. N., Gribble F. M., and Woods C. G. (2006) An SCN9A channelopathy causes congenital inability to experience pain. Nature 444, 894–898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Akopian A. N., Sivilotti L., and Wood J. N. (1996) A tetrodotoxin resistant voltage-gated sodium channel expressed by sensory neurons. Nature 379, 257–262 [DOI] [PubMed] [Google Scholar]
- 42. Sangameswaran L., Delgado S. G., Fish L. M., Koch B. D., Jakeman L. B., Stewart G. R., Sze P., Hunter J. C., Eglen R. M., and Herman R. C. (1996) Structure and function of a novel voltage-gated, tetrodotoxin-resistant sodium channel specific to sensory neurons. J. Biol. Chem. 271, 5953–5956 [DOI] [PubMed] [Google Scholar]
- 43. Joshi S. K., Mikusa J. P., Hernandez G., Baker S., Shieh C. C., Neelands T., Zhang X. F., Niforatos W., Kage K., Han P., Krafte D., Faltynek C., Sullivan J. P., Jarvis M. F., and Honore P. (2006) Involvement of the TTX-resistant sodium channel Nav1.8 in inflammatory and neuropathic, but not post-operative, pain states. Pain 123, 75–82 [DOI] [PubMed] [Google Scholar]
- 44. Cestèle S., Qu Y., Rogers J. C., Rochat H., Scheuer T., and Catterall W. A. (1998) Voltage sensor-trapping: enhanced activation of sodium channels by β-scorpion toxin bound to the S3-S4 loop in domain II. Neuron 21, 919–931 [DOI] [PubMed] [Google Scholar]
- 45. Kahlig K. M., Lepist I., Leung K., Rajamani S., and George A. L. (2010) Ranolazine selectively blocks persistent current evoked by epilepsy-associated Nav1.1 mutations. Br. J. Pharmacol. 161, 1414–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Porsolt R. D., Le Pichon M., and Jalfre M. (1977) Depression: a new animal model sensitive to antidepressant treatments. Nature 266, 730–732 [DOI] [PubMed] [Google Scholar]
- 47. Galeotti N., Ghelardini C., Caldari B., and Bartolini A. (1999) Effect of potassium channel modulators in mouse forced swimming test. Br. J. Pharmacol. 126, 1653–1659 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.