

Abstract
The Cas family scaffolding protein p130Cas is a Src substrate localized in focal adhesions (FAs) and functions in integrin signaling to promote cell motility, invasion, proliferation, and survival. p130Cas targeting to FAs is essential for its tyrosine phosphorylation and downstream signaling. Although the N-terminal SH3 domain is important for p130Cas localization, it has also been reported that the C-terminal region is involved in p130Cas FA targeting. The C-terminal region of p130Cas or Cas family homology domain (CCHD) has been reported to adopt a structure similar to that of the focal adhesion kinase C-terminal focal adhesion-targeting domain. The mechanism by which the CCHD promotes FA targeting of p130Cas, however, remains unclear. In this study, using a calorimetry approach, we identified the first LD motif (LD1) of the FA-associated protein paxillin as the binding partner of the p130Cas CCHD (in a 1:1 stoichiometry with a Kd ∼4.2 μm) and elucidated the structure of the p130Cas CCHD in complex with the paxillin LD1 motif by X-ray crystallography. Of note, a comparison of the CCHD/LD1 complex with a previously solved structure of CCHD in complex with the SH2-containing protein NSP3 revealed that LD1 had almost identical positioning of key hydrophobic and acidic residues relative to NSP3. Because paxillin is one of the key scaffold molecules in FAs, we propose that the interaction between the p130Cas CCHD and the LD1 motif of paxillin plays an important role in p130Cas FA targeting.
Keywords: focal adhesion, nuclear magnetic resonance (NMR), PTK2 protein tyrosine kinase 2 (PTK2) (focal adhesion kinase) (FAK), scaffold protein, X-ray crystallography, p130Cas/BCAR1, paxillin
Introduction
p130 Crk-associated substrate (Cas)2 (also known as breast cancer antiestrogen resistance 1 (BCAR1)) is a Src substrate that colocalizes with paxillin in FAs and is involved in cell migration, survival, transformation, invasion, and cancer (1, 2). p130Cas was first identified as a prominent tyrosine-phosphorylated protein in cells that were transformed by oncogenes v-crk and v-src (2, 3). p130Cas is not an enzymatic protein but a docking and scaffolding protein (3), which can either mediate protein-protein interactions or directly interact with other proteins through its various domains and motifs. For example, FAK is a key component of FAs that interacts with p130Cas (4). It is well established that the SH3 domain of p130Cas interacts with FAK polyproline motifs. Similarly to FAK, p130Cas localizes to FAs and is phosphorylated by tyrosine kinases in response to adhesion signals (5–7). Within FAs, p130Cas signaling is known to regulate actin engagement and dynamics, leading to enhanced cell motility (8, 9). A role for p130Cas during tamoxifen resistance in estrogen receptor-positive breast cancer has been demonstrated (10). Therefore, human p130Cas is also known as BCAR1. In addition to its signaling role in FAs, p130Cas also plays an important role in promoting cell proliferation and survival in cancer cells that are tamoxifen-resistant (1, 11–13).
Although the importance of p130Cas localization and phosphorylation is well established, how p130Cas is targeted to FAs has not been fully elucidated. Previous studies have demonstrated that the N-terminal SH3 domain is critical for p130Cas localization via its interactions with FAK. FA localization of p130Cas compared with wild type was reduced 6-fold when the SH3 domain of p130Cas was deleted (9). The SH3 domain also serves to bind p130Cas to vinculin in a tyrosine phosphorylation-dependent manner. When p130Cas is tyrosine-phosphorylated on Tyr-12 (part of the SH3 domain), the p130Cas SH3 domain interacts with the polyproline motif PPKP on vinculin and thereby acts as a second route by which p130Cas can be targeted to FAs via the SH3 domain (14). In addition, deletion of the CCHD leads to a 3-fold decrease in FA localization of p130Cas (9). The CCHD consists of four α-helices similar to the FAT domain of FAK (15). How the p130Cas CCHD promotes FA localization, however, is unclear. In this study, we identified that paxillin, which plays a major role in targeting FAK to FAs, also binds the CCHD of p130Cas directly. Furthermore, we determined the structure of p130Cas CCHD binding with the LD1 motif of paxillin, demonstrating the mechanism by which the CCHD promotes p130Cas FA targeting.
Experimental procedures
Protein expression and purification
Mouse p130Cas cDNA was cloned in a previous study (4). Solubility and expression of CCHD alone (residues 738–874) were severely limited. Therefore, we generated a series of expanded constructs based on secondary structure predictions. By testing the solubility and stability of these constructs, we ultimately selected an expanded sequence known as p130Cas(665–874) (residues 665–874) (Fig. 1). This construct contained the full-length CCHD sequence (residues 738–874; p130Cas(738–874)) and an N-terminal 69-amino acid extension (residues 665–737), which dramatically enhanced the solubility of the protein in solution. We also made cysteine mutations to prevent potential disulfide bond formation (C755A and C824S) and introduced a thrombin cleavage site between the CCHD and the N-terminal extension by mutation (G735R, R736G, and T737S), which allowed for removal of the N-terminal extension during protein purification. We subcloned the p130Cas(665–874) construct into a pET28 vector (Novagen) fused with an N-terminal His6 tag. Incorporation was verified by sequencing using T7 terminator primer. The His-tagged p130Cas(665–874) construct was transformed into Escherichia coli strain BL21 (DE3). E. coli containing the construct were cultured in 100 ml of LB medium with 30 μg/ml kanamycin at 37 °C in a rotary shaker at 250 rpm overnight. Then a 10-ml starting culture was inoculated into each of six 1-liter aliquots of LB medium containing 30 μg/ml kanamycin, and the 1-liter cultures were allowed to grow at 37 °C at 250 rpm for 4–5 h until the A600 reached 0.6–0.8. Protein expression was initiated by induction of T7 RNA polymerase with the addition of isopropyl β-d-thiogalactopyranoside to a final concentration of 1 mm. Induced cultures were then incubated overnight at a reduced temperature and shaker speed of 18 °C and 200 rpm, respectively, to allow protein expression. Isotope-labeled protein was prepared using MOPS-buffered medium containing 15NH4Cl (1 g/liter) and [13C6]glucose (3.6 g/liter). The purification of p130Cas(665–874) and p130Cas(738–874) was performed using standard protein purification procedures, including Ni2+ histidine affinity chromatography, optional thrombin cleavage (for p130Cas(738–874)), and size exclusion chromatography. Cell lysates from 6 liters of overnight cultures were loaded manually on to a prepacked 5-ml Ni2+ histidine affinity column with a flow rate of ∼1 ml/min. The resins were then washed with 10 column volumes of lysis buffer, and the His-p130Cas(665–874) was eluted with a solution containing 50 mm phosphate buffer, 500 mm sodium chloride, 0.02% CHAPS, and 200 mm imidazole. The purified p130Cas(665–874) protein was dialyzed in 50 mm HEPES, pH 7.8, 300 mm potassium chloride, and 0.02% CHAPS and further purified by gel filtration.
Figure 1.

Sequence analysis and secondary structure prediction of CCHD and the LD motifs of paxillin. A, secondary structure prediction of the C-terminal region of p130Cas suggests that the CCHD consists of four α helices, indicated here by green cylinders, which is similar to CCHD helices observed in the CCHD/NSP3 crystal complex structure (Protein Data Bank code 3T6G, chain B). In addition, a putative α helix located N-terminally to the CCHD was predicted. This potential α helix, indicated here in gray schematic, stabilizes the CCHD protein during expression and purification. The construct p130Cas(665–874) containing p130Cas(738–874) (CCHD) and the putative helix-containing region p130Cas(665–737) was generated. The sequence colored in gold has been mutated to a secondary thrombin cleavage site for removing the p130Cas(665–737) during purification. In addition, naturally occurring cysteine residues 755 and 824, colored in green and underlined, were mutated to alanine and serine, respectively. Residues colored in red indicate non-identical residues comparing the mouse and human species. Within the CCHD, the sequence identity is more than 98% with only two semiconserved differences between them. B, paxillin is highly conserved between species and composed of multiple protein-binding motifs, including the N-terminal LD motifs, the C-terminal LIM domains, and several phosphotyrosine-SH2 domain-docking sites. Shown in figure are the five LD peptides derived from the LD motifs of paxillin. The conserved LD residues are highlighted in red.
p130Cas(738–874) was produced by removing the N-terminal His tag and non-CCHD portion of Ni2+-purified p130Cas(665–874) using thrombin cleavage. Thrombin was added to His-p130Cas(665–874) at a mass ratio of 1:100 and CaCl2 was added to a final concentration of 2.5 mm for overnight cleavage at 4 °C. The His tag along with the N-terminal extension, p130Cas(665–737), were thereby cleaved from the target region, p130Cas(738–874). The mixture was then concentrated in the presence of a 5× excess of paxillin LD1 peptide to prevent precipitation of the CCHD and to aid removal of unwanted p130Cas(665–737). The concentrated p130Cas(738–874)/LD1 peptide complex was further purified by size exclusion chromatography (26/60 Superdex 75 HR column, GE Healthcare) using an ÄKTA FPLC system. The gel filtration and final protein storage buffer was 20 mm HEPES, pH 7.5, and 100 mm potassium chloride.
Peptide synthesis
Paxillin-derived peptides LD1 (Gallus; residues 1–15), LD2 (Gallus; residues 139–162), LD3 (Gallus; residues 217–237), LD4 (Gallus; residues 261–276), and LD5 (Gallus; residues 29–324) were chemically synthesized and purified by high-pressure liquid chromatography (HPLC) at the Hartwell Center of Bioinformatics and Biotechnology (St. Jude Children's Research Hospital). All peptide stocks were prepared at a concentration of 2 mm in 50 mm HEPES, pH 7.8, 300 mm potassium chloride, and 0.02% CHAPS.
Isothermal titration calorimetry (ITC) studies
Isothermal titration calorimetry experiments were performed using a MicroCal Auto-ITC200 instrument (GE Healthcare). The sample cell of the calorimeter was loaded with p130CAS(665–874) at a concentration of 25 μm in 50 mm HEPES, pH 7.8, 300 mm potassium chloride, and 0.02% CHAPS. The syringe of the calorimeter was loaded with LD peptides at a concentration of 250 μm in the same buffer with p130Cas(665–874). Prepared solutions were centrifuged for 10 min at 14,000 × g for degassing. All the titration experiments were performed at 25 °C with injection volumes of 2 μl and a spacing of 120 s. For the Kd determinations, the data were fit using a one-site binding model in Origin ITC data analysis software.
Circular dichroism (CD) studies
Circular dichroism spectra were obtained with an Aviv 202-01DS CD spectrometer (Aviv, Lakewood, NJ) and processed using Origin 6.1. Experiments were performed using a quartz cuvette with a 0.1-cm path length and 300-μl volume. CD spectra were taken at 25 °C in 50 mm HEPES, pH 7.8, 100 mm potassium chloride, and 0.02% CHAPS. The stability tests of p130Cas(665–874) and p130Cas(738–874)/LD1 complex were carried out by monitoring the CD signal at 222 nm while increasing the sample temperature from 25 to 95 °C.
NMR spectroscopy
p130Cas(738–874) was 15N/13C-double labeled and expressed for NMR studies using the p130Cas(665–874) construct. The proteins were purified as described previously for non-labeled proteins with final buffer 20 mm HEPES, pH 7.5, 100 mm potassium chloride, and 5% (v/v) D2O. The final p130Cas(738–874)/LD1 sample was concentrated down to 0.25 mm with the addition of LD1 peptide at a molar ratio of 1:5. All NMR samples were ∼500 μl, and the NMR spectra for structure determination were acquired at 25 °C with a Bruker Avance 600-MHz spectrometer equipped with a cryoprobe.
Crystallization, structure determination, and model quality
The purified p130Cas(738–874)/paxillin LD1 peptide complex was crystallized at 18 °C by sitting-drop batch crystallization. Prior to setup, additional LD1 peptide was added to the protein/peptide sample. The final 5-μl crystallization drop contained 0.5 mm protein and 2 mm peptide. The drop and 500-μl reservoir solution both contained 20 mm HEPES, pH 7.5, and 50 mm KCl. Crystals were cryopreserved by slowly increasing the glycerol concentration from 15 (v/v) to 30% (v/v) with 85 (v/v) and 70% (v/v) well solution, respectively. Native data were collected at the Southeast Regional Collaborative Access Team BM beamline. Data were integrated and scaled to 2.0 Å with HKL2000 (16). Molecular replacement was performed using the previously published human p130Cas C-terminal domain crystal structure (Protein Data Bank code 3T6G, chain B) as the search model (17). The crystals belong to space group C2 with three 1:1 protein/peptide complexes in the asymmetric unit (chains A/D, B/E, and C/F). Model building was performed using Coot (18). Structure refinement, which included use of non-crystallographic symmetry restraints until the final stages, was performed using Phenix (19). Moreover, 5% of the data were sequestered for the calculation of Rfree. Structure validation was performed with MolProbity (20). Ramachandran statistics showed that 99.75 and 0.25% of the residues were in the preferred and allowed regions, respectively. Structural figures were generated with PyMOL (Schrödinger, LLC).
Results
Paxillin binds to the mouse p130Cas CCHD through the LD1 motif
Structural information on the CCHD and relevant binding partners is necessary to fully understand the detailed mechanism of p130Cas localization to FAs. The structure of CCHD alone has not been determined to date. However, a crystal structure exists for CCHD in complex with the novel SH2-containing protein (NSP) known as NSP3 (15). Studies of the NSP3/CCHD complex reveal that CCHD adopts a similar structural fold as the FAT domain of FAK and displays FAT-like domain behavior biochemically and in vivo (15), suggesting its potential ability to interact with FA members such as paxillin. However, the CCHD alone does not form a stable protein in solution. This precluded us from obtaining suitable protein for apo-CCHD structure determination. Furthermore, expressing and purifying the CCHD to perform a pulldown assay for FA binding partners were not feasible. Hence, we identified an N-terminally extended but minimally expanded CCHD construct, which would improve the biophysical behavior of the resultant protein. This construct included a portion of the Src-binding motif that may potentially form a helix as predicted with the program ExPASy (www.expasy.org) (Fig. 1A). Introducing this region into the CCHD construct dramatically increased the stability and solubility of the CCHD. Using this approach, we successfully expressed and purified the elongated construct p130Cas(665–874) and tested its ability to bind paxillin.
Paxillin consists of N-terminal LD motifs and a C-terminal Lim domain (21, 22). It has been recognized as an adaptor protein important for signal transduction in FAs. Previous studies have shown that the LD motifs of paxillin can interact with multiple FA proteins such as FAK, GIT1, and PYK2 (23). All these proteins contain a FAT or FAT-like domain, allowing them to interact with paxillin LD motifs (24–26). To investigate whether the FA targeting properties of the CCHD of p130Cas previously observed (9) may be explained by a direct interaction with paxillin, we performed ITC studies using p130Cas(665–874) and paxillin motif peptides. Paxillin LD1, LD2, LD3, LD4, and LD5 (Fig. 1B) were synthesized and tested for binding with purified p130Cas(665–874) by ITC titrations. The results show that paxillin can interact with the CCHD but only through the binding of the LD1 motif. A representative titration is shown in Fig. 2. The experimental data of LD1 with p130Cas(665–874) demonstrate a 1:1 binding stoichiometry with a Kd of ∼4.2 μm. Other LD peptides (LD2–LD5) have very weak or no binding affinity to p130Cas(665–874). These data suggest that paxillin interacts with p130Cas through the association of the LD1 motif with the CCHD.
Figure 2.
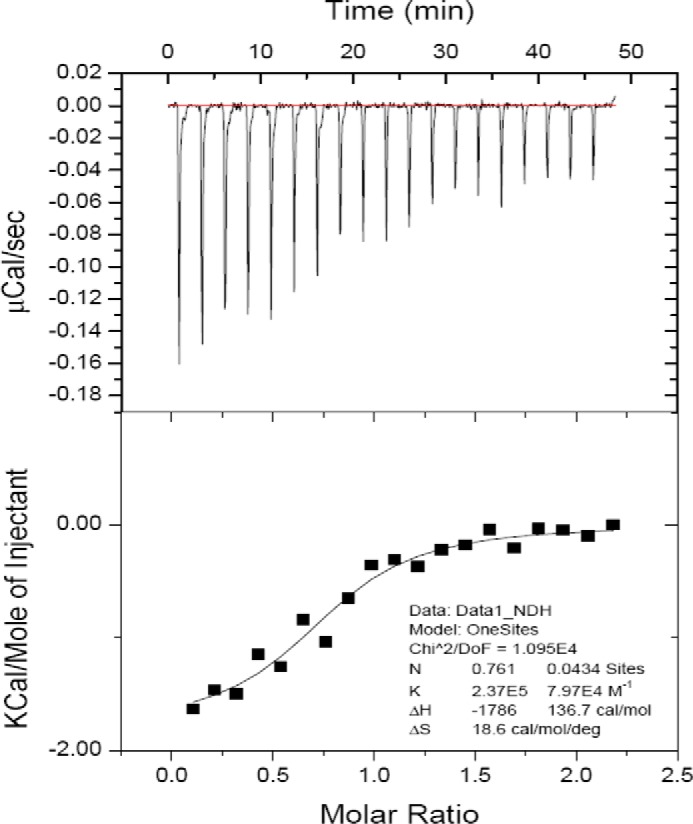
ITC of paxillin LD1 binding to CCHD of p130Cas. Synthetic LD1 peptide was titrated into purified p130Cas(665–874) at 25 °C. The upper panel shows raw data with integration baseline (red). The lower panel shows data after peak integration and subtraction of blank titrations. The black boxes in the lower panel represent the fit to a one-site model with an estimated Kd of 4.2 μm. deg, degrees; DoF, degrees of freedom.
NMR spectrometry shows p130Cas CCHD stabilized in complex form with paxillin LD1
In our previous protein purification, we were able to express and purify p130Cas(665–874) and cleave the unwanted p130Cas(665–737). Initial efforts to further isolate the desired CCHD portion (p130Cas(738–874)) from the thrombin cleavage solution were unsuccessful. However, inclusion of paxillin LD1 in the entire purification procedure greatly increased the yield of soluble CCHD (p130Cas(738–874)) in a complex form with LD1. These results indicate that LD1 acts to stabilize the CCHD in solution, and this was confirmed by a CD melting study comparing the stability of p130Cas(665–874) versus CCHD with LD1 peptide (Fig. 3).
Figure 3.
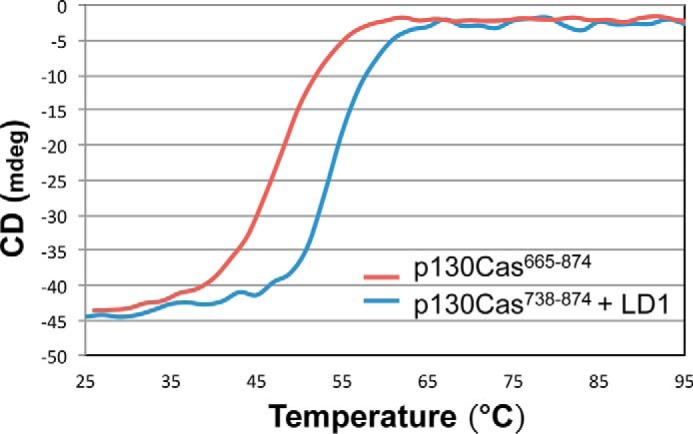
CD spectra of p130Cas(665–874) versus CCHD with LD1 paxillin peptide. CD melting curves for p130Cas(665–874) (red) and p130Cas(738–874) with LD1 (blue) are shown. The intensity of the CD signal at 222 nm as a function of increasing temperature (25–95 °C) was measured as an indicator of loss of helical content upon unfolding. The significant shift in protein thermal melting temperature in the presence of LD1 suggests that complex formation enhances stability. mdeg, millidegrees.
Next we tested the quality of the LD1-stabilized CCHD on a molecular level. As NMR spectroscopy has been widely used to evaluate the quality of the protein samples, the acquisition of 15N HSQC spectra was performed. A good quality protein sample that can be used for structural study must display a 15N HSQC spectrum with well dispersed peaks of uniform shape and consistent peak numbers characterizing a well folded protein. The p130Cas(738–874)/LD1 complex showed significant improvement in 15N HSQC spectra as compared with p130Cas(665–874) or the cut p130Cas(665–737)/p130Cas(738–874) mixture (Fig. 4). Taken together, these spectra suggest that the CCHD portion is likely well folded, whereas the p130Cas(665–737) region may be partially unfolded or dynamic.
Figure 4.
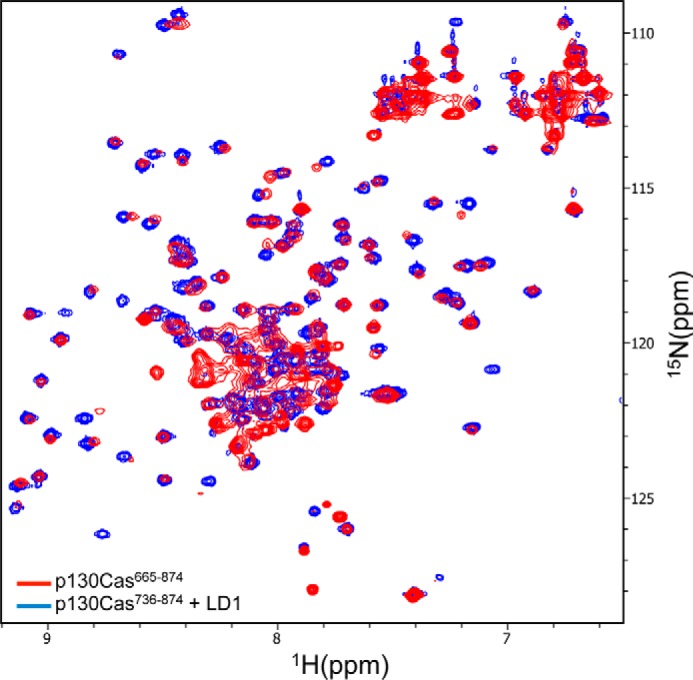
1H-15N HSQC spectra of p130Cas CCHD complex with paxillin LD1. Superposition of 1H-15N HSQC spectra of 15N-labeled p130Cas(738–874) complex with unlabeled LD1 peptide (blue) and the apoprotein (red). Unlike the apoprotein, the peaks in the spectrum of the complex are uniform in shape and evenly dispersed, which indicates the CCHD domain is only well folded upon complex formation with LD1.
Crystal structure of the CCHD in complex with LD1
To further examine the interaction between the p130Cas CCHD and LD1 of paxillin, we solved the crystal structure of the complex at a resolution of 2.0 Å. Data collection and refinement statistics are provided in Table 1. The asymmetric unit of the crystal contains three CCHD/LD1 complexes (Fig. 5A). In agreement with our ITC characterization, the crystal structure shows that the CCHD and LD1 peptide form a 1:1 molecular arrangement and confirms that the p130Cas CCHD forms a functional FAT-like domain.
Table 1.
Data collection and refinement statistics
The data set was collected from a single crystal. r.m.s., root mean square.
| p130Cas(738–874)/LD1 | |
|---|---|
| Data collection | |
| Space group | C2 |
| Cell dimensions | |
| a, b, c (Å) | 84.3, 37.7, 152.5 |
| α, β, γ (°) | 90.0, 100.9, 90.0 |
| Resolution (Å) | 2.0 (2.07–2.00)a |
| Rsym (%) | 6.0 (37.6) |
| I/σI | 30.3 (2.5) |
| Completeness (%) | 97.3 (77.1) |
| Redundancy | 5.7 (3.3) |
| Refinement | |
| Resolution (Å) | 30.0–2.0 |
| No. reflections | 31,396 |
| Rwork/Rfree | 19.6/23.7 |
| No. atoms | |
| Protein | 2,877 |
| Peptide | 247 |
| Water | 181 |
| B-factors | |
| Protein | 44.6 |
| Peptide | 46.4 |
| Water | 47.3 |
| r.m.s. deviations | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 0.009 |
a Values in parentheses are for the highest resolution shell.
Figure 5.

Crystal structure of p130Cas CCHD complex with paxillin LD1. A, the CCHD/LD1 complex crystal contains three protein/peptide complexes in the asymmetric unit that are clearly defined by electron density. As shown, the CCHD protein and LD1 form a 1:1 molecular arrangement. The hydrophobic core residues of CCHD are shown by the yellow balls. B, the key residues that form the hydrophobic core of CCHD are colored in yellow. These residues are extremely conserved among species and play an important role in maintaining the four-helix bundle structure of FAT and FAT-like domains. C, representative electron density for p130Cas(738–874)/LD1. Fo − Fc simulated annealing omit maps contoured at 3σ are shown for a few key p130Cas(738–874) residues (blue density, gray sticks) and LD1 peptide (green density, gold sticks). The protein/peptide complex corresponding to chains A/D is shown.
The CCHD forms a compact rodlike domain structure with well defined termini. The four-helix bundle has a right-handed up and down topology with a height of ∼59 Å. Approximate measurements for individual H1–H4 helices are 48, 41, 45, and 42 Å, corresponding to residues 741–777 for H1, 778–807 for H2, 809–841 for H3, and 843–873 for H4. Given the extensive hydrophobic core formed by interlacing side chains from all four helices, it remains unclear why an N-terminal extension was instrumental in the initial expression and purification of soluble CCHD-containing protein. One can only speculate that these residues (665–737) may be important for initial structure formation or aid in the dynamic stabilization of the protein in the absence of a binding partner. A similar strategy was reported to aid in obtaining suitable CCHD-containing constructs for p130Cas/NSP3 studies (15). A structure-guided sequence comparison of p130Cas CCHD with other FAT and FAT-like domains that adopt a similar four-helix bundle structure (the FAT domain of FAK and PYK2 and the protein-binding domain of GIT1) shows that hydrophobic residues in the core regions are well conserved. In p130Cas CCHD, there are a total of 17 leucines; 14 of them face toward the central hydrophobic core to provide strong hydrophobic interactions. For example, Leu-762 from helix 1, Leu-796 from helix 2, Leu-823 from helix 3, and Leu-857 from helix 4 form a leucine cluster that dramatically stabilizes the central hydrophobic core (Fig. 5B).
FAT and FAT-like domains function as targeting domains in focal adhesion signaling, interacting with binding partners by recruiting helical motifs to surface grooves between their antiparallel helices (24, 26, 27). In our crystal structure, we observed that the LD1 motif of paxillin can interact with CCHD of p130Cas through hydrophobic interaction.
The LD1 motif of paxillin binds to the p130Cas CCHD at the H2/H3 surface
The binding between the p130Cas CCHD and an FA binding partner is essential to localize the p130Cas to the FA (9). Although the function of the CCHD in targeting p130Cas to FAs had been proposed, the detailed mechanism has remained unknown. Our complex structure provides a tool to address this question at the molecular level. It has been shown that paxillin LD motifs create five binding surfaces for the recruitment of FA proteins (28). It has also been reported that the LD1 motif can interact with actopaxin (29), vinculin (30), and the integrin-linked kinase (31). LD2 mediates the binding to vinculin, FAK, and PYK2 (25). LD4 binds to actopaxin, FAK, and PYK2 (25, 29). Thus far, no binding partners of LD3 and LD5 have been reported.
Our crystal study reveals how the CCHD recognizes paxillin, an interaction mediated by the paxillin LD1 motif. The details of the interaction are clearly defined by the electron density as shown in representative simulated annealing omit maps (Fig. 5C). The binding site of LD1 is located exclusively in the central region of the H2/H3 solvent-exposed surface; leucine residues from LD1 insert into the hydrophobic grooves formed between α helices 2 and 3 of the CCHD. In addition, charged residues Asp-2 and Asp-10 of LD1 form salt bridges with Arg-828 and Lys-835 from helix 3 of CCHD, respectively, providing both favorable and specific anchor points for LD1 compared with other paxillin LD motifs. These structural observations are consistent with our ITC data whereby only LD1 shows detectable binding to p130Cas CCHD. Specifically, sequence alignment of paxillin LD motifs reveals that Asp-2 of LD1 is substituted by Ser in LD2, LD3, and LD5 and Arg in LD4. Asp-10 of LD1 is replaced by Ser in LD3, LD4, and LD5. Lastly, although hydrophobic in nature, the interaction between Met-1 of LD1 and Phe-798 from CCHD helix 2 helps distinguish LD1 from other paxillin LD motifs; the N-terminal Met-1 of LD1 fits perfectly into a partially hydrophobic crevice partially formed by Phe-798, Val-797, His-794, and Asp-801 from H2 of CCHD. Therefore, it follows that the N terminus of paxillin is optimally suited for binding to p130Cas CCHD compared with downstream paxillin LD motifs (Fig. 6). Furthermore, the combined shape and hydrophobic properties of Met-1 are better suited for this surface than Leu of LD2, Thr of LD4, Gly of LD5, and particularly Glu of LD3.
Figure 6.
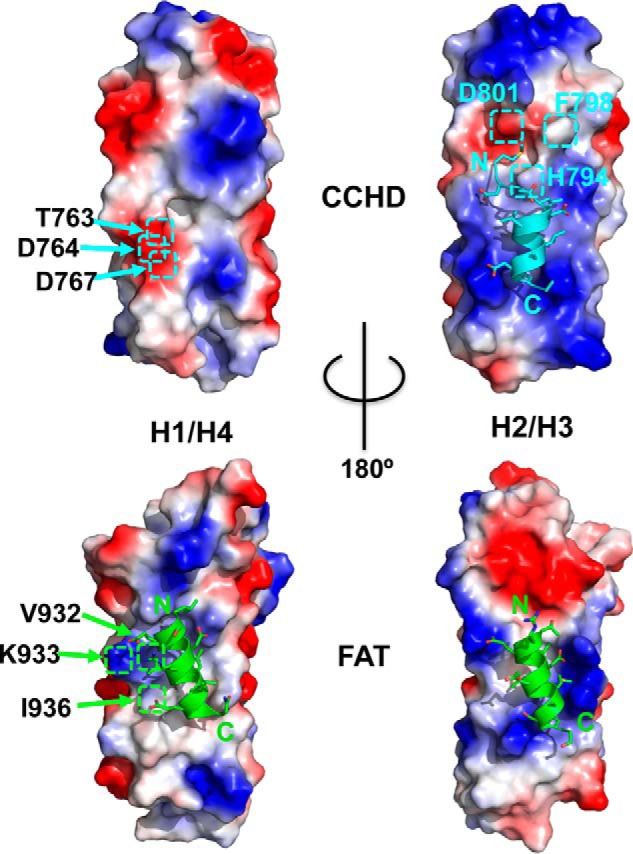
Elucidation of differences between the H1/H4 site of FAT and CCHD. Both CCHD and FAT molecular surfaces, positive (colored blue), negative (red), and non-polar or hydrophobic (35), expose two hydrophobic patches formed by helices H1/H4 (H1/H4 site) and helices H2/H3 (H2/H3 site), respectively. These central hydrophobic patches are surrounded by charged residues, which in FAT also contribute to binding of LD motifs. However, unlike FAT, CCHD does not demonstrate an ability to bind paxillin LD motifs at its H1/H4 site likely due to its reduced hydrophobic character. Distal H1 residues Lys-933, Val-932, and Ile-936 of FAT, which are in hydrophobic contact with leucines LDXLLXLL of the paxillin LD2 motif (Protein Data Bank code 1OW8), are replaced by structurally equivalent residues Asp-764, Thr-763, and Asp-767 in CCHD. The paxillin LD1 peptide schematic is colored cyan on the H2/H3 surface of CCHD. Paxillin LD2 and LD4 peptide schematics are colored green for both H1/H4 and H2/H3 surfaces, respectively, corresponding to FAT/LD peptide complex structures 1OW8 and 1OW7.
Comparison of p130Cas CCHD with FAK FAT domain
Although the FAK FAT domain has two hydrophobic surfaces that serve as paxillin LD motif-binding sites, p130Cas CCHD has only one such surface. As detailed in Fig. 6, comparison of electrostatically charged molecular surfaces provides some insight. Only FAT has been shown to support paxillin binding at H1/H4 (27, 32). As the paxillin LDXLLXLL consensus motif implies, the hydrophobic interaction of several LD motif leucines with FAT is important for robust binding. However, CCHD has reduced hydrophobic character relative to FAT at the H1/H4 surface. Specifically, distal H1 residues Lys-933, Val-932, and Ile-936 of FAT, which are observed in hydrophobic contact with leucines LDXLLXLL of the paxillin LD2 motif (Protein Data Bank code 1OW8), are replaced by structurally equivalent residues Asp-764, Thr-763, and Asp-767 in CCHD.
CCHD- and FAT H2/H3-binding sites bear the most similarity and support the binding of LD1 and LD4 (27), respectively. Within the CCHD, the side chains from H2 and H3 form two positively charged ridges sandwiching an elongated hydrophobic groove that covers about two-thirds of the four-helix bundle length. Lys-788, Arg-828, and Lys-835 help define the central ridge portion of the pocket and are conserved in all organisms for which sequences for p130Cas are available. Moreover, these residues are also conserved in the FAK FAT domain where they similarly have been shown to support binding of paxillin LD4 (Protein Data Bank code 1OW7) (33). However, when considering the portion of these surfaces responsible for binding the N-terminal regions of their respective LD motifs, they exhibit marked differences in shape and charge, reflecting their individual preferences for paxillin motif binding.
The H2/H3 surface also plays a key role in the interaction between p130Cas CCHD and NSP3 (15). As shown in Fig. 7, the superposition of CCHD/LD1 with an equivalent portion of the CCHD/NSP3 complex reveals almost identical positioning of key hydrophobic and acidic residues for LD1 relative to NSP3. However, the extended NSP3 helix runs parallel to the H2 helix of CCHD versus an antiparallel arrangement for paxillin LD1. Specifically, LD1 and NSP3 structurally equivalent residues include Asp-2/Glu-624, Leu-4/Leu-623, Leu-7/Leu-620, Leu-8/Leu-619, Asp-10/Glu-617, and Leu-11/Val-616. The CCHD hydrophobic area key in forming both complexes at H2/H3 may indeed be a general feature utilized by other yet identified CCHD ligands. As observed by Mace et al. (15), NSP3 also recognizes the H1/H2 surface of CCHD. Overall, this affords a 2.1-fold increase in the total buried surface area of CCHD/NSP3 compared with CCHD/LD1, contributing to a 140-fold stronger affinity (reported Kd of 30 nm for CCHD/NSP3 versus 4.2 μm for CCHD/LD1). However, the H2/H3 portion alone is similarly utilized among them with a total buried surface area of 1269.9 Å2 for CCHD/NSP3 versus 1123.8 Å2 for CCHD/LD1 as measured using PISA (34).
Figure 7.
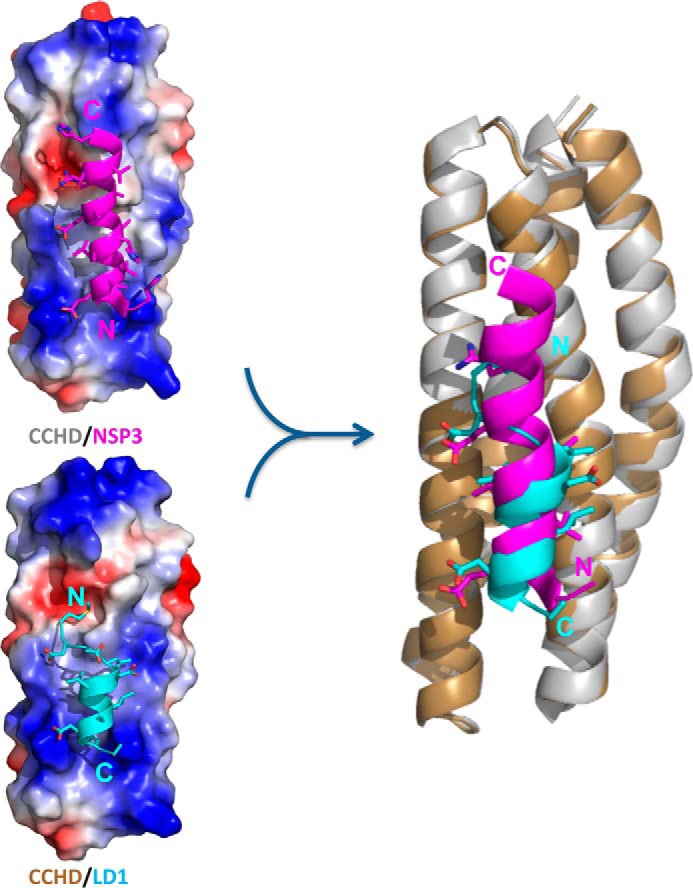
Comparison of the CCHD/LD1 structure with CCHD/NSP3. Shown on the left are molecular surface renderings colored by electrostatic potential of CCHD bound to LD1 (bottom) and a similar helical portion of NSP3 (top) for comparison. The schematic rendering on the right is a superposition of the two CCHD complexes. The CCHD and the NSP3-binding portion of the CCHD/NSP3 structure (Protein Data Bank code 3T6G, chain B and chain A) are colored gray and magenta, and the CCHD and LD1 from our CCHD/LD1 structure are colored gold and cyan, respectively. NSP3 and paxillin LD1 bind in a parallel and antiparallel orientation, respectively, relative to the H2 helix of CCHD. Despite this difference, the chemical nature and conformation of their side-chain interactions with CCHD are remarkably similar.
Discussion
In this study, we attempt to address the significance of the CCHD for p130Cas FA localization as well as the importance of p130Cas function in FAs and its role in disease. It has long been known that the SH3 domain of p130Cas interacts with FAK (4), but a recent study showed that the CCHD located near the C-terminal end of p130Cas plays a role in FA targeting equal to that of the SH3 domain (9). Our current study further supports the importance of the CCHD for p130Cas targeting. Furthermore, we provide the first mechanistic insight into how the CCHD facilitates FA targeting on a molecular level.
From our data, paxillin, an abundant protein in focal adhesions, plays an important role in recruiting p130Cas into the focal adhesion complex. The LD1 motif of paxillin binds to the H2/H3 site of the p130Cas CCHD, which is unlike the interaction made by paxillin with the FAT domain of FAK that is achieved through LD2 and LD4 interacting with H1/H4 and H2/H3, respectively. This variation illustrates the selective ability of paxillin LD motifs to interact with different focal adhesion proteins. Furthermore, these data suggest paxillin could be in complex with both the FAT domain of FAK and the CCHD of p130Cas simultaneously through the binding of LD2 and LD4 with FAT and the binding of LD1 with CCHD.
With two LD-binding sites, the FAK FAT domain should have a much stronger binding affinity with paxillin than the p130Cas CCHD, which has only one LD-binding site (Kd = 4.2 μm). The relatively weak interaction may not be sufficient for p130Cas targeting to FAs, which is why both the SH3 domain and CCHD of p130Cas are necessary for p130Cas to efficiently localize to focal adhesions. This mechanism could eliminate the interaction between p130Cas and FAK outside of the focal adhesion complex where both proteins could have other independent signaling functions.
Previous studies indicated that p130Cas is targeted to FAs through the binding of its SH3 domain to FAK with FAK localizing in FAs through the interaction of the FAT domain with LD2 and LD4 motifs of paxillin. Now our study reveals how the CCHD of p130Cas can have an important role in targeting p130Cas to FAs by binding with the LD1 motif of paxillin. We hypothesize that a FAK/p130Cas/paxillin supercomplex forms from the simultaneous associations of the p130Cas SH3 domain with FAK polyproline motifs, the p130Cas CCHD with the paxillin LD1 motif, and FAK FAT domain with paxillin LD2/LD4 motifs.
Author contributions
J. J. Z. and S. K. H. conceived the study, and J. J. Z. coordinated the study. C. Z., D. J. M., D. M. D., S. K. H., and J. J. Z. wrote the paper. C. Z. performed most of the experimental and computational studies. D. J. M. designed and executed most of the X-ray crystallization analysis. C. D. G. helped with protein preparation. All authors reviewed the results and approved the final version of the manuscript.
Acknowledgments
Data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) 22-BM beamline at the Advanced Photon Source, Argonne National Laboratory. Use of the Advanced Photon Source was supported by the United States Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract W-31-109-Eng-38. We thank Dr. Weixing Zhang and Dr. Christy R. R. Grace for assistance with NMR experiments.
This work was supported by National Institutes of Health Grant GM100909 and by Research to Prevent Blindness. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
The atomic coordinates and structure factors (code 5W93) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- Cas
- Crk-associated substrate
- BCAR1
- breast cancer antiestrogen resistance 1
- CCHD
- Cas family homology domain
- FA
- focal adhesion
- FAK
- focal adhesion kinase
- FAT
- focal adhesion targeting
- ITC
- isothermal titration calorimetry
- SH
- Src homology
- NSP
- novel SH2-containing protein
- HSQC
- heteronuclear single quantum coherence.
References
- 1. Cabodi S., Tinnirello A., Di Stefano P., Bisarò B., Ambrosino E., Castellano I., Sapino A., Arisio R., Cavallo F., Forni G., Glukhova M., Silengo L., Altruda F., Turco E., Tarone G., et al. (2006) p130Cas as a new regulator of mammary epithelial cell proliferation, survival, and HER2-neu oncogene-dependent breast tumorigenesis. Cancer Res. 66, 4672–4680 [DOI] [PubMed] [Google Scholar]
- 2. Bouton A. H., Riggins R. B., and Bruce-Staskal P. J. (2001) Functions of the adapter protein Cas: signal convergence and the determination of cellular responses. Oncogene 20, 6448–6458 [DOI] [PubMed] [Google Scholar]
- 3. Sakai R., Iwamatsu A., Hirano N., Ogawa S., Tanaka T., Mano H., Yazaki Y., and Hirai H. (1994) A novel signaling molecule, p130, forms stable complexes in vivo with v-Crk and v-Src in a tyrosine phosphorylation-dependent manner. EMBO J. 13, 3748–3756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Polte T. R., and Hanks S. K. (1995) Interaction between focal adhesion kinase and Crk-associated tyrosine kinase substrate p130Cas. Proc. Natl. Acad. Sci. U.S.A. 92, 10678–10682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Petch L. A., Bockholt S. M., Bouton A., Parsons J. T., and Burridge K. (1995) Adhesion-induced tyrosine phosphorylation of the p130 src substrate. J. Cell Sci. 108, 1371–1379 [DOI] [PubMed] [Google Scholar]
- 6. Vuori K., and Ruoslahti E. (1995) Tyrosine phosphorylation of p130Cas and cortactin accompanies integrin-mediated cell adhesion to extracellular matrix. J. Biol. Chem. 270, 22259–22262 [DOI] [PubMed] [Google Scholar]
- 7. Nojima Y., Tachibana K., Sato T., Schlossman S. F., and Morimoto C. (1995) Focal adhesion kinase (pp125FAK) is tyrosine phosphorylated after engagement of α4β1 and α5β1 integrins on human T-lymphoblastic cells. Cell. Immunol. 161, 8–13 [DOI] [PubMed] [Google Scholar]
- 8. Honda H., Oda H., Nakamoto T., Honda Z., Sakai R., Suzuki T., Saito T., Nakamura K., Nakao K., Ishikawa T., Katsuki M., Yazaki Y., and Hirai H. (1998) Cardiovascular anomaly, impaired actin bundling and resistance to Src-induced transformation in mice lacking p130Cas. Nat. Genet. 19, 361–365 [DOI] [PubMed] [Google Scholar]
- 9. Donato D. M., Ryzhova L. M., Meenderink L. M., Kaverina I., and Hanks S. K. (2010) Dynamics and mechanism of p130Cas localization to focal adhesions. J. Biol. Chem. 285, 20769–20779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brinkman A., van der Flier S., Kok E. M., and Dorssers L. C. (2000) BCAR1, a human homologue of the adapter protein p130Cas, and antiestrogen resistance in breast cancer cells. J. Natl. Cancer Inst. 92, 112–120 [DOI] [PubMed] [Google Scholar]
- 11. Cabodi S., Moro L., Baj G., Smeriglio M., Di Stefano P., Gippone S., Surico N., Silengo L., Turco E., Tarone G., and Defilippi P. (2004) p130Cas interacts with estrogen receptor α and modulates non-genomic estrogen signaling in breast cancer cells. J. Cell Sci. 117, 1603–1611 [DOI] [PubMed] [Google Scholar]
- 12. Riggins R. B., Thomas K. S., Ta H. Q., Wen J., Davis R. J., Schuh N. R., Donelan S. S., Owen K. A., Gibson M. A., Shupnik M. A., Silva C. M., Parsons S. J., Clarke R., and Bouton A. H. (2006) Physical and functional interactions between Cas and c-Src induce tamoxifen resistance of breast cancer cells through pathways involving epidermal growth factor receptor and signal transducer and activator of transcription 5b. Cancer Res. 66, 7007–7015 [DOI] [PubMed] [Google Scholar]
- 13. Soni S., Lin B. T., August A., Nicholson R. I., and Kirsch K. H. (2009) Expression of a phosphorylated p130(Cas) substrate domain attenuates the phosphatidylinositol 3-kinase/Akt survival pathway in tamoxifen resistant breast cancer cells. J. Cell. Biochem. 107, 364–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Janoštiak R., Brábek J., Auernheimer V., Tatárová Z., Lautscham L. A., Dey T., Gemperle J., Merkel R., Goldmann W. H., Fabry B., and Rösel D. (2014) CAS directly interacts with vinculin to control mechanosensing and focal adhesion dynamics. Cell. Mol. Life Sci. 71, 727–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mace P. D., Wallez Y., Dobaczewska M. K., Lee J. J., Robinson H., Pasquale E. B., and Riedl S. J. (2011) NSP-Cas protein structures reveal a promiscuous interaction module in cell signaling. Nat. Struct. Mol. Biol. 18, 1381–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 17. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta. Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 19. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brown M. C., Perrotta J. A., and Turner C. E. (1996) Identification of LIM3 as the principal determinant of paxillin focal adhesion localization and characterization of a novel motif on paxillin directing vinculin and focal adhesion kinase binding. J. Cell Biol. 135, 1109–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brown M. C., Curtis M. S., and Turner C. E. (1998) Paxillin LD motifs may define a new family of protein recognition domains. Nat. Struct. Biol. 5, 677–678 [DOI] [PubMed] [Google Scholar]
- 23. Brown M. C., and Turner C. E. (2004) Paxillin: adapting to change. Physiol. Rev. 84, 1315–1339 [DOI] [PubMed] [Google Scholar]
- 24. Zhang Z. M., Simmerman J. A., Guibao C. D., and Zheng J. J. (2008) GIT1 paxillin-binding domain is a four-helix bundle, and it binds to both paxillin LD2 and LD4 motifs. J. Biol. Chem. 283, 18685–18693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vanarotti M. S., Miller D. J., Guibao C. D., Nourse A., and Zheng J. J. (2014) Structural and mechanistic insights into the interaction between Pyk2 and paxillin LD motifs. J. Mol. Biol. 426, 3985–4001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu G., Guibao C. D., and Zheng J. (2002) Structural insight into the mechanisms of targeting and signaling of focal adhesion kinase. Mol. Cell. Biol. 22, 2751–2760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bertolucci C. M., Guibao C. D., and Zheng J. (2005) Structural features of the focal adhesion kinase-paxillin complex give insight into the dynamics of focal adhesion assembly. Protein Sci. 14, 644–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lorenz S., Vakonakis I., Lowe E. D., Campbell I. D., Noble M. E., and Hoellerer M. K. (2008) Structural analysis of the interactions between paxillin LD motifs and α-parvin. Structure 16, 1521–1531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nikolopoulos S. N., and Turner C. E. (2000) Actopaxin, a new focal adhesion protein that binds paxillin LD motifs and actin and regulates cell adhesion. J. Cell Biol. 151, 1435–1448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Turner C. E., Brown M. C., Perrotta J. A., Riedy M. C., Nikolopoulos S. N., McDonald A. R., Bagrodia S., Thomas S., and Leventhal P. S. (1999) Paxillin LD4 motif binds PAK and PIX through a novel 95-kD ankyrin repeat, ARF-GAP protein: a role in cytoskeletal remodeling. J. Cell Biol. 145, 851–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nikolopoulos S. N., and Turner C. E. (2001) Integrin-linked kinase (ILK) binding to paxillin LD1 motif regulates ILK localization to focal adhesions. J. Biol. Chem. 276, 23499–23505 [DOI] [PubMed] [Google Scholar]
- 32. Hayashi I., Vuori K., and Liddington R. C. (2002) The focal adhesion targeting (FAT) region of focal adhesion kinase is a four-helix bundle that binds paxillin. Nat. Struct. Biol. 9, 101–106 [DOI] [PubMed] [Google Scholar]
- 33. Hoellerer M. K., Noble M. E., Labesse G., Campbell I. D., Werner J. M., and Arold S. T. (2003) Molecular recognition of paxillin LD motifs by the focal adhesion targeting domain. Structure 11, 1207–1217 [DOI] [PubMed] [Google Scholar]
- 34. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 35. Wang C., Grey M. J., Palmer A. G. 3rd (2001) CPMG sequences with enhanced sensitivity to chemical exchange. J. Biomol. NMR 21, 361–366 [DOI] [PubMed] [Google Scholar]