

Abstract
Alzheimer's disease is one of the most devastating neurodegenerative diseases without effective therapies. Immunotherapy is a promising approach, but amyloid antibody structural information is limited. Here we simulate the recognition of monomeric, oligomeric, and fibril amyloid-β (Aβ) by three homologous antibodies (solanezumab, crenezumab, and their chimera, CreneFab). Solanezumab only binds the monomer, whereas crenezumab and CreneFab can recognize different oligomerization states; however, the structural basis for this observation is not understood. We successfully identified stable complexes of crenezumab with Aβ pentamer (oligomer model) and 16-mer (fibril model). It is noteworthy that solanezumab targets Aβ residues 16–26 preferentially in the monomeric state; conversely, crenezumab consistently targets residues 13–16 in different oligomeric states. Unlike the buried monomeric peptide in solanezumab's complementarity-determining region, crenezumab binds the oligomer's lateral and edge residues. Surprisingly, crenezumab's complementarity-determining region loops can effectively bind the Aβ fibril lateral surface around the same 13–16 region. The constant domain influences antigen recognition through entropy redistribution. Different constant domain residues in solanezumab/crenezumab/chimera influence the binding of Aβ aggregates. Collectively, we provide molecular insight into the recognition mechanisms facilitating antibody design.
Keywords: Alzheimer disease, amyloid, amyloid-beta (AB), antibody, immunotherapy, protein dynamic
Introduction
Alzheimer's disease (AD)3 is one of the most devastating neurodegenerative diseases without effective therapies (1). The amyloid-β (Aβ) hypothesis (2) argues that accumulation of Aβ leads to tangles and plaques in AD-affected brains, which cause neuron loss. Several strategies have been developed to reduce the Aβ production, inhibit the Aβ aggregation, or directly enhance the Aβ clearance. Immunotherapy exploiting antibodies and antibody fragments is a major approach (3–5), and active and passive anti-Aβ immunotherapies can clear brain Aβ deposits. For example, aducanumab (7, 8), a human monoclonal antibody, selectively targets aggregated Aβ, reduces brain Aβ in a dose- and time-dependent manner, and slows clinical decline (9). Decreasing Aβ concentration in blood could also be an effective therapeutic strategy based on the peripheral sink hypothesis (10). The peripheral sink hypothesis proposed that drug molecules, which bind plasma Aβ, can sequester brain Aβ into peripheral tissues without crossing the blood–brain barrier and reduce the accumulation of Aβ in the CNS. Based on this hypothesis, the Aβ-binding properties of various biologics (11, 12) (e.g. therapeutic antibodies, albumins (13–16), and transferrins (17)) provide the basis for treating AD. Therefore, antibodies targeting various states of Aβ peptide have been actively studied. However, therapies aimed at reducing protein processing and clearance in AD have been unsuccessful in clinical trials, due to unfavorable pharmacokinetics, difficulty in crossing the blood–brain barrier, and potential neurotoxicity. The mechanisms underlying amyloid-based immunotherapy are complex (18, 19) and not fully understood (20). Antibody and other proteins can bind Aβ peptide in different oligomerization states and on different Aβ regions. For example, the three domains of apo-albumin recognized the C-terminal hydrophobic residues (13, 15). Despite the many crystal structures of monomeric amyloidogenic peptides in complex with antibodies, there is a lack of structural information on antibody recognition of any aggregated protein.
Among the several therapeutic antibodies for AD, solanezumab (Lilly) and crenezumab (Genentech) are two leading humanized monoclonal antibodies interacting with the mid-region of the toxic Aβ aggregates. Solanezumab (21) used IgG1 as template and mainly recognizes monomeric soluble Aβ with picomolar affinity, whereas crenezumab (22) used IgG4 as template and can recognize monomers, oligomers, and fibrils. Solanezumab and crenezumab have the same number of residues in the CDR loops as well as in the fragment antigen-binding (Fab) region, with 11 residues on CDR loops (L1, H1, and H2) and 11 on the constant domain (Table 1). The constant domains of the light chain of the two mAbs have identical amino acid sequences, whereas the constant domains of the heavy chain displayed different residues in the CH1–1 loop and in another loop, close to the C terminus. Sequence alignment showed that only <6% of the Fab residues are different between solanezumab and crenezumab (supplemental Table S1), making them an excellent pair for comparing antibodies with similar sequences but different specificities and raising the question of how these few residues (supplemental Fig. S1) differentially influence Aβ aggregate recognition.
Table 1.
Summary of the properties of the two therapeutic antibodies solanezumab and crenezumab
| Solanezumab | Crenezumab | |
|---|---|---|
| Epitope | Aβ12–28 | Aβ12–23 |
| Aβ form | Monomeric | Monomeric, oligomers, and fibrils |
| Affinity | Picomolar level | Nanomolar level |
| Template | IgG1 | IgG4 |
| CDR-L1 | RSSQSLIYSDGNAYLHTFL | RSSQSLVYSNGDTYLHTYL |
| CDR-L2 | KVSNRFS | KVSNRFS |
| CDR-L3 | SQSTHVPWT | SQSTHVPWT |
| CDR-H1 | GFTFSRYSMS | GFTFSSYGMS |
| CDR-H2 | QINSVGNSTYYPDTVKGRFT | SINSNGGSTYYPDSVKGRFT |
| CDR-H3 | GDY | GDY |
The crystal structure of the complex between solanezumab and Aβ12–28 has been resolved (11), revealing that Aβ16–26 forms extensive contacts and hydrogen bonds with solanezumab and that the Phe19-Phe20 hydrophobic core is buried. Recently, Ultsch et al. (12) reported the structure of the complex of Aβ11–25 with an engineered crenezumab (CreneFab), which has crenezumab's variable domain and solanezumab's constant domain (23). In the engineered CreneFab complex, like the solanezumab complex, Aβ's Phe19/Phe20 form hydrophobic contacts with crenezumab. A broken essential salt bridge affects the organization of the Aβ oligomer. However, these co-crystal structures still provide limited information to answer why crenezumab recognized more Aβ species. Several recent studies suggested that the constant domain also plays an important role in the antigen recognition (24–28). There is solid evidence for distant communication between the variable and constant domains (29). Evidence also suggests allosteric effects during antibody–antigen recognition (30).
Here we selected three homologous antibodies and compared their structure and dynamics upon binding to Aβ peptides in monomeric, oligomeric, and fibril forms. Based on crystal structures of solanezumab-Aβ and CreneFab·Aβ complexes (supplemental Fig. S2), we systematically examined the structures of the solanezumab and crenezumab with the soluble, oligomeric, and fibril forms of Aβ using homology modeling, molecular docking, and molecular dynamics simulations. Using this protocol, we screened possible complexes between crenezumab and Aβ aggregates (monomeric, pentamer, and fibril-like structures). We investigated the structure, energy, and dynamics related to the roles of the CDR loops as well as the constant domain in amyloid recognition. Besides the CDR loops, the constant domain loops are also highly correlated with Aβ, indicating their role in Aβ recognition. The flexibility of the constant domain CH1–1 and H4 loops (residues 400–410) changed in response to the Aβ binding. We interpreted the response and correlation as reflecting entropy transfer and release from the antibody–antigen interface to the constant domains. Collectively, our results provide atomic level structure and dynamics information which may facilitate antibody design in AD.
Results
Solanezumab is conformationally more flexible than crenezumab in the apo form
As can be seen in supplemental Table S1, solanezumab and crenezumab differ by 5, 2, and 4 residues on CDR-L1, H1, and H3 loops, respectively, whereas other CDR loops are identical. Besides CDRs, the light chains have three different N-terminal residues, and the heavy chains have 4 different residues on the CH1 loop and 5 different residues near the C-terminal, distant from the CDR loops. There is no crystal structure of “wild-type” crenezumab. To gain structural insight into Aβ recognition by crenezumab, we compare the crystal structures of the solanezumab·Aβ complex with CreneFab (models Smab1, CHmab0, and CHmab1; Table 2) (23). Superimposition of the three crystal structures (supplemental Fig. S2 and Table S3) on the V domain (RMSD < 0.8 Å) and C domain (RMSD < 0.65 Å) suggested that they are very similar locally, but the orientations of the C-V domains differ significantly (>1.3 Å) if the whole Fab structures are used, especially for 5VZX and 5VZY, considering that the V domain and C domain are only 0.382 and 0.575 Å locally but 4.045 Å with full Fab.
Table 2.
List of simulated antibody–antigen complexes
| Model | Antibody | Antigen | PDB template |
|---|---|---|---|
| Smab0 | Solanezumab | NAa | 4XXD |
| Smab1 | Solanezumab | Aβ12–28 monomer | 4XXD |
| Smab5 | Solanezumab | Aβ11–42 pentamer | 4XXD |
| Smab16 | Solanezumab | Aβ11–42 16mer | 4XXD |
| Cmab0a | Crenezumab | NA | 5KMV |
| Cmab0b | Crenezumab | NA | 5KNA |
| Cmab1 | Crenezumab | Aβ12–28 monomer | 5KNA |
| Cmab5 | Crenezumab | Aβ11–42 pentamer | 5KNA |
| Cmab16 | Crenezumab | Aβ11–42 16-mer | 5KNA |
| CHmab0a | Crenezumab chimera | NA | 5KMV |
| CHmab0b | Crenezumab chimera | NA | 5KNA |
| CHmab1 | Crenezumab chimera | Aβ12–28 monomer | 5KNA |
| CHmab5 | Crenezumab chimera | Aβ11–42 pentamer | 5KNA |
| CHmab16 | Crenezumab chimera | Aβ11–42 16-mer | 5KNA |
a NA, not applicable.
We modeled the apo structure of solanezumab (model Smab0) by removing the Aβ antigen from the solanezumab·Aβ complex. Two possible conformers of the apo form of the crenezumab Fab structure were modeled based on the crystal structures of CreneFab apo (PDB code 5VZX) and CreneFab·Aβ (PDB code 5VZY) (models Cmab0a and Cmab0b, Table 2). 100–200-ns simulations were performed for both solanezumab and crenezumab apo structures. (As only the bound structure of solanezumab was reported, we simulated the apo structure for 200 ns to extend the conformational search space.) First, we compared all of the sampled structures of solanezumab (Smab0), crenezumab (Cmab0), and the CreneFab (CHmab0) by 2D RMSD and conformation clustering. To obtain their overall fluctuations, the populations of solanezumab, crenezumab, and the CreneFab are divided into clusters (based on whole Fab backbone RMSD of 4 Å) by superimposing their V domains (Fig. 1a). The two sets of apo structures of crenezumab were combined and analyzed. Apo-crenezumab and apo-CreneFab are homogeneous, with 99 and 98% of the population within 4 Å of RMSD, respectively. The structures of apo-solanezumab are more diverse, with three conformational clusters with 84, 12, and 4% of the population (Fig. 1b).
Figure 1.

The structure and dynamics of the three Fabs in their apo form suggest that when unbound to antigens, solanezumab is more conformationally diverse than the other two Fabs. a, 2D RMSDs of the structures sampled in the MD simulation. The structures of crenezumab and CreneFab are taken from PDB entries 4KMV (A) and 4KNA (B), respectively. b, cluster analysis, with backbone RMSD = 4 Å, used to define the cluster. Clusters are colored blue, red, and gray, respectively. c, motion correlation among the residues of the three Fabs. Residues with highly correlated and anti-correlated motion are red and blue, respectively. d, RMSFs of the three Fabs from five independent MD simulations. The RMSFs of Smab0, CHmab0a, CHmab0b, Cmab0a, and Cmab0b are colored black, red, pink, blue, and cyan, respectively.
Cross-talk between the antibody subdomains helps antibody–antigen recognition (28, 31–33). The dynamical covariance matrix plots of the apo structures of solanezumab and crenezumab showed that the motions of Fab subdomains are correlated with four major local correlations along the diagonal, representing the four subdomains, VL, CL, VH, and CH1 (Fig. 1c). There are couplings between distant parts of the complex in some systems. The motion correlation analysis suggested that apo-solanezumab has a much larger negative intersubdomain motion, especially between VH and CH-1 and between CL and CH1 (Fig. 1c), which resulted in a more diverse conformational population.
Second, we investigated the local residue fluctuations by using the RMSFs of individual residues (Fig. 1d). The residue RMSFs of the light chain loops (including CDRs) of solanezumab are slightly higher than crenezumab/CreneFab, whereas those of the heavy chain loops (including CDRs) of solanezumab are much larger than crenezumab/CreneFab. We compared the sequence and loop flexibility in their apo forms. There is an interchain disulfide bond between the light and heavy chains of crenezumab (Cmab0) but not in solanezumab/CreneFab (Smab0 and CHmab0). This interchain disulfide bond, which constrains the loop fluctuation, is more flexible in the CH1-1 in apo-solanezumab/CreneFab than in crenezumab. On the heavy chain around residue 410, the chimera apo structure (CHmab0) showed an even higher peak than solanezumab. This analysis suggested that solanezumab and crenezumab/CreneFab have different structural and dynamic properties in their apo forms. Without antigen binding, solanezumab is conformationally more flexible than crenezumab.
The recognition mechanism of solanezumab/crenezumab/chimera and Aβ monomer
Both solanezumab and crenezumab can bind soluble Aβ monomer with similar epitopes, but solanezumab showed stronger binding affinity than crenezumab (34). The crystal structure of the complex of solanezumab and Aβ12–28 showed that Aβ12–28 adopted a helical structure and formed extensive contacts with solanezumab (34). The crystal structure of the complex between the chimera and Aβ11–25 also showed that Aβ formed extensive contacts with the chimera Fab with a similar helical structure (Fig. 2a). Therefore, we first mutated the C domain of the chimera Fab back to crenezumab sequence with V domain and Aβ (model Cmab1 is derived from model CHmab1).
Figure 2.

The V domains of crenezumab and CreneFab can recognize a more diverse Aβ ensemble than solanezumab. a, the two Aβ-bound crystal structures superimposed on the Aβ and V domains. 4XXD and 4KNA are colored red and lime, respectively. b, clustered conformations of Aβ12–28 in complex with solanezumab, crenezumab, and CreneFab (RSMD for clustering is 4 Å). c, secondary structure components of Aβ12–28. Helical, β-strand, turn, and random coil structures are colored black, red, blue, and green, respectively. d, contact preference on the Fabs–Aβ interface from the Aβ and Fab sides. The Fab amino acid preference was obtained by summation of the contacts based on the Fab side contact preference.
Crenezumabs can recognize diverse Aβ conformation
During the 200-ns simulation of the solanezumab·Aβ12–28 complex, the structure of Aβ12–28, especially the helical structures between Ala21 and Asn27 in all (100%) conformers (RMSD < 5 Å), were well maintained (Fig. 2b). In the crenezumab system, the Aβ12–28 structure largely fluctuates and leads to diverse conformations. Cluster analysis suggested that there is one major cluster accounting for 86% of the conformations, whereas the other three account for 10, 2, and 2%, respectively. In the CreneFab system, the Aβ12–28 structures became more diverse. Besides the major cluster (76%), the other three account for 18, 3, and 3%, respectively. We measured the secondary structure distribution in the three complexes (Fig. 2c). The central region of the peptide Aβ20–25 mainly (>95%) adopted a helical structure in solanezumab, whereas in the system of crenezumab and CreneFab, Aβ20–25 adopted more diverse structures, including ∼70% turn structure, ∼20% helical structure, and ∼10% random coil. Therefore, whereas solanezumab mainly recognizes helical Aβ12–28, crenezumab and CreneFab can recognize other secondary structure features. As the helical structure is only highly populated in soluble Aβ species, this may explain the specificity of solanezumab recognition of soluble Aβ monomer and the diverse recognition of crenezumab of monomer, oligomers, and fibrils.
Fab structures show similar conformational trends. We clustered the Fab structures from the three complexes (Smab1, Cmab1, and CHmab1) by backbone RMSD of 4 Å. When in complex with Aβ12–28, 100% of the population of solanezumab is in one cluster (Fig. 3a). For crenezumab, the conformations are in two clusters with one dominant (98%). For the CreneFab, the conformations become more diverse, with three clusters with populations of 79, 16, and 5%, respectively. In the apo form, solanezumab has higher conformational diversity than crenezumab/CreneFab while being more uniform in complex with Aβ12–28.
Figure 3.

Solanezumab is less conformationally diverse than the other two Fabs when bound to Aβ12–28. a, cluster analysis of the structures. Backbone RMSD = 4 Å is used to define the cluster. Clusters are colored blue, red, and gray, respectively. b, motion correlation among the residues of the three Fabs. Residues with highly correlated and anti-correlated motion are red and blue, respectively. c and d, RMSFs (c) and order parameters S2 (d) of the three Fabs in complex with Aβ12–28. The location of CDRs and important constant domain loops are boxed. The values of solanezumab, crenezumab, and CreneFab are colored black, blue, and red, respectively.
We calculated the binding energies of the three antibodies binding the Aβ12–28 monomer. That of the solanezumab·Aβ12–28 complexes is −74.3 ± 3.7 kcal/mol, whereas crenezumab·Aβ12–28 complexes are slightly less favorable (−66.0 ± 3.7 kcal/mol) (Table 3), and CreneFab·Aβ12–28 complexes are around −17.8 ± 3.7 kcal/mol. Whereas the absolute energy difference from molecular mechanics is not directly comparable with experimental binding affinities, the relative energy binding preferences from the calculations agree with the experimental trend. Therefore, in terms of the energy landscape, all conformations of solanezumab are optimized for monomer recognition, whereas crenezumab can adapt to a diversified binding environment with some penalty for monomer-binding affinity.
Table 3.
Binding energy between Fab and Aβ in the forms of monomer, oligomer, and fibrils
The binding energy is calculated by the equation, 〈Ebind〉 = 〈Ecomplex〉 − 〈EFab〉 − 〈Epeptide〉.
| Antibody | Aβ chains | Binding energy | Total residue number | Normalized binding energy |
|---|---|---|---|---|
| kcal/mol | kcal/mol/residue | |||
| Solanezumab | 1-mer | −74.3 ± 3.7 | 451 | −0.165 |
| 5-mer | −3.9 ± 4.5 | 594 | −0.007 | |
| 16-mer | −41.2 ± 6.3 | 946 | −0.044 | |
| Crenezumab | 1-mer | −66.0 ± 3.7 | 451 | −0.146 |
| 5-mer | −39.8 ± 4.4 | 594 | −0.067 | |
| 16-mer | −195.8 ± 6.3 | 946 | −0.207 | |
| Crenezumab chimera | 1-mer | −17.8 ± 3.7 | 451 | −0.039 |
| 5-mer | −6.4 ± 4.5 | 594 | −0.011 | |
| 16-mer | −85.2 ± 6.0 | 946 | −0.090 |
Solanezumab and crenezumab have different epitope preferences
Aβ formed extensive contacts in the complex of solanezumab·Aβ12–28 (Smab1), with an average contact area of 1889.7 Å2 (supplemental Table S4). There are more interactions with heavy chain residues than light chain residues. In the crenezumab·Aβ12–28 complex (Cmab1), the total contact area was 1785.2 Å2 with VL and VH contributing similarly. The CreneFab·Aβ12–28 interactions (CHmab1) are similar to the crenezumab·Aβ12–28 complex. Essentially, the numbers of hydrogen bonds, hydrophobic and electrostatic interactions revealed that all three antibodies have similar contacts with the Aβ12–28 monomer.
However, the contact frequency between residues from Aβ and Fabs during the simulation revealed that solanezumab and crenezumab have different epitope preferences (Fig. 2d). In the complex of solanezumab·Aβ12–28 (Smab1), almost all of the residues from Aβ12–28 made contacts, and Phe19, Lys16, and Phe20 ranked as the top 3. The CDR-H1 and -H3 loops of solanezumab contributed most of the contacts. In the complex of crenezumab·Aβ12–28 (Cmab1), Aβ12–28 made considerable contacts; however, the preferred interacting residues shifted to the N terminus. Lys16, Phe20, and Leu17 ranked as the top 3. Lys16 becomes the most preferred residue, whereas the contacts from Phe19/20 were reduced about 50%. In the complex of chimera·Aβ12–28 (CHmab1), the preferred interacting residues further shifted to the N terminus, with Lys16 becoming the most preferred residue and Phe19/20 reduced to about 80%.
To further identify the essential residues of Fabs in the recognition, antibody–antigen contacts within 3 Å of each other were monitored during the simulation and normalized by the total contact time. A contact value of 1.0 means that a certain Fab residue always contacts an Aβ residue during the simulation, and a value >1.0 indicates that the residue contacts at least two Aβ residues.
The underlying driving force in epitope differentiation is embedded in the slight difference in the CDR region. There are more hydrophobic residues in contact with Aβ in solanezumab than in crenezumab/CreneFab. We identified the essential residues for the recognition by monitoring the change of accumulated antigen contacts at specific sites, especially where the residues of the two mAbs differ (supplemental Table S5). Four sites were identified with large contact change (Δcontact > 0.25) with mutations between the two mAbs. Arg250(31H), Ser252(33H), Phe41(36L), and Gln269(50H) in solanezumab were mutated to Ser, Gly, Tyr, and Ser in crenezumab, with 2.15, 0.93, 0.89, and 0.31 contacts lost. These mutations also resulted in neighboring residue contact change (e.g. Tyr54, Glu220, Ser61, and Ser272).
The constant domains contribute to Aβ-Fab recognition by entropy dissipation
The binding energy difference of crenezumab·Aβ12–28 complex (Cmab1) and CreneFab·Aβ12–28 complex (CHmab1) suggested that even with the same sequence and initial conformation of the V domain, the binding affinity can be different when mutating the C domain back to the solanezumab sequence. This suggested that motion correlations between the variable domain and constant domain as well as between the antigen and constant domain influence Fab-Aβ recognition (Fig. 3b). Loop residues present stronger correlation with Aβ12–28 than other residues.
To identify important distant residues in Aβ-Fab recognition, we compared the RMSF change of three Fabs upon binding to Aβ (Fig. 3c). For the V domains of those Fabs, the RMSFs are very similar, with only slight variations, whereas for the C domains, the flexibility varied upon binding Aβ12–28. The L4 loop of the CL domain (around residue 205) of CreneFab showed significant flexibility (Fig. 3c). The distant CH1-1 loop region of solanezumab is highly flexible compared with crenezumab and CreneFab, with the chimera showing larger RMSF than solanezumab and crenezumab in the remainder of the CH1 domain. The conformational changes of solanezumab and Aβ12–28 upon recognition indicate a larger entropy cost for stabilizing both solanezumab and Aβ12–28, suggesting an entropy dissipation mechanism. The larger fluctuations of the distant loop may help in transferring vibrational entropy. Thus, the mutations in these distant loops appear important in antigen recognition.
There is a good correlation between backbone entropy and the order parameter obtained from MD simulations (35). To check all residues, including proline, we calculated the generalized order parameter S2 of the C=O bond of each individual residue of the antibodies (Fig. 3d). We found that on the heavy chain, which mainly interacts with Aβ, solanezumab and crenezumab showed higher S2 on the CDRs, whereas they showed lower S2 than CreneFab on the constant domain CH1-1 loop. This suggests a transfer of entropy from CDRs to CH1-1 when Aβ and solanezumab/crenezumab form a stable complex. The diversified and longer communication pathways between the antigen epitopes and distant antibody residues may also reflect constant domain contributions. We examined the signals from the Aβ monomer to the constant domain of the three antibodies, especially the regions with strong response and bearing mutation sites. In most cases, there are longer optimal pathways from the Phe20/Lys16 of Aβ to C domain residues in solanezumab than crenezumab (Table 4 and supplemental Table S8). There are no communications between Aβ and C domain residues in CreneFab. Therefore, the solanezumab constant domain has less effect on antigen recognition, whereas in crenezumab, the constant domain has a larger effect.
Table 4.
The shortest communication pathway distance from distant sites to Lys16 of peptide in the Fab/peptide 1-mer, 5-mer, and 16-mer complexes
| System | Residuea | Name | Chain | Lys16 |
||
|---|---|---|---|---|---|---|
| Solanezumab | Crenezumab | Chimera | ||||
| 1-mer | 205 | Gly/Glyb | L | 461 | 407 | NAc |
| 347 | Lys/Arg | H | 507 | 371 | NA | |
| 410 | Gln/Lys | H | 315 | 258 | NA | |
| 351 | Gly/Glu | H | 431 | 313 | NA | |
| 352 | Gly/Ser | H | 371 | 311 | NA | |
| 413 | Ile/Thr | H | 287 | 245 | NA | |
| 417 | Asn/Asp | H | 262 | 222 | NA | |
| 428 | Lys/Arg | H | 295 | 252 | NA | |
| 431 | Pro/Ser | H | 388 | 349 | NA | |
| 433 | Ser/Tyr | H | 454 | 425 | NA | |
| 434 | Cys/Gly | H | 443 | NA | NA | |
| 5-mer | 205 | Gly/Gly | L | 391 | 236 | 317 |
| 347 | Lys/Arg | H | 408 | 140 | 234 | |
| 410 | Gln/Lys | H | 316 | 105 | 197 | |
| 351 | Gly/Glu | H | 376 | 147 | 233 | |
| 352 | Gly/Ser | H | 349 | 134 | 232 | |
| 413 | Ile/Thr | H | 300 | 91 | 190 | |
| 417 | Asn/Asp | H | 281 | 75 | 176 | |
| 428 | Lys/Arg | H | 304 | 94 | 193 | |
| 431 | Pro/Ser | H | 390 | 106 | 203 | |
| 433 | Ser/Tyr | H | 426 | 135 | 245 | |
| 434 | Cys/Gly | H | 389 | 142 | 235 | |
| 16-mer | 205 | Gly/Gly | L | 294 | 131 | 171 |
| 347 | Lys/Arg | H | 199 | 101 | 142 | |
| 410 | Gln/Lys | H | 152 | 91 | 122 | |
| 351 | Gly/Glu | H | 182 | 96 | 170 | |
| 352 | Gly/Ser | H | 174 | 94 | 153 | |
| 413 | Ile/Thr | H | 144 | n/a | 118 | |
| 417 | Asn/Asp | H | 129 | 79 | 109 | |
| 428 | Lys/Arg | H | 147 | 90 | 119 | |
| 431 | Pro/Ser | H | 161 | 94 | 127 | |
| 433 | Ser/Tyr | H | 182 | 98 | 150 | |
| 434 | Cys/Gly | H | NA | 100 | 155 | |
a Residue number in the MD simulation.
b The first and second residue is the corresponding residue in solanezumab and crenezumab, respectively.
c NA, not available.
The recognition mechanism of crenezumab and Aβ oligomer
Identification of stable structures of antibody·Aβ oligomer complexes
Based on the populations of the crenezumab Fabs, ∼90 and ∼10% of the conformations fall into two clusters, and the V domains are very similar in all. This ensemble led us to select the crenezumab Fab based on the crystal structure of the chimera for molecular docking with Aβ12–28.
The polymorphic nature of the Aβ oligomers and fibrils makes it difficult to obtain their crystal structures in complex with antibodies. Aβ1–42/1–40 fibril structures have been resolved by ssNMR (36–39) and by computations (40–51). In this work, a pentamer and 16-mer of Aβ11–42 obtained from ssNMR (PDB code 2MXU) (51) were selected to represent the oligomers and fibrils, respectively. In the isolated states, without antibody interactions, the overall structure and the β structure of Aβ11–42 5-mer and 16-mer were well maintained during the 200-ns simulation, whereas the Aβ12–28 lost its initial helical structure (Fig. 4). Therefore, we used the Aβ11–42 5- and 16-mer for searching a potential antibody recognition pattern by antibody docking and subsequent MD simulations of the docked complexes.
Figure 4.

Flexibility and structural analysis of Aβ monomer, oligomers, and fibrils. a, RMSFs of the three Aβ aggregates. Shown are structures or clustered structures (b) and secondary structure components (c) of Aβ monomer, 5-mer, and 16-mer, respectively. Helical, β, turn, and random coil structures are colored black, red, blue, and green, respectively.
Initial docking between Aβ pentamer and crenezumab pointed to two possible patches, His13-His14-Gln15 and Ala21-Glu22-Asp23, of Aβ as antibody-binding sites. We selected five models (three with the 21–23 and two with the 13–15 at the interface; supplemental Fig. S3) for further refinement. The Aβ pentamer dissociated from crenezumab within 30–70 ns for models with the 21–23 patch as epitope, whereas complexes with the 13–15 epitope were stable through the 200-ns simulation (supplemental Fig. S4). In the stable complexes, crenezumab mainly recognizes Aβ N-terminal residues 11–22 with some contact with C-terminal residue Ile32 as well. The RMSDs of the two stable complexes with the 13–15 epitope were ∼5 Å throughout the 200-ns simulation, whereas the three complexes with the 21–23 epitope quickly reached >8 Å in ∼50 ns (supplemental Fig. S4a). The binding energy (supplemental Table S6) also indicated that crenezumab prefers to bind the positively charged 13–16 region (13HHQK16) with a binding energy of −37.2 to −39.8 kcal/mol rather than the 20–21 hydrophobic core 20FF21 patch +2.4 to +78.2 kcal/mol) of the pentamer.
The recognition mechanism of crenezumab and Aβ oligomer
Using the most stable crenezumab-Aβ oligomer model Cmab5 (−39.8 kcal/mol) as representative, we also similarly examined a possible solanezumab-Aβ oligomer (Smab5) and a chimera-Aβ oligomer (CHmab5) recognition. After 200 ns, the Aβ pentamer interaction with solanezumab rotated from the 13–16 patch to the 19FFAE22 and 32IGL34 regions, with a small binding energy (−3.9 ± 4.5 kcal/mol). Our simulations confirmed that solanezumab prefers to bind monomeric Aβ, whereas crenezumab prefers oligomeric Aβ, agreeing with the experimental results. Crenezumab can maintain a consistent binding epitope around Aβ13–15, although with different conformation, whereas solanezumab is only able to stay around Aβ21–23. Two binding modes have similar contact surface areas; the stable crenezumab·Aβ11–42 oligomer complex has a contact area of 1505.5 Å2. VL (46.7%) and VH (53.3%) bind to the Aβ pentamer with a similar contribution. The corresponding solanezumab·Aβ11–42 5-mer complex has a contact area of 1456.8 Å2. Aβ pentamer preferred to bind VH (61.4%) rather than VL (38.6%).
What are the factors responsible for the binding preference switch between monomer/oligomer for crenezumab and solanezumab? Fig. 5a highlights residues with cumulative contacts > 1, and Fig. 5b shows the residue components on the interface. Ser, Tyr, Thr, and Asn are the top ranking residues on the crenezumab-Aβ interfaces, whereas solanezumab uses Arg, Asn, and Val for the Aβ binding. Arg contributes to the binding in most of the solanezumab·Aβ pentamer complexes, whereas in the crenezumab·Aβ pentamer complex, there is only a small contribution from Arg. Hydrophobic residues and Leu contribute to the contacts in the solanezumab·Aβ pentamer complex, whereas in crenezumab·Aβ pentamer complexes, most of the preferred contacts are from hydrophilic and electrostatic interactions. Overall, crenezumab CDR-L1 and H1 contribute to the binding more than other CDRs, with very minor hydrophobic contributions (0.6 hydrophobic interactions, 13.8 hydrogen bonds, and 2.1 electrostatic interactions on the interface). The hydrogen bonds were reduced to 9.2, whereas hydrophobic interactions increased to 8.2.
Figure 5.

The epitope of Aβ oligomer shifted to the N-terminal hydrophilic and cationic residues when in complex with crenezumab and CreneFab compared with solanezumab. a, molecular details of the Aβ oligomer-crenezumab complex. Residues with cumulative contacts >1.0 are represented by sticks. Crenezumab residues Tyr251H and Asp319H (alanine scanning using SPR (23)) are highlighted by beads, whereas other important residues, Ser250H and Asn33L, which differ between crenezumab and solanezumab, are also underlined. Residues from Aβ, light chain, and heavy chain are indicated by Aβ, L, and H, respectively. Light chain, heavy chain, and Aβ oligomer are colored pink, lime, and ice blue, respectively. Hydrophobic, hydrophilic, cationic, and anionic residues are colored white, green, blue, and red, respectively. b, contact preference on the Fabs–Aβ interface from Aβ side and Fab side. Fab amino acids preference was obtained by summation of the contacts based on the Fab side contact preference.
Several CDR residues, which differed in solanezumab and crenezumab, dramatically changed the contact behavior (Fig. 5b and supplemental Table S7). In the crenezumab·Aβ pentamer complexes, Ser250(31H) interacts with the Aβ cationic region of 13HHQKL17, whereas in the solanezumab·Aβ pentamer complex, this residue was mutated to cationic Arg, causing electrostatic repulsion on the interface and a shift to 19FFAE22. Clearly, The Arg250(31H) in solanezumab prevents interaction with the highly positive charged 13HHQKL17 patch and leads solanezumab to the negatively charged Aβ 19FFAE22 patch. Other residue differences enhanced the hydrophobic interactions or hydrogen bonds at the solanezumab interface. For example, in the crenezumab·Aβ complexes, Asn273(53H) and Gly275(55H) made no contacts with the antigen; however, in the solanezumab·Aβ complex, the corresponding Val273(53H) and Asn275(55H) interact with Aβ 32IGL34. Although these two mutations increase the hydrophobic interactions and hydrogen bonds, they potentially weaken the electrostatic interactions with Aβ 13HHQKL17. The change of cumulative contacts at specific sites between solanezumab and crenezumab and the Aβ oligomer is shown in supplemental Table S7. Many residues have large contact change (Δcontact > 0.25). For example, Arg250(31H) and Asp33(33H) in solanezumab were mutated to Ser and Asn in crenezumab, leading to 1.82 and 1.75, respectively. These mutations also resulted in neighboring residue contact change (e.g. Thr247, Tyr251, Tyr31, and Gly34).
Constant domain contribution
We simulated possible structures of the CreneFab·Aβ pentamer complex, starting with a conformation identical to that of the crenezumab·Aβ pentamer complex. It was unchanged during the 100-ns simulation. The CreneFab·Aβ11–42 5-mer complex has a contact area of 1724.5 Å2. VH binding to the Aβ pentamer is preferred (60.7%) over that of VL (39.3%). Hydrogen bonds were reduced to 9.4, whereas hydrophobic interactions increased to 7.6 (Table 4). Even with identical variable domains, the binding energy of CreneFab·Aβ11–42 oligomer complex was reduced significantly compared with the wild-type crenezumab (−6.4 ± 4.5 kcal/mol; Table 3), suggesting that constant domain mutations can influence the Aβ recognition.
We clustered the Fab structures from the three complexes (Smab5, Cmab5, and CHmab5) by backbone RMSD of 4 Å. As shown in Fig. 6a, both solanezumab and CreneFab (which share the constant domains) become relatively rigid. The conformations of solanezumab form five clusters with one dominant (86%). For CreneFab, the conformations are categorized into four clusters with one dominant (94%). The conformations become more diverse for crenezumab, which has six clusters with one dominant (56%). Fig. 6c compares the RMSFs of the complexes of the three Fabs. Residues 1–110 (especially CDR-L1) in crenezumab light chain are stabilized, whereas the 120–178 fragment is more flexible compared with solanezumab. All RMSFs of the heavy chain CH1 domain are higher for crenezumab than solanezumab. We observed that crenezumab and CreneFab have lower S2 in the constant domain of light chain (Fig. 6d). These features indicated that crenezumab has tight variable domain binding and higher constant domain motion for entropy dissipation, as in the case of monomer binding.
Figure 6.

Partially ordered Aβ oligomers induce the subdomain reorientation of the crenezumab Fab to transfer the entropy upon stable antibody–antigen interface formation. a, cluster analysis of the structures. Backbone RMSD = 4 Å is used to define the cluster. Clusters are colored blue, red, gray, yellow, and orange, respectively. b, motion correlation among the residues of the three Fabs and Aβ oligomer. Residues with highly correlated or anti-correlated motion are red or blue. c and d, RMSFs (c) and order parameters S2 (d) of the three Fabs in complex with Aβ oligomer. The locations of CDRs and important constant domain loops are boxed. The curves of solanezumab, crenezumab, and CreneFab are colored black, blue, and red, respectively.
Is the motion correlation pattern found in monomer recognition conserved in oligomer binding? Compared with the solanezumab/crenezumab·Aβ monomer complexes, the Fab subdomains in solanezumab/crenezumab·Aβ oligomer complexes showed much stronger positive or negative correlation (Fig. 6b). In the crenezumab·Aβ complexes (Cmab5), there are strong negative motion correlations among VL, CL, and VH domains, whereas these correlations are reduced in the solanezumab·Aβ and CreneFab·Aβ complexes. As crenezumab has the most favorable binding energy with the Aβ oligomer, it seems that the subdomain fluctuations or flexibility is necessary for Fab to recognize larger Aβ aggregates. Moreover, in the complex of Aβ pentamer with crenezumab, there are strong positive correlations between crenezumab CDRs and the contacting residues from Aβ, whereas those Aβ residues that are far from the interface showed negative correlation with CDRs. However, in the complex of Aβ pentamer and CreneFab, both the positive and negative motion correlations were greatly reduced. We further examine the signals from the Aβ oligomer to the constant domain, especially the regions with strong response and mutations, in the three antibodies (supplemental Table S7). In most cases, there are shorter optimal pathways from Aβ Lys16 to C domain residues in crenezumab than solanezumab or CreneFab.
Which antibody structural features recognize the Aβ fibril?
We screened the binding modes of the Aβ 16-mer/crenezumab complex with the epitope of the Aβ13–15 region based on the simulations of crenezumab·Aβ pentamer. Considering the linear morphology of the Aβ fibril, three different binding orientations were tested (supplemental Fig. S5). Our simulations indicated that in models a and b, the Aβ 16-mer dissociated from crenezumab within 160 and ∼20 ns, respectively, whereas in model c, Aβ 16-mer and crenezumab form a stable complex throughout the 200-ns simulation. This indicated that the Aβ13–15 region disfavored the light chain of crenezumab. The net charge of the crenezumab light and heavy chain is +2 and 0, respectively, whereas the net charge of Aβ13–15 is +1 (His13 and His14 might also be protonated); thus, repulsion might prevent the binding between the light chain and Aβ13–15. Through extensive screening of possible complexes of Aβ fibril and crenezumab, we obtained a stable complex structure of Aβ 16-mer·crenezumab (supplemental Fig. S5), with a binding energy of −195.8 ± 6.3 kcal/mol.
The recognition mechanism of crenezumab and Aβ fibril
The stable crenezumab·Aβ11–42 16-mer complex has a contact area of 1667 Å2, and VL and VH contributed 34.1 and 65.9% contact area to the interface, mostly from CDR-H1 and H2. In this model, Crenezumab can bind the Aβ fibril on the lateral surface around residues 10–16, which constitute also the epitopes in oligomer and monomer binding. The interactions are mostly salt bridge and hydrogen bonds (Table 4 and Fig. 7a): 1) salt bridges between Lys16Aβ and Asp35L (Asn in solanezumab), Asn33L and Glu1H of crenezumab; 2) hydrogen bonds between Glu11Aβ and Asn273H (Val in solanezumab) of crenezumab; 3) multiple hydrogen bonds between Gln15Aβ and Tyr251H.
Figure 7.
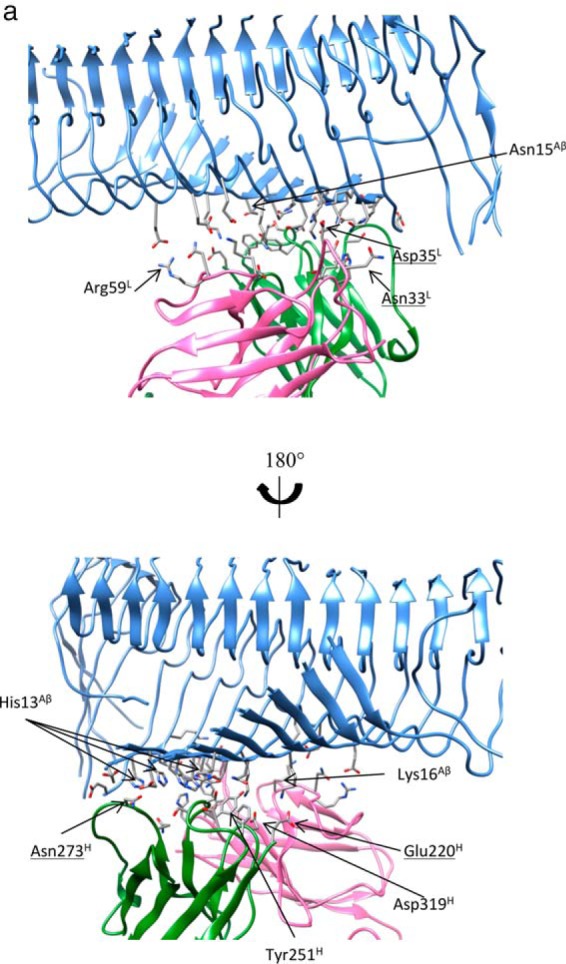

The array of N-terminal hydrophilic and cationic residues of Aβ fibrils were recognized by crenezumab with dominant salt bridges and hydrogen bonds. a, molecular details of the Aβ fibrils-crenezumab complex. Residues with cumulative contacts >1.0 are represented by sticks. Crenezumab residues Tyr251H (alanine scanning using SPR (23)) are further highlighted by beads, whereas Asn273H, Asp35L, and Asn33L, which differ between crenezumab and solanezumab, are also underlined. Residues from Aβ, light chain, and heavy chain are indicated by Aβ, L, and H, respectively. Light chain, heavy chain, and Aβ oligomer are colored pink, lime, and ice blue, respectively. Hydrophobic, hydrophilic, cationic, and anionic residues are colored white, green, blue, and red, respectively. b, contact preference on the Fabs–Aβ interface from the Aβ side and Fab side. Fab amino acid preference was obtained by summation of the contacts based on the Fab side contact preference.
This most stable conformation was used to test whether solanezumab Fab can bind Aβ fibrils. After 200-ns simulations, solanezumab is indeed able to maintain interaction with the Aβ fibril. Hydrophobic residues (e.g. Val) contribute to the contacts in solanezumab·Aβ monomer complex, whereas in crenezumab·Aβ oligomer complexes, most of the preferred contacts were from hydrophilic interactions (Fig. 7b). Although the contact area between solanezumab·Aβ11–42 16-mer (1818.7 Å2) is larger than between crenezumab·Aβ 16-mer, the binding energy is only −41.2 ± 6.3 kcal/mol, much less than the corresponding binding energy of −195.8 ± 6.3 kcal/mol of crenezumab (Table 3). The binding energy indicated that crenezumab can bind Aβ fibril better, agreeing with experimental results. Three mutations D33N(28L), V273N(53H), and N35D(30L) from solanezumab to crenezumab are potentially important in differentiating between the recognition of the Aβ fibril by the two Fabs (Table 6). Clearly, these mutations enhanced antibody-Aβ fibril interactions for crenezumab.
The expected increasing flexibility of the H1 and H2 loops due to more Gly residues in crenezumab is reflected in the Aβ fibril recognition. As shown in Fig. 8c, the RMSFs of CDR H1 and H2 loops as well as L2, L3, and L4 are larger in crenezumab than in solanezumab. The higher RMSFs in most loops may imply that higher flexibility helps in binding the amyloid fibril.
Figure 8.

Recognition of highly ordered Aβ fibrils required more flexible and dynamic Fabs to transfer the entropy from antibody–antigen complex formation. a, cluster analysis of the structures. Backbone RMSD = 4 Å is used to define the cluster. Clusters are colored blue, red, and gray, respectively. b, motion correlation among the residues of the three Fabs and Aβ fibril. Residues with highly correlated and anti-correlated motion are red and blue. c and d, RMSFs (c) and order parameters S2 (d) of the three Fabs in complex with Aβ fibril. The locations of CDRs and important constant domain loops are boxed. The curves of solanezumab, crenezumab, and CreneFab are colored black, blue, and red, respectively.
Constant domain contribution in amyloid fibril recognition
We simulated possible structures of CreneFab·Aβ 16-mer complex, with a starting conformation identical to that of the crenezumab·Aβ 16-mer complex. The complex is stable throughout the 100-ns simulation, with a much smaller contact area (887.1 Å2) and fewer hydrogen bonds (Table 4). VH binding to the Aβ 16-mer is dominantly preferred to VL's (94% versus 6%). The binding energy is lower than crenezumab but higher than solanezumab (−85.2 ± 6.0 kcal/mol; Table 3), indicating an interplay between the variable and constant domains in antigen recognition. When forming a complex with Aβ 16-mer, the population of solanezumab conformations is stably distributed, and all are in one cluster (Fig. 8a). For crenezumab, the conformations are in four clusters with one dominant (80%). For the CreneFab, the conformations fall into three clusters with populations of 81, 11, and 8%, respectively.
The motion correlations between Fabs and Aβ aggregates were also studied (Fig. 8b). The interdomain motion correlation is enhanced in the Aβ 5-mer complex and is stronger in crenezumab than in solanezumab and CreneFab in complex with Aβ 16-mer. There is a stronger Aβ-Fab correlation in crenezumab than in solanezumab/CreneFab.
The order parameter S2 showed that when binding to Aβ 16-mer, crenezumab showed higher S2 on CDRs and a lower one on constant domain loops, especially on the light chain. As can be seen in Fig. 9b, crenezumab has a regular shift of increasing S2 in CDR loops and decreasing S2 in light chain constant domain from the apo form to complexes with Aβ monomer, Aβ 5-mer, and Aβ 16-mer.
Figure 9.

Analysis of RMSFs (a) and order parameters S2 of Fab residues (b) in the different simulation systems suggested the transfer of entropy from CDRs to constant domain loops. The RMSFs/order parameters of Fabs in apo form and in complex with Aβ monomer, pentamer, and 16-mer are colored black, red, green, and blue, respectively.
We finally examined the signals from the Aβ fibril to the constant domain, especially the regions with strong response and mutations (Table 4). In most cases, like the Aβ oligomer, there are shorter optimal pathways from Aβ Lys16 to C domain residues in crenezumab than in solanezumab and CreneFab (Table 4).
Discussion
Development of high affinity recombinant anti-Aβ monoclonal antibodies (mAbs) has shown promising results (3–5). However, the key question of how antibodies recognize protein aggregates is still elusive. Available structures of monomeric Aβ-Fab complexes can be classified into three categories, depending on the targeted regions of Aβ: N-terminal (positions 2–7), mid-region (positions 12–24), and C-terminal (positions 30–40). These mAbs recognize Aβ aggregates with different tertiary structures. Only Aβ monomer-antibody structures have been solved (11, 18, 53–59), but Aβ oligomers/fibrils are the toxic species, and Aβ protofibrils are internalized by microglia more extensively than monomers (23, 60).
To elucidate the recognition mechanism of different oligomerization states in atomic detail, here we search for potential complexes between Aβ oligomers/fibrils and solanezumab/crenezumab and systematically evaluate their stabilities. We found that crenezumab consistently recognizes exposed Aβ11–16 epitopes in different oligomerization states. The sequence similarity between solanezumab and crenezumab suggested that both exploit similar epitopes in the recognition of Aβ monomers and oligomers (11, 23). The new structure of the CreneFab·Aβ monomer complex indicated that crenezumab binds to Aβ13–24, whereas solanezumab prefers Aβ16–26 (23). Consistently, our docking, modeling, and energy evaluation indicated that crenezumab recognized Aβ11–16, especially in complex with Aβ oligomers/fibrils rather than 16–26 as in solanezumab. Experimentally, crenezumab binds with aggregated Aβ species with ∼10 times lower KD than with the monomer (23). The secondary structure and exposed residues in aggregated states differ from the monomer state, all suggesting that crenezumab might recognize slightly different epitopes in the Aβ species.
Among the available ssNMR structures of Aβ fibrils (36–39), Aβ residues (Phe19, Phe20, Ser26, and Asn27) were almost buried inside the hydrophobic core (PDB code 2NAO) or in the turn region (PDB codes 2M4J and 2LMP) and thus cannot be recognized by antibodies. The Aβ 13HHQK16 region is usually exposed to bulk solution with higher flexibility compared with Aβ 19FFAEDVGSN27. Thus, the nature of the Aβ fibrils also determines the recognizable regions. Moreover, cationic residues on the antigen (e.g. Lys and His) are easier to recognize than anionic residues on the antibody–antigen interface.4 The preference of cationic residues further favored Aβ 13HHQK16. There are other antibodies and biologics specifically recognizing C-terminal residues. For example, anti-Aβ42 oligomeric VIA antibody was raised against VIAVIA peptide (61), which corresponds to the Aβ40–42 residues.
Aβ oligomer and fibril structures are highly polymorphic (46), and antibodies should have the ability to adjust their conformation to recognize them. Our analysis of Aβ12–28 in complex with three different Fabs indicated that whereas crenezumab and CreneFab can bind heterogeneous Aβ12–28 conformations, this is not the case for solanezumab. This is in line with our observation that solanezumab has stronger binding affinity to monomeric Aβ12–28 in helical conformation and fails to adapt to other conformations in Aβ oligomers and fibrils.
Essential variable domain residues shift crenezumab's epitope preference. Alanine-scanning mutagenesis of crenezumab suggested that Y251(32H)A, G318(95H)A, and D319(101H)A abolish binding (23). The three residues are the same in solanezumab and crenezumab. This suggests that the three residues are important for monomer binding, but not other Aβ species. However, several mutations (R250S(H) and S252G(H)) shift Aβ recognition from the middle to the N-terminal region. As to Aβ oligomer/fibril·crenezumab complexes, Asp35(L) and Asn273(H) make considerable contacts with Aβ 13HHQK16, whereas the corresponding residue Asn and Val in solanezumab undermines the interaction with Aβ. These residues were anionic (Asp) or hydrogen bond–forming (Asn, Ser, and Tyr) in crenezumab recognizing Aβ 13HHQK16, whereas they were cationic (Arg) or hydrophobic (Val and Phe) in solanezumab recognizing Aβ 19FFAEDVGSN27. These residues, which differ in solanezumab and crenezumab, directly interact with the Aβ aggregates.
The constant domain may affect antigen recognition through an entropy redistribution mechanism. The recognition is associated with structural transitions of the inherently flexible antibody (32, 62, 63). The variable domains, especially CDRs, mainly modulate the specificity and affinity (64), whereas the constant domains control the isotype/effector (65). However, recent studies indicated that besides the variable domains, the constant domain is also implicated in antigen binding (24–28). Communications between the variable domains of the light and heavy chains (66) as well as between the variable and constant domains (29) were unraveled, consistent with allostery-elicited conformational changes in antibodies (67). In our previous work involving a short prion peptide and an antibody with IgG2 template (31), we found that oxidation of the intermolecular disulfide bond dramatically undermines the binding affinity. In this work, we observed that CreneFab, which combines the crenezumab V domain and solanezumab C domain, showed decreased binding affinity to all Aβ species compared with wild-type crenezumab, demonstrating that the C domain influences antigen recognition. In crenezumab, there is an interchain disulfide bond between the CH1–1 loop and C terminus of the light chain but not in solanezumab and CreneFab. This disulfide bond allosterically influences prion recognition (31), suggesting its essential role in antigen conformer recognition. Two H4 loop residues, which differ between solanezumab and crenezumab (Asn-417 → Asp(H) and Gln-410 → Lys(H)), are negatively correlated (reverse synchronous motion) with the Aβ antigen and may act in the recognition.
Although there is no uniform structural change upon binding to Aβ species by solanezumab or crenezumab, there are significant light chain 160–180 and 210–220 and heavy chain 415–425 changes. Considering the RMSF, overall conformational diversities, and order parameter change, it is evident that upon the binding of Aβ, the Fab entropy is redistributed. The high entropy in the flexible CDR loops and peptide needs to be transferred to the solvent or another antibody region to avoid entropy penalty upon ligand binding. In Fabs or in the full antibody, the entropy might be transferred to the loops in the constant domain or further to the Fc region facilitating receptor binding.
The correlated variations of mutations, conformational diversities, and communication pathways suggest that binding to larger Aβ oligomers requires balance of paratope–epitope interaction, allosteric loop response, and antibody conformational dynamics.
Conclusions
Complementary to the known antibody·Aβ monomer crystal structures, for the first time we identified interaction patterns of therapeutic antibodies in recognizing Aβ oligomers/fibrils. In Aβ oligomers/fibrils, the antibodies prefer epitope residues 13–16. Crenezumab anionic and hydrogen bond–forming residues in the CDR loop are responsible for the interaction. For the larger and more organized Aβ aggregates, a conformational reorganization takes place among Fab subdomains. In solanezumab and crenezumab, constant domain loops' residues respond differently, indicating a role in Aβ aggregate recognition. Entropy transfer to these constant domain loops upon antigen binding might trigger a change in flexibility. Understanding how an antibody can recognize simultaneously a peptide monomer and oligomer has been puzzling. Unlike a previously suggested mechanism that monomers are sequestered from oligomers to enable oligomer recognition, our results suggest that the flexibility of the antibody CDR region is the key in recognition of the same epitope region with different antibody conformations (i.e. conformational selection (18, 68–71) is a decisive factor).
Experimental procedures
System preparation
The structures of the bound and apo form of the CreneFab were directly obtained from the crystal structures of 5VZX and 5VZY (23). For solanezumab, as there are two complexes packed in the asymmetric unit of the crystal (PDB code 4XXD) of solanezumab·Aβ complex with slight structure variation (11), we considered both complexes. The unbound (apo) form of solanezumab was obtained by manually removing the Aβ peptide in the bound (PDB code 4XXD) structure.
Although there is no structural information available for crenezumab, as it showed high sequence similarity (>97%) (Table 2) compared with CreneFab, the structures of the bound and apo form of the crenezumab were modeled by template-based homology modeling using the SWISS-MODEL server with the CreneFab structures as templates. The missing residues were modeled by template-based homology modeling using the SWISS-MODEL server (72).
Solid-state NMR structures (PDB code 2MXU) (51) were used as two representative fibril (16-mer)/oligomeric (5-mer) structures of Aβ. To search the potential interfaces between Aβ oligomers/fibrils and the three Fabs, we performed automated molecular docking of Aβ oligomers/fibrils and Fabs with the program HADDOCK version 2.1 web server (73). HADDOCK can use various kinds of ambiguous interaction restraints to guide the docking process. In this work, the epitope of Aβ oligomers is 12–28, whereas the paratopes of solanezumab/crenezumab were set as CDR loops. In the HADDOCK docking, 12–28 of Aβ and CDR loops of Fabs were input as active residues; Aβ oligomers/fibrils were set rigid, whereas the residues of CDR loops of Fabs, which were in the loop conformation according to the crystal structures (i.e. position 26–37, 55–58, 95–102, 245–253, 270–277, and 318–320) were set flexible.
For the crenezumab·Aβ oligomer/fibril complex, the analysis of the final 200 HADDOCK models of complexes resulted in five and three clusters for crenezumab·Aβ-5-mer and crenezumab·Aβ-16-mer, respectively. To further refine these complexes between Fabs and Aβ in different states, the initial poses from HADDOCK were locally perturbed by Rosetta docking (74–77). The poses with larger cluster size, lower Z-score and total_score/I_sc were considered promising candidates. The complexes between Aβ oligomers/fibrils and solanezumab or CreneFab were obtained by mutations of crenezumab back to the solanezumab or CreneFab sequences using the crenezumab·Aβ oligomer/fibril complex.
MD simulation protocols
The conserved disulfide bonds were constructed according to the specific IgG subtypes. As the non-sequential Kabat numbering scheme is used in the crystal structures, we renumber the residues for convenience in the simulation (see supplemental Table S2). For the light chain, the heavy chain, and the peptide, the N termini and C termini were charged as NH3+ and COO− groups, respectively. The crystal water molecules in the crystal structures were kept. The systems were then solvated by TIP3P water molecules, and sodium and chlorides were added to neutralize the system and to achieve a total concentration of ∼150 mm. The resulting solvated systems were energy-minimized for 5000 conjugate gradient steps, where the protein was fixed and water molecules and counterions could move, followed by an additional 5000 conjugate gradient steps, where all atoms could move. In the equilibration stage, each system was gradually relaxed by performing a series of dynamic cycles, in which the harmonic restraints on proteins were gradually removed to optimize the protein–water interactions. In the production stage, all simulations were performed using the NPT ensemble at 310 K. All MD simulations were performed using the NAMD software (78) with CHARMM36 force field (79). MD trajectories were saved by every 2 ps for analysis. A summary of all simulation systems is given in supplemental Table S6 and supplemental Table S9.
MD simulation analysis
To identify the essential interactions between amyloid and Fabs, all atoms within 3 Å between amyloid and the Fabs during the last 100-ns simulation were considered as input into PROTMAP2D (80), which can calculate the accumulated contact map by summing up all of the frames during simulations.
To evaluate the binding energy between Fab and the prion peptide, the trajectory for each bound and apo system was extracted from the last 20 ns of explicit solvent MD without water molecules and ions. The solvation energies of all systems were calculated using the generalized Born method with molecular volume (GBMV) (81) after 500 steps of energy minimization to relax the local geometries caused by the thermal fluctuations that occurred in the MD simulations. In the GBMV calculation, the dielectric constant of water is set to 80, and no distance cutoff is used. The binding energy between the two Fabs and the Aβ species was calculated by the equation, 〈Ebind〉 = 〈Ecomplex〉 − 〈EFab〉 − 〈EAβ〉. Binding energy is summarized in supplemental Fig. S6.
Correlations between all the residues in the 12 systems were analyzed for the entire 100-ns MD trajectory (25,000 frames) using the normalized covariance to characterize the correlation in motion of protein residues (82–85), ranging from −1 to 1. If two residues move in the same (opposite) direction in most the frames, the motion is considered as (anti-)correlated, and the correlation value is close to −1 or 1. If the correlation value between two residues is close to zero, they are generally uncorrelated. The correlation evaluation was performed by using CARMA (52). The weighted network, optimal/suboptimal paths in Fab/peptide systems were analyzed using the NetworkView (6) module in VMD.
Generalized order parameter S2 of the carbonyl group of each individual antibody residue in the 12 systems was analyzed for the last 20-ns MD trajectory (5000 frames) using the CHARMM NMR analysis module, ranging from 0 to 1. Higher value indicated a more ordered structure.
Author contributions
J. Z. performed experiments and wrote the paper. R. N. wrote the paper. B. M. conceived and coordinated the study and wrote the paper. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
All simulations were performed using the high-performance computational facilities of the Biowulf PC/Linux cluster at the National Institutes of Health (Bethesda, MD).
This work was supported in whole or in part by Federal funds from the NCI, National Institutes of Health, under Contract HHSN261200800001E. This work was also supported in part by the Intramural Research Program of the NCI, National Institutes of Health, Center for Cancer Research. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains supplemental Figs. S1–S6 and Tables S1–S9.
M. Wang, D. Zhu, J. Zhu, R. Nussinov, and B. Ma, unpublished data.
- AD
- Alzheimer's disease
- Aβ
- amyloid-β
- Fab
- antigen-binding fragment
- MD
- molecular dynamics
- V domain
- antibody variable domain
- C domain
- antibody constant domain
- VL
- light chain variable domain
- CL
- light chain constant domain
- VH
- heavy chain variable domain
- CH1
- heavy chain constant domain-1
- GBMV
- generalized Born method with molecular volume
- RMSD
- root mean square deviation
- RMSF
- root mean square fluctuation
- PDB
- Protein Data Bank
- ssNMR
- solid-state NMR.
References
- 1. Ballard C., Gauthier S., Corbett A., Brayne C., Aarsland D., and Jones E. (2011) Alzheimer's disease. Lancet 377, 1019–1031 [DOI] [PubMed] [Google Scholar]
- 2. Hardy J. A., and Higgins G. A. (1992) Alzheimer's disease: the amyloid cascade hypothesis. Science 256, 184–185 [DOI] [PubMed] [Google Scholar]
- 3. De Genst E., Messer A., and Dobson C. M. (2014) Antibodies and protein misfolding: from structural research tools to therapeutic strategies. Biochim. Biophys. Acta 1844, 1907–1919 [DOI] [PubMed] [Google Scholar]
- 4. Herrmann A., and Spires-Jones T. (2015) Clearing the way for tau immunotherapy in Alzheimer's disease. J. Neurochem. 132, 1–4 [DOI] [PubMed] [Google Scholar]
- 5. Hardy J., Bogdanovic N., Winblad B., Portelius E., Andreasen N., Cedazo-Minguez A., and Zetterberg H. (2014) Pathways to Alzheimer's disease. J. Intern. Med. 275, 296–303 [DOI] [PubMed] [Google Scholar]
- 6. Eargle J., and Luthey-Schulten Z. (2012) NetworkView: 3D display and analysis of protein·RNA interaction networks. Bioinformatics 28, 3000–3001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dubois B., Feldman H. H., Jacova C., Cummings J. L., Dekosky S. T., Barberger-Gateau P., Delacourte A., Frisoni G., Fox N. C., Galasko D., Gauthier S., Hampel H., Jicha G. A., Meguro K., O'Brien J., et al. (2010) Revising the definition of Alzheimer's disease: a new lexicon. Lancet Neurol. 9, 1118–1127 [DOI] [PubMed] [Google Scholar]
- 8. Albert M. S., DeKosky S. T., Dickson D., Dubois B., Feldman H. H., Fox N. C., Gamst A., Holtzman D. M., Jagust W. J., Petersen R. C., Snyder P. J., Carrillo M. C., Thies B., and Phelps C. H. (2011) The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 7, 270–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sevigny J., Chiao P., Bussière T., Weinreb P. H., Williams L., Maier M., Dunstan R., Salloway S., Chen T., Ling Y., O'Gorman J., Qian F., Arastu M., Li M., Chollate S., et al. (2016) The antibody aducanumab reduces Aβ plaques in Alzheimer's disease. Nature 537, 50–56 [DOI] [PubMed] [Google Scholar]
- 10. Mangialasche F., Solomon A., Winblad B., Mecocci P., and Kivipelto M. (2010) Alzheimer's disease: clinical trials and drug development. Lancet Neurol. 9, 702–716 [DOI] [PubMed] [Google Scholar]
- 11. Crespi G. A., Hermans S. J., Parker M. W., and Miles L. A. (2015) Molecular basis for mid-region amyloid-β capture by leading Alzheimer's disease immunotherapies. Sci. Rep. 5, 9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ultsch M., Li B., Maurer T., Mathieu M., Adolfsson O., Muhs A., Pfeifer A., Pihlgren M., Bainbridge T. W., Reichelt M., Ernst J. A., Eigenbrot C., Fuh G., Atwal J. K., Watts R. J., and Wang W. (2016) Structure of Crenezumab complex with Aβ shows loss of β-hairpin. Sci. Rep. 6, 39374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Algamal M., Milojevic J., Jafari N., Zhang W., and Melacini G. (2013) Mapping the interactions between the Alzheimer's Aβ-peptide and human serum albumin beyond domain resolution. Biophys. J. 105, 1700–1709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Milojevic J., Raditsis A., and Melacini G. (2009) Human serum albumin inhibits Aβ fibrillization through a “monomer-competitor” mechanism. Biophys. J. 97, 2585–2594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Milojevic J., and Melacini G. (2011) Stoichiometry and affinity of the human serum albumin-Alzheimer's Aβ peptide interactions. Biophys. J. 100, 183–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Milojevic J., Costa M., Ortiz A. M., Jorquera J. I., and Melacini G. (2014) In vitro amyloid-β binding and inhibition of amyloid-β self-association by therapeutic albumin. J. Alzheimers Dis. 38, 753–765 [DOI] [PubMed] [Google Scholar]
- 17. Raditsis A. V., Milojevic J., and Melacini G. (2013) Aβ association inhibition by transferrin. Biophys. J. 105, 473–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ma B., Zhao J., and Nussinov R. (2016) Conformational selection in amyloid-based immunotherapy: survey of crystal structures of antibody-amyloid complexes. Biochim. Biophys. Acta 1860, 2672–2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhao J., Ma B., and Nussinov R. (2016) Compilation and analysis of enzymes, engineered antibodies, and nanoparticles designed to interfere with amyloid-β aggregation. Isr. J. Chem. 57, 622–633 [Google Scholar]
- 20. Helms L. R., and Wetzel R. (1996) Specificity of abnormal assembly in immunoglobulin light chain deposition disease and amyloidosis. J. Mol. Biol. 257, 77–86 [DOI] [PubMed] [Google Scholar]
- 21. Legleiter J., Czilli D. L., Gitter B., DeMattos R. B., Holtzman D. M., and Kowalewski T. (2004) Effect of different anti-Aβ antibodies on Aβ fibrillogenesis as assessed by atomic force microscopy. J. Mol. Biol. 335, 997–1006 [DOI] [PubMed] [Google Scholar]
- 22. Adolfsson O., Pihlgren M., Toni N., Varisco Y., Buccarello A. L., Antoniello K., Lohmann S., Piorkowska K., Gafner V., Atwal J. K., Maloney J., Chen M., Gogineni A., Weimer R. M., Mortensen D. L., et al. (2012) An effector-reduced anti-β-amyloid (Abeta) antibody with unique Aβ binding properties promotes neuroprotection and glial engulfment of Aβ. J. Neurosci. 32, 9677–9689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wall J., Schell M., Murphy C., Hrncic R., Stevens F. J., and Solomon A. (1999) Thermodynamic instability of human λ6 light chains: correlation with fibrillogenicity. Biochemistry 38, 14101–14108 [DOI] [PubMed] [Google Scholar]
- 24. Adachi M., Kurihara Y., Nojima H., Takeda-Shitaka M., Kamiya K., and Umeyama H. (2003) Interaction between the antigen and antibody is controlled by the constant domains: normal mode dynamics of the HEL–HyHEL-10 complex. Protein Sci. 12, 2125–2131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pritsch O., Hudry-Clergeon G., Buckle M., Petillot Y., Bouvet J. P., Gagnon J., and Dighiero G. (1996) Can immunoglobulin C (H) 1 constant region domain modulate antigen binding affinity of antibodies? J. Clin. Invest. 98, 2235–2243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dam T. K., Torres M., Brewer C. F., and Casadevall A. (2008) Isothermal titration calorimetry reveals differential binding thermodynamics of variable region-identical antibodies differing in constant region for a univalent ligand. J. Biol. Chem. 283, 31366–31370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tudor D., Yu H., Maupetit J., Drillet A.-S., Bouceba T., Schwartz-Cornil I., Lopalco L., Tuffery P., and Bomsel M. (2012) Isotype modulates epitope specificity, affinity, and antiviral activities of anti–HIV-1 human broadly neutralizing 2F5 antibody. Proc. Natl. Acad. Sci. U.S.A. 109, 12680–12685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li T., Tracka M. B., Uddin S., Casas-Finet J., Jacobs D. J., and Livesay D. R. (2015) Rigidity emerges during antibody evolution in three distinct antibody systems: evidence from QSFR analysis of Fab fragments. PLoS Comput. Biol. 11, e1004327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Janda A., and Casadevall A. (2010) Circular dichroism reveals evidence of coupling between immunoglobulin constant and variable region secondary structure. Mol. Immunol. 47, 1421–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sela-Culang I., Kunik V., and Ofran Y. (2013) The structural basis of antibody-antigen recognition. Front. Immunol. 4, 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhao J., Nussinov R., and Ma B. (2017) Allosteric control of antibody-prion recognition through oxidation of a disulfide bond between the CH and CL chains. Protein Eng. Des. Sel. 30, 67–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Keskin O. (2007) Binding induced conformational changes of proteins correlate with their intrinsic fluctuations: a case study of antibodies. BMC Struct. Biol. 7, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. James L. C., Roversi P., and Tawfik D. S. (2003) Antibody multispecificity mediated by conformational diversity. Science 299, 1362–1367 [DOI] [PubMed] [Google Scholar]
- 34. Panza F., Logroscino G., Imbimbo B. P., and Solfrizzi V. (2014) Is there still any hope for amyloid-based immunotherapy for Alzheimer's disease? Curr. Opin. Psychiatry 27, 128–137 [DOI] [PubMed] [Google Scholar]
- 35. Sharp K. A., O'Brien E., Kasinath V., and Wand A. J. (2015) On the relationship between NMR-derived amide order parameters and protein backbone entropy changes. Proteins 83, 922–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lührs T., Ritter C., Adrian M., Riek-Loher D., Bohrmann B., Döbeli H., Schubert D., and Riek R. (2005) 3D structure of Alzheimer's amyloid-β(1–42) fibrils. Proc. Natl. Acad. Sci. U.S.A. 102, 17342–17347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Petkova A. T., Yau W. M., and Tycko R. (2006) Experimental constraints on quaternary structure in Alzheimer's β-amyloid fibrils. Biochemistry 45, 498–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lu J. X., Qiang W., Yau W. M., Schwieters C. D., Meredith S. C., and Tycko R. (2013) Molecular structure of β-amyloid fibrils in Alzheimer's disease brain tissue. Cell 154, 1257–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wälti M. A., Ravotti F., Arai H., Glabe C. G., Wall J. S., Böckmann A., Güntert P., Meier B. H., and Riek R. (2016) Atomic-resolution structure of a disease-relevant Aβ(1–42) amyloid fibril. Proc. Natl. Acad. Sci. U.S.A. 113, E4976–E4984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Perfetti V., Casarini S., Palladini G., Vignarelli M. C., Klersy C., Diegoli M., Ascari E., and Merlini G. (2002) Analysis of V(λ)-J(λ) expression in plasma cells from primary (AL) amyloidosis and normal bone marrow identifies 3r (λIII) as a new amyloid-associated germline gene segment. Blood 100, 948–953 [DOI] [PubMed] [Google Scholar]
- 41. Abraham R. S., Geyer S. M., Price-Troska T. L., Allmer C., Kyle R. A., Gertz M. A., and Fonseca R. (2003) Immunoglobulin light chain variable (V) region genes influence clinical presentation and outcome in light chain-associated amyloidosis (AL). Blood 101, 3801–3808 [DOI] [PubMed] [Google Scholar]
- 42. Solomon A., Frangione B., and Franklin E. C. (1982) Bence Jones proteins and light chains of immunoglobulins. Preferential association of the V λ VI subgroup of human light chains with amyloidosis AL (λ). J. Clin. Invest. 70, 453–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ozaki S., Abe M., Wolfenbarger D., Weiss D. T., and Solomon A. (1994) Preferential expression of human λ-light-chain variable-region subgroups in multiple myeloma, AL amyloidosis, and Waldenstrom's macroglobulinemia. Clin. Immunol. Immunopathol. 71, 183–189 [DOI] [PubMed] [Google Scholar]
- 44. Baden E. M., Owen B. A., Peterson F. C., Volkman B. F., Ramirez-Alvarado M., and Thompson J. R. (2008) Altered dimer interface decreases stability in an amyloidogenic protein. J. Biol. Chem. 283, 15853–15860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. del Pozo Yauner L., Ortiz E., Sánchez R., Sánchez-López R., Güereca L., Murphy C. L., Allen A., Wall J. S., Fernández-Velasco D. A., Solomon A., and Becerril B. (2008) Influence of the germline sequence on the thermodynamic stability and fibrillogenicity of human λ6 light chains. Proteins 72, 684–692 [DOI] [PubMed] [Google Scholar]
- 46. Miller Y., Ma B., and Nussinov R. (2010) Polymorphism in Alzheimer Aβ amyloid organization reflects conformational selection in a rugged energy landscape. Chem. Rev. 110, 4820–4838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Poshusta T. L., Sikkink L. A., Leung N., Clark R. J., Dispenzieri A., and Ramirez-Alvarado M. (2009) Mutations in specific structural regions of immunoglobulin light chains are associated with free light chain levels in patients with AL amyloidosis. PLoS One 4, e5169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Olsen K. E., Sletten K., and Westermark P. (1998) Extended analysis of AL-amyloid protein from abdominal wall subcutaneous fat biopsy: κIV immunoglobulin light chain. Biochem. Biophys. Res. Commun. 245, 713–716 [DOI] [PubMed] [Google Scholar]
- 49. Hooft C., and van Acker K. J. (1973) Effects of long-term treatment with anticonvulsant drugs. J. Pediatr. 82, 1097–1098 [DOI] [PubMed] [Google Scholar]
- 50. NCBI Resource Coordinators (2016) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 44, D7–D19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xiao Y., Ma B., McElheny D., Parthasarathy S., Long F., Hoshi M., Nussinov R., and Ishii Y. (2015) Aβ(1–42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer's disease. Nat. Struct. Mol. Biol. 22, 499–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Glykos N. M. (2006) Software news and updates carma: a molecular dynamics analysis program. J. Comput. Chem. 27, 1765–1768 [DOI] [PubMed] [Google Scholar]
- 53. Gardberg A. S., Dice L. T., Ou S., Rich R. L., Helmbrecht E., Ko J., Wetzel R., Myszka D. G., Patterson P. H., and Dealwis C. (2007) Molecular basis for passive immunotherapy of Alzheimer's disease. Proc. Natl. Acad. Sci. U.S.A. 104, 15659–15664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Miles L. A., Wun K. S., Crespi G. A., Fodero-Tavoletti M. T., Galatis D., Bagley C. J., Beyreuther K., Masters C. L., Cappai R., McKinstry W. J., Barnham K. J., and Parker M. W. (2008) Amyloid-β–anti-amyloid-β complex structure reveals an extended conformation in the immunodominant B-cell epitope. J. Mol. Biol. 377, 181–192 [DOI] [PubMed] [Google Scholar]
- 55. Basi G. S., Feinberg H., Oshidari F., Anderson J., Barbour R., Baker J., Comery T. A., Diep L., Gill D., Johnson-Wood K., Goel A., Grantcharova K., Lee M., Li J., Partridge A., et al. (2010) Structural correlates of antibodies associated with acute reversal of amyloid β-related behavioral deficits in a mouse model of Alzheimer disease. J. Biol. Chem. 285, 3417–3427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Miles L. A., Crespi G. A., Doughty L., and Parker M. W. (2013) Bapineuzumab captures the N-terminus of the Alzheimer's disease amyloid-β peptide in a helical conformation. Sci. Rep. 3, 1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. La Porte S. L., Bollini S. S., Lanz T. A., Abdiche Y. N., Rusnak A. S., Ho W. H., Kobayashi D., Harrabi O., Pappas D., Mina E. W., Milici A. J., Kawabe T. T., Bales K., Lin J. C., and Pons J. (2012) Structural basis of C-terminal β-amyloid peptide binding by the antibody ponezumab for the treatment of Alzheimer's disease. J. Mol. Biol. 421, 525–536 [DOI] [PubMed] [Google Scholar]
- 58. Bohrmann B., Baumann K., Benz J., Gerber F., Huber W., Knoflach F., Messer J., Oroszlan K., Rauchenberger R., Richter W. F., Rothe C., Urban M., Bardroff M., Winter M., Nordstedt C., and Loetscher H. (2012) Gantenerumab: a novel human anti-Aβ antibody demonstrates sustained cerebral amyloid-β binding and elicits cell-mediated removal of human amyloid-β. J. Alzheimers Dis. 28, 49–69 [DOI] [PubMed] [Google Scholar]
- 59. Feinberg H., Saldanha J. W., Diep L., Goel A., Widom A., Veldman G. M., Weis W. I., Schenk D., and Basi G. S. (2014) Crystal structure reveals conservation of amyloid-β conformation recognized by 3D6 following humanization to bapineuzumab. Alzheimers Res. Ther. 6, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kim Y., Wall J. S., Meyer J., Murphy C., Randolph T. W., Manning M. C., Solomon A., and Carpenter J. F. (2000) Thermodynamic modulation of light chain amyloid fibril formation. J. Biol. Chem. 275, 1570–1574 [DOI] [PubMed] [Google Scholar]
- 61. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 [DOI] [PubMed] [Google Scholar]
- 62. Thielges M. C., Zimmermann J., Yu W., Oda M., and Romesberg F. E. (2008) Exploring the energy landscape of antibody- antigen complexes: protein dynamics, flexibility, and molecular recognition. Biochemistry 47, 7237–7247 [DOI] [PubMed] [Google Scholar]
- 63. Li T., Tracka M. B., Uddin S., Casas-Finet J., Jacobs D. J., and Livesay D. R. (2014) Redistribution of flexibility in stabilizing antibody fragment mutants follows Le Chatelier's principle. PLoS One 9, e92870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mian I. S., Bradwell A. R., and Olson A. J. (1991) Structure, function and properties of antibody binding sites. J. Mol. Biol. 217, 133–151 [DOI] [PubMed] [Google Scholar]
- 65. Torres M., and Casadevall A. (2008) The immunoglobulin constant region contributes to affinity and specificity. Trends Immunol. 29, 91–97 [DOI] [PubMed] [Google Scholar]
- 66. Pellequer J. L., Chen S. W., Roberts V. A., Tainer J. A., and Getzoff E. D. (1999) Unraveling the effect of changes in conformation and compactness at the antibody VL-VH interface upon antigen binding. J. Mol. Recognit. 12, 267–275 [DOI] [PubMed] [Google Scholar]
- 67. Sela-Culang I., Alon S., and Ofran Y. (2012) A systematic comparison of free and bound antibodies reveals binding-related conformational changes. J. Immunol. 189, 4890–4899 [DOI] [PubMed] [Google Scholar]
- 68. Ma B., Kumar S., Tsai C. J., and Nussinov R. (1999) Folding funnels and binding mechanisms. Protein Eng. 12, 713–720 [DOI] [PubMed] [Google Scholar]
- 69. Ma B., Shatsky M., Wolfson H. J., and Nussinov R. (2002) Multiple diverse ligands binding at a single protein site: a matter of pre-existing populations. Protein Sci. 11, 184–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ma B., and Nussinov R. (2012) Selective molecular recognition in amyloid growth and transmission and cross-species barriers. J. Mol. Biol. 421, 172–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nussinov R., Ma B., and Tsai C. J. (2014) Multiple conformational selection and induced fit events take place in allosteric propagation. Biophys. Chem. 186, 22–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Schwede T., Kopp J., Guex N., and Peitsch M. C. (2003) SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31, 3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. de Vries S. J., van Dijk M., and Bonvin A. M. (2010) The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 5, 883–897 [DOI] [PubMed] [Google Scholar]
- 74. Gray J. J., Moughon S., Wang C., Schueler-Furman O., Kuhlman B., Rohl C. A., and Baker D. (2003) Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J. Mol. Biol. 331, 281–299 [DOI] [PubMed] [Google Scholar]
- 75. Wang C., Schueler-Furman O., and Baker D. (2005) Improved side-chain modeling for protein-protein docking. Protein Sci. 14, 1328–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang C., Bradley P., and Baker D. (2007) Protein-protein docking with backbone flexibility. J. Mol. Biol. 373, 503–519 [DOI] [PubMed] [Google Scholar]
- 77. Chaudhury S., Berrondo M., Weitzner B. D., Muthu P., Bergman H., and Gray J. J. (2011) Benchmarking and analysis of protein docking performance in Rosetta v3.2. PLoS One 6, e22477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Kale L., Skeel R., Bhandarkar M., Brunner R., Gursoy A., Krawetz N., Phillips J., Shinozaki A., Varadarajan K., and Schulten K. (1999) NAMD2: greater scalability for parallel molecular dynamics. J. Comput. Phys. 151, 283–312 [Google Scholar]
- 79. MacKerell A. D., Bashford D., Bellott M., Dunbrack R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T. K., Mattos C., et al. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 [DOI] [PubMed] [Google Scholar]
- 80. Pietal M. J., Tuszynska I., and Bujnicki J. M. (2007) PROTMAP2D: visualization, comparison and analysis of 2D maps of protein structure. Bioinformatics 23, 1429–1430 [DOI] [PubMed] [Google Scholar]
- 81. Lee M. S., Feig M., Salsbury F. R. Jr., and Brooks C. L. 3rd (2003) New analytic approximation to the standard molecular volume definition and its application to generalized Born calculations. J. Comput. Chem. 24, 1348–1356 [DOI] [PubMed] [Google Scholar]
- 82. Ichiye T., and Karplus M. (1991) Collective motions in proteins: a covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins 11, 205–217 [DOI] [PubMed] [Google Scholar]
- 83. Hünenberger P. H., Mark A. E., and van Gunsteren W. F. (1995) Fluctuation and cross-correlation analysis of protein motions observed in nanosecond molecular dynamics simulations. J. Mol. Biol. 252, 492–503 [DOI] [PubMed] [Google Scholar]
- 84. Young M. A., Gonfloni S., Superti-Furga G., Roux B., and Kuriyan J. (2001) Dynamic coupling between the SH2 and SH3 domains of c-Src and Hck underlies their inactivation by C-terminal tyrosine phosphorylation. Cell 105, 115–126 [DOI] [PubMed] [Google Scholar]
- 85. Tai K., Shen T., Börjesson U., Philippopoulos M., and McCammon J. A. (2001) Analysis of a 10-ns molecular dynamics simulation of mouse acetylcholinesterase. Biophys. J. 81, 715–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.