

Abstract
Infections account for a major cause of death throughout the developing world. This is mainly due to the emergence of newer infectious agents and more specifically due to the appearance of antimicrobial resistance. With time, the bacteria have become smarter and along with it, massive imprudent usage of antibiotics in clinical practice has resulted in resistance of bacteria to antimicrobial agents. The antimicrobial resistance is recognized as a major problem in the treatment of microbial infections. The biochemical resistance mechanisms used by bacteria include the following: antibiotic inactivation, target modification, altered permeability, and “bypass” of metabolic pathway. Determination of bacterial resistance to antibiotics of all classes (phenotypes) and mutations that are responsible for bacterial resistance to antibiotics (genetic analysis) are helpful. Better understanding of the mechanisms of antibiotic resistance will help clinicians regarding usage of antibiotics in different situations. This review discusses the mechanism of action and resistance development in commonly used antimicrobials.
Keywords: Antibiotics, antimicrobial resistance, bacterial cell wall, mechanism of action
Introduction
The struggle of mankind against infectious diseases is well known. The discovery of antibiotics led to optimism that infections can be controlled and prevented. However, infections are still the leading cause of death in developing world. This is due to the emergence of new disease, reemergence of diseases once controlled and more specifically due to the appearance of antimicrobial resistance. It appears that the emergence of antimicrobial resistance is inevitable to almost every new drug, and it is recognized as a major problem in the treatment of microbial infections in both hospitals and community. This review intends to discuss the mechanism of action and resistance development in commonly used antimicrobials. For this purpose, we need to know the basic anatomy of bacterial cell, classification of antibiotics based on their mechanism of action, mechanisms of antibiotic resistance, and individual antibiotics with their common mechanism of resistance.
Basic Anatomy of Bacterial Cell
The Gram-positive bacteria consists of cytoplasmic membrane surrounded by a tough and rigid mesh called cell wall. In contrast, Gram-negative bacteria consist of thin cell wall that is surrounded by second lipid membrane called outer membrane (OM). The space between the OM and cytoplasmic membrane is referred as periplasm [Figure 1]. The OM is an additional protective layer in Gram-negative bacteria and prevents many substances from entering into the bacterium. However, this membrane contains channels called porins, which allow the entry of various molecules such as drugs.[1] The cell wall is a tough layer that gives bacterium a characteristic shape and prevents it from osmotic and mechanical stresses. The cytoplasmic membrane prevents ions from flowing into or out of the cell and maintains the cytoplasmic and bacterial components in a defined space.
Figure 1.
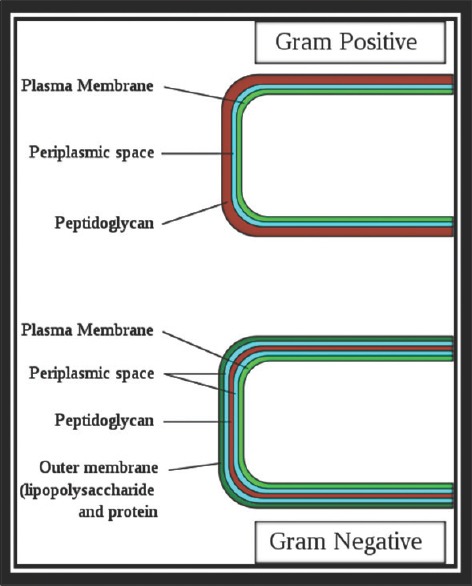
Structure of bacterial cell envelope
Classification of Antibiotics on the Basis of Mechanism of Action
The antibiotics are classified on the basis of mechanism of action as described in Figure 2.
Figure 2.
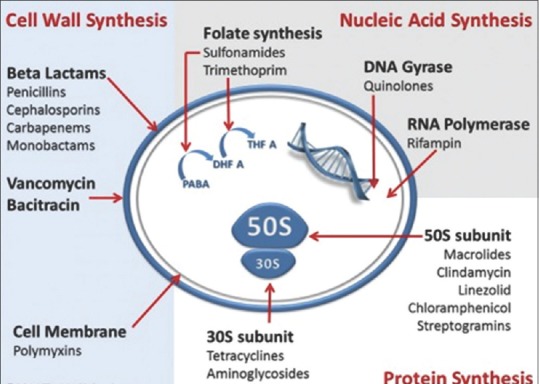
Mechanism of action of antibiotics
Antibiotics targeting cell wall
Bacterial cells are surrounded by a cell wall made of peptidoglycan, which consists of long sugar polymers. The peptidoglycan undergoes cross-linking of the glycan strands by the action of transglycosidases, and the peptide chains extend from the sugars in the polymers and form cross links, one peptide to another.[2] The D-alanyl-alanine portion of peptide chain is cross linked by glycine residues in the presence of penicillin binding proteins (PBPs).[3] This cross-linking strengthens the cell wall. β-lactams and the glycopeptides inhibit cell wall synthesis.
Beta-lactam antibiotics
The primary targets of the β-lactam agents are the PBPs. It has been hypothesized that the β-lactam ring mimics the D-alanyl D-alanine portion of peptide chain that is normally bound by PBP. The PBP interacts with β-lactam ring and are not available for the synthesis of new peptidoglycan. The disruption of peptidoglycan layer leads to the lysis of bacterium [Figure 3].[4]
Figure 3.
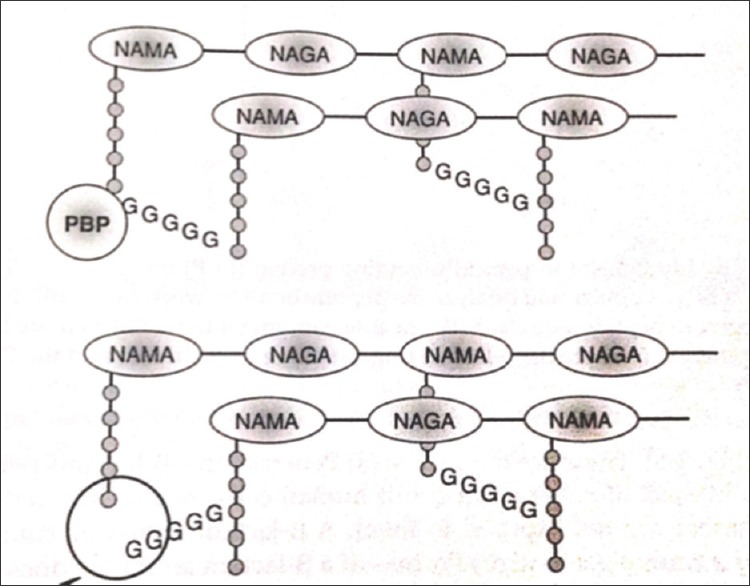
Mechanism of action of β-lactam antibiotics
Glycopeptides
The glycopeptides binds to D-alanyl D-alanine portion of peptide side chain of the precursor peptidoglycan subunit. The large drug molecule vancomycin prevents binding of this D-alanyl subunit with the PBP, and hence inhibits cell wall synthesis [Figure 3].[4,5]
Inhibitors of protein biosynthesis
First the information in bacterial DNA is used to synthesize an RNA molecule referred to as messenger RNA (m-RNA) a process known as transcription [Figure 4]. Then, the macromolecular structure called ribosome synthesizes proteins present in m-RNA, a process called translation. Protein biosynthesis is catalyzed by ribosomes and cytoplasmic factors. The bacterial 70S ribosome is composed of two ribonucleoprotein subunits, the 30S and 50S subunits.[6] Antimicrobials inhibit protein biosynthesis by targeting the 30S or 50S subunit of the bacterial ribosome.[7,8]
Figure 4.

Site of action of protein biosynthesis inhibitors
Inhibitors of 30S subunit
Aminoglycosides
The aminoglycosides (AG's) are positively-charged molecules which attach to the OM which is negatively charged leading to formation of large pores, and thus allow antibiotic penetration inside the bacterium. The main target of action is bacterial ribosome; to enter, there it must pass through cytoplasmic membrane requiring energy dependent active bacterial transport mechanism, which requires oxygen and an active proton motive force. For these reasons, AG work in aerobic conditions and have poor activity against anaerobic bacteria. These AG have synergism with those antibiotics, which inhibit cell wall synthesis (such as β-lactam and glycopeptides) as it allows greater penetration of AG within the cell and at low dosages. AG's interact with the 16S r-RNA of the 30S subunit near the A site through hydrogen bonds. They cause misreading and premature termination of translation of mRNA.
Tetracyclines
Tetracyclines, such as tetracycline, chlortetracycline, doxycycline, or minocycline, act upon the conserved sequences of the 16S r-RNA of the 30S ribosomal subunit to prevent binding of t-RNA to the A site.[6,9]
Inhibitors of 50S subunit
Chloramphenicol
It interacts with the conserved sequences of the peptidyl transferase cavity of the 23S r-RNA of the 50S subunit. Hence, it inhibits the protein synthesis by preventing binding of t-RNA to the A site of the ribosome.[6,7]
Macrolides
These affect the early stage of protein synthesis, namely translocation, by targeting the conserved sequences of the peptidyl transferase center of the 23S r-RNA of the 50S ribosomal subunit.[6,9] This results in a premature detachment of incomplete peptide chains. Macrolides, lincosamides, and streptogramins B show a similar mechanism of action.
Oxazolidinones
Linezolid is a recently approved member of novel class of antibiotic of this group which is completely synthetic. Oxazolidinones interfere with protein synthesis at several stages: (i) inhibit protein synthesis by binding to 23Sr RNA of the 50S subunit and (ii) suppress 70S inhibition and interact with peptidyl-t-RNA.[10,11]
Inhibitors of DNA replication
Quinilones
The fluoroquinolones (FQ) inhibit the enzyme bacterial DNA gyrase, which nicks the double-stranded DNA, introduces negative supercoils and then reseals the nicked ends. This is necessary to prevent excessive positive supercoiling of the strands when they separate to permit replication or transcription. The DNA gyrase consists of two A subunits and two B subunits. A subunit carries out the nicking of DNA, B subunit introduces negative supercoils, and then A subunit reseal the strands. The FQ's bind to A subunit with high affinity and interfere with its strand cutting and resealing function. In Gram-positive bacteria, the major target of action is topoisomerase IV which nicks and separate's daughter DNA strand after DNA replication. Greater affinity for this enzyme may confer higher potency against Gram-positive bacteria. In place of DNA gyrase or topoisomerase IV, mammalian cells possess topoisomerase II, which has very low affinity for FQ-hence low toxicity to cells.[6,9,12]
Folic acid metabolism inhibitors
Sulfonamides and trimethoprim
Each of these drugs inhibits distinct steps in folic acid metabolism. A combination of sulpha drugs and trimethoprim acting at distinct steps on the same biosynthetic pathway shows synergy and a reduced mutation rate for resistance.[6] Sulfonamides inhibit dihydropteroate synthase in a competitive manner with higher affinity for the enzyme than the natural substrate, p-amino benzoic acid. Agents such as trimethoprim act at a later stage of folic acid synthesis and inhibit the enzyme dihydrofolate reductase.[6]
Mechanisms of Antimicrobial Resistance
Prevention of accumulation of antimicrobials either by decreasing uptake or increasing efflux of the antimicrobial from the cell i.e Changes in outer membrane permeability
Drug molecules to a cell can be transferred by diffusion through porins, diffusion through the bilayer and by self-uptake. The porin channels are located in OM of Gram-negative bacteria. The small hydrophilic molecules (β-lactams and quinolones) can cross the OM only through porins. The decrease in number of porin channels, lead to decreased entry of β-lactam antibiotics and FQ into the cell, hence resistance to these classes of antibiotics. Acquired resistance to all antibiotic classes in Pseudomonas aeruginosa is due to low-OM permeability.
Efflux pumps
Membrane proteins that export antibiotics from the cell and maintain their low-intracellular concentrations are called efflux pumps.[4] At the same speed, where these antimicrobials are entering the cell, efflux mechanisms are pumping them out again, before they reach their target.[9] These pumps are present in the cytoplasmic membrane, unlike porins which are present in OM. Antibiotics of all classes except polymyxin are susceptible to the activation of efflux systems.[13] Efflux pumps can be specific to antibiotics. Most of them are multidrug transporters that are capable to pump a wide range of unrelated antibiotics – macrolides, tetracyclines, and FQ – and thus significantly contribute to multidrug resistant organisms.[4]
Modification of target molecule
Natural variations or acquired changes in the target sites of antimicrobials that prevent drug binding is a common mechanism of resistance. Target site changes often result from spontaneous mutation of a bacterial gene on the chromosome. Since antibiotic interaction with target molecule is generally quite specific, minor alteration of the target molecule can have important effect on antibiotic binding.
Alteration in the 30S subunit or 50S subunit: Of the ribosome leads to resistance to drugs that affect the protein synthesis, i.e., macrolides, tetracycline, chloramphenicol, and AG's. AG's bind to the 30S ribosomal subunit,[13] whereas chloramphenicol, macrolides, lincosamides, and streptogramin B bind to the 50S ribosomal subunit to suppress protein synthesis[14]
Alteration in PBP: Modification of the PBP is a favored mechanism of resistance to Gram-positive bacteria, whereas production of β-lactamases is a mechanism for the development of resistance to Gram-negative bacteria. The presence of mutation in penicillin-binding protein leads to a reduced affinity to β-lactam antibiotics. The resistance of Enterococcus faecium to ampicillin and Streptococcus pneumoniae to penicillin is by this mechanism. Similarly, in Staphylococcus aureus, the resistance to methicillin and oxacillin is associated with integration of a mobile genetic element – “staphylococcal cassette chromosome mec” – into the chromosome of S.aureus that contains resistance gene mec A.[4,15,16] mec A gene encodes PBP2a protein, a new penicillin-binding protein, that is required to change a native staphylococcal PBP. PBP2a shows a high resistance to β-lactam antibiotics. S. aureus strains resistant to methicillin can be cross resistant to all β-lactam antibiotics, streptomycin, and tetracycline and in some cases to erythromycin[5]
Altered cell wall precursors: Cell wall synthesis in Gram-positive bacteria can be inhibited by glycopeptides, e.g., vancomycin or teicoplanin, by their binding to D-alanyl-D-alanine residues of peptidoglycan precursors. D-alanyl-alanine is changed to D-alanyl-lactate as a result of which glycopeptides do not cross link with them, hence resistance to them develops.[4,5] E. faecium and Enterococcus faecalis strains have high resistance to vancomycin and teicoplanin (Van A-type resistance). Van B and Van C type resistance show resistance to vancomycin but is sensitive to teicoplanin[17]
Mutated-DNA gyrase and topoisomerase IV leads to FQ resistance: Quinolones bind to DNA gyrase A subunit. The mechanism of resistance involves the modification of two enzymes: DNA gyrase (coded by genes gyr A and gyr B) and topoisomerase IV (coded by genes par C and par E).[18] Mutations in genes gyr A and par C leads to the replication failure and as a result FQ cannot bind
Ribosomal protection mechanisms imparting resistance to tetracyclines
RNA polymerase mutations conferring resistance to rifampicin.
Antibiotic inactivation
There are three main enzymes that inactivate antibiotics such as β-lactamases, aminoglycoside-modifying enzymes, and chloramphenicol acetyltransferases (AACs).[19]
Beta-lactamases
β-lactamases hydrolyze nearly all β-lactams that have ester and amide bond, e.g., penicillins, cephalosporins, monobactams, and carbapenems. About 300 β-lactamases are known till date. β-lactamases are broadly prevalent enzymes that are classified using two main classification systems: Ambler (structural) and Bush–Jacoby–Medeiros (functional).[15] Ambler classification system is described below:
Class A β-lactamases: Also referred as penicillinase; these are clavulanic acid susceptible. Two commonly encountered Class A β-lactamases found in members of Enterobacteriaceae are designated as TEM-1, SHV-1. These are penicillinase with little or no activity against cephalosporin.[20] These are progenitors of extended-spectrum β-lactamases (ESBL). ESBL are enzymes that have changed substrate profile because of amino-acid substitution allowing hydrolysis of most cephalosporins. ESBL are resistant to penicillins, third-generation cephalosporins (e.g., ceftazidime, cefotaxime, ceftriaxone), aztreonam, cefamandole, cefoperazone, but are sensitive to methoxy-cephalosporins, e.g., cephamycins and carbapenems and are inhibited by inhibitors of β-lactamases, e.g., clavulanic acid, sulbactam, or tazobactam[21,22,23]
Class B β-lactamases: These are metallo-β-lactamases. These require enzymes such as zinc or heavy metals for catalysis and their activity is inhibited by chelating agents. These classes of enzymes are resistant to inactivation by clavulanate, sulbactam, aztreonam, and carbapenems. E.g., New Delhi metallo-β-lactamase[24]
Class C β-lactamases: These are also called cephalosporinases. These are produced by all Gram-negative bacteria with exception of Salmonella and Klebsiella. Class C hydrolyzes cephalosporins including extended spectrum cephalosporins, in comparison to class A β-lactamases, these have large cavities, and as a result, they are able to bind the bulky extended spectrum penicillin. An example of this type is Amp C β-lactamases. This class of enzymes is resistant to all β-lactams except carbapenems. They are not inhibited by clavulanate[25,26]
Class D β-lactamases: These are oxacillin hydrolyzing enzymes – found most commonly in Enterobacteriaceae and in P. aeruginosa. Oxacillin-hydrolyzing enzymes confer resistance to penicillin, cloxacillin, oxacillin, and methicillin. They are weakly inhibited by clavulanic acid but are inhibited by sodium chloride.[27]
Aminoglycoside modifying enzymes (AGE's)
AG are neutralized by specific enzymes: Phosphoryl-transferases, nucleotidyl-transferases or adenylyl-transferases, and AACs. These aminoglycoside-modifying enzymes (AMEs) reduce affinity of a modified molecule, impede binding to the 30S ribosomal subunit,[28] and provide extended spectrum resistance to AG's and FQ.[29] AMEs are identified in S. aureus, E. faecalis, and S. pneumoniae strains.
Chloramphenicol-acetyl-transferases
Few Gram-positive and Gram-negative bacteria and some of Haemophilus influenzae strains are resistant to chloramphenicol, and they have an enzyme chloramphenicol transacetylase that acetylates hydroxyl groups of chloramphenicol. Modified chloramphenicol is unable to bind to a ribosomal 50S subunit properly.[30]
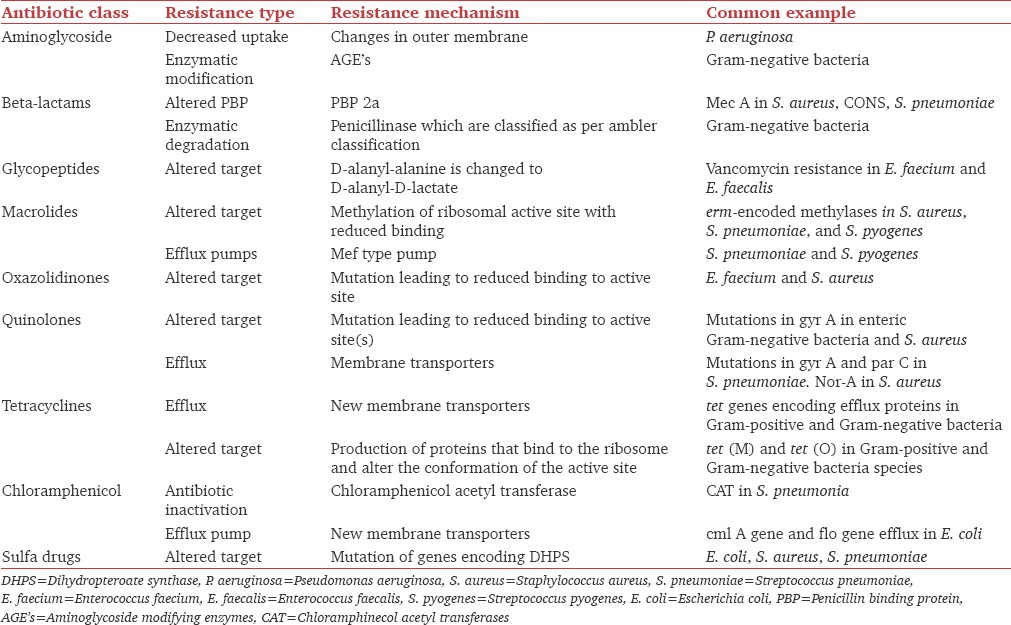
Resistance mechanism of various antibiotics is described in Table 1.
Table 1.
Resistance mechanism of individual antibiotics
Conclusion
The discovery of antibiotics led to sigh of relief, that now no bacteria will reside in this planet. With time, the bacteria have become smarter, and along with it, massive usage of antibiotics in clinical practice has resulted in resistance of bacteria to antimicrobial agents. The following biochemical types of resistance mechanisms are used by bacteria: Antibiotic inactivation, target modification, altered permeability, and “bypass” metabolic pathway. Determination of bacterial resistance to antibiotics of all classes (phenotypes) and mutations that are responsible for bacterial resistance to antibiotics (genetic analysis) are helpful. Better understanding of the mechanisms of antibiotic resistance, will help clinicians regarding usage of antibiotics in different situations.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Hauser AR, editor. Cell envelope. Antibiotic Basic for Clinicians. 2nd ed. New Delhi: Wolters Kluwer (India) Pvt. Ltd; 2015. pp. 3–5. [Google Scholar]
- 2.Kahne D, Leimkuhler C, Lu W, Walsh C. Glycopeptide and lipoglycopeptide antibiotics. Chem Rev. 2005;105:425–48. doi: 10.1021/cr030103a. [DOI] [PubMed] [Google Scholar]
- 3.Reynolds PE. Structure, biochemistry and mechanism of action of glycopeptide antibiotics. Eur J Clin Microbiol Infect Dis. 1989;8:943–50. doi: 10.1007/BF01967563. [DOI] [PubMed] [Google Scholar]
- 4.Džidic S, Šuškovic J, Kos B. Antibiotic resistance mechanisms in bacteria: Biochemical and genetic aspects. Food Technol Biotechnol. 2008;46:11–21. [Google Scholar]
- 5.Grundmann H, Aires-de-Sousa M, Boyce J, Tiemersma E. Emergence and resurgence of meticillin-resistant Staphylococcus aureus as a public-health threat. Lancet. 2006;368:874–85. doi: 10.1016/S0140-6736(06)68853-3. [DOI] [PubMed] [Google Scholar]
- 6.Yoneyama H, Katsumata R. Antibiotic resistance in bacteria and its future for novel antibiotic development. Biosci Biotechnol Biochem. 2006;70:1060–75. doi: 10.1271/bbb.70.1060. [DOI] [PubMed] [Google Scholar]
- 7.Vannuffel P, Cocito C. Mechanism of action of streptogramins and macrolides. Drugs. 1996;51(Suppl 1):20–30. doi: 10.2165/00003495-199600511-00006. [DOI] [PubMed] [Google Scholar]
- 8.Johnston NJ, Mukhtar TA, Wright GD. Streptogramin antibiotics: Mode of action and resistance. Curr Drug Targets. 2002;3:335–44. doi: 10.2174/1389450023347678. [DOI] [PubMed] [Google Scholar]
- 9.Wise R. A review of the mechanisms of action and resistance of antimicrobial agents. Can Respir J. 1999;6(Suppl A):20A–2A. [PubMed] [Google Scholar]
- 10.Lambert PA. Bacterial resistance to antibiotics: Modified target sites. Adv Drug Deliv Rev. 2005;57:1471–85. doi: 10.1016/j.addr.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 11.Bozdogan B, Appelbaum PC. Oxazolidinones: Activity, mode of action, and mechanism of resistance. Int J Antimicrob Agents. 2004;23:113–9. doi: 10.1016/j.ijantimicag.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 12.Higgins PG, Fluit AC, Schmitz FJ. Fluoroquinolones: Structure and target sites. Curr Drug Targets. 2003;4:181–90. doi: 10.2174/1389450033346920. [DOI] [PubMed] [Google Scholar]
- 13.Lambert PA. Mechanisms of antibiotic resistance in Pseudomonas aeruginosa. J R Soc Med. 2002;95(Suppl 41):22–6. [PMC free article] [PubMed] [Google Scholar]
- 14.Tenover FC. Mechanisms of antimicrobial resistance in bacteria. Am J Med. 2006;119(6 Suppl 1):S3–10. doi: 10.1016/j.amjmed.2006.03.011. [DOI] [PubMed] [Google Scholar]
- 15.Alekshun MN, Levy SB. Molecular mechanisms of antibacterial multidrug resistance. Cell. 2007;128:1037–50. doi: 10.1016/j.cell.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 16.Hiramatsu K, Cui L, Kuroda M, Ito T. The emergence and evolution of methicillin-resistant Staphylococcus aureus. Trends Microbiol. 2001;9:486–93. doi: 10.1016/s0966-842x(01)02175-8. [DOI] [PubMed] [Google Scholar]
- 17.Giedraitiene A, Vitkauskiene A, Naginiene R, Pavilonis A. Antibiotic resistance mechanisms of clinically important bacteria. Medicina (Kaunas) 2011;47:137–46. [PubMed] [Google Scholar]
- 18.Kim YH, Cha CJ, Cerniglia CE. Purification and characterization of an erythromycin esterase from an erythromycin-resistant Pseudomonas sp. FEMS Microbiol Lett. 2002;210:239–44. doi: 10.1111/j.1574-6968.2002.tb11187.x. [DOI] [PubMed] [Google Scholar]
- 19.Dockrell HM, Goering RV, Roitt I, Wakelin D, Zuckerman M. In: Attacking the enemy: Antimicrobial agents and chemotherapy. Medical Microbiology. Mims C, Dockrell HM, Goering RV, Roitt I, Wakelin D, Zuckerman M, editors. Netherlands: Elsevier Mosby; 2004. pp. 473–507. [Google Scholar]
- 20.Rice LB, Sahm D, Bonomo R. Mechanisms of resistance to antibacterial agents. In: Murray PR, editor. Manual of Clinical Microbiology. 8th ed. Washington, DC: ASM Press; 2003. pp. 1084–7. [Google Scholar]
- 21.Jacoby GA, Munoz-Price LS. The new beta-lactamases. N Engl J Med. 2005;352:380–91. doi: 10.1056/NEJMra041359. [DOI] [PubMed] [Google Scholar]
- 22.Ma L, Chang FY, Fung CP, Chen TL, Lin JC, Lu PL, et al. Variety of TEM-, SHV-, and CTX-M-type beta-lactamases present in recent clinical isolates of Escherichia coli, Klebsiella pneumoniae, and Enterobacter cloacae from Taiwan. Microb Drug Resist. 2005;11:31–9. doi: 10.1089/mdr.2005.11.31. [DOI] [PubMed] [Google Scholar]
- 23.Bonnet R. Growing group of extended-spectrum beta-lactamases: The CTX-M enzymes. Antimicrob Agents Chemother. 2004;48:1–14. doi: 10.1128/AAC.48.1.1-14.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rasmussen BA, Bush K. Carbapenem-hydrolyzing beta-lactamases. Antimicrob Agents Chemother. 1997;41:223–32. doi: 10.1128/aac.41.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crichlow GV, Kuzin AP, Nukaga M, Mayama K, Sawai T, Knox JR. Structure of the extended-spectrum class C beta-lactamase of Enterobacter cloacae GC1, a natural mutant with a tandem tripeptide insertion. Biochemistry. 1999;38:10256–61. doi: 10.1021/bi9908787. [DOI] [PubMed] [Google Scholar]
- 26.Lobkovsky E, Billings EM, Moews PC, Rahil J, Pratt RF, Knox JR. Crystallographic structure of a phosphonate derivative of the Enterobacter cloacae P99 cephalosporinase: Mechanistic interpretation of a beta-lactamase transition-state analog. Biochemistry. 1994;33:6762–72. doi: 10.1021/bi00188a004. [DOI] [PubMed] [Google Scholar]
- 27.Naas T, Nordmann P. OXA-type beta-lactamases. Curr Pharm Des. 1999;5:865–79. [PubMed] [Google Scholar]
- 28.Strateva T, Yordanov D. Pseudomonas aeruginosa – A phenomenon of bacterial resistance. J Med Microbiol. 2009;58(Pt 9):1133–48. doi: 10.1099/jmm.0.009142-0. [DOI] [PubMed] [Google Scholar]
- 29.Maurice F, Broutin I, Podglajen I, Benas P, Collatz E, Dardel F. Enzyme structural plasticity and the emergence of broad-spectrum antibiotic resistance. EMBO Rep. 2008;9:344–9. doi: 10.1038/embor.2008.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tolmasky ME. Bacterial resistance to aminoglycosides and beta-lactams: The Tn1331 transposon paradigm. Front Biosci. 2000;5:D20–9. doi: 10.2741/tolmasky. [DOI] [PubMed] [Google Scholar]