

Abstract
Agouti-related protein (AGRP) is a potent orexigenic peptide that antagonizes the melanocortin-3 and -4 receptors (MC3R and MC4R). While the C-terminal domain of AGRP, AGRP(87-132), is equipotent to the full-length peptide, further truncation decreases potency at the MC3R and MC4R. Herein, we report AGRP-derived peptides designed to mimic the active β-hairpin secondary structure that contains the hypothesized Arg-Phe-Phe pharmacophore. The most potent scaffold, c[Pro-Arg-Phe-Phe-Asn-Ala-Phe-DPro], comprised the hexa-peptide β-hairpin loop from AGRP cyclized through a DPro-Pro motif. A 20 compound library was synthesized from this scaffold for further structure-activity relationship studies. The most potent peptide from this library was an Asn to diaminopropionic acid substitution that possessed sub-nM antagonist activity at the mMC4R and was greater than 160-fold selective for the mMC4R versus the mMC3R. The reported ligands may serve as probes to characterize the melanocortin receptors in vivo and leads in the development of novel therapeutics.
Keywords: GPCR, melanocortin receptors, AGRP, β-hairpin mimetics, structure-activity relationship
TOC image
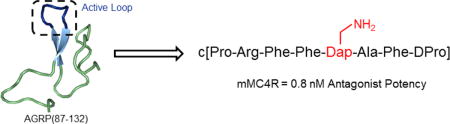
Introduction
The melanocortin system is known to participate in numerous physiological processes, including skin pigmentation,1,2 steroidogenesis,3 and energy homeostasis.4 The melanocortin system consists of five receptors (MC1-5R)5–12 belonging to the family of G-protein coupled receptors (GPCRs). Endogenous ligands for these receptors include agonists derived from the proopiomelanocortin (POMC) gene transcript13 and two antagonists, Agouti and Agouti-related protein (AGRP).14–16 The MC3R and MC4R, POMC agonists, and AGRP have all been shown to directly regulate food intake, obesity, and weight homeostasis.4,17–20 As an antagonist to both the MC3R and MC4R, AGRP elicits an increase in food intake when delivered via an intracerebroventricular (i.c.v.) injection.16,17,19 Overexpression of AGRP in transgenic mice results in an obesity phenotype; the amount of weight gain correlates to the expression level of AGRP.21 Studying AGRP and AGRP derivatives may therefore lead to new insights into the etiology and potential treatment of obesity. Additionally, as a potent orexigenic neuropeptide, AGRP may also be useful in the development of selective melanocortin antagonists for the treatment of negative energy balance disorders such as anorexia nervosa and cachexia.22,23 Since AGRP is an inverse agonist at the MC4R,24,25 AGRP-derived compounds may have unique pharmacology compared to other melanocortin antagonist templates.
Human AGRP is a 132 amino acid protein containing a putative signal sequence, an acidic middle portion with unknown function, and a cysteine-rich C–terminal domain.15 Truncation products containing the C-terminal domain have previously been shown to exhibit equipotent binding and activity compared to the mature peptide.15,26 Within the C-terminal domain, mutagenesis studies identified AGRP(111-113) residues Arg-Phe-Phe as the key residues critical for receptor binding.27 The necessity of the Arg-Phe-Phe was further validated by substitution with Ala-Ala-Ala28 or inverting residue chirality within the Arg-Phe-Phe tripeptide,29 resulting in diminished to no observable antagonist activity in subsequent structure-activity relationship (SAR) studies. The substitution of the Arg-Phe-Phe into the potent agonist NDP-MSH resulted in agonist peptides,30 while substitution of the potent agonist sequence His-DPhe-Arg-Trp into the AGRP Arg-Phe-Phe domain also resulted in agonist activity,31,32 supporting the functional significance of this region of AGRP. The NMR structure of AGRP(87-132) indicated this tripeptide motif is located on an exposed beta-hairpin loop in a well-defined fold of the molecule stabilized by five disulfide bonds.33,34 As an engineered minimized derivative of AGRP(87-132) designed with 4 disulfide bonds and the C-terminal 12 residues removed, AGRP(87-120) displayed similar antagonist activity and binding to AGRP(87-132).35 The NMR analysis of this “miniAGRP” indicated a similar overall folding compared to AGRP(87-132), especially in the critical Arg-Phe-Phe beta-hairpin loop.35 Attempts to further truncate AGRP have resulted in compounds with diminished binding affinity and potency. The minimal sequence with detectable antagonist activity was comprised of AGRP(110-117), a disulfide bonded cyclic octapeptide Ac-c[Cys-Arg-Phe-Phe-Asn-Ala-Phe-Cys]-NH2.27 While no detectable binding was observed at the hMC3R, it possessed an IC50 binding affinity of 127 nM at the hMC4R, approximately 36-fold lower than the C-terminal AGRP(83-132) peptide utilized in that study. Elongation of this minimal fragment by one tyrosine on both terminal Cys to generate the decapeptide AGRP(109-118) (Tyr-c[Cys-Arg-Phe-Phe-Asn-Ala-Phe-Cys]-Tyr) recovered binding affinity at the hMC3R (~1.9 μM) and slightly increased binding affinity at the hMC4R (57 nM).27 When tested at the mMC3R and mMC4R, this decapeptide possessed detectable but greatly reduced antagonist activity at the mMC4R compared to hAGRP(87-132) (pA2 = 6.8 and 9.4 respectively) and no activity up to 10 μM at the mMC3R.24,36 To recover antagonist activity at the mMC3R, further elongation was required. Substitution of Cys(108) and/or Cys(119) with Ala and adding residues to either terminal resulted in hAGRP(107-120) (Thr-Ala-Tyr-c[Cys-Arg-Phe-Phe-Asn-Ala-Phe-Cys]-Tyr-Ala-Arg-NH2; pA2 = 6.1 at the mMC3R) or hAGRP(109-122) (Tyr-c[Cys-Arg-Phe-Phe-Asn-Ala-Phe-Cys]-Tyr-Ala-Arg-Lys-Leu; pA2 = 6.2 at the mMC3R) and were demonstrated to be the minimal sequences to retain activity at the mMC3R [hAGRP(87-132) pA2 = 8.9 at the mMC3R].37 A bicyclic peptide derived from hAGRP(101-122) was a more potent antagonist at both the mMC3R and mMC4R (pA2 = 6.8 and 7.5, respectively) compared to hAGRP(109-118), though still diminished compared to the longer hAGRP(87-132).38
Since the shorter cyclic analogues containing the Arg-Phe-Phe pharmacophore are significantly less active than AGRP(87-132), it is hypothesized that the beta-hairpin structure containing the Arg-Phe-Phe is critical for potent antagonist activity. In attempts to generate new, more potent analogues of AGRP, six peptides containing beta-hairpin inducing motifs were synthesized and characterized to probe their effects on increasing the potency of the AGRP active loop. The most potent scaffold, which contained the Arg-Phe-Phe-Asn-Ala-Phe active loop of AGRP grafted onto a beta-hairpin mimetic induced by a DPro-Pro cyclization, was further studied to generate more potent and selective antagonists.
Results
Peptide Synthesis and Characterization
The reported peptides were synthesized manually using standard fluorenylmethoxycarbonyl (Fmoc) chemistry.39,40 The peptides were purified using semi-preparative reverse-phase high-pressure liquid chromatography (RP-HPLC). The purity of the peptides (>95%) was assessed by analytical HPLC in two diverse solvent systems (Table 1). The correct peptide molecular weight was confirmed through MALDI mass spectrometry (University of Florida Protein Core Facility). The compounds were assayed for agonist activity using a cAMP based beta-galactosidase reporter assay at the mouse melanocortin 1, 3, 4, and 5 receptors.41 Antagonist activity was evaluated at the mMC3 and mMC4 receptors through a Schild analysis using the nonselective and potent synthetic peptide MTII as the agonist [pA2 = −log(Ki)].42,43 For comparison, the previously reported hAGRP(87-132), hAGRP(87-120), hAGRP(109-118) and SHU911944 are included. Due to the inherent error within the assay, compounds that were within a 3-fold potency range were considered to be equipotent.
Table 1.
Analytical Data for the Peptides Synthesized in This Studya
| Peptide | Structure | HPLC k′ (system 1) | HPLC k′ (system 2) | M+1 (calcd) | mass spectral analysis (M+1), purity % |
|---|---|---|---|---|---|
|
| |||||
| 1 |
|
5.9 | 10.6 | 1771.8 | 1771.9 (>98%) |
| 2 |
|
5.6 | 10.7 | 1831.8 | 1831.9 (>98%) |
| 3 |
|
5.5 | 10.5 | 1833.7 | 1832.9 (>98%) |
| 4 | c[Pro-Tyr-Ala-Arg-Phe-Phe-Asn-Ala-Phe-Ala-Tyr-DPro] | 7.7 | 12.9 | 1445.7 | 1445.6 (>98%) |
| 5 | c[Pro-Tyr-Thr-Arg-Phe-Phe-Asn-Ala-Phe-Thr-Tyr-DPro] | 7.9 | 13.5 | 1505.7 | 1505.6 (>98%) |
| 6 | c[Pro-Arg-Phe-Phe-Asn-Ala-Phe-DPro] | 7.4 | 5.5 | 977.5 | 977.6 (>95%) |
| 7 | c[Pro-Arg-Bip-Phe-Asn-Ala-Phe-DPro] | 9.0 | 15.0 | 1054.2 | 1053.6 (>95%) |
| 8 | c[Pro-Arg-Tic-Phe-Asn-Ala-Phe-DPro] | 7.2 | 12.6 | 990.1 | 990.3 (>98%) |
| 9 | c[Pro-Arg-Phg-Phe-Asn-Ala-Phe-DPro] | 7.0 | 11.9 | 964.1 | 964.3 (>98%) |
| 10 | c[Pro-Arg-hPhe-Phe-Asn-Ala-Phe-DPro] | 7.9 | 13.3 | 992.1 | 991.5 (>95%) |
| 11 | c[Pro-Arg-Anc-Phe-Asn-Ala-Phe-DPro] | 7.7 | 12.6 | 1000.1 | 1000.0 (>95%) |
| 12 | c[Pro-Arg-Nal(1′)-Phe-Asn-Ala-Phe-DPro] | 8.4 | 14.1 | 1028.2 | 1027.8 (>98%) |
| 13 | c [Pro-Arg-Nal(2′)-Phe-Asn-Ala-Phe-DPro] | 8.5 | 14.5 | 1028.2 | 1028.0 (>98%) |
| 14 | c[Pro-Arg-Phe-Bip-Asn-Ala-Phe-DPro] | 8.6 | 15.1 | 1054.2 | 1057.1 (>95%) |
| 15 | c[Pro-Arg-Phe-Tic-Asn-Ala-Phe-DPro] | 8.3 | 13.0 | 990.1 | 989.2 (>98%) |
| 16 | c[Pro-Arg-Phe-Phg-Asn-Ala-Phe-DPro] | 6.9 | 11.9 | 964.1 | 964.2 (>98%) |
| 17 | c[Pro-Arg-Phe-hPhe-Asn-Ala-Phe-DPro] | 7.9 | 13.5 | 992.1 | 991.6 (>95%) |
| 18 | c[Pro-Arg-Phe-Anc-Asn-Ala-Phe-DPro] | 8.3 | 13.1 | 1000.1 | 999.8 (>95%) |
| 19 | c[Pro-Arg-Phe-Nal(1′)-Asn-Ala-Phe-DPro] | 8.3 | 14.4 | 1028.2 | 1028.1 (>98%) |
| 20 | c[Pro-Arg-Phe-Nal(2′)-Asn-Ala-Phe-DPro] | 8.5 | 14.2 | 1028.2 | 1028.2 (>98%) |
| 21 | c[Pro-Arg-Phe-Phe-Gly-Ala-Phe-DPro] | 7.7 | 5.9 | 920.5 | 920.8 (>98%) |
| 22 | c[Pro-Arg-Phe-Phe-Dap-Ala-Phe-DPro] | 3.2 | 5.6 | 949.6 | 950.0 (>98%) |
| 23 | c[Pro-Arg-Phe-Phe-Dab-Ala-Phe-DPro] | 6.9 | 5.6 | 963.5 | 964.4 (>98%) |
| 24 | c[Pro-Arg-Phe-Phe-Orn-Ala-Phe-DPro] | 7.1 | 5.5 | 977.6 | 977.9 (>95%) |
| 25 | c[Pro-Arg-Phe-Phe-Lys-Ala-Phe-DPro] | 7.1 | 5.5 | 991.6 | 991.9 (>95%) |
| 26 | c[Pro-Arg-Phe-Phe-Arg-Ala-Phe-DPro] | 7.2 | 12.6 | 1019.6 | 1020.5 (>98%) |
HPLC k′ = [(peptide retention time – solvent retention time)/solvent retention time] in solvent system 1 (10% acetonitrile in 0.1% trifluoroacetic acid/water and a gradient to 90% acetonitrile over 35 min) or solvent system 2 (10% methanol in 0.1% trifluoroacetic acid/water and a gradient to 90% methanol over 35 min). An analytical Vydac C18 column (Vydac 218TP104) was used with a flow rate of 1.5 mL/min. The peptide purity was determined by HPLC at a wavelength of 214 nm.
Template Scaffolds Design and Biological Evaluation
Two scaffold series were designed and evaluated for activity at the melanocortin receptors, summarized in Table 2. One series was derived from the hAGRP(107-120) peptide previously reported to be the minimal sequence resulting in antagonist activity at both the mMC3R and the mMC4R.37 Since the mMC3R and mMC4R both are involved in energy and metabolic homeostasis, it may be desirable to start with a peptide that is active at both receptors. The reported hAGRP(107-120) involved substitution of two native Cys(108) and Cys(119) with Ala, as these Cys form disulfide bonds with other Cys outside of this fragment. Peptide 1 contained the same residues as the previously reported hAGRP(107-120) and additionally was acetylated at the N-terminus. Consistent with the previous report, 1 possessed μM agonist potency at the mMC1R, no activity at the mMC5R, and weak antagonist activity at both the mMC3R (pA2 = 6.1; Ki = 790 nM) and mMC4R [pA2 = 6.5, Ki = 320 nM; the free amine N-terminal hAGRP(107-120) reported pA2 = 6.1 and 6.6 at the mMC3R and mMC4R, respectively].37
Table 2.
Pharmacology of Truncated hAGRP Peptide Analogues at the Mouse Melanocortin Receptors (Ki=10-pA2).a
| Peptide | Structure | mMC1R | mMC3R | mMC4R | mMC5R | ||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Agonist EC50 (μM) |
Antagonist pA2 |
Ki (nM) |
Antagonist pA2 |
Ki (nM) |
Agonist EC50 (μM) |
||
|
| |||||||
| hAGRP (87–132)* |
|
>100,000 | 8.9±0.2 | 1.3 | 9.4±1.0 | 0.40 | >100,000 |
| miniAGRP (87-120)* |
|
>100,000 | 8.1±0.1 | 7.9 | 8.5±0.1 | 3.2 | >100,000 |
| hAGRP (109-118)* |
|
5.1±3.0 | >100,000 | 6.8±0.2 | 160 | >100,000 | |
| SHU9119* | Ac-Nle-c[Asp-His-DNal(2′)-Arg-Trp-Lys]-NH2 | 0.64±0.26 nM | 9.5 | 0.32 | 10.4 | 0.040 | 2.31±0.45nM |
| 1 |
|
6.1±1.8 | 6.1±0.3 | 790 | 6.5±0.2 | 320 | >100,000 |
| 2 |
|
11.7±4.0 | 6.2±0.2 | 630 | 6.8±0.2 | 160 | >100,000 |
| 3 |
|
7.3±3.1 | 6.2±0.2 | 630 | 7.2±0.1 | 63 | >100,000 |
| 4 | c[Pro-Tyr-Ala-Arg-Phe-Phe-Asn-Ala-Phe-Ala-Tyr-DPro] | 17.6±10.1 | >100,000 | >100,000 | >100,000 | ||
| 5 | c[Pro-Tyr-Thr-Arg-Phe-Phe-Asn-Ala-Phe-Thr-Tyr-DPro] | 11.6±5.0 | 5.9±0.1 | 1,300 | 6.8±0.2 | 160 | >100,000 |
| 6 | c[Pro-Arg-Phe-Phe-Asn-Ala-Phe-DPro] | 4.5±0.2 | 6.4±0.2 | 400 | 7.7±0.1 | 20 | >100,000 |
The indicated errors represent the standard error of the mean determined from at least three independent experiments >100,000 indicates that the compound was examined but lacked agonist activity at up to 100 μM concentrations. (*) The values indicated for hAGRP(87-132), miniAGRP, hAGRP(109-118) and SHU9119 have previously been reported.28,70
In attempts to improve the antagonist activity, Ala at positions 108 and 119 (AGRP numbering) were substituted with Thr, generating 2. Threonine was selected because beta-branched amino acids such as Thr are more likely to be found in beta-sheets when solved protein crystal structures are analyzed.45,46 Additionally, substitution of Thr into a guest position within a beta-sheet in either an IgG-binding domain from protein G47,48 or a zinc finger peptide49 was found to increase the propensity for beta-sheet formation. Therefore it was hypothesized that substituting in a Thr side chain would help stabilize the purported beta-hairpin structure, increasing the stability of the beta-strands at the base of the loop, and potentially resulting in a more potent antagonist. Similar to 1, 2 resulted in μM activity at the mMC1R, antagonist activity at the mMC3R and mMC4R that were within the inherent experimental error of the assay, as compared to 1, and no activity up to 100 μM at the mMC5R.
Residues 108 and 119 were also replaced with Cys, and a second, unnatural disulfide bond between Cys(108) and Cys(119) resulted in the more constrained 3. It was further hypothesized that the addition of a second ring constraining disulfide bond would further limit the flexibility of the peptide, potentially locking the active Arg-Phe-Phe pharmacophore into a more “bioactive” conformation. Activity at the mMC1R, mMC3R, and mMC5R were similar as compared to 1, while a 5-fold increase in antagonist potency at the mMC4R resulted.
The second peptide scaffold series was synthesized through a head-to-tail cyclization of the active loop through a DPro-Pro motif. This heterochiral DPro-Pro dipeptide has previously been used to cyclize a loop from the interferon γ receptor,50 a protruding loop from the platelet-derived growth factor B,51 and the L3 and H2 hypervariable loops from an antibody,52 generating beta-hairpin loop mimetics in a relatively chemically facile manner. Therefore, it was hypothesized that grafting the active loop of AGRP onto the DPro-Pro template would result in active, conformationally constrained beta-hairpin peptides that could be utilized in further SAR studies. Two of the peptides (4 and 5) were designed based on the Tyr-c[Cys-Arg-Phe-Phe-Asn-Ala-Phe-Cys]-Tyr fragment of AGRP [AGRP(109-118)] that demonstrated binding at both the hMC3R and hMC4R27 fixed into the DPro-Pro motif. The Cys [Cys(110) and Cys(117)] were replaced with Ala in 4, resulting in μM agonist activity at the mMC1R and no activity at the mMC3-5R up to 100 μM. Substitution of the Ala with the postulated beta-sheet stabilizing Thr (described above) resulted in a similar μM activity at the mMC1R and no activity at the mMC5R, while recovering modest antagonist potency at the mMC3R and mMC4R.
Peptide 6 was designed to contain only the protruding Arg-Phe-Phe-Asn-Ala-Phe hexapeptide loop cyclized through the DPro-Pro residues. This octapeptide was a μM agonist at the mMC1R and was unable to stimulate the mMC5R up to 100 μM. Interestingly, despite being the smallest template studied, 6 was the most potent antagonist at the mMC3Rand mMC4R. Also noteworthy was the lack of agonist activity at the mMC3R that is commonly observed in small peptide antagonists such as SHU9119 at the MC3R (Figure 1).44 As possessing the highest activity at the mMC4R [50-fold less potent than hAGRP(87-132) and 6-fold less potent than the more easily synthesized miniAGRP analogue of hAGRP(87-132)], 6 was selected for further SAR studies.
Figure 1.
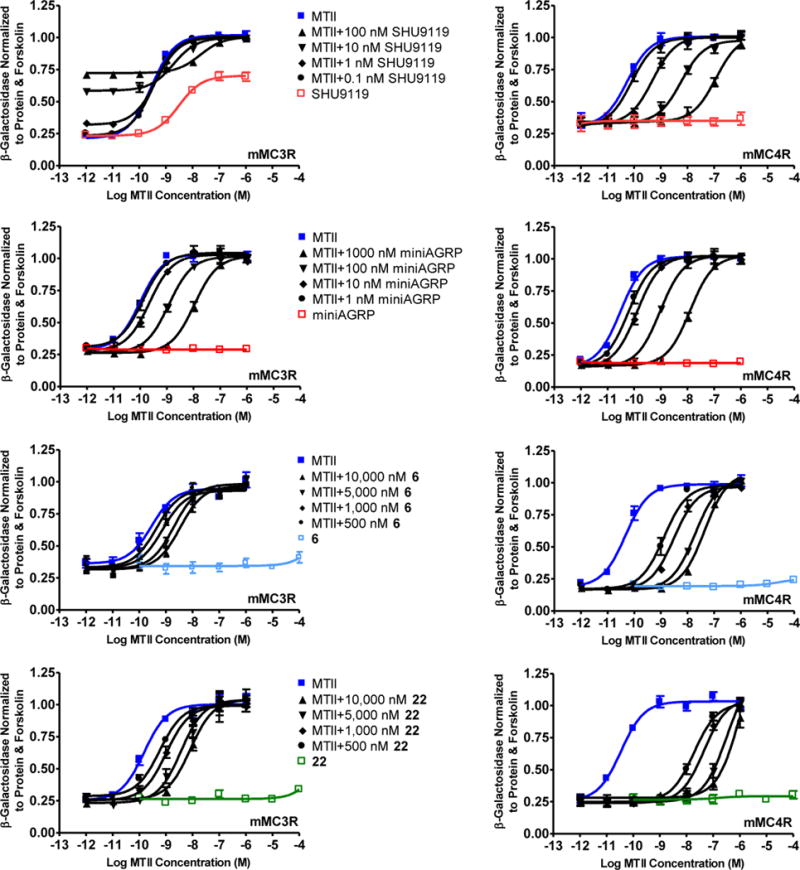
Illustrations of the in vitro antagonist pharmacology of SHU9119, miniAGRP, 6, and 22 at the mMC3R and mMC4R. A Schild antagonist experimental design was applied and the agonist MTII was utilized in these studies.
Structure-Activity Relationship Studies of 6 c[Pro-Arg-Phe-Phe-Asn-Ala-Phe-Pro]
Three residues were selected for further studies, summarized in Table 3: Phe(112), Phe(113), and Asn(114) (AGRP numbering). Residues Phe(112) and Phe(113) were selected as these amino acids are postulated to be part of the pharmacophore of AGRP.27 It was hypothesized that substitution of the Phe amino acids with more constrained aromatic amino acids would result in more potent and selective peptides. The unnatural amino acids used in this study, shown in Figure 2 along with their abbreviations, were previously incorporated into the potent melanocortin tetrapeptide sequence Ac-His-DPhe-Arg-Trp-NH2 and were shown to mediate differential activities between the mMC3R and mMC4R.53–55 Replacement of Phe(112) with Bip (peptide 7) resulted in a 5-fold more potent agonist at the mMC1R compared to lead 6, similar potency at the mMC3R, and 8-fold decreased antagonist potency at the mMC4R. Peptide 8, containing Tic, did not possess any agonist or antagonist activity at every receptor subtype tested up to 100 μM. Substitution of Phg resulted in peptide 9, which displayed agonist activity at the mMC1R (13.6 μM), but no activity was observed at either the mMC3R or mMC4R. Incorporation of the hPhe residue (10) resulted in an equipotent agonist at the mMC1R (1.5 μM), no activity at the mMC3R, and a 5-fold potency loss at the mMC4R. Peptide 11, containing the constrained Anc bicyclic ring, maintained μM agonist activity at the mMC1R, but possessed no activity at the mMC3R or mMC4R at up to 100 μM concentrations. Incorporation of the naphthalene ring system Nal(1′) in 12 maintained equipotent activity at all three receptors compared to 6. The regioisomer Nal(2′) (13) had similar agonist activity at the mMC1R compared to 6, equipotent antagonist potency at the mMC3R, and a 5-fold antagonist potency loss at the mMC4R. All Phe(112) substitutions were unable to stimulate the mMC5R up to 100 μM.
Table 3.
Pharmacology of Beta-hairpin Based Ligands at the Mouse Melanocortin Receptors (Ki=10−pA2).a
| Peptide | Structure | mMC1R | mMC3R | mMC4R | mMC5R | ||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Agonist EC50 (μM) | Antagonist pA2 | Ki(nM) | Antagonist pA2 | Ki(nM) | Agonist EC50 (μM) | ||
|
| |||||||
| 6 | c[Pro-Arg-Phe-Phe-Asn-Ala-Phe-DPro] | 4.5±0.2 | 6.4±0.2 | 400 | 7.7±0.1 | 20 | >100,000 |
| 7 | c[Pro -Arg-Bip -Phe -Asn-Ala-Phe-DPro] | 0.8±0.2 | 6.2±0.1 | 630 | 6.8±0.1 | 160 | >100,000 |
| 8 | c[Pro-Arg-Tic-Phe-Asn-Ala-Phe-DPro] | >100,000 | >100,000 | >100,000 | >100,000 | ||
| 9 | c[Pro-Arg-Phg-Phe-Asn-Ala-Phe-DPro] | 13.6±6.6 | >100,000 | >100,000 | >100,000 | ||
| 10 | c[Pro-Arg-hPhe-Phe-Asn-Ala-Phe-DPro] | 1.5±0.6 | >100,000 | 7.0±0.1 | 100 | >100,000 | |
| 11 | c[Pro-Arg-Anc-Phe-Asn-Ala-Phe-DPro] | 2.5±1.1 | >100,000 | >100,000 | >100,000 | ||
| 12 | c[Pro -Arg-Nal(1′)-Phe-Asn-Ala-Phe-DPro] | 2.5±1.0 | 6.1±0.1 | 790 | 7.8±0.1 | 16 | >100,000 |
| 13 | c[Pro -Arg-Nal(2′)-Phe-Asn-Ala-Phe-DPro] | 8.8±2.6 | 6.0±0.2 | 1,000 | 7.0±0.1 | 100 | >100,000 |
| 14 | c[Pro-Arg-Phe-Bip - Asn-Ala-Phe-DPro] | 0.5±0.3 | >100,000 | 6.8±0.2 | 160 | >100,000 | |
| 15 | c[Pro-Arg-Phe-Tic-Asn-Ala-Phe-DPro] | 3.9±1.0 | >100,000 | >100,000 | >100,000 | ||
| 16 | c[Pro-Arg-Phe-Phg-Asn-Ala-Phe-DPro] | 21.0±9.7 | >100,000 | 5.9±0.1 | 1,300 | >100,000 | |
| 17 | c[Pro-Arg-Phe-hPhe-Asn-Ala-Phe-DPro] | 20.3±8.0 | >100,000 | 8.2±0.1 | 6.3 | >100,000 | |
| 18 | c[Pro-Arg-Phe-Anc-Asn-Ala-Phe-DPro] | 12.1±3.7 | >100,000 | >100,000 | 28±4 | ||
| 19 | c[Pro -Arg-Phe-Nal(1′) - Asn-Ala-Phe-DPro] | 12.7±3.8 | 5.6±0.1 | 2,500 | 7.8±0.2 | 16 | >100,000 |
| 20 | c[Pro -Arg-Phe-Nal(2′) - Asn-Ala-Phe-DPro] | 19.6±6.4 | >100,000 | 7.2±0.1 | 63 | >100,000 | |
| 21 | c[Pro-Arg-Phe-Phe-Gly-Ala-Phe-DPro] | 50% at 100μM | 7.6±0.5 | 25 | 8.4±0.1 | 4.0 | >100,000 |
| 22 | c[Pro-Arg-Phe-Phe-Dap-Ala-Phe-DPro] | 75% at 100μM | 6.9±0.1 | 130 | 9.1±0.1 | 0.79 | >100,000 |
| 23 | c[Pro-Arg-Phe-Phe-Dab-Ala-Phe-DPro] | 50% at 100μM | 7.0±0.1 | 100 | 8.4±0.2 | 4.0 | >100,000 |
| 24 | c[Pro-Arg-Phe-Phe-Orn-Ala-Phe-DPro] | 30% at 10μM | 6.8±0.1 | 160 | 8.3±0.1 | 5.0 | >100,000 |
| 25 | c[Pro-Arg-Phe-Phe-Lys-Ala-Phe-DPro] | >100,000 | 7.0±0.1 | 100 | 8.3±0.3 | 5.0 | >100,000 |
| 26 | c[Pro-Arg-Phe-Phe-Arg-Ala-Phe-DPro] | >100,000 | 6.7±0.2 | 200 | 8.4±0.4 | 4.0 | >100,000 |
The indicated errors represent the standard error of the mean determined from at least three independent experiments. The antagonistic pA2 values were determined using the Schild analysis and the antagonist MTII. >100,000 indicates that the compound was examined but lacked agonist or antagonist properties at up to 100 μM concentrations. A percentage denotes the percent maximal stimulatory response observed at 100 μM concentrations but not enough stimulation was observed to determine an EC50 value.
Figure 2.
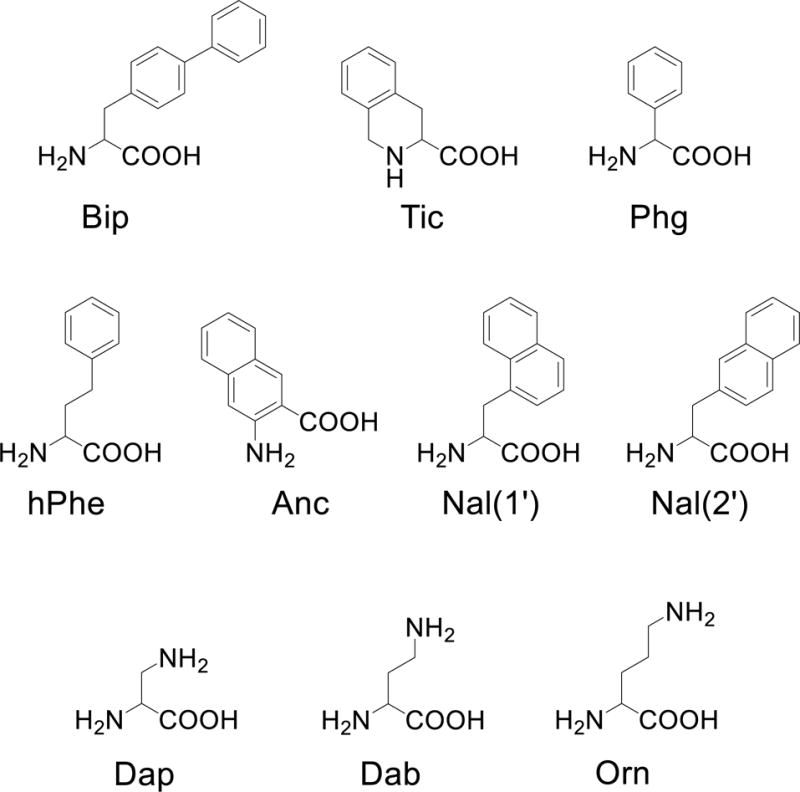
Structures of the unnatural amino acids used in the to replace Phe(112), Phe(113) and Asn(114).
The same aromatic amino acids were substituted at Phe(113). Peptide 14, which contained the biphenyl Bip, resulted in a 10-fold more potent mMC1R agonist compared to 6, no activity at the mMC3R at up to 100 μM, and 8-fold decreased potency at the mMC4R. The Tic derivative 15, was equipotent to 6 as an mMC1R agonist, but possessed no activity at the remaining selected receptor subtypes up to 100 μM. Peptide 16, containing the Phg residue, possessed a modest 4-fold decreased potency compared to 6 at the mMC1R, possessed no activity at the mMC3R up to 100 μM, and resulted in 65-fold decreased antagonist potency at the mMC4R. The hPhe containing peptide 17, modestly decreased potency at the mMC1R 4-fold, decreased antagonist potency at the mMC3R 5-fold, and was slightly more potent at the mMC4R compared to 6. Peptide 18, containing the constrained Anc residue, was as potent an agonist as 6 at the mMC1R with no activity observed at up to 100 μM at the mMC3R and mMC4R. This Anc substitution resulted in the only peptide in this study that possessed agonist activity at the mMC5R. The naphthalene Nal(1′) was incorporated into 19 and resulted in an equipotent mMC1R agonist, 6-fold decreased antagonist potency at the mMC3R, and similar potency at the mMC4R compared to 6. The regioisomer Nal(2′) (20) possessed similar agonist potency at the mMC1R, no activity at the mMC3R at up to 100 μM, and was equipotent at the mMC4R.
The AGRP Asn(114) amino acid was selected for substitution because a docking model of AGRP(87-132) with the mMC4R indicated this residue might be in close proximity to the Asp(181) of the mMC4R and could potentially be exploited to form a novel ligand-receptor salt bridge.28 Positively charged amino acids were utilized with varying side chain lengths to probe for this postulated ligand-receptor interaction with the Gly amino acid serving as a neutral control. All peptides containing an Asn substitution resulted in either some partial agonist activity or no activity at the mMC1R and no agonist activity at the mMC5R at up to 100 μM. Surprisingly, peptide 21 containing the Gly control resulted in 16-fold increased mMC3R antagonist potency and 5-fold increased potency at the mMC4R. The shortest charged residue assayed, Dap (22), resulted in equivalent antagonist potency to 6 at the mMC3R and 25-fold increased potency at the mMC4R (Figure 1). Similar to 6, 22 did not result in partial agonist activity at the mMC3R. Elongating the side chain by one methylene to Dab (23), resulted in modest 4-fold increased antagonist potency at the mMC3R and 5-fold increased potency at the mMC4R. Further extension by one or two methylenes for the Orn or Lys derivatives 24 and 25 resulted in equipotent antagonists at the mMC3R and modest 4-fold increased potency at the mMC4R compared to 6. Substitution with Arg (26) resulted in an equipotent antagonist at the mMC3R compared to 6 and 5-fold increased potency at the mMC4R.
Discussion and Conclusions
It has long been recognized that there remains a need for potent and selective ligands for the melanocortin receptors.56 Potent antagonists for the MC3R and MC4R have previously been reported. The most well recognized is SHU9119, possessing nM and sub-nM antagonist potency at the MC3R and MC4R, respectively.44 Administration of SHU9119 through an i.c.v. injection into mice induces significant increased feeding in fasted mice,18 suggesting an important role for MC3R and/or MC4R antagonism in negative energy homeostasis disease states and obesity. Complicating the interpretation of the biological response is that SHU9119 also possesses partial agonist activity at the MC3R (Figure 1), as well as agonist activity at the mMC1R and mMC5R.44 The mixed pharmacology and high potency at multiple melanocortin receptor subtypes for SHU9119 limits its use in characterizing the role of the specific receptors in the observed induction of appetite. This limitation may be overcome by the development of selective and potent antagonists. Relatively selective MC4R peptide antagonists have been previously reported; peptides HS014, HS024, and HS131 were reported to be approximately 20-fold selective for the MC4R compared to the MC3R, and maintained μM or lower agonist activity at the MC1R and MC5R.57–59 A 90-fold selectivity in binding affinity was reported for the JKC-363 ligand for the MC4R over the MC3R.60 An even more potent peptide antagonist, MBP10, was reported to be 125-fold functionally selective for the MC4R compared to the MC3R.61 Interestingly, all previously reported selective, potent MC4R antagonists are derived from modifications to the endogenous linear agonists α-MSH and β-MSH. To date, a small potent peptide has not been developed from AGRP, despite AGRP being the endogenous antagonist for both the MC3R and MC4R. Additionally, in a panel of central administered antagonists in a mouse cachexia model, AGRP was best able to ameliorate the anorectic effects of IL-1β following a single low dose, indicating antagonists derived from AGRP may be preferential in developing novel therapeutics.62
Therapeutics derived from AGRP may also benefit from the many ascribed functions of AGRP. In addition to being an antagonist at the MC3R and MC4R, AGRP has also been demonstrated to be an inverse agonist at the MC4R.24,25 Additionally, AGRP has been reported to be a biased agonist for the MC4R. A small body of evidence has suggested that AGRP can also signal through non-Gs pathways, including Gi/o signaling,63 the MAPK cascade,64 and most recently through the Kir7.1channel.65 These additional purported roles may result in AGRP derived ligands possessing unique pharmacology that could be beneficial in the development of AGRP-based therapeutics.
Therefore the goal of this project was to design an AGRP derived small peptide that could potentially be further developed into selective and potent antagonists for the MC3R and MC4R. In this study, it was hypothesized that stabilization of the purported AGRP beta-hairpin scaffold might generate an antagonist that could be furthered modified for desirable activity. Peptides 1-6 were therefore synthesized to examine different beta-hairpin loop mimic strategies. The N-terminal de-acetylated form of 1 was previously described as one of the minimal fragments of AGRP that retained antagonist activity at both mMC3R and mMC4R, with the acetylated form 1 showing similar activity.37 The Thr side chain has a greater probability of being found in beta-sheets45,46 and increases the propensity of beta-sheet formation in two different model proteins.47–49 Therefore, herein two Ala amino acids were replaced with the Thr side chain to generate 2 in attempts to stabilize the beta-strands at the base of the hairpin loop. This approach did not result in any significant gain in potency at either the mMC3R or mMC4R. The same Ala residues from 1 were also replaced with two Cys with concurrent formation of a second, unnatural Cys(108-119) disulfide bond resulted in the peptide 3. The more contained peptide 3 resulted in 5-fold increased antagonist potency at the mMC4R. Interestingly, peptide 3 possessed similar potency as a previously reported 22 residue bicyclic peptide derived from hAGRP(101-122) at both the mMC3R and mMC4R, despite possessing eight fewer amino acids.38 This observation supported the working hypothesis that controlling the confirmation of the active loop may impart increased potency without having to elongate the peptide.
To further investigate a different confirmation of the active loop, a series of peptides were synthesized and cyclized head-to-tail through a DPro-Pro sequence. This beta-hairpin mimic design has previously been used in the study of hairpin loops derived from antibodies, platelet-derived growth factor B, and the integrin γ receptor.50–52,66 The synthesis of a beta-hairpin library utilizing the DPro-Pro mimic suggests that it is amenable to SAR studies.51 Peptide 4, cyclizing hAGRP(109-118) through DPro-Pro residues with concurrent substitution of Cys(110,117) to Ala, did not possess antagonist activity at the mMC3R or mMC4R at up to 100 μM concentrations. Replacement of these Ala residues with Thr to generate 5 recovered activity at both receptors, perhaps because of the presence of the beta-strand stabilizing Thr residues. Grafting the Arg-Phe-Phe-Asn-Ala-Phe hexapeptide loop of AGRP cyclized through the DPro-Pro motif resulted in 6, the smallest scaffold peptide with the highest antagonist potency tested at the mMC3R and mMC4R. To our knowledge, this is the first peptide containing only residues from the active loop of AGRP that possesses nM potency at the mMC4R while retaining antagonist activity at the mMC3R. Strikingly, unlike SHU9119, 6 possesses mMC3R antagonist activity without any partial agonist activity (Figure 1), and therefore may be useful in the development of mMC3R selective antagonists without mixed pharmacology.67
The relatively high activity and chemical simplicity of 6 led to further structure-activity relationship studies. Two AGRP based residues, Phe(112) and Phe(113), were substituted with unnatural aromatic amino acids (Figure 2) that had previously been incorporated at varying positions in the potent agonist tetrapeptide sequence Ac-His-DPhe-Arg-Trp-NH2.53–55 It was hypothesized that the potency and selectivity could be increased by incorporating these residues into the 6 scaffold, similar to the 4700-fold agonist selectivity observed when His was replaced by Anc in the tetrapeptide library.54 Unfortunately no peptide was significantly more potent at the mMC3R or mMC4R. Peptides 7 and 12 were equipotent at the mMC3R, while many did not possess activity at up to 100 μM concentrations. Three peptides (12, 17, and 19) maintained the potency of 6 at the mMC4R, while diminished or no observable activity up to 100 μM was seen with the remainder. Substitution at the Phe positions did not result in mMC3R selective peptides, and 17 had the highest mMC4R to mMC3R selectivity of these derivatives (>100-fold compared to 32-fold for 6).
The AGRP Asn(114) residue was also selected for further analysis. The binding affinity of a recombinant Asn(114)Ala hAGRP mutant was minimally affected when compared to the wild-type protein, indicating this residue was not critical for antagonist function.27 Additionally, a docking model of hAGRP(87-132) to the mMC4R suggested that Asn(114) may be in close proximity to Asp(181) of the mMC4R.28 Based upon these observations, it was hypothesized that this position was amendable to substitution and incorporation of a basic amino acid might result in a novel ligand-receptor salt bridge that had potential to increase potency. To explore this hypothesis, a series of basic amino acids were substituted at the AGRP Asn(114) position in the 6 template with varying side chain lengths to probe the potential distance requirements for this purported ligand-receptor salt bridge. An uncharged Gly residue was designed to serve as a negative control. Interestingly, every substitution resulted in increased antagonist potency at the mMC3R and mMC4R and diminished agonist activity at the mMC1R compared to 6. While the increased potency of the basic amino acids at the mMC4R supported the salt bridge model, the Gly substitution was just as potent; further substitutions at this position may be used to clarify this observation. Unlike SHU9119, no partial agonist activity was observed at the mMC3R (Figure 1). This suggests that SHU9119 and 6 may have unique interactions at the mMC3R resulting in the different receptor pharmacological profiles. Unique ligand-receptor interactions have previously been demonstrated at the mMC4R, where specific mMC4R mutations (E92K, F253S) had minimal effects on SHU9119 binding but were unable to bind AGRP(83-132).68 Another mutation (F176K) resulted in a 6-fold loss of SHU9119 antagonist potency and a complete loss of activity for AGRP(83-132).68 These results indicate specific mMC4R residues that may be interacting with AGRP(83-132) but not SHU9119. Similar studies involving SHU9119, 6, and the mMC3R may help elucidate the specific mMC3R residues responsible for the partial agonist activity of SHU9119 and could allow the rationale design of specific mMC3R antagonists that lack agonist activity.
Peptide 22 (Asn to Dap substitution) is particularly exciting as it is as potent as the hAGRP(87-132), within experimental error, at the mMC4R despite being 38 residues smaller. The drastic increase in the mMC4R activity results in 22 possessing an over 160-fold selectivity for the mMC4R over the mMC3R while retaining sub-nM activity at the mMC4R. This high level of selectivity combined with no observable activity at the mMC5R and partial agonist activation observed at the mMC1R may permit 22 to be a valuable tool in elucidating the specific biological effects of MC4R antagonism in vivo.
Experimental
Peptide synthesis
All peptides were synthesized using standard Fmoc methodology.39,40 The coupling reagents [benzotriazol-1-yl-oxy-tris(dimethylamino) phosphonium hexafluorophosphate (BOP) and 1-hydroxybenzotriazole (HOBt)] and amino acids Fmoc-Thr(tBu), Fmoc-Ala, Fmoc-Tyr(tBu), Fmoc-Cys(Trt), Fmoc-Arg(Pbf), Fmoc-Phe, Fmoc-Asn(Trt), Fmoc-Cys(Acm), Fmoc-DPro, Fmoc-phenylglycine (Phg), Fmoc-3-(1-naphthyl)alanine [Nal(1′)], Fmoc-3-(2-naphthyl)alanine [Nal(2′)], Fmoc-Gly, Nα-Fmoc-Nβ-Boc-2,3-diaminopropionic acid (Dap), Nα-Fmoc-Nβ-Boc-2,4-diaminobutyric acid (Dab), Nα-Fmoc-Nδ-Boc-ornithine (Orn), and Fmoc-Lys(Boc) were purchased from Peptides International (Louisville, KY). The Fmoc-4-phenyl-phenylalanine (Bip), Fmoc-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid (Tic), and Fmoc-homo-phenylalanine (hPhe) amino acids were purchased from Synthetech (Albany, OR). The amino acid 3-Fmoc-napthalene-2-carboxylic acid (Anc) was purchased from Pharma Core (High Point, NC). Dichloromethane (DCM), glacial acetic acid, methanol, acetonitrile, and anhydrous ethyl ether were purchased from Fisher (Fair Lawn, NJ). N,N-dimethylformamide (DMF) was purchased from Burdick and Jackson (McGaw Park, IL). Trifluoroacetic acid (TFA), dimethyl sulfoxide (DMSO), piperidine, phenol, and 1,2-ethanedithiol (EDT) were purchased from Sigma (St. Louis, MO). N,N-diisopropylethylamine (DIEA) and triisopropylsilane (TIS) were purchased from Aldrich (Milwaukee, WI). All reagents and chemicals were ACS grade or better and were used without further purification.
Peptides 1, 2, and 3 were manually synthesized on a Rink-amide-MBHA resin (0.4 meq/g substitution) purchased from Peptides International. The syntheses (0.2 mmol scale) consisted of the following steps: (i) removal of the Fmoc group by 20% piperidine in DMF (1×2 min, 1×20 min) (ii) single 2 h coupling of Fmoc-amino acid (3 equiv) using BOP (3 equiv), HOBt (3 equiv), and DIEA (6 equiv) in DMF and repeated until the peptide synthesis was complete. The presence or absence of the free N-α-amino group was monitored using the Kaiser test.69 After completion of the syntheses, peptides were cleaved from the resin and side chain deprotected with a cleavage cocktail consisting of 91:3:3:3 TFA:H2O:TIS:EDT for 2 h. The solution was concentrated, and the peptide precipitated and washed using cold (4 °C) anhydrous ethyl ether. Peptides containing a single disulfide bridge were diluted to 1 mg/mL and oxidized in aqueous solution with 20% DMSO at room temperature and the reaction monitored using analytical RP-HPLC. To ensure the correct disulfide formation for bicyclic peptide 3, paired thiol groups were protected with either the acid labile trityl groups or Acm groups. Following purification of the Acm-protected linear peptide, oxidation of the first disulfide occurred in 20% DMSO in water. After the oxidation reaction was complete, the solution was lyophilized and the peptide was used without further purification. The second disulfide bridge was formed by oxidation with iodine (I2). The peptide was dissolved in AcOH:H2O (4:1) to a final concentration of 2 mg/mL, and I2 (10 equiv) in MeOH was added to the dissolved peptide. The reaction mixture was mixed in the dark at 20 °C for 2 h. The mixture was diluted to twice the volume with water and excess iodine was removed by extraction with an equivalent volume of carbon tetrachloride (repeated five times). The AcOH:H2O solution was then dried to yield the crude, cyclized peptide.
The DPro-Pro containing peptides (0.15 mmol) were manually synthesized on a preloaded H-Pro-2-Chlorotrityl resin (0.67 meq/g substitution) purchased from Peptides International (Louisville, KY), as described above. After the completed synthesis, peptides were cleaved from the resin using 1% TFA/DCM for 5 min. The solution was concentrated and protected peptides were precipitated and washed using cold (4 °C), anhydrous diethyl ether. Cyclization was performed overnight in DMF with a peptide concentration of 1 mg/mL using BOP (3 equiv), HOBt (3 equiv), and DIEA (6 equiv). The DMF was removed with an Alltech Extract Clean reverse phase SPE column (Grace, Columbia MD) and final side chain deprotection was performed in TFA:TIS:H2O (95:2.5:2.5).
All peptides were purified by reversed-phase HPLC using a Shimadzu chromatography system with a photodiode array detector and a semipreparative RP-HPLC C18 bonded silica column (Vydac 218TP1010, 1.0×25 cm). The purified peptides were at least >95% pure as determined by RP-HPLC in two diverse solvent systems and had the correct molecular mass (University of Florida Protein Core Facility).
cAMP Based Functional Bioassay
HEK-293 cells stably expressing the melanocortin receptors were transfected with 4 μg CRE/β-galactosidase reporter gene as previously described.41 Briefly, 5000 to 15000 post-transfection cells were plated into 96-well Primera plates (Falcon) and incubated overnight. Forty-eight hours post-transfection the cells were stimulated with 100 μL of peptide (10−4 to 10−12 M) or forskolin (10−4 M) control in assay medium (DMEM containing 0.1 mg/mL BSA and 0.1 mM isobutylmethylxanthine) for 6 h. The assay media was aspirated and 50 μL of lysis buffer (250 mM Tris-HCl pH 8.0 and 0.1% Triton X-100) was added. The plates were stored at −80 °C overnight. The plates containing the cell lysates were thawed the following day. Aliquots of 10 μL were taken from each well and transferred to another 96-well plate for relative protein determination. To the cell lysate plates, 40 μL of phosphate-buffered saline with 0.5% BSA was added to each well. Subsequently, 150 μL of substrate buffer (60 mM sodium phosphate, 1 mM MgCl2, 10 mM KCl, 5 mM β-mercaptoethanol, 2 mg/mL ONPG) was added to each well and the plates were incubated at 37 °C. The sample absorbance, OD405, was measured using a 96-well plate reader (Molecular Devices). The relative protein was determined by adding 200 μL of 1:5 dilution Bio Rad G250 protein dye:water to the 10 μL cell lysate sample taken previously, and the OD595 was measured on a 96-well plate reader (Molecular Devices). Data points were normalized both to the relative protein content and non-receptor dependent forskolin stimulation. The antagonistic properties of these compounds were evaluated by the ability of these ligands to competitively displace the MTII agonist (Bachem) in a dose-dependent manner, at up to 10 μM concentrations. The pA2 values were generated using the Schild analysis method [pA2 = −log(Ki)].42
Data Analysis
The EC50 and pA2 values represent the mean of duplicate replicates performed in at least three independent experiments. The EC50 and pA2 estimates, and their associated standard errors (SEM), were determined by fitting the data to a nonlinear least-squares analysis using the PRISM program (v4.0, GraphPad Inc.). The ligands were assayed as TFA salts and not corrected for peptide context.
Acknowledgments
This work has been supported by NIH Grants R01DK64250 and R01DK091906. The table of content graphic was generated using the PyMOL Molecular Graphics System, version 1.7.0.5, Schrӧdinger, LLC with the computing resources of the University of Minnesota Supercomputing Institute. Dr. A. Wilczynski was a recipient of an American Heart Association Postdoctoral Fellowship.
Abbreviations
- AGRP
Agouti-related protein
- MC3R
melanocortin 3 receptor
- MC4R
melanocortin 4 receptor
- POMC
proopiomelanocortin
- RP-HPLC
reverse-phase high-pressure liquid chromatography
References
- 1.Allen BM. The results of extirpation of the anterior lobe of the hypophysis and of the thyroid of rana pipiens larvae. Science. 1916;44:755–758. doi: 10.1126/science.44.1143.755. [DOI] [PubMed] [Google Scholar]
- 2.Smith PE. Experimental ablation of the hypophysis in the frog embryo. Science. 1916;44:280–282. doi: 10.1126/science.44.1130.280. [DOI] [PubMed] [Google Scholar]
- 3.Haynes RC, Jr, Berthet L. Studies on the mechanism of action of the adrenocorticotropic hormone. J Biol Chem. 1957;225:115–124. [PubMed] [Google Scholar]
- 4.Huszar D, Lynch CA, Fairchild-Huntress V, Dunmore JH, Fang Q, Berkemeier LR, Gu W, Kesterson RA, Boston BA, Cone RD, Smith FJ, Campfield LA, Burn P, Lee F. Targeted disruption of the melanocortin-4 receptor results in obesity in mice. Cell. 1997;88:131–141. doi: 10.1016/s0092-8674(00)81865-6. [DOI] [PubMed] [Google Scholar]
- 5.Chhajlani V, Wikberg JE. Molecular cloning and expression of the human melanocyte stimulating hormone receptor cDNA. FEBS Lett. 1992;309:417–420. doi: 10.1016/0014-5793(92)80820-7. [DOI] [PubMed] [Google Scholar]
- 6.Mountjoy KG, Robbins LS, Mortrud MT, Cone RD. The cloning of a family of genes that encode the melanocortin receptors. Science. 1992;257:1248–1251. doi: 10.1126/science.1325670. [DOI] [PubMed] [Google Scholar]
- 7.Roselli-Rehfuss L, Mountjoy KG, Robbins LS, Mortrud MT, Low MJ, Tatro JB, Entwistle ML, Simerly RB, Cone RD. Identification of a receptor for gamma melanotropin and other proopiomelanocortin peptides in the hypothalamus and limbic system. Proc Natl Acad Sci USA. 1993;90:8856–8860. doi: 10.1073/pnas.90.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gantz I, Konda Y, Tashiro T, Shimoto Y, Miwa H, Munzert G, Watson SJ, DelValle J, Yamada T. Molecular cloning of a novel melanocortin receptor. J Biol Chem. 1993;268:8246–8250. [PubMed] [Google Scholar]
- 9.Gantz I, Miwa H, Konda Y, Shimoto Y, Tashiro T, Watson SJ, DelValle J, Yamada T. Molecular cloning, expression, and gene localization of a fourth melanocortin receptor. J Biol Chem. 1993;268:15174–15179. [PubMed] [Google Scholar]
- 10.Chhajlani V, Muceniece R, Wikberg JE. Molecular cloning of a novel human melanocortin receptor. Biochem Biophys Res Commun. 1993;195:866–873. doi: 10.1006/bbrc.1993.2125. [DOI] [PubMed] [Google Scholar]
- 11.Gantz I, Shimoto Y, Konda Y, Miwa H, Dickinson CJ, Yamada T. Molecular cloning, expression, and characterization of a fifth melanocortin receptor. Biochem Biophys Res Commun. 1994;200:1214–1220. doi: 10.1006/bbrc.1994.1580. [DOI] [PubMed] [Google Scholar]
- 12.Griffon N, Mignon V, Facchinetti P, Diaz J, Schwartz JC, Sokoloff P. Molecular cloning and characterization of the rat fifth melanocortin receptor. Biochem Biophys Res Commun. 1994;200:1007–1014. doi: 10.1006/bbrc.1994.1550. [DOI] [PubMed] [Google Scholar]
- 13.Nakanishi S, Inoue A, Kita T, Nakamura M, Chang AC, Cohen SN, Numa S. Nucleotide sequence of cloned cDNA for bovine corticotropin-beta-lipotropin precursor. Nature. 1979;278:423–427. doi: 10.1038/278423a0. [DOI] [PubMed] [Google Scholar]
- 14.Blanchard SG, Harris CO, Ittoop OR, Nichols JS, Parks DJ, Truesdale AT, Wilkison WO. Agouti antagonism of melanocortin binding and action in the B16F10 murine melanoma cell line. Biochemistry. 1995;34:10406–10411. doi: 10.1021/bi00033a012. [DOI] [PubMed] [Google Scholar]
- 15.Ollmann MM, Wilson BD, Yang YK, Kerns JA, Chen YR, Gantz I, Barsh GS. Antagonism of central melanocortin receptors in vitro and in vivo by Agouti-related protein. Science. 1997;278:135–138. doi: 10.1126/science.278.5335.135. [DOI] [PubMed] [Google Scholar]
- 16.Fong TM, Mao C, MacNeil T, Kalyani R, Smith T, Weinberg D, Tota MR, VanderPloeg LHT. ART (protein product of Agouti-related transcript) as an antagonist of MC-3 and MC-4 receptors. Biochem Biophys Res Commun. 1997;237:629–631. doi: 10.1006/bbrc.1997.7200. [DOI] [PubMed] [Google Scholar]
- 17.Irani BG, Xiang ZM, Yarandi HN, Holder JR, Moore MC, Bauzo RM, Proneth B, Shaw AM, Millard WJ, Chambers JB, Benoit SC, Clegg DJ, Haskell-Luevano C. Implication of the melanocortin-3 receptor in the regulation of food intake. Eur J Pharmacol. 2011;660:80–87. doi: 10.1016/j.ejphar.2010.10.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fan W, Boston BA, Kesterson RA, Hruby VJ, Cone RD. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature. 1997;385:165–168. doi: 10.1038/385165a0. [DOI] [PubMed] [Google Scholar]
- 19.Ebihara K, Ogawa Y, Katsuura G, Numata Y, Masuzaki H, Satoh N, Tamaki M, Yoshioka T, Hayase M, Matsuoka N, Aizawa-Abe M, Yoshimasa Y, Nakao K. Involvement of agouti-related protein, an endogenous antagonist of hypothalamic melanocortin receptor, in leptin action. Diabetes. 1999;48:2028–2033. doi: 10.2337/diabetes.48.10.2028. [DOI] [PubMed] [Google Scholar]
- 20.Poggioli R, Vergoni AV, Bertolini A. ACTH-(1-24) and Alpha-MSH antagonize feeding-behavior stimulated by kappa opiate agonists. Peptides. 1986;7:843–848. doi: 10.1016/0196-9781(86)90104-x. [DOI] [PubMed] [Google Scholar]
- 21.Graham M, Shutter JR, Sarmiento U, Sarosi I, Stark KL. Overexpression of AGRT leads to obesity in transgenic mice. Nat Genet. 1997;17:273–274. doi: 10.1038/ng1197-273. [DOI] [PubMed] [Google Scholar]
- 22.Adan RAH, Hillebrand JJG, De Rijke C, Nijenhuis W, Vink T, Garner KM, Kas MJH. Melanocortin system and eating disorders. Ann N Y Acad Sci. 2003;994:267–274. doi: 10.1111/j.1749-6632.2003.tb03189.x. [DOI] [PubMed] [Google Scholar]
- 23.Kas MJH, van Dijk G, Scheurink AJW, Adan RAH. Agouti-related protein prevents self-starvation. Mol Psychiatr. 2003;8:235–240. doi: 10.1038/sj.mp.4001206. [DOI] [PubMed] [Google Scholar]
- 24.Haskell-Luevano C, Monck EK. Agouti-related protein functions as an inverse agonist at a constitutively active brain melanocortin-4 receptor. Regul Pept. 2001;99:1–7. doi: 10.1016/s0167-0115(01)00234-8. [DOI] [PubMed] [Google Scholar]
- 25.Nijenhuis WAJ, Oosterom J, Adan RAH. AgRP(83-132) acts as an inverse agonist on the human-melanocortin-4 receptor. Mol Endocrinol. 2001;15:164–171. doi: 10.1210/mend.15.1.0578. [DOI] [PubMed] [Google Scholar]
- 26.Yang YK, Thompson DA, Dickinson CJ, Wilken J, Barsh GS, Kent SBH, Gantz I. Characterization of Agouti-related protein binding to melanocortin receptors. Mol Endocrinol. 1999;13:148–155. doi: 10.1210/mend.13.1.0223. [DOI] [PubMed] [Google Scholar]
- 27.Tota MR, Smith TS, Mao C, MacNeil T, Mosley RT, Van der Ploeg LHT, Fong TM. Molecular interaction of Agouti protein and Agouti-related protein with human melanocortin receptors. Biochemistry. 1999;38:897–904. doi: 10.1021/bi9815602. [DOI] [PubMed] [Google Scholar]
- 28.Wilczynski A, Wang XS, Joseph CG, Xiang ZM, Bauzo RM, Scott JW, Sorensen NB, Shaw AM, Millard WJ, Richards NG, Haskell-Luevano C. Identification of putative Agouti-related protein(87-132)-melanocortin-4 receptor interactions by homology molecular modeling and validation using chimeric peptide ligands. J Med Chem. 2004;47:2194–2207. doi: 10.1021/jm0303608. [DOI] [PubMed] [Google Scholar]
- 29.Joseph CG, Wang XS, Scott JW, Bauzo RM, Xiang ZM, Richards NG, Haskell-Luevano C. Stereochemical studies of the monocyclic agouti-related protein (103-122) Arg-Phe-Phe residues: Conversion of a melanocortin-4 receptor antagonist into an agonist and results in the discovery of a potent and selective melanocortin-1 agonist. J Med Chem. 2004;47:6702–6710. doi: 10.1021/jm0492756. [DOI] [PubMed] [Google Scholar]
- 30.Joseph CG, Wilczynski A, Holder JR, Xiang ZM, Bauzo RM, Scott JW, Haskell-Luevano C. Chimeric NDP-MSH and MTII melanocortin peptides with agouti-related protein (AGRP) Arg-Phe-Phe amino acids possess agonist melanocortin receptor activity. Peptides. 2003;24:1899–1908. doi: 10.1016/j.peptides.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 31.Wilczynski A, Wilson KR, Scott JW, Edison AS, Haskell-Luevano C. Structure-activity relationships of the unique and potent agouti-related protein (AGRP)-melanocortin chimeric Tyr-c[beta-Asp-His-DPhe-Arg-Trp-Asn-Ala-Phe-Dpr]-Tyr-NH2 peptide template. J Med Chem. 2005;48:3060–3075. doi: 10.1021/jm049010r. [DOI] [PubMed] [Google Scholar]
- 32.Jackson PJ, Yu B, Hunrichs B, Thompson DA, Chai B, Gantz I, Millhauser GL. Chimeras of the agouti-related protein: insights into agonist and antagonist selectivity of melanocortin receptors. Peptides. 2005;26:1978–1987. doi: 10.1016/j.peptides.2004.12.036. [DOI] [PubMed] [Google Scholar]
- 33.McNulty JC, Thompson DA, Bolin KA, Wilken J, Barsh GS, Millhauser GL. High-resolution NMR structure of the chemically-synthesized melanocortin receptor binding domain AGRP(87-132) of the agouti-related protein. Biochemistry. 2001;40:15520–15527. doi: 10.1021/bi0117192. [DOI] [PubMed] [Google Scholar]
- 34.Bolin KA, Anderson DJ, Trulson JA, Thompson DA, Wilken J, Kent SBH, Gantz I, Millhauser GL. NMR structure of a minimized human agouti related protein prepared by total chemical synthesis. FEBS Lett. 1999;451:125–131. doi: 10.1016/s0014-5793(99)00553-0. [DOI] [PubMed] [Google Scholar]
- 35.Jackson PJ, McNulty JC, Yang YK, Thompson DA, Chai B, Gantz I, Barsh GS, Millhauser GL. Design, pharmacology, and NMR structure of a minimized cystine knot with agouti-related protein activity. Biochemistry. 2002;41:7565–7572. doi: 10.1021/bi012000x. [DOI] [PubMed] [Google Scholar]
- 36.Haskell-Luevano C, Monck EK, Wan YP, Schentrup AM. The agouti-related protein decapeptide (Yc[CRFFNAFC]Y) possesses agonist activity at the murine melanocortin-1 receptor. Peptides. 2000;21:683–689. doi: 10.1016/s0196-9781(00)00194-7. [DOI] [PubMed] [Google Scholar]
- 37.Joseph CG, Bauzo RM, Xiang Z, Shaw AM, Millard WJ, Haskell-Luevano C. Elongation studies of the human agouti-related protein (AGRP) core decapeptide (Yc[CRFFNAFC]Y) results in antagonism at the mouse melanocortin-3 receptor. Peptides. 2003;24:263–270. doi: 10.1016/s0196-9781(03)00030-5. [DOI] [PubMed] [Google Scholar]
- 38.Wilczynski A, Wang XS, Bauzo RM, Xiang Z, Shaw AM, Millard WJ, Richards NG, Edison AS, Haskell-Luevano C. Structural characterization and pharmacology of a potent (Cys101-Cys119, Cys110-Cys117) bicyclic agouti-related protein (AGRP) melanocortin receptor antagonist. J Med Chem. 2004;47:5662–5673. doi: 10.1021/jm049620r. [DOI] [PubMed] [Google Scholar]
- 39.Carpino LA, Han GY. 9-Fluorenylmethoxycarbonyl function, a new base-sensitive amino-protecting group. J Am Chem Soc. 1970;92:5748–5749. [Google Scholar]
- 40.Carpino LA, Han GY. The 9-fluorenylmethoxycarbonyl amino-protecting group. J Org Chem. 1972;37:3404–3409. [Google Scholar]
- 41.Chen WB, Shields TS, Stork PJS, Cone RD. A colorimetric assay for measuring activation of G(s)-coupled and G(q)-coupled signaling pathways. Anal Biochem. 1995;226:349–354. doi: 10.1006/abio.1995.1235. [DOI] [PubMed] [Google Scholar]
- 42.Schild HO. pA, a new scale for the measurement of drug antagonism. Br J Pharmacol Chemother. 1947;2:189–206. doi: 10.1111/j.1476-5381.1947.tb00336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Al-Obeidi F, O’Connor SD, Job C, Hruby VJ, Pettitt BM. NMR and quenched molecular dynamics studies of superpotent linear and cyclic alpha-melanotropins. J Pept Res. 1998;51:420–431. doi: 10.1111/j.1399-3011.1998.tb00640.x. [DOI] [PubMed] [Google Scholar]
- 44.Hruby VJ, Lu DS, Sharma SD, Castrucci AD, Kesterson RA, Alobeidi FA, Hadley ME, Cone RD. Cyclic lactam alpha-melanotropin analogs of Ac-Nle(4)-cyclo Asp(5),D-Phe(7),Lys(10) alpha-melanocyte-stimulating hormone-(4-10)-NH2 with bulky aromatic-amino-acids at position-7 show high antagonist potency and selectivity at specific melanocortin receptors. J Med Chem. 1995;38:3454–3461. doi: 10.1021/jm00018a005. [DOI] [PubMed] [Google Scholar]
- 45.Chou PY, Fasman GD. Conformational parameters for amino-acids in helical beta sheet and random coil regions calculated from proteins. Biochemistry. 1974;13:211–222. doi: 10.1021/bi00699a001. [DOI] [PubMed] [Google Scholar]
- 46.Fasman GD. The development of the prediction of protein structure. In: Fasman GD, editor. Prediction of Protein Structure and the Principles of Protein Conformation. Plenum; New York: 1989. pp. 193–316. [Google Scholar]
- 47.Minor DL, Jr, Kim PS. Measurement of the beta-sheet-forming propensities of amino acids. Nature. 1994;367:660–663. doi: 10.1038/367660a0. [DOI] [PubMed] [Google Scholar]
- 48.Smith CK, Withka JM, Regan L. A thermodynamic scale for the beta-sheet forming tendencies of the amino-acids. Biochemistry. 1994;33:5510–5517. doi: 10.1021/bi00184a020. [DOI] [PubMed] [Google Scholar]
- 49.Kim CA, Berg JM. Thermodynamic beta-sheet propensities measured using a zinc-finger host peptide. Nature. 1993;362:267–270. doi: 10.1038/362267a0. [DOI] [PubMed] [Google Scholar]
- 50.Spath J, Stuart F, Jiang LY, Robinson JA. Stabilization of a beta-hairpin conformation in a cyclic peptide using the templating effect of a heterochiral diproline unit. Helv Chim Acta. 1998;81:1726–1738. [Google Scholar]
- 51.Jiang LJ, Moehle K, Dhanapal B, Obrecht D, Robinson JA. Combinatorial biomimetic chemistry: Parallel synthesis of a small library of beta-hairpin mimetics based on loop III from human platelet-derived growth factor B. Helv Chim Acta. 2000;83:3097–3112. [Google Scholar]
- 52.Favre M, Moehle K, Jiang LY, Pfeiffer B, Robinson JA. Structural mimicry of canonical conformations in antibody hypervariable loops using cyclic peptides containing a heterochiral diproline template. J Am Chem Soc. 1999;121:2679–2685. [Google Scholar]
- 53.Holder JR, Bauzo RM, Xiang ZM, Haskell-Luevano C. Structure-activity relationships of the melanocortin tetrapeptide Ac-His-DPhe-Arg-Trp-NH2 at the mouse melanocortin receptors: Part 2 modifications at the Phe position. J Med Chem. 2002;45:3073–3081. doi: 10.1021/jm010524p. [DOI] [PubMed] [Google Scholar]
- 54.Holder JR, Bauzo RM, Xiang ZM, Haskell-Luevano C. Structure-activity relationships of the melanocortin tetrapeptide Ac-His-DPhe-Arg-Trp-NH2 at the mouse melanocortin receptors. 1. Modifications at the His position. J Med Chem. 2002;45:2801–2810. doi: 10.1021/jm0104872. [DOI] [PubMed] [Google Scholar]
- 55.Holder JR, Xiang ZM, Bauzo RM, Haskell-Leuvano C. Structure-activity relationships of the melanocortin tetrapeptide Ac-His-D-Phe-Arg-Trp-NH2 at the mouse melanocortin receptors. 4. Modifications at the Trp position. J Med Chem. 2002;45:5736–5744. doi: 10.1021/jm020296e. [DOI] [PubMed] [Google Scholar]
- 56.Hruby VJ, Han G. The molecular pharmacology of alpha-melanocyte stimulating hormone. In: Cone RD, editor. The Melanocortin Receptors. Humana Press Inc; Totowa, NJ: 2000. pp. 239–262. [Google Scholar]
- 57.Kask A, Mutulis F, Muceniece R, Pahkla R, Mutule I, Wikberg JES, Rago L, Schioth HB. Discovery of a novel superpotent and selective melanocortin-4 receptor antagonist (HS024): Evaluation in vitro and in vivo. Endocrinology. 1998;139:5006–5014. doi: 10.1210/endo.139.12.6352. [DOI] [PubMed] [Google Scholar]
- 58.Schioth HB, Mutulis F, Muceniece R, Prusis P, Wikberg JE. Discovery of novel melanocortin4 receptor selective MSH analogues. Br J Pharmacol. 1998;124:75–82. doi: 10.1038/sj.bjp.0701804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schioth HB, Kask A, Mutulis F, Muceniece R, Mutule I, Mutule I, Mandrika I, Wikberg JE. Novel selective melanocortin 4 receptor antagonist induces food intake after peripheral administration. Biochem Biophys Res Commun. 2003;301:399–405. doi: 10.1016/s0006-291x(02)03065-6. [DOI] [PubMed] [Google Scholar]
- 60.Kim MS, Small CJ, Russell SH, Morgan DG, Abbott CR, alAhmed SH, Hay DL, Ghatei MA, Smith DM, Bloom SR. Effects of melanocortin receptor ligands on thyrotropin-releasing hormone release: evidence for the differential roles of melanocortin 3 and 4 receptors. J Neuroendocrinol. 2002;14:276–282. doi: 10.1046/j.1365-2826.2002.00769.x. [DOI] [PubMed] [Google Scholar]
- 61.Bednarek MA, MacNeil T, Kalyani RN, Tang R, Van der Ploeg LHT, Weinberg DH. Selective, high affinity peptide antagonists of alpha-melanotropin action at human melanocortin receptor 4: Their synthesis and biological evaluation in vitro. J Med Chem. 2001;44:3665–3672. doi: 10.1021/jm010165y. [DOI] [PubMed] [Google Scholar]
- 62.Joppa MA, Ling N, Chen C, Gogas KR, Foster AC, Markison S. Central administration of peptide and small molecule MC4 receptor antagonists induce hyperphagia in mice and attenuate cytokine-induced anorexia. Peptides. 2005;26:2294–2301. doi: 10.1016/j.peptides.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 63.Buch TRH, Heling D, Damm E, Gudermann T, Breit A. Pertussis toxin-sensitive signaling of melanocortin-4 receptors in hypothalamic GT1-7 cells defines Agouti-related protein as a biased agonist. J Biol Chem. 2009;284:26411–26420. doi: 10.1074/jbc.M109.039339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mo XL, Tao YX. Activation of MAPK by inverse agonists in six naturally occurring constitutively active mutant human melanocortin-4 receptors. Biochim Biophys Acta -Mol Basis Dis. 2013;1832:1939–1948. doi: 10.1016/j.bbadis.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 65.Ghamari-Langroudi M, Digby GJ, Sebag JA, Millhauser GL, Palomino R, Matthews R, Gillyard T, Panaro BL, Tough IR, Cox HM, Denton JS, Cone RD. G-protein-independent coupling of MC4R to Kir71 in hypothalamic neurons. doi: 10.1038/nature14051. [Online Early Access] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Robinson JA. Beta-hairpin peptidomimetics: design, structures and biological activities. Acc Chem Res. 2008;41:1278–1288. doi: 10.1021/ar700259k. [DOI] [PubMed] [Google Scholar]
- 67.Doering SR, Todorovic A, Haskell-Luevano C. Melanocortin antagonist tetrapeptides with minimal agonist activity at the mouse melanocortin-3 receptor. ACS Med Chem Lett. doi: 10.1021/ml500340z. [Online Early Access] Published Online: December 29, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Haskell-Luevano C, Cone RD, Monck EK, Wan YP. Structure activity studies of the melanocortin-4 receptor by in vitro mutagenesis: Identification of agouti-related protein (AGRP), melanocortin agonist and synthetic peptide antagonist interaction determinants. Biochemistry. 2001;40:6164–6179. doi: 10.1021/bi010025q. [DOI] [PubMed] [Google Scholar]
- 69.Kaiser E, Colescott RL, Bossinger CD, Cook PI. Color test for detection of free terminal amino groups in the solid-phase synthesis of peptides. Anal Biochem. 1970;34:595–598. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]
- 70.Haskell-Luevano C, Lim S, Yuan W, Cone RD, Hruby VJ. Structure activity studies of the melanocortin antagonist SHU9119 modified at the 6, 7, 8, and 9 positions. Peptides. 2000;21:49–57. doi: 10.1016/s0196-9781(99)00167-9. [DOI] [PubMed] [Google Scholar]