

Abstract
Previous studies have shown that acute kidney injury (AKI) is exacerbated in C-reactive protein transgenic mice (CRP Tg) and alleviated in Smad3 knockout mice (Smad3 KO). The present study used CRP Tg, Smad3 KO, and CRP Tg/Smad3 KO mice to investigate the signaling mechanisms by which CRP promotes AKI. As previously reported, we found that the elevation of serum creatinine and the extent of tubular epithelial cell (TEC) necrosis following ischemia-reperfusion injury (IRI) induced AKI was greater for CRP Tg than wild type (Wt). In contrast in Smad3 KO mice, whether CRP Wt or CRP Tg, these responses were blunted. Exacerbation of AKI in CRP Tg was associated with increased TGF-β/Smad3 signaling, i.e. increased expression of the cyclin kinase inhibitor p27, suppression of phosphorylated CDK2, and decreased expression of cyclin E. Concomitantly, TEC proliferation was arrested at the G1 phase in CRP Tg, as evidenced by fewer BrdU+ and PCNA+ cells. On the other hand protection from AKI in Smad3 KO mice was associated with decreased expression of p27 and promotion of CDK2/cyclin E-dependent G1/S transition of TECs. In vitro studies using human TEC showed that (i) CRP activates Smad3 via both TGF-β-dependent and ERK/MAPK crosstalk mechanisms, (ii) Smad3 binds directly to p27, and (iii) blockade of Smad3 or the Fc receptor CD32 prevents CRP-induced p27-dependent G1 cell cycle arrest. In vivo, treatment of CRP Tg with a Smad3 inhibitor improved AKI outcomes. In their sum these data suggest that in CRP Tg mice undergoing AKI, impaired TEC regeneration is likely the result of CD32-Smad3-p27 driven inhibition of the CDK2/cyclin E complex. Targeting Smad3 may offer a new treatment approach for AKI.
Keywords: CRP, AKI, Smad3, p27, cell cycle arrest
Introduction
Increasing evidence shows that inflammation plays a critical role in the triggering and progression of acute kidney injury (AKI).1,2 Thus innate and adaptive immunity, pro-inflammatory cytokines and chemokines, oxidative stress molecules, damage–associated molecular patterns, and pathogen–associated molecular patterns have all been shown to be important in AKI.1,2 However, a thorough understanding of the pathogenic mechanisms of AKI has not yet been achieved and AKI remains a major cause of chronic kidney disease.3,4 Thus, research into AKI needs to continuously look for new pathogenic mechanisms with the hopes of developing new therapies for AKI.5
C-reactive protein (CRP) is an acute phase protein, i.e. its blood levels are raised rapidly and robustly in response to various inflammatory states. Besides its use as a biomarker of inflammation, CRP has been recognized as a pathogenic mediator in angiotensin II-induced hypertensive heart disease,6 diabetic kidney disease,7 and obstructive nephropathy.8 Recent studies done using mice that express human CRP (CRP Tg) showed that CRP promotes AKI.9 We subsequently reported that serum levels of CRP are highly elevated in patients with AKI and that CRP promotion of AKI in CRP Tg mice is linked to impaired tubular epithelial cell (TEC) regeneration.10 However, the signaling mechanisms by which CRP exacerbates AKI or impairs the regeneration of TEC during AKI remain unknown.
Increasing evidence shows that activation of TGF-β signaling in TECs can promote AKI by inhibiting TEC proliferation and increasing renal inflammation.11–13 Similarly we have previously shown that CRP promotion of inflammation and fibrosis in renal and cardiac diseases is also associated with activation of TGF-β/Smad3 signaling.6–8 It is now well accepted that TGF-β, via a Smad-dependent mechanism, causes cell cycle arrest at the G1 phase and thereby potently inhibits cell proliferation.14–16 A recent study showing that mice lacking Smad3 are protected against renal ischemia-reperfusion injury (IRI) underlined the importance of Smad3 in AKI.17 However, the causal mechanisms linking Smad3 to AKI pathogenesis remain unknown. TECs have been identified as the main targets of renal ischemia and of nephrotoxic drugs, and the degree of TEC regeneration or repair largely determines the progression or repair of AKI.18,19
Since CRP has been shown to activate Smad3 signaling in TEC,6–8 and because phosphorylated Smad3 can translocate into the nucleus and transcriptionally regulate target genes that regulate the cell cycle,16 we hypothesized that CRP promotion of AKI might involve a Smad3-dependent mechanism. We tested this hypothesis in vivo using CRP Tg mice in which the Smad3 gene was deleted and in vitro using cultured TEC. Finally, we tested in vivo whether targeted inhibition of Smad3 was a viable therapeutic approach for AKI.
Results
CRP induces Smad3 activation in vivo and in vitro
It has been shown that CRP activates TGF-β/Smad signaling in cardiac and chronic kidney diseases.6–8 Herein we examined whether CRP is able to induce Smad3 activation in vivo using a mouse model of AKI and in vitro using human TEC (HK-2 cells). As shown in Figure 1 (A, B), Western blots and immunohistochemistry revealed that activated (phosphorylated) Smad3 was found in increased abundance in the kidneys of mice subjected to AKI, with the increase being significantly enhanced for AKI kidneys from CRP Tg mice. Similarly, HK-2 cells cultured in the absence of TGF-β1 showed rapid and sustained Smad3 phosphorylation in response to CRP (Figure 1C). CRP-induced Smad3 phosphorylation in HK-2 cells was dose-dependent and could be blocked by inclusion of a neutralizing anti-CD32 antibody (Figure 1D), suggesting that CRP activation of Smad3 might be via FcγRII.
Figure 1. CRP activates Smad3 signaling in the injured kidney and HK-2 TECs in vitro.
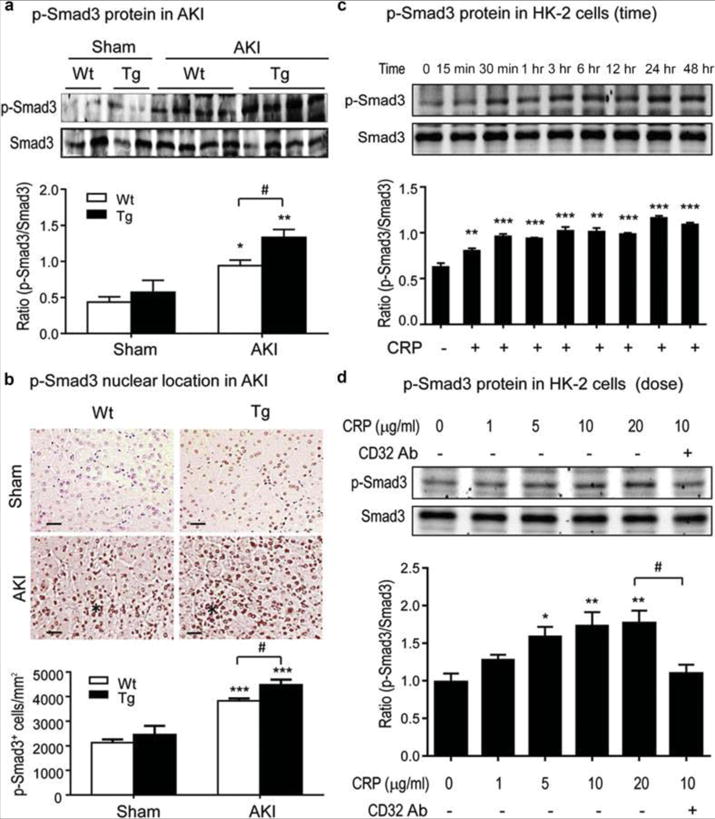
Western blot of phospho (p)-Smad3 protein (A) and its nuclear localization (B) in the kidneys after renal IRI. The asterisks in panel B mark areas of damage where Smad3 appears to exhibit a nuclear staining pattern. Scale bar= 50μM. (C, D) Western blots demonstrate that CRP induces phosphorylation of Smad3 in HK-2 cells in a time and dosage-dependent manner, being significant as early as 15–30 mins and peaking at 24 h. CRP-induced p-Smad3 at 30 mins is blocked by a neutralizing anti-CD32 antibody (5μg/ml). Each bar represents the mean ± SEM from groups of 6 mice or at least 3 independent experiments in vitro. *P<0.05, **P<0.01, ***P<0.001 versus sham group or time/dosage 0. # P<0.05 as indicated.
Deletion of Smad3 prevents CRP-induced exacerbation of renal dysfunction and injury in CRP Tg mice subjected to AKI
We next tested the functional importance of Smad3 in the amplification of AKI seen in transgenic mice expressing human CRP. By breeding CRP Tg mice (C57BL/6) to Smad3 heterozygous mice (also C57BL/6) we generated the following 4 mouse genotypes: CRP wild-type (Wt)/Smad3 Wt, CRP Wt/Smad3 KO, CRP Tg/Smad3 Wt, and CRP Tg/Smad3 KO. We then induced AKI in these mice using a well-established renal ischemic-reperfusion (IR) approach that involved bilateral clamping of the renal arteries for 40 mins.10 Twenty-four hours after renal IR, CRP Tg/Smad3 Wt mice developed more severe renal injury with higher serum creatinine levels and more severe ischemic necrosis in tubular epithelial cells (TECs) compared to treatment-matched CRP Wt/Smad3 Wt mice (Figure 2). In stark contrast, deletion of Smad3 prevented both the elevation of serum creatinine and the induction of TEC necrosis in CRP Wt and CRP Tg mice undergoing AKI (Figure 2). Importantly, these results show that the AKI exacerbating effect of CRP requires Smad3 expression.
Figure 2. Smad3 deficiency protects against renal dysfunction and tubular necrosis and eliminates the exacerbation of AKI in CRP Tg mice.
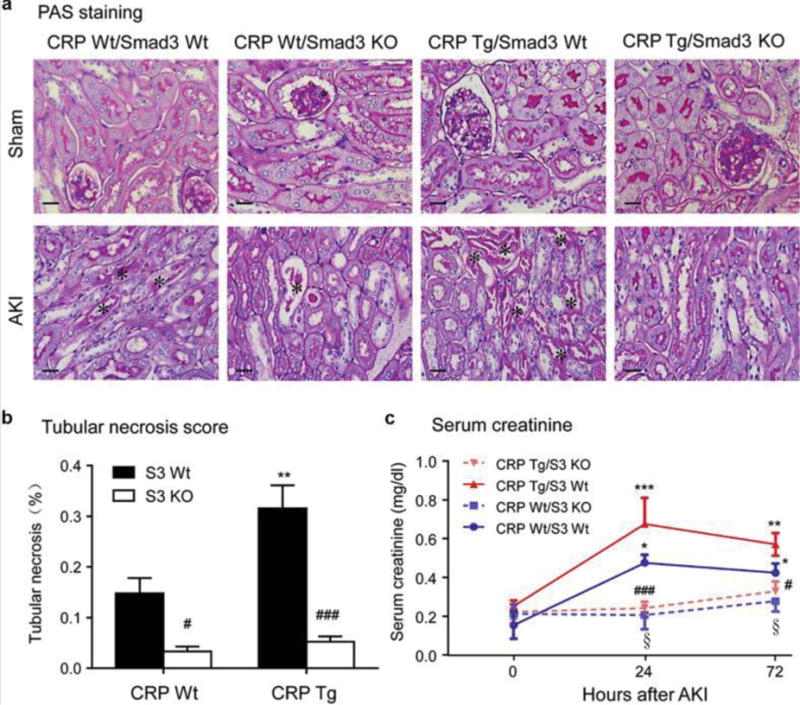
(A and B) PAS-staining in the sham and AKI kidneys and quantitation of tubular necrosis. The asterisks in panel A indicates necrotic tubules. (C) Serum creatinine levels after AKI. Each bar represents the mean ± SEM for groups of 6 mice. *P<0.05, **P<0.01,**P<0.001 versus CRP Wt/Smad3 Wt mice or normal controls (0); ﹟P<0.05, ###P<0.001 versus the CRP Tg/Smad3 Wt mice at the same time point; §P<0.05 versus CRP Wt/Smad3 Wt mice at the same time point. Scale bar, 50μM (A).
Mice lacking Smad3 are protected from CRP–induced inhibition of TEC regeneration and G1/S cell cycle arrest during the repair process after AKI
As TEC regeneration is crucial to the AKI repair process, we used BrdU uptake and PCNA staining to assess and compare proliferation of TEC after AKI amongst the four CRP/Smad3 genotypes. The results showed that AKI was accompanied by an increase in the number of BrdU+ and PCNA+ TECs in CRP Wt/Smad3 Wt mice, but not in CRP Tg/Smad3 Wt mice (Figure 3). In the absence of Smad3 expression however the kidneys were largely protected from AKI-induced TEC necrosis, as indicated by the many BrdU+ and PCNA+ cells in both CRP Wt/Smad3 KO and CRP Tg/Smad3 KO kidneys after AKI (Figure 3, Figure S1). These observations suggest that CRP might foster an abnormal repair process in AKI by promoting G1 cell cycle arrest via a Smad3-dependent mechanism. Notably, the few BrdU+ cells seen in CRP Tg/Smad3 Wt AKI kidneys suggests that CRP might inhibit TEC regeneration by preventing cell entry into S phase. Smad3 deficiency is expected to prevent this inhibitory effect by enhancing TEC entering into the S-phase cell cycle. Smad3 may thus act as a key negative regulator of the G1/S transition during TEC regeneration after AKI.
Figure 3. Deficiency of Smad3 promotes tubular epithelial cell regeneration in CRP Tg mice with AKI.
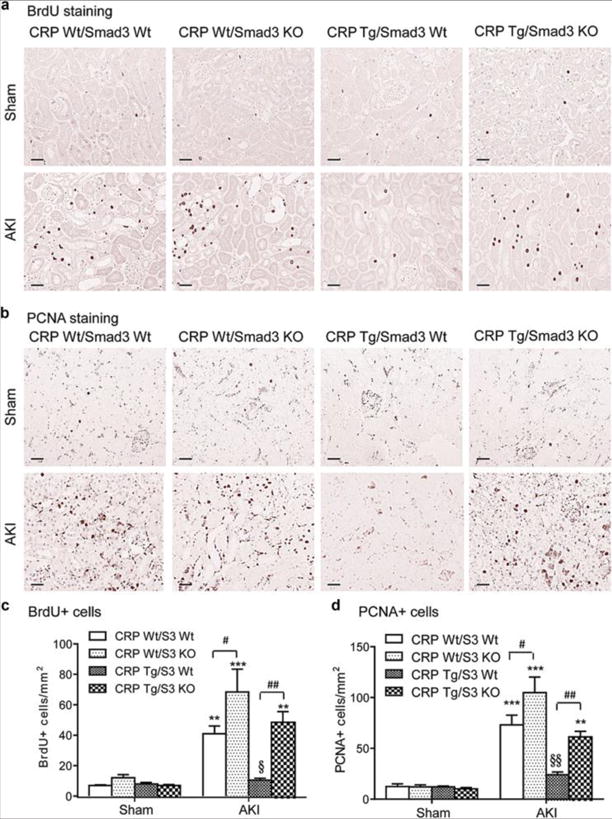
(A, C) Immmunohistochemical staining and quantitation of BrdU+ cells in sham and AKI kidneys. (B, D) Immmunohistochemical staining and quantitation of PCNA+ cells in sham and AKI kidneys. Each bar represents the mean ± SEM for groups of 6 mice. *P<0.05,**P<0.01, ***P<0.001 versus sham group; ﹟P<0.05,##P<0.01 as indicated; §P<0.05, §§ P<0.01 versus CRP Wt/Smad3 Wt mice. Scale bars= 50μM.
It has been shown using many cell types including renal cells,16, 20–22 that TGF-β1 arrests the cell cycle in the middle to late G1 phase via induction of p27 and inhibition of CDK2/cyclin E-dependent signaling. Therefore we next examined if CRP inhibition of TEC regeneration after AKI might involve Smad3-mediated p27-dependent inhibition of CDK2-cyclin E. As shown in Figure 4, following AKI the kidneys from CRP Wt/Smad3 Wt mice showed reduced expression of p27 and increased levels of p-CDK2 and cyclin E, and these changes were exaggerated in CRP Wt/Smad3 KO mice. In contrast, following AKI in CRP Tg/Smad3 Wt mice the expression of p27 and the levels of p-CDK2 and cyclin E in the kidneys were virtually unchanged compared to sham-treated mice. Importantly, the wild-type pattern of p27 expression and p-CDK2 and cyclin-E was observed in CRP Tg mice lacking Smad3. These results are consistent with CRP inhibition of TEC regeneration by arresting cells at the G1 phase via a Smad3-dependent mechanism.
Figure 4. Deletion of Smad3 inhibits p27 but promotes p-CDK2 and cyclin E in CRP Tg mice with AKI.
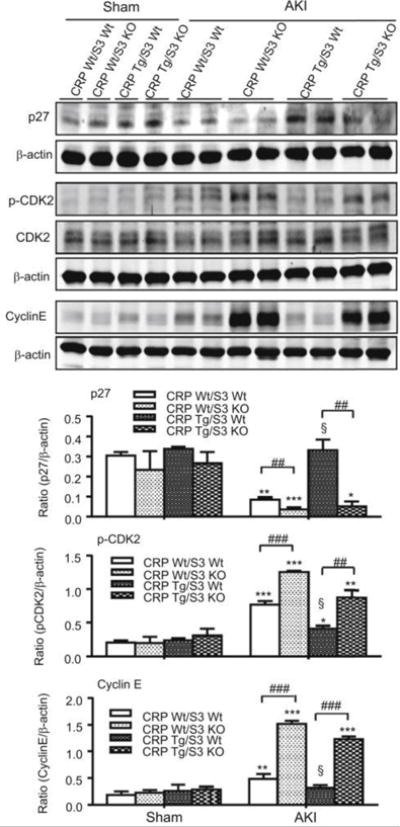
Western blot analysis detects a significant upregulation of p27 but downregulation of phospho (p)-CDK2 and cyclin E after AKI in CRP Tg when compared to the CRP Wt genotype. This pattern is reversed when Smad3 gene is deleted. Each bar represents the mean ± SEM for groups of 6 mice. *P<0.05,**P<0.01, ***P<0.001 versus sham group; ##P<0.01, ###P<0.001 as indicated; §P<0.05 versus CRP Wt/Smad3 Wt mice.
CRP induces Smad3 signaling in TECs via both TGF-β-dependent and TGF-β -independent mechanisms
We next examined the intracellular signaling mechanism by which CRP activates Smad3 in TECs. As shown above (see Figure 1C), in human HK-2 TEC the addition of CRP induced a rapid phosphorylation of Smad3. This action was paralleled by the rapid phosphorylation of the ERK1/2 and p38 MAP kinases (Figure S2A). To further dissect the relationship between Smad3 and MAP kinases, an ERK 1/2 inhibitor (PD98059, 10μg/ml) or a p38 inhibitor (SB203580, 10μg/ml) was added to TEC cultures prior to CRP stimulation. Interestingly, inhibition of either ERK1/2 or p38 effectively blocked the CRP-induced phosphorylation of Smad3, expression of p27, and inhibition of p-CDK2 and cyclin E at 30 min (Figure S2B). CRP thus appears to activate Smad3 signaling via an early ERK/p38 MAPK-Smad3 crosstalk signaling pathway. Since CRP was also able to activate Smad3 signaling at 24 h after its addition to cultures (Figure.1C), we also examined whether this late Smad3 phosphorylation is TGF-β dependent. To determine this we used a neutralizing TGF-β1 antibody and we over-expressed a dominant negative form of the TGF-β receptor II (DN-TGF-βRII) on rat NRK52E TEC. As shown in Supplementary Figure 3, CRP-induced Smad3 phosphorylation at 24 h was blocked by an anti-TGF-β antibody and by overexpressing DN-TGF-βRII, suggesting a TGF-β1-depenent mechanism for CRP-induced late Smad3 signaling.
CRP arrests TEC regeneration at the G1 phase through a TGF-β/Smad3-dependent mechanism in vitro.
We next dissected the specific role of TGF-β/Smad3 signaling in CRP-induced inhibition of TEC proliferation in rat NRK52E cells. First we determined if CRP induces TEC arrest at the G1 phase via a TGF-β-dependent mechanism. We found that addition of a neutralizing anti-TGF-β1 antibody, but not an isotype-matched mouse IgG1, was able to block CRP-induced upregulation of p27 and downregulation of p-CDK2 and cyclin E (Figure 5A). Moreover, overexpression of DN-TGF-βRII also inhibited CRP-induced loss of p-CDK2 and therefore blocked CRP-induced inhibition of TEC proliferation at 24 h as determined by MMT assay (Figure 5, B and C). Flow cytometry also revealed that addition of CRP was able to induce the G1 cell cycle arrest on TECs at 24 h, which was TGF-β-independent since CRP-induced TEC arrest at the G1 phase was not altered by overexpressing DN-TGF-βRII (Figure S4A, B). However, CRP-induced TEC arrest at the G1 cell cycle at 48 h was inhibited by overexpressing DN-TGF-βRII (Figure S4C), suggesting a TGF-β-dependent mechanism.
Figure 5. Blockade of TGF-β signaling inhibits CRP-induced TEC proliferation by downregulating p27 and upregulating p-CDK2 and cyclin E.
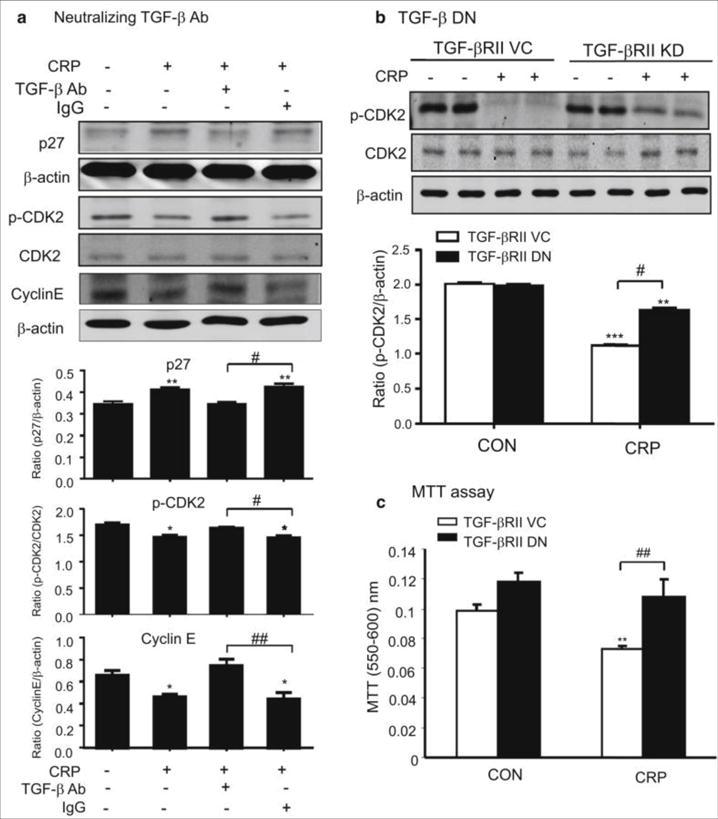
(A and B) Western blot analysis demonstrates that CRP (10μg/ml) induced upregulation of p27 and downregulation of p-CDK2 and cyclin E in NRK52E TECs at 24 h is blocked by a neutralizing anti-TGF-β1 antibody (10μg/ml) (A) and by overexpression of DN-TGF-βRII (B). (C) MTT assay shows that NRK52E TECs overexpressing DN-TGF-βRII are protected against CRP-induced inhibition of cell proliferation. Each bar represents the mean ± SEM for at least three independent experiments (A and B) or six independent experiments (C). *P<0.05, **P<0.01, ***P<0.001 versus control (CON), ﹟P<0.05, ##P<0.01 as indicated.
We then examined the intracellular TGF-β signaling response associated with CRP-induced TEC cycle arrest. Importantly, we found that Smad3 was capable of binding to the p27 promoter in response to CRP, as demonstrated by ChIP assay (Figure 6A). By using the siRNA technique, we could show that Smad3 was substantially knocked down in both NRK52E and HK-2 TECs (Figure 6B and Figure S5A), which resulted in prevention of CRP-induced upregulation of p27 and CRP-induced downregulation of p-CDK2/cyclin E at both the protein and mRNA levels (Figure 6C, D and Figure S5B) thereby abolishing the inhibitory effect of CRP on HK-2 TEC proliferation as detected by MTT assay (Figure S5C). In addition, blocking the binding of CRP to CD32 with a neutralizing anti-CD32 antibody was also capable of inhibiting CRP-induced upregulation of p27 while restoring the levels of p-CDK2 and cyclin E in HK-2 TEC (Figure S5B), suggesting that CRP inhibits TEC proliferation by causing the G1 cell cycle arrest via the TGF-β/Smad3-p27-dependent mechanism.
Figure 6. CRP-induced upregulation of p27 and downregulation of p-CDK2 and cyclin E are Smad3 dependent in TECs.
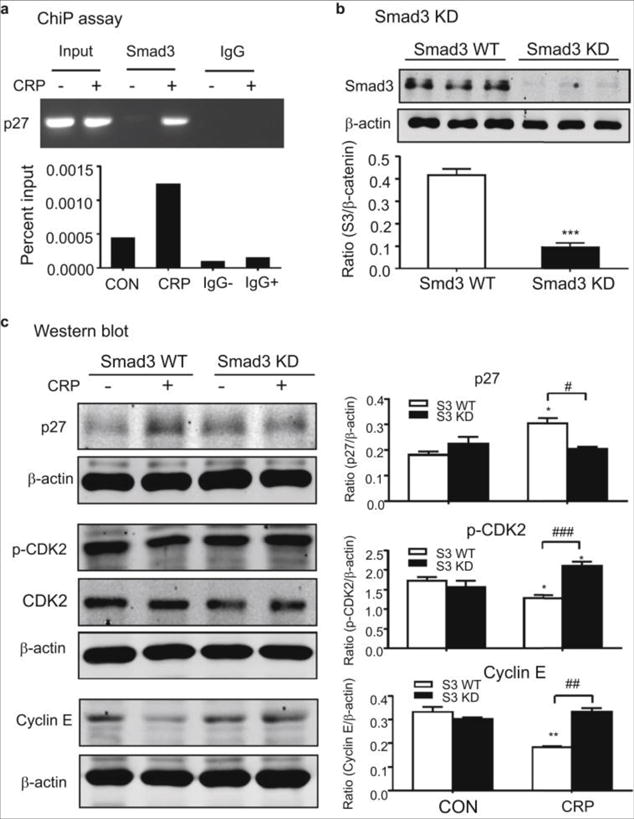
(A) ChIP assay showing that binding of Smad3 to the p27 promoter is enhanced by CRP (10μg/ml). (B)Western blot analysis of Smad3 protein in NRK52E TECs subjected to Smad3 knockdown (KD) (C) Western blots revealed that CRP (10μg/ml) induced upregulation of p27 and downregulation of p-CDK2 and cyclin E at 24 h are inhibited in NRK52E cells by stable expression of Smad3 shRNA. Each bar represents the mean ± SEM for at least three independent experiments. *P<0.05, **P<0.001, ***P<0.001 versus CON or S3 WT, ﹟P<0.05, ##P<0.01, ###P<0.001 as indicated.
Smad3 inhibition improves renal function and ameliorates tubular necrosis in CRP Tg mice with AKI
To further confirm a role for Smad3 in CRP-mediated exacerbation of AKI and to test a potential new therapeutic approach for AKI, we treated CRP Tg/Wt mice with the Smad3 inhibitor SIS3.23 Compared to CRP-Wt mice, in which serum creatinine was elevated and tubular necrosis was moderate 72h after AKI, CRP Tg mice developed higher levels of serum creatinine and more pronounced tubular necrosis (Figure 7). In both CRP Wt/Smad3 Wt and CRP Tg/Smad3 Wt mice subjected to AKI, treatment with SIS3 significantly attenuated progressive renal functional and histological injury compared treatment with DMSO (Figure 7).
Figure 7. Treatment with SIS3 improves renal function and ameliorates tubular necrosis in CRP Wt/Smad3 Wt and CRP Tg/Smad3 Wt mice with AKI.
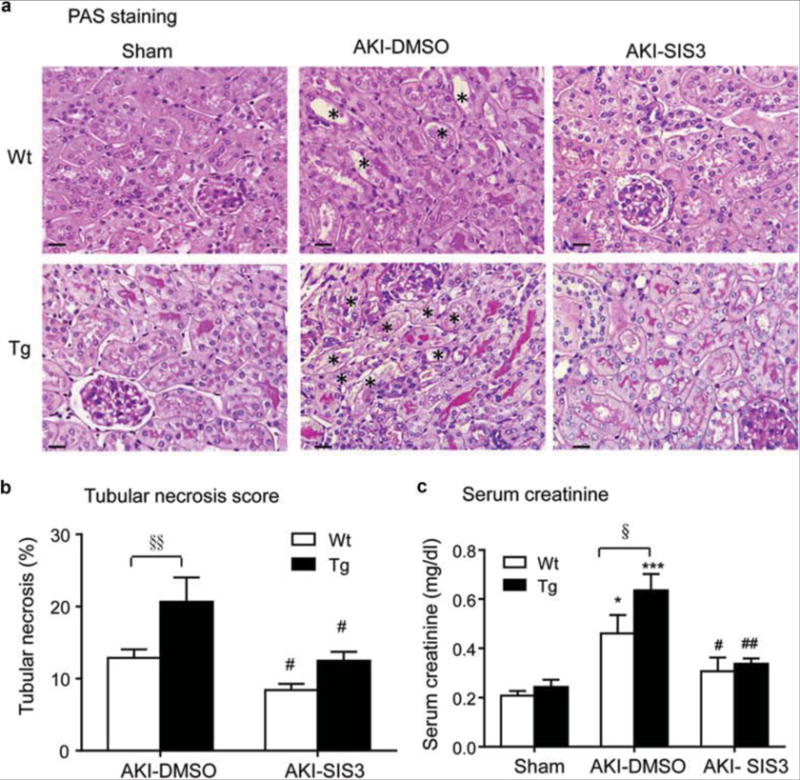
(A) PAS-staining showing tubular necrosis (*) in the sham and AKI kidneys induced in CRP Wt and CRP Tg mice treated with or without SIS3 as described in the Methods. (B) Quantitation of tubular necrosis. (C) Serum creatinine levels. Each bar represents the mean ± SEM for groups of 6 mice. *P<0.05, ***P<0.001 versus sham group; ﹟P<0.05, ##P<0.01 versus the DMSO control-treated group (AKI-DMSO); §P<0.05,§§P<0.01 as indicated. Scale bar, 50μM (A).
Treatment with SIS3 improves tubular epithelial cell regeneration in CRP Tg mice with AKI by inhibiting Smad3-p27 dependent G1 cell cycle arrest
Similar to the results observed in CRP Tg/Smad3KO mice (see Figures 3 and 4), pharmacological inhibition of Smad3 with SIS3 largely protected against TEC death in CRP Tg/Smad3 Wt mice by promoting TEC proliferation with great numbers of BrdU+ and PCNA+ cells (Figure 8). This protective effect was largely associated with the inhibition of Smad3-p27 signaling while largely enhancing p-CDK2 and cyclin E activities (Figures 9 and 10), thereby preventing the G1 cell cycle arrest and promoting TEC G1/S transition as demonstrated by a large member of BrdU+ cells (Figure 8).
Figure 8. Treatment with SIS3 improves tubular epithelial cell proliferation in CRP Wt/Tg mice with AKI.
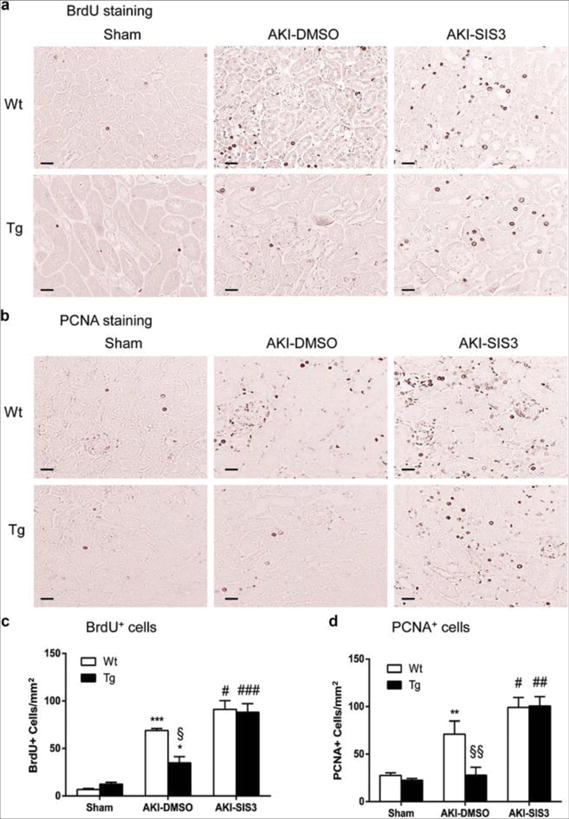
(A, C) BrdU immunostaining and quantitation of BrdU+ cells. (B, D) PCNA immunostaining and quantitation of PCNA+ cells. Each bar represents the mean ± SEM for groups of 6 mice. *P<0.05,**P<0.01, ***P<0.001 versus sham group; ﹟P<0.05, ##P<0.01, ###P<0.001 versus DMSO treated animals (AKI-DMSO); §P<0.05, §§ P<0.01 versus DMSO treated CRP Wt mice. Scale bar, 50μM (A, B).
Figure 9. Treatment with SIS3 inhibits Smad3 signaling in CRP Wt/Tg mice with AKI.
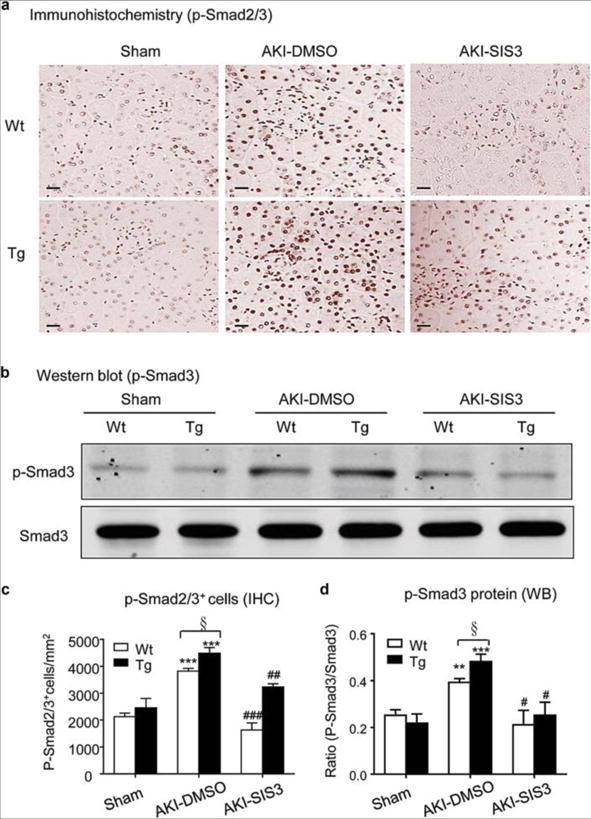
(A) Immunohistochemistry shows an inhibitory effect of SIS3 treatment on levels of phospho-Smad3 nuclear location in the AKI kidney. (B) Western blots show an inhibitory effect of SIS3 treatment on levels of phospho-Smad3 protein in the AKI kidney. (C) Quantitative analysis of p-Smad2/3+ cells by immunohistochemistry (IHC). (D) Quantitative analysis of phosphorylation of Smad3 by Western blots (WB). Each bar represents the mean ± SEM for groups of 6 mice. **P<0.01, ***P<0.001 versus sham group; ﹟P<0.05, ##P<0.01, ###P<0.001 versus DMSO treated group (AKI-DMSO); §P<0.05 as indicated. Scale bar, 50μM (A).
Figure 10. Treatment with SIS3 downregulates p27 and increases p-CDK2 and cyclin E expression in CRP Wt/Tg mice with AKI.
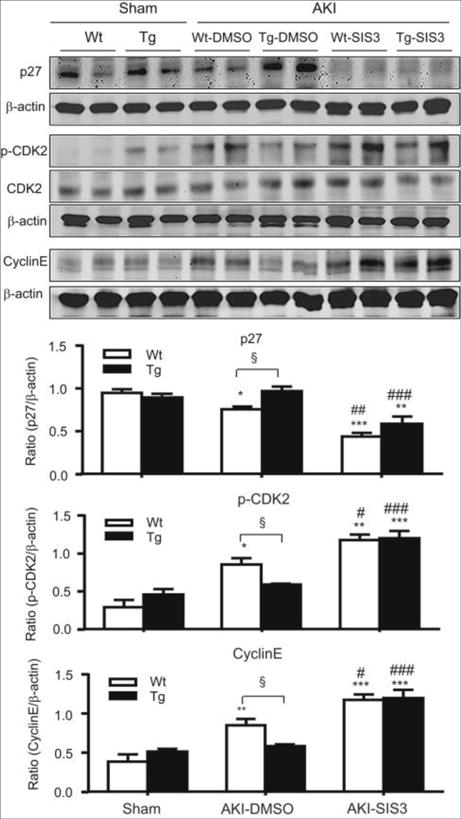
Western blot analysis showing that blockade of Smad3 signaling with SIS3 inhibits p27 but upregulates pCDK2 and cyclin E expression in the AKI kidney in CRP Wt/Tg mice. Each bar represents the mean ± SEM for groups of 6 mice. *P<0.05,**P<0.01, ***P<0.001 versus sham group; ﹟P<0.05,##P<0.01,###P<0.001 versus DMSO treated group (AKI- DMSO); §P<0.05 as indicated.
Discussion
Previously we showed that CRP-mediated exacerbation of AKI involves inhibition of TEC regeneration.10 In the present study we replicate this finding and we define an underlying mechanism by which this effect is potentially realized, i.e. we show that CRP promotion of AKI involves induction of G1 cell cycle arrest via the Smad3-mediated p27-dependent inhibition of CDK2-cyclin E. This finding is novel and is supported by our evidence showing that severely impaired TEC regeneration after AKI in CRP Tg/Smad3 Wt mice is associated with over-activation of TGF-β/Smad3 signaling and increased p27 but reduced CDK2-cyclin E activities. The functional importance of Smad3 in AKI was further demonstrated by our finding that deletion of Smad3 or its pharmacological inhibition protected kidneys from AKI by enhancing TEC regeneration, which was tightly associated with inhibition of p27 while enhancing CDK2-cyclin E activities. Moreover, findings from this study also provided the first evidence for targeting Smad3 as a novel therapy for AKI.
The finding that CRP promotion of AKI involves Smad3-dependent signaling is both novel and significant. We found that CRP could activate Smad3 in TECs directly via the ERK/p38 MAP kinase crosstalk pathway. It has been reported that CRP is capable of signaling through ERK1/2 to induce VEGF by inflammatory macrophages and MMP-10 by cardiomyocytes.24, 25 We have also demonstrated that activation of ERK12/p38 MAP kinases in response to angiotensin II or advanced glycation-end products is able to induce Smad3 signaling.26–28 The present study adds to these findings and suggests that CRP induces rapid Smad3 activation in TEC via a CD32-ERK1/2 and p38 MAP kinase-crosstalk pathway, since blockade of CRP signaling with a neutralizing CD32 antibody or ERK1/2 and p38 signaling with inhibitors is capable of inhibiting CRP-induced phosphorylation of Smad3. In addition, we also found that CRP activates delayed activation of Smad3 in TECs via a TGF-β-dependent manner, as blocking TGF-β with a neutralizing TGF-β antibody or by overexpressing dominant negative TGF-β receptor II (TβRII) was also able to attenuate CRP-induced Smad3 signaling at 24 h. It has been reported that enhanced TGFβ1 signaling in TECs by specifically over-expressing active TGF-β1 is sufficient to cause AKI,11 while conditional deletion of TβRII or inhibition of TGF-β signaling with an ALK5 antagonist can improve AKI by promoting TEC proliferation12,13 revealing an important role of TGF-β signaling in the pathogenesis of AKI. In the present study, we added new information that activation of TGF-β/Smad3 signaling may be an essential mechanism through which CRP mediates AKI. This was supported by the finding that Smad3 was markedly activated in CRP Tg mice with progressive AKI and disrupted Smad3 gene or inhibition of Smad3 with an inhibitor were able to protect CRP Tg mice from AKI. This finding is also consistent with previous studies that genetic deficiency of Smad3 confers structural and functional protection against AKI.17
We also provide initial evidence that CD32 is required by CRP to promote G1 cell cycle arrest in TEC, which was mediated via the Smad3-p27 dependent mechanism. Indeed, In CRP Tg mice, CD32b is highly upregulated in TECs after AKI.10 During the abnormal repair process, the arrest in the G2/M phase of the cell cycle results in severe or sustained kidney injury by facilitating the generation of TGF-β1 and CTGF.29 In the present study we found that CRP induced TEC arrest at the G1 phase of the cell cycle, and we provided evidence that this action is achieved via a CD32-Smad3-p27 dependent pathway. Thus, blocking CRP interaction with FcγRII using an anti-CD32 antibody, and genetic deletion or pharmacological inhibition of Smad3 using SIS3, both enabled TEC G1/S transition. Furthermore, we showed in vivo and in vitro that the rescuing effect of antibody blockade and SIS3 treatment was accompanied by suppression of p27 and enhancement of CDK2- cyclin E activities. It is well known that p27 is a CDK inhibitor, forming quaternary complexes with CDKs and thereby preventing cell cycle progression via cell-cycle arrest at the G1 phase30. Like CRP, TGF-β1 can also induce p27 protein expression while inhibiting CDK2 and cyclin E.20–22, 31 In the present study using ChIP assays we showed that Smad3, a key member of the TGF-β signaling family of molecules, was capable of binding to the p27 promoter in response to CRP stimulation. Thus, CRP can activate Smad3 via both TGF-β-dependent and ERK MAP kinase crosstalk mechanisms, thus inducing p27 expression which may inhibit the CDK2-cyclin E-dependent regeneration of TECs. Accordingly in the absence of Smad3 or in the presence of the Smad3 inhibitor, Smad3 signaling was suppressed and therefore CRP was unable to induce p27 protein expression, thus protecting TECs from the G1 cell cycle arrest. Our findings may also point to activation of Smad3 as the underlying mechanism through which TGF-β reportedly potently inhibits cell proliferation.24, 25
It has previously been shown that inhibition of TGF-β signaling, achieved via targeting of the type-1 receptor with an ALK5 antagonist, can promote the AKI repair process by enhancing TEC proliferation.13 In this study we showed that Smad3 may also be a viable target for therapeutic intervention in AKI. Thus treatment of CRP Tg/Smad3 Wt mice with the Smad3 inhibitor SIS3 resulted in a greater preservation of renal function and reduced the histological signs of renal injury following AKI. This protective effect was associated with the ability of SIS3 to block p27-dependent inhibition of CDK2-cyclin E activities, thereby enhancing the repair process after AKI by promoting the G1/S transition as shown by a large number of BrdU+ TECs in the SIS3-treated AKI kidney.
In conclusion, our data suggests that CRP interacting with TECs (likely via FcγRII) and acting via Smad3, drives p27-dependant inhibition of the CDK2/Cyclin E pathway required for the G1/S transition (summarized in Figure 11). While there are probably multiple reasons for CRP-mediated exacerbation of AKI,7–9 the results from the present study suggest that one of the major means by which CRP promotes AKI is by direct interaction with TECs. Genetic deletion or pharmacological inhibition of Smad3 is able to block this pathway, rescue TEC regeneration, and and thereby protect against AKI. Thus, targeting Smad3 may represent a novel and effective therapy for AKI.
Figure 11. Potential mechanisms of CRP promote AKI.
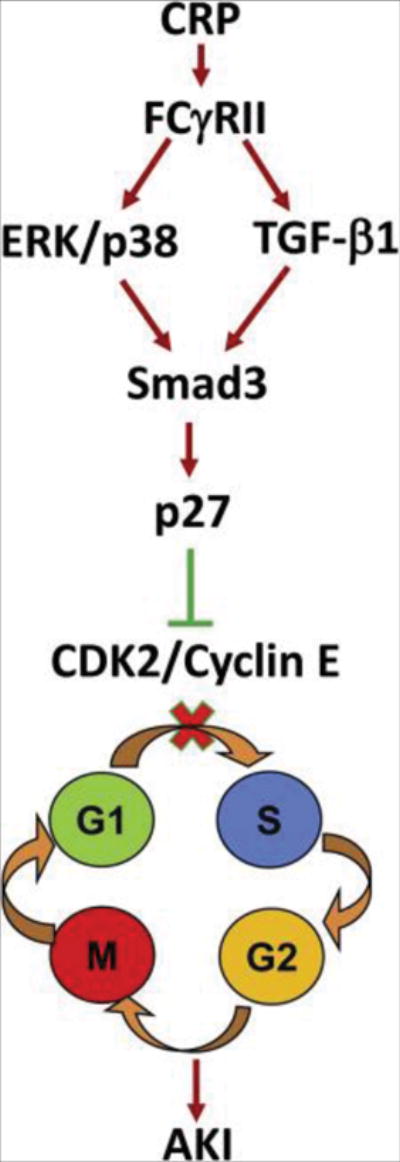
CRP activates Smad3 via both TGF-β1 or ERK/p38 MAP kinase-dependent mechanisms, which upregulates p27 to cause TEC growth arrest at the G1 cell cycle by suppressing CDK2/cyclin E activities.
METHODS
Mouse model of AKI
Human CRP Tg and Smad3 KO mice were used; both phenotypes have been previously described.32, 33 We bred CRP Tg mice (C57BL/6) with Smad3 KO heterozygous mice (C57BL/6), and the resultant CRP Tg/Smad3 KO heterozygous mice were back-crossed to Smad3 KO heterozygous mice to generate four genotypes of mice for the study; CRP Wt/Smad3 Wt, CRP Wt/Smad3 KO, CRP Tg/Smad3 Wt, and CRP Tg/Smad3 KO. Bilateral AKI was induced under anesthesia with i. p. injection of ketamine/xyline in groups of 6–8 mice by clamping the renal arteries with vascular clips to induce ischemia for 40 mins, followed by removal of the clips to allow for reperfusion as previously described.10 Mice were sacrificed at 24h or 72 h after AKI surgery.
To test the effectiveness of Smad3 inhibition on AKI, groups of 6 CRP Tg/Smad3 Wt mice were treated with SIS3 at a dose of 5ug/g/day i.p. for 3 days immediately after surgery. SIS3 has been shown to specifically inhibit Smad3 phosphorylation in vitro23 and the dose used in this study was based on a previous report.34 SIS3 was from Sigma-Aldrich (St Louis, MO, USA) and dissolved in DMSO for use. Control mice received daily i.p. DMSO for 3 days. Mice were sacrificed at 72 h after SIS3 or DMSO treatment.
To identify cells entering S phase, mice received i.p an injection of BrdU (Sigma-Aldrich) at a dose of 50 mg per kg body weight 2 h prior to being sacrificed. Cells entering into the S phase were identified by BrdU positivity by immunohistochemistry. All experimental procedures were approved by the Animal Ethics Committee of The Chinese University of Hong Kong.
Cell culture
Renal tubular epithelial cell lines from both human (HK-2 cells) and rat (NRK52E) were obtained from the American Type Culture Collection (Manassas, VA, USA) and were cultured in DMEM-F12 medium supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA) until sub-confluent. The cells were exposed to recombinant human CRP (R&D Systems, Minneapolis, MN, USA) at concentrations of 0, 1, 5, 10, and 20μg/ml for periods of 15, 30, and 60 min and 3, 6, 12, 24 and 48 h in the presence or absence of a neutralizing polyclonal goat antibody to CD32 or control IgG1 (5μg/ml, R&D system).
To define the crosstalk pathway of CRP-induced Smad3 signaling via ERK/p38 MAP kinases, specific inhibitors to ERK1/2 (PD98059, 10 μg/ml) and p38 (SB203580, 10 μg/ml) from Calbiochem (La Jolla, CA, USA) were added into the culture 1 h before CRP (10μg/ml) stimulation. In addition, CRP-induced Smad3 signaling via the TGF-β-dependent mechanism was examined in a stable dominant negative TGF-βRII (DN-TGFβRII) -expressing NRK52E cell line or by a neutralizing rabbit polyclonal TGF-β1 antibody or control IgG (10μg/ml, R&D system) as previously described.34
To dissect the role of Smad3 signaling in CRP-induced TEC arrest, a stable Smad3 gene knockdown NRK52E TEC line was used34–36. We also generated a transient Smad3 knockdown HK-2 TECs by transfecting cells with siRNA targeting Smad3 and negative control siRNA (scrambled siRNA) using the siPORTTM NeoFXTM Transfection Agent according to the manufacturer’s instructions. The sequences of the siRNA Smad3 used in this study were AGGUCUGCGUGAAUCCUATT (sense strand) and UAGGGAUUCACGCAGACCUCG (antisense strand). Cells with Smad3 knockdown were cultured with or without CRP (10μg/ml) and the effect of Smad3 on CRP-induced cell cycle arrest and proliferation was determined.
Renal function and histology
Blood was collected from each AKI mouse via clipping tails at 0, 24 h and 72h after AKI surgery. Serum creatinine was determined by the quantitative enzymatic method (Stanbio Laboratory, Texas, USA). Kidney tissues were fixed in methyl Carnoy’s for periodic acid–Schiff (PAS) staining. Percentage of tubular necrosis was scored from 400 cortical tubules as previously reported.10
Western blot analysis
Proteins from the kidney cortex and cells were extracted with RIPA lysis buffer and subjected to Western blot analysis with a rabbit polyclonal antibody to phospho-CDK2 (Thr160), CDK2, and phospho-Smad3 (Cell Signaling, CST, MA); a rabbit polyclonal antibody to p27 (Santa Cruz Biotechnology, CA), a rabbit polyclonal antibody to Smad3 (Invitrogen), a mouse monoclonal antibody to cyclin E, β-actin or GAPDH (Santa Cruz) as described previously.10 After being washed extensively, the membranes were incubated with an IRDyeTM800-conjugated secondary antibody (Rockland Immunochemicals, Gilbertsville, PA) for 1 h at room temperature in 1% BSA/TBST. The signals were visualized by the Odyssey Infrared Imaging System (San Diego, CA) and quantitatively analyzed by normalizing to β-actin or GAPDH using the Image J software (NIH, Bethesda, MD).
MTT assay
Cells were cultured in 96 wells plates and the MTT solution (0.25mg/ml) was added into culture medium for 3~4h until purple precipitate was visible. Medium was then removed and DMSO solution was added to resolved precipitate. The absorbance was measured at 550nm–600nm according to the manufacturer’s instructions. At least four -independent studies were performed.
Cell cycle analysis
To examine the direct effect of CRP on cell cycle, a rat TEC cell line (NRK52E) that stably expresses DN-TGFβRII was used.34 NRK52E TECs with or without overexpressing DN-TGFβRII were seeded in a 6-well plate (3 × 105/well) and cultured in serum-free medium overnight, followed by treatment with CRP (10μg/ml) for 24 h or 48 h. After CRP treatment, the cells were collected and fixed in 70% ethanol overnight at 4°C. Then the cells were stained in PBS containing propidium iodide (PI) (10 μg/ml) and RNase A (50 μg/ml), and analyzed by a FACSort flow cytometer (Becton Dickinson, San Jose, CA).
Immunohistochemistry
Immunohistochemistry was performed on paraffin tissue sections (4μm) using a microwave-based antigen retrieval technique37. The antibody used in this study including sheep polyclonal antibody to BrdU (Abcam, Cambridge, UK), rabbit polyclonal antibody to PCNA or p-Smad2/3 (Santa Cruz).
ChIP Analysis
ChIP was performed by Chromatin IP kit (Cell Signaling) according to the manufacturer’s instructions. In brief, NRK52 cells treated with CRP for 3h were cross-linked with 1% formaldehyde for 10 mins at room temperature, quenched with glycine, and then sonicated using a Bioruptor (Diagenode, Liege, Belgium) to generate 300- to 600-bp DNA fragments. Immunoprecipitation was performed with the antibody against Smad3 (Cell Signaling), and a mouse IgG was used as a control. Precipitated DNAs were detected by PCR and qPCR using specific primers to detect the binding of Smad3 to p27: Forward 5′- GGTGGACCAAATGCCTGACT and reverse 5′- GCAAAGAGGAGCTACGGAGA.
Statistical Analysis
Each experiment was repeated at least three times. Data were expressed as means ± SEM and analyzed using one-way ANOVA with Newman-Keuls comparison program from GraphPad Prism 6.0 (GraphPad Software, San Diego, CA, USA).
Supplementary Material
Acknowledgments
We thank Dr Chuxia Deng from the NIH for providing Smad3 KO mice. This study was supported by The Research Grants Council of Hong Kong (GRF 468711, CUHK3/CRF/12R), the Focused Investment Scheme A and CRF matching fund from Chinese University of Hong Kong; National Natural Scientific Foundation of China (81500512); the Basic Research Grant of Traditional Chinese Medicine Bureau of Guangdong Province (20151166); and the Clinical Medicine Special Funding of Chinese Medical Association (14050490586). AJS is supported by NIH grant R01 DK099092.
Footnotes
Competing interests
The authors have declared that no competing interests exist.
References
- 1.Bonventre JV, Zuk A. Ischemic acute renal failure: an inflammatory disease? Kidney Int. 2004;66:480–485. doi: 10.1111/j.1523-1755.2004.761_2.x. [DOI] [PubMed] [Google Scholar]
- 2.Rabb H, Griffin MD, McKay DB, et al. Acute Dialysis Quality Initiative Consensus XIII Work Group: Inflammation in AKI: Current Understanding, Key Questions, and Knowledge Gaps. J Am Soc Nephrol. 2015 doi: 10.1681/ASN.2015030261. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Coca SG, Singanamala S, Parikh CR. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int. 2012;81:442–448. doi: 10.1038/ki.2011.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferenbach DA, Bonventre JV. Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat review Nephrol. 2015;11:264–76. doi: 10.1038/nrneph.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Humphreys BD, Cantaluppi V, Portilla D, et al. Acute Dialysis Quality Initiative (ADQI) XIII Work Group: Targeting Endogenous Repair Pathways after AKI. J Am Soc Nephrol. 2015 doi: 10.1681/ASN.2015030286. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang R, Zhang YY, Huang XR, et al. C-reactive protein promotes cardiac fibrosis and inflammation in angiotensin II-induced hypertensive cardiac disease. Hypertension. 2010;55:953–960. doi: 10.1161/HYPERTENSIONAHA.109.140608. [DOI] [PubMed] [Google Scholar]
- 7.Liu F, Chen HY, Huang XR, et al. C-reactive protein promotes diabetic kidney disease in a mouse model of type 1 diabetes. Diabetologia. 2011;54:2713–2723. doi: 10.1007/s00125-011-2237-y. [DOI] [PubMed] [Google Scholar]
- 8.Li ZI, Chung AC, Zhou L, et al. C-reactive protein promotes acute renal inflammation and fibrosis in unilateral ureteral obstructive nephropathy in mice. Lab Invest. 2011;91:837–851. doi: 10.1038/labinvest.2011.42. [DOI] [PubMed] [Google Scholar]
- 9.Pegues MA, McCrory MA, Zarjou A, Szalai AJ. C-reactive protein exacerbates renal ischemia-reperfusion injury. Am J Physiol-Renal Physiol. 2013;304:F1358–1365. doi: 10.1152/ajprenal.00476.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang Y, Huang XR, Lv J, et al. C-reactive protein promotes acute kidney injury by impairing G1/S-dependent tubular epithelium cell regeneration. Clin sci. 2014;126:645–659. doi: 10.1042/CS20130471. [DOI] [PubMed] [Google Scholar]
- 11.Gentle ME, Shi S, Daehn I, et al. Epithelial cell TGFβ signaling induces acute tubular injury and interstitial inflammation. J Am Soc Nephrol. 2013;24:787–799. doi: 10.1681/ASN.2012101024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gewin L, Vadivelu S, Neelisetty S, et al. Deleting the TGF-β receptor attenuates acute proximal tubule injury. J Am Soc Nephrol. 2012;23:2001–2011. doi: 10.1681/ASN.2012020139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Geng H, Lan R, Wang G, et al. Inhibition of autoregulated TGFbeta signaling simultaneously enhances proliferation and differentiation of kidney epithelium and promotes repair following renal ischemia. Am J Pathol. 2009;174:1291–308. doi: 10.2353/ajpath.2009.080295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309. doi: 10.1016/s0092-8674(00)00121-5. [DOI] [PubMed] [Google Scholar]
- 15.Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat genetics. 2001;29:117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 16.Matsuura I, Denissova NG, Wang G, et al. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature. 2004;430:226–231. doi: 10.1038/nature02650. [DOI] [PubMed] [Google Scholar]
- 17.Nath KA, Croatt AJ, Warner GM, Grande JP. Genetic deficiency of Smad3 protects against murine ischemic acute kidney injury. Am J Physiol-Renal Physiol. 2011;301:F436–442. doi: 10.1152/ajprenal.00162.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Humphreys BD, Valerius MT, Kobayashi A, et al. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell. 2008;2:284–291. doi: 10.1016/j.stem.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 19.Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121:4210–4221. doi: 10.1172/JCI45161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagahara H, Ezhevsky SA, Vocero-Akbani AM, et al. Transforming growth factor beta targeted inactivation of cyclin E:cyclin-dependent kinase 2 (Cdk2) complexes by inhibition of Cdk2 activating kinase activity. Proc Natl Acad Sci U S A. 1999;96:14961–14966. doi: 10.1073/pnas.96.26.14961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Robson CN, Gnanapragasam V, Byrne RL, et al. Transforming growth factor-beta1 up-regulates p15, p21 and p27 and blocks cell cycling in G1 in human prostate epithelium. J Endocrinol. 1999;160:257–266. doi: 10.1677/joe.0.1600257. [DOI] [PubMed] [Google Scholar]
- 22.Schoecklmann HO, Rupprecht HD, Zauner I, Sterzel RB. TGF-beta1-induced cell cycle arrest in renal mesangial cells involves inhibition of cyclin E-cdk 2 activation and retinoblastoma protein phosphorylation. Kidney Int. 1997;51:1228–1236. doi: 10.1038/ki.1997.168. [DOI] [PubMed] [Google Scholar]
- 23.Jinnin M, Ihn H, Tamaki K. Characterization of SIS3, a novel specific inhibitor of Smad3, and its effect on transforming growth factor-beta1-induced extracellular matrix expression. Mol Pharmacol. 2006;69:597–607. doi: 10.1124/mol.105.017483. [DOI] [PubMed] [Google Scholar]
- 24.Bello G, Cailotto F, Hanriot D, et al. C-reactive protein (CRP) increases VEGF-A expression in monocytic cells via a PI3-kinase and ERK 1/2 signaling dependent pathway. Atherosclerosis. 2008;200:286–293. doi: 10.1016/j.atherosclerosis.2007.12.046. [DOI] [PubMed] [Google Scholar]
- 25.Cui C, Shi Q, Zhang X, et al. CRP promotes MMP-10 expression via c-Raf/MEK/ERK and JAK1/ERK pathways in cardiomyocytes. Cellular signalling. 2012;24:810–818. doi: 10.1016/j.cellsig.2011.11.019. [DOI] [PubMed] [Google Scholar]
- 26.Li JH, Huang XR, Zhu HJ, et al. Advanced glycation end products activate Smad signaling via TGF-beta-dependent and independent mechanisms: implications for diabetic renal and vascular disease. FASEB J. 2004;18:176–178. doi: 10.1096/fj.02-1117fje. [DOI] [PubMed] [Google Scholar]
- 27.Wang W, Huang XR, Canlas E, et al. Essential role of Smad3 in angiotensin II-induced vascular fibrosis. Circ Res. 2006;98:1032–1039. doi: 10.1161/01.RES.0000218782.52610.dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang F, Chung AC, Huang XR, Lan HY. Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-beta-dependent and -independent Smad pathways: the role of Smad3. Hypertension. 2009;54:877–884. doi: 10.1161/HYPERTENSIONAHA.109.136531. [DOI] [PubMed] [Google Scholar]
- 29.Yang L, Besschetnova TY, Brooks CR, et al. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16:535–543. doi: 10.1038/nm.2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roy A, Banerjee S. p27 and leukemia: cell cycle and beyond. J Cell Physiol. 2015;230:504–509. doi: 10.1002/jcp.24819. [DOI] [PubMed] [Google Scholar]
- 31.Carlson ME, Hsu M, Conboy IM. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature. 2008;454:528–532. doi: 10.1038/nature07034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ciliberto G, Arcone R, Wagner EF, Rüther U. Inducible and tissue-specific expression of human C-reactive protein in transgenic mice. EMBO J. 1987;20(6):4017–4022. doi: 10.1002/j.1460-2075.1987.tb02745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang X, Letterio JJ, Lechleider RJ, et al. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. The EMBO J. 1999;18:1280–1291. doi: 10.1093/emboj/18.5.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chung AC, Zhang H, Kong YZ, et al. Advanced glycation end-products induce tubular CTGF via TGF-beta-independent Smad3 signaling. J Am Soc Nephrol. 2010;21:249–260. doi: 10.1681/ASN.2009010018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang F, Huang XR, Chung AC, et al. Essential role for Smad3 in angiotensin II-induced tubular epithelial-mesenchymal transition. J Pathol. 2010;221:390–401. doi: 10.1002/path.2721. [DOI] [PubMed] [Google Scholar]
- 36.Meng XM, Huang XR, Chung AC, et al. Smad2 protects against TGF-beta/Smad3-mediated renal fibrosis. J Am Soc Nephrol. 2010;21:1477–1487. doi: 10.1681/ASN.2009121244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lan HY, Hutchinson P, Tesch GH, et al. A novel method of microwave treatment for detection of cytoplasmic and nuclear antigens by flow cytometry. J Immunol Method. 1996;190:1–10. doi: 10.1016/0022-1759(95)00233-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.