

Abstract
Background
Acute pulmonary exacerbations are associated with progressive lung function decline and increased mortality in cystic fibrosis (CF). The role of pulmonary vascular disease in pulmonary exacerbations is unknown. We investigated the association between pulmonary artery enlargement (PA:A>1), a marker of pulmonary vascular disease, and exacerbations.
Methods
We analyzed clinical, computed tomography (CT), and prospective exacerbation data in a derivation cohort of 74 adult CF patients, measuring the PA:A at the level of the PA bifurcation. We then replicated our findings in a validation cohort of 190 adult CF patients. Patients were separated into groups based on the presence or absence of a PA:A>1 and were followed for 1-year in the derivation cohort and 2-years in the validation cohort. The primary endpoint was developing ≥1 acute pulmonary exacerbation during follow-up. Linear and logistic regression models were used to determine associations between clinical factors, the PA:A ratio, and pulmonary exacerbations. We used Cox regression to determine time to first exacerbation in the validation cohort.
Findings
We found that PA:A>1 was present in n=37/74 (50%) of the derivation and n=89/190 (47%) of the validation cohort. In the derivation cohort, n=50/74 (68%) had ≥1 exacerbation at 1 year and n=133/190 (70%) in the validation cohort had ≥1 exacerbation after 2 years. PA:A>1 was associated with younger age in both cohorts and with elevated sweat chloride (100.5±10.9 versus 90.4±19.9mmol/L, difference between groups 10.1mmol/L [95%CI 2.5–17.7], P=0.017) in the derivation group. PA:A>1 was associated with exacerbations in the derivation (OR 3.49, 95%CI 1.18–10.3, P=0.023) and validation (OR 2.41, 95%CI 1.06–5.52, P=0.037) cohorts when adjusted for confounders. Time to first exacerbation was shorter in PA:A>1 versus PA:A<1 [HR 1.66 (95%CI 1.18–2.34), P=0.004] in unadjusted analysis, but not when adjusted for sex, BMI, prior exacerbation, positive Pseudomonas status, and FEV1/FVC [HR 1.14 (95%CI 0.80–1.62), P=0.82]).
Interpretation
PA enlargement is prevalent in adult CF patients and is associated with acute pulmonary exacerbation risk in two well-characterized cohorts. PA:A may be a predictive marker in CF.
Introduction
Cystic fibrosis (CF) is the most common genetic cause of chronic lung disease, affecting 30,000 children and adults in the United States and 70,000 world-wide1. CF is characterized by defects in chloride transport through the cystic fibrosis transmembrane conductance regulator (CFTR) protein that lead to bacterial infection, progressive airway obstruction, and ultimately bronchiectasis; extra-pulmonary manifestations also occur in the pancreas and gastrointestinal tract1. Pulmonary exacerbations are critical events characterized by increased dyspnea, rapidly progressive lung function loss, and intense metabolic demands2, and are thought to drive progressive loss of lung function3. Currently, risk factors for pulmonary exacerbations are not well characterized and are avidly sought to identify patients at greatest risk for hospitalization and lung function decline which could be used to guide chronic therapy or monitoring4,5.
Pulmonary vascular disease is becoming increasingly recognized as a major pathologic complication of many chronic lung diseases, including chronic obstructive pulmonary disease (COPD) and CF6,7. Development of pulmonary hypertension in both diseases is an independent predictor of morbidity and mortality8,9. While the gold standard for diagnosis of pulmonary vascular disease is right heart catheterization, non-invasive techniques, specifically computed tomography (CT), are increasingly being used as diagnostic tools. Pulmonary artery enlargement characterized by an elevated pulmonary artery to ascending aorta diameter ratio (PA:A ratio >1) is a robust predictor of exacerbations in COPD, including those events that require hospitalization, and outperformed traditional clinical markers of COPD exacerbation risk, including a history of prior exacerbations10. Further, the PA:A ratio is superior to echocardiography in identifying pulmonary hypertension as confirmed with right heart catheterization in COPD11. Therefore, we hypothesized that pulmonary vascular disease detected on CT would be predictive of CF pulmonary exacerbations in an adult CF population. To test this, we analyzed data from two cohorts of adult CF patients.
Methods
Patient populations
We built our derivation cohort using extracted data collected from placebo treated patients in a prospective clinical trial and built the validation cohort using historic data from a tertiary care center. The derivation cohort was built using de-identified patient data from the “Study of Ataluren (PTC124TM) in Cystic Fibrosis” (ClinicalTrials.gov identifier NCT00803205) enrolled between Sept 8, 2009 and Nov 30, 2010. The full clinical trial protocol is described elsewhere12. All patients in the derivation cohort had a confirmed diagnosis of CF and at least one CFTR nonsense mutation as per inclusion criteria to the parent clinical trial. We included all subjects randomized to the placebo arm with age ≥18 years old at the time of trial enrollment who had baseline CT images available for interpretation as outlined in Figure 1A. We restricted our analysis to adults since the PA:A has not been characterized in the pediatric population and thus it could be less reliable. The occurrence of exacerbations were recorded prospectively and repeat CT scans were performed at 12 months after randomization. We chose to test our hypothesis in this population due to the unique nature of this CF cohort in that it included CT imaging and prospective analysis, and we validated our findings in a “real-world” setting. We built our validation cohort using de-identified patient data collected by the Prince Charles Hospital (TPCH) Adult Cystic Fibrosis Centre, Brisbane) for patients who were evaluated and had CT imaging performed between 2002 and 2014 as outlined in Figure 1B. Details regarding this cohort, CFTR genotyping methods, and spirometry are provided in the Supplementary Methods. The median (IQR) time between spirometry and CT was 2 (15) days. The Institutional Review Board at the University of Alabama at Birmingham and the Human Research Ethics and Research Governance Offices at the TPCH approved the conduct this analysis (N130131003 and SSA/15/QPCH/15; HREC/14/QPCH/173). For both cohorts, positive Pseudomonas status is recorded if chronic infection was present on airway cultures13.
Figure 1. Disposition of subject data used in the study.

(A) The derivation cohort was developed using de-identified data from subjects enrolled in the placebo arm of a clinical trial. A total of 74 subjects were included. (B) The validation cohort consisted of de-identified data from patients enrolled at the Adult Cystic Fibrosis Centre, Prince Charles Hospital Brisbane, Queensland, Australia. A total of 190 subjects were included.
CT imaging
In the derivation cohort, low-dose non-contrast enhanced CT images were acquired during full inspiration as part of the parent clinical trial. In the validation cohort, chest CT scans were performed for the following indications: clinical deterioration (n=67/197, 35%), lung transplant assessment (n=64/197, 32%), hemoptysis (n=44/197, 22%), and investigation of NTM infection (n=22/197, 11%). Clinical deterioration may have included investigation of new infiltrates on chest radiographs, unexplained lung function decline, isolation of a new pathogen, and follow-up of allergic bronchopulmonary aspergillosis. The PA diameter was measured at the level of the main pulmonary artery bifurcation and the average of two perpendicular measurements of the ascending aorta diameter (A) were taken on the same CT image using mediastinal windows, as previously described10,11. The average PA, A, and PA:A ratio between the two readers were used for analysis in the derivation cohort and the measurements taken by a single reader were used in the validation cohort. Investigators were blind to all clinical data, lung parenchymal appearance, and to the time of acquisition of CT scan (baseline or follow-up). A representative image of PA enlargement is shown in Figure 2.
Figure 2. Representative measurements of the pulmonary artery and aortic diameters by CT scan.
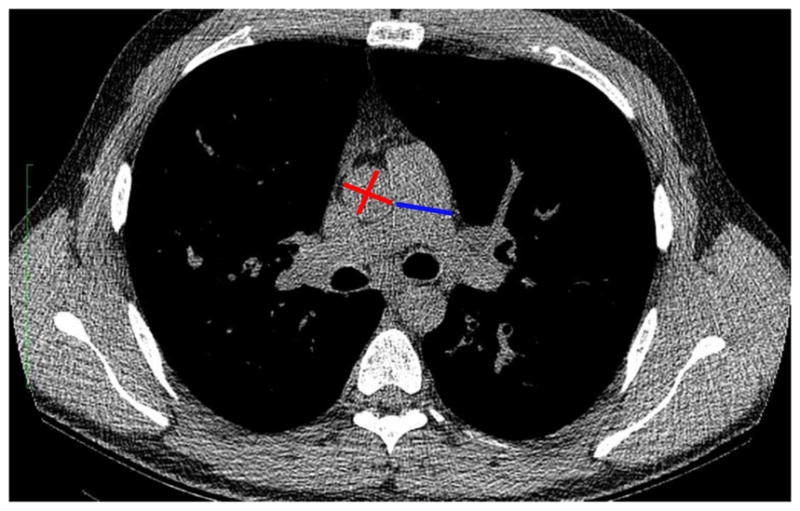
Non-contrast enhanced chest CT images depict the site of PA (blue) and A (red) diameters, measured at the level of the PA bifurcation. Aortic diameters were averaged values of two perpendicular measurements.
Pulmonary exacerbations
In the derivation cohort, pulmonary exacerbations were determined by comprehensive respiratory event data forms, hospitalization records, and investigator-defined pulmonary exacerbation. Events meeting the modified Fuchs’ criteria (at least 4 of 12 Fuchs’ signs and symptoms with or without intravenous antibiotic treatment)14 were counted as exacerbations for the 48-week follow-up period per the parent trial design. In the validation cohort, acute pulmonary exacerbations were defined by an event requiring intravenous antibiotics or hospitalization for respiratory indications as reported previously15,16. Exacerbations were recorded 12-months prior to index CT through 24-months post index CT scan, with prior exacerbations defined by events that occurred within 12-months prior to the index CT and prospective exacerbations as events occurring 1- and 2-years after the index CT.
Statistical analysis
Baseline data are expressed as means with standard deviations for normally distributed values or median with interquartile range for non-Gaussian distributed values. Bivariate analyses were conducted with the use of a two-tailed Fisher’s exact test for categorical data and two tailed t-tests or a Wilcoxon rank-sum test for continuous data when appropriate. Linear regression models were used to identify correlations between patient characteristics and the PA:A ratio as a continuous variable, adjusting for age, sweat chloride, and FEV1% predicted or age, sweat chloride, and BMI based on associations (P<0.10) on univariable analysis. We used logistic regression models to determine associations between clinical, laboratory, and radiologic characteristics and the occurrence of a pulmonary exacerbation during study follow-up. Variables associated with pulmonary exacerbations on univariate logistic analysis (at P<0.10) plus sex and BMI were included in stepwise backward multivariable logistic models to adjust for confounders. We used Cox regression analysis to determine time to first exacerbation in the validation cohort. Performance characteristics of the PA:A ratio were measured by receiver operating characteristic (ROC) analysis. Cohen’s kappa was calculated to identify the intra-observer and inter-observer agreement for the presence of a PA:A>1 and Bland-Altman analysis was used to evaluate the inter-observer agreement for PA and PA:A ratio measurements or change in PA:A ratio over time in the derivation cohort. We used complete-case analysis and did not impute values for missing data. All analyses were performed with the use of SPSS software, version 23.0, and P values <0.05 were considered statistically significant.
Role of the funding source: The funding sponsors did not have a role in study design, analysis, or interpretation of the data nor in the writing of the report. PTC Therapeutics provided blinded, de-identified data used in building the derivation cohort. The corresponding authors (JMW and SMR) had full access to all of the data and the final responsibility to submit for publication.
Results
Reproducibility of PA:A measurements
Two independent, blinded investigators measured the PA and A diameters to assess the reproducibility of the PA:A measurements in CF in the derivation cohort. There was good correlation of agreement between readers in regards to PA:A ratio >1 (Kappa 0.80, 95% CI 0.70–0.90, p<0.05). Using Bland-Altman analysis, the mean differences between PA diameters between the two readers were 0.03 cm and 0.008 for the PA:A ratio (Figure S1).
Study subjects and the PA:A ratio
For the derivation cohort, 74 adult CF patients had baseline CT imaging available and were included in the analysis. The patients were 28±8 (mean±SD) years old, 97% white, 57% male, and had a mean predicted forced expiratory volume in 1- second (FEV1) of 57±14%. The mean PA diameter was 2.55±0.30cm, the mean A diameter was 2.57±0.39cm, and the mean PA:A was 1.01±0.12. Baseline patient characteristics of all adult subjects with CT data in the derivation cohort (n=140, omitting trial randomization status) are shown in Table S2. The distributions of PA:A in both study cohorts are shown in Figure S2. Patients were separated into subgroups based on the presence or absence of PA enlargement defined by a PA:A>1, with 37/74 (50%) having a PA:A>1 in the derivation cohort and 89/190 (47%) in the validation cohort as outlined in Table 1. With the exception of higher Pseudomonas infection rates (87% versus 42%) and lower FEV1 (50±21% versus 57±14%), characteristics of patients in the validation cohort were similar to those in the derivation group. Of note, the median FEV1 for the patients with CT scans in the validation cohort was 10% lower than the median FEV1 for the entire TPCH CF registry, a reflection of acquiring CT images for the clinical implications listed in the Methods. Thus, the patients with CT scans in the validation cohort may be at higher risk for exacerbation than the general CF population. There were no within cohort differences in regards to sex, race, height, weight, body mass index (BMI), FEV1, ameliorating CFTR mutation status, and Pseudomonas infection status for subjects with PA:A>1 compared to PA:A≤1 in both cohorts. Patients in the validation cohort with a PA:A>1 were younger, had higher rates of Pseudomonas infection, and higher rates of previous acute pulmonary exacerbations. Patients in the derivation cohort with a PA:A>1 had higher sweat chloride (100.5±10.9 versus 90.4±19.9mmol/L, difference between groups 10.1mmol/L [95%CI 2.5–17.7], P=0.017). This failed to meet statistical significance in the validation cohort (105.9±24.7 versus 88.2±23.1, difference between groups 17.7mmol/L [95%CI -0.6–36.7], P=0.057), though the number of subjects with sweat chloride data available was limited (n=27). As an exploratory analysis, we found a modest correlation between sweat chloride and the PA:A ratio (Pearson’s r=0.25, P=0.001) in a pooled analysis of all subjects with sweat chloride data available (n=156), suggesting a relationship with CFTR function. Among subjects in the derivation cohort, age and higher sweat chloride were linearly correlated to PA:A, with age (β=−0.34,p=0.007) remaining significant in an adjusted model as outlined in Table 2A. In the validation cohort, age, BMI, and sweat chloride were linearly correlated to PA:A, but only sweat chloride remained significant in adjusted models (Table 2B). Patients with a PA:A>1 were younger and had higher rates of Pseudomonas infection than those with a PA:A ratio ≤1 in the validation cohort (Table 1). We explored the relationship between CFTR genotype and PA enlargement by dichotomizing CFTR mutations as functional or nonfunctional (Supplementary Appendix) and the prevalence of functional mutations is shown in Table 1. There were no statistical differences in CFTR functional status in our study based on the low prevalence of functional mutations in these cohorts.
Table 1.
Baseline characteristics of the two cohorts.
| Derivation Cohort | Validation Cohort | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||
| Total (n=74) |
PA:A ≤ 1 (n=37) |
PA:A >1 (n=37) |
Point Estimate |
95%CI | P-value | All (n=190) | PA:A≤1 (n=101) |
PA:A>1 (n=89) |
Point Estimate |
95%CI | P-value | |
|
| ||||||||||||
| Age, years† | 28±8 | 29±10 | 26±6 | −3 | −6.9–0.9 | 0.06 | 29±9 | 30±11 | 26±8 | −4 | −7 – −2 | <0.001 |
|
| ||||||||||||
| White race | 72/74 | 37/37 | 35/37 | −5% | −18 – 5 | 0.16 | 190/190 | 101/101 | 89/89 | n/a | n/a | n/a |
| (97%) | (100%) | (95%) | (100%) | (100%) | (100%) | |||||||
|
| ||||||||||||
| Male sex | 42/74 | 22/37 | 20/37 | −5% | −27 – 16 | 0.64 | 97/190 | 54/101 | 43/89 | −5% | −20 – 9 | 0.56 |
| (57%) | (59%) | (54%) | (51%) | (53%) | (48%) | |||||||
|
| ||||||||||||
| BMI, kg/m2 | 22.0±2. | 22.6±2. | 21.5±2.3 | −1.1 | −2.3– 0.1 | 0.06 | 21.2±3.6 | 21.4±3.9 | 20.9±3.3 | −0.5 | −1.7– 0.5 | 0.27 |
|
| ||||||||||||
| Pseudomonas positive | 31/74 | 17/37 | 14/37 | −8% | −29 – 14 | 0.49 | 166/190 | 82/101 | 84/89 | 13% | 4 – 22 | 0.008 |
| (42%) | (46%) | (38%) | (87%) | (81%) | (94%) | |||||||
|
| ||||||||||||
| Functional CFTR genotypea | 3/74 | 2/37 | 1/37 | −2% | −15 − 9 | 0.56 | 11/164 | 7/84 (8%) | 4/80 (5%) | −3% | −12 – 4 | 0.37 |
| (4%) | (5%) | (3%) | (7%) | |||||||||
|
| ||||||||||||
| Sweat chloride, mmol/L | 95.5±16.7 | 90.4±19.9 | 100.5±10.9 | 10.1 | 2.5–17.7 | 0.017 | 97.9±25.2 | 88.2±23.1 | 105.9±24.7b | 17.7 | −0.6 – 36 | 0.057 |
|
| ||||||||||||
| FEV1% predicted | 57±14 | 58±15 | 56±12 | −2 | −8 –4 | 0.38 | 50±21 | 50±22 | 50±20 | 0 | −6 – 6 | 0.86 |
|
| ||||||||||||
| FEV1/FVC | 0.64±0. 10 | 0.64±0. 11 | 0.65±0.10 | 0.01 | −0.01– 0.03 | 0.67 | 0.62±0.24 | 0.63±0.31 | 0.61±0.13 | −0.02 | −0.09–0.05 | 0.95 |
|
| ||||||||||||
| PA, cm | 2.55±0. 30 | 2.51±0. 34 | 2.59±0.25 | 0.08 | 0.01–0.15 | 0.032 | 2.57±0.32 | 2.48±0.28 | 2.67±0.33 | 0.19 | 0.10–0.28 | <0.001 |
|
| ||||||||||||
| A, cm | 2.57±0. 39 | 2.77±0. 38 | 2.36±0.28 | −0.41 | −0.57–−0.26 | <0.001 | 2.59±0.34 | 2.72±0.32 | 2.45±0.30 | −0.27 | −0.36–−0.19 | <0.001 |
|
| ||||||||||||
| PA:A ratio | 1.01±0. 12 | 0.91±0. 07 | 1.10±0.08 | 0.19 | 0.16–0.23 | <0.001 | 0.99±0.11 | 0.91±0.06 | 1.09±0.08 | 0.18 | 0.16–0.20 | <0.001 |
|
| ||||||||||||
| Prior exacerbation | -- | -- | -- | 120/190 | 56/101 | 64/89 | 17% | 3 – 30 | 0.02 | |||
| (63%) | (55%) | (72%) | ||||||||||
|
| ||||||||||||
| Inhaled antibiotic use | 53/74 | 26/37 | 27/37 | 3% | −17 – 23 | 0.80 | -- | -- | -- | |||
| (72%) | (70%) | (73%) | ||||||||||
|
| ||||||||||||
| Inhaled DNAse use | 55/74 | 27/37 | 28/37 | 3% | −17 – 22 | 0.79 | -- | -- | -- | |||
| (74%) | (73%) | (76%) | ||||||||||
Data are mean±SD or n(%). Abbreviations: FEV1 = forced expiratory volume in 1 second, FVC = forced vital capacity, PA = pulmonary artery diameter, A = ascending aortic diameter, prior exacerbation = at least one acute pulmonary exacerbation within the year prior to index CT scan, 95%CI = 95% confidence interval.
Functional CFTR genotype defined by the presence of one mutation with residual function.
Sweat chloride values are only available for a subset of subjects [n=29 total; n=13 PA:A≤1; n=16 PA:A>1] in the validation cohort. Data on exacerbation prior to index CT scan is unavailable for the derivation cohort.
Table 2.
Unadjusted and adjusted linear regression models for the association between clinical factors and the PA:A Ratio
| A. Derivation cohort | ||||
|---|---|---|---|---|
| PA:A Ratio | ||||
| Unadjusted | Adjusteda | |||
| Beta | P-value | Beta | P-value | |
| Age, years | −0.32 | 0.005 | −0.34 | 0.007 |
| BMI, kg/m2 | −0.08 | 0.46 | ||
| Sweat chloride, mmol/L | 0.25 | 0.05 | 0.21 | 0.08 |
| FEV1% predicted | 0.04 | 0.76 | ||
| FEV1/FVC | 0.14 | 0.22 | ||
| B. Validation cohort | ||||||
|---|---|---|---|---|---|---|
| PA:A Ratio | ||||||
| Unadjusted | Adjustedb | Adjustedc | ||||
| Beta | P-value | Beta | P-value | Beta | P-value | |
| Age, years | −0.28 | <0.001 | ||||
| BMI, kg/m2 | −0.15 | 0.05 | ||||
| Sweat chloride, mmol/L | 0.37 | 0.05 | 0.32 | 0.11 | 0.36 | 0.06 |
| FEV1% predicted | −0.04 | 0.63 | ||||
| FEV1/FVC | 0.07 | 0.33 | ||||
Model adjusted for age, sweat chloride, FEV1% predicted. The R2 for the adjusted model is 0.17 (P=0.004). Abbreviations: BMI = body mass index, FEV1 = forced expiratory volume in 1 second, FVC = forced vital capacity.
Model adjusted for age, sweat chloride, FEV1% predicted with R2 = 0.17 (P=0.10).
Model adjusted for age, sweat chloride, BMI with R2 = 0.13 (P=0.07).
Abbreviations: BMI = body mass index, FEV1 = forced expiratory volume in 1 second, FVC = forced vital capacity.
Relationship to acute pulmonary exacerbations
To determine if PA enlargement was associated with acute pulmonary exacerbations, we analyzed the longitudinal follow-up data to identify factors associated with CF acute pulmonary exacerbations. Figure 3 shows the relationship between the PA:A ratio and exacerbations in both cohorts at 1 and 2-years of follow-up. In the derivation cohort, 50/74 (68%) participants had ≥1 exacerbation (median 1, IQR 3) at 1-year, including 30/37 (81%) in the PA:A>1 and 20/37 (54%) in the PA:A≤1 group (P=0.013) . Sweat chloride, FEV1% predicted, and PA:A>1 (OR 3.54, 95% CI 1.28–10.4, p=0.015) were associated with the occurrence of a pulmonary exacerbation upon univariate analysis (Table 3A). The AUC was 0.70 (95%CI 0.57–0.83, Figure S4), indicating the PA:A had fair accuracy for predicting a pulmonary exacerbation. We incorporated sex, BMI, sweat chloride, FEV1% predicted, and PA:A>1 into a multivariate logistic regression model to determine if the PA:A>1 was independently associated with acute pulmonary exacerbations. Indeed, we found that the PA:A>1 (OR 3.49, 95%CI 1.18–10.3, p=0.02) and FEV1% predicted (OR 0.96, 95%CI 0.92–0.99, P=0.03) were independently associated with an acute pulmonary exacerbation at 1-year follow-up (Table 3A). The AUC of the multivariable model increased from 0.70 (0.57–0.83, P=0.005) to 0.76 (0.64–0.87, P<0.001; Figure S4) with addition of PA:A. There were no associations between age, race, sex, BMI, Pseudomonas colonization, forced vital capacity (FVC), or FEV1/FVC with pulmonary exacerbations in the derivation cohort.
Figure 3. Relationship between PA enlargement and acute pulmonary exacerbations in adults with cystic fibrosis.

We found an association between PA:A>1 and acute pulmonary exacerbations at (A) 12-months of follow-up in our derivation cohort. We recapitulated these findings in a validation cohort when followed for (B) 12-months and (C) 24-months. *P<0.05.
Table 3.
Factors associated with an acute pulmonary exacerbation during follow-up
| A. Derivation cohort | ||||
|---|---|---|---|---|
| ≥1 Acute pulmonary exacerbation at 1-year of follow-up | ||||
| Univariate | Multivariatea | |||
| OR (95% CI) | p-value | OR (95% CI) | p-value | |
| Age, year | 0.97 (0.92–1.03) | 0.32 | ||
| White race | 2.13 (0.13–35.5) | 0.60 | ||
| Male sex | 0.54 (0.20–1.49) | 0.54 | 0.40 (0.12–1.28) | 0.12 |
| BMI, kg/m2 | 0.97 (0.79––1.16) | 0.65 | 1.09 (0.87–1.37) | 0.45 |
| Positive Pseudomonas status | 1.01 (0.38–2.72) | 0.98 | 1.28 (0.42–3.88) | 0.66 |
| Sweat chloride, mmol/L | 1.04 (1.003–1.08) | 0.033 | ||
| PA:A>1 | 3.64 (1.28–10.4) | 0.015 | 3.49 (1.18–10.3) | 0.023 |
| FEV1% predicted | 0.96 (0.92–0.99) | 0.021 | 0.96 (0.92–0.99) | 0.032 |
| FVC% predicted | 0.98 (0.94–1.02) | 0.27 | ||
| FEV1/FVC | 0.01 (0.00–1.92) | 0.09 | ||
| Inhaled antibiotic use | 1.42 (0.49–4.10) | 0.51 | ||
| DNAse use | 2.40 (0.82–7.06) | 0.11 | ||
| B. Validation cohort (1-year follow-up) | ||||
|---|---|---|---|---|
| ≥1 Acute pulmonary exacerbation at 1-year of follow-up | ||||
| Univariate | Multivariateb | |||
| OR (95% CI) | p-value | OR (95% CI) | p-value | |
| Age, year | 1.01 (0.98–1.04) | 0.45 | ||
| Male sex | 0.94 (0.53–1.68) | 0.94 | 0.76 (0.32–1.83) | 0.54 |
| BMI, kg/m2 | 0.99 (0.91–1.08) | 0.99 | 1.02 (0.90–1.15) | 0.74 |
| Positive Pseudomonas status | 2.29 (0.96–5.46) | 0.06 | 1.96 (0.58–6.66) | 0.28 |
| Sweat chloride, mmol/L | 1.01 (0.98–1.05) | 0.39 | ||
| PA:A>1 | 2.05 (1.13–3.72) | 0.02 | 1.43 (0.63–3.29) | 0.40 |
| FEV1% predicted | 0.99 (0.98–1.01) | 0.22 | ||
| FEV1/FVC | 0.15 (0.02–0.91) | 0.04 | 0.35 (0.02–6.65) | 0.48 |
| Exacerbation in the previous year | 30.4 (13.4–68.7) | <0.001 | 21.0 (9.07–48.9) | <0.001 |
| C. Validation cohort (2-year follow-up) | ||||
|---|---|---|---|---|
| ≥1 Acute pulmonary exacerbation at 2-years of follow-up | ||||
| Univariate | Multivariatec | |||
| OR (95% CI) | p-value | OR (95% CI) | p-value | |
| Age, year | 1.01 (0.98–1.05) | 0.40 | ||
| Male sex | 1.37 (0.73–2.55) | 0.33 | 1.15 (0.49–2.73) | 0.75 |
| BMI, kg/m2 | 0.97 (0.86–1.07) | 0.57 | 1.00 (0.89–1.13) | 0.96 |
| Positive Pseudomonas status | 2.69 (1.13–6.42) | 0.03 | 2.42 (0.77–7.60) | 0.13 |
| Sweat chloride, mmol/L | 1.02 (0.98–1.05) | 0.33 | ||
| PA:A>1 | 3.12 (1.59–6.1) | 0.001 | 2.41 (1.06–5.52) | 0.037 |
| FEV1% predicted | 0.99 (0.98–1.01) | 0.30 | ||
| FEV1/FVC | 0.07 (0.01–0.57) | 0.02 | 0.15 (0.07–3.24) | 0.23 |
| Exacerbation in the previous year | 13.9 (6.56–29.6) | <0.001 | 8.21 (3.66–18.4) | <0.001 |
Multivariate logistic regression model includes: sex, BMI, FEV1% predicted, PA:A>1 (R2=0.20, P=0.003).
Multivariate logistic regression model includes: sex, BMI, positive Pseudomonas status, FEV1/FVC, PA:A>1, exacerbation in the year prior to index CT scan (R2=0.32, P<0.001).
Multivariate logistic regression model includes: sex, BMI, positive Pseudomonas status, FEV1/FVC, PA:A>1, exacerbation in the year prior to index CT scan (R2=0.19, P<0.001).
Abbreviations: FEV1 = forced expiratory volume in 1 second, FVC = forced vital capacity, PA = pulmonary artery diameter, A = ascending aortic diameter. Positive Pseudomonas status indicates a history of either an acute infection or chronic colonization with Pseudomonas.
We validated these findings in our second cohort with 1- and 2-year follow-up. At one-year, 113/190 (59%) patients had ≥1 exacerbation (median 1, IQR 3), including 61/89 (69%) in the PA:A>1 and 52/101 (50%) in the PA:A≤1 group (P=0.017). On univariate logistic regression analysis, the PA:A>1 and a previous exacerbation within 1 year prior to index CT were associated with an acute pulmonary exacerbation at 1 year of follow-up (Table 3B). We included sex, BMI, Pseudomonas Infection, FEV1/FVC, PA:A>1, and previous exacerbation in a multivariable logistic regression model and found that only an exacerbation in the year prior to the index CT scan was independently associated with a subsequent exacerbation (OR 21.0, 95%CI 9.07–48.9, P<0.001). After 2-years of follow-up, 133/190 (70%) patients had ≥1 exacerbation (median 2, IQR 4), with 73/89 (82%) in the PA:A>1 and 60/101 (59%) in the PA:A≤1 group (P=0.001). We found significant associations between Pseudomonas infection, PA:A>1, FEV1/FVC, and prior exacerbation with development of an acute pulmonary exacerbation within 2-years of follow-up on univariate analysis. When these factors are incorporated into a multivariate model, only PA:A>1 (OR 2.41, 95% CI 1.06–5.52, P=0.03) and prior exacerbation (OR 8.21, 95% CI 3.66–18.4, P<0.001) remain significant as shown in Table 3C. In this cohort, adding the PA:A>1 changed the AUC for detecting an exacerbation at 1-year from 0.55 (95%CI 0.46–0.65, P=0.23) to 0.60 (95%CI 0.51–0.69, P=0.03) and at 2-year from 0.56 (95%CI 0.46–0.66, P=0.23) to 0.64 (95% CI 0.54–0.73, P=0.006). In a sensitivity analysis, increased PA:A was also associated with the occurrence of CF exacerbations in 12 months prior to the CT scan (OR 2.06, 95%CI 1.12–3.77, P=0.02). In a second sensitivity analysis, patients who did not have index CT scans performed in the setting of an acute respiratory event (n=93/190) yielded similar results as seen in Supplementary Table S3. Patients with a PA:A>1 had a shorter median (IQR) time-to-first exacerbation compared to patients with a PA:A≤1 [214 (390) versus 339 (720), P=0.028], with an unadjusted HR 1.66 (95%CI 1.18–2.34, P=0.004), but this failed to remain significant when adjusted for positive Pseudomonas status, prior exacerbation, and FEV1/FVC [adjusted HR 1.14 (95%CI 0.80–1.62, P=0.82)], as shown in Figure 4.
Figure 4. Time to first acute pulmonary exacerbation in the validation cohort.

Time to first exacerbation was shorter in PA:A>1 versus PA:A<1 [HR 1.66 (95%CI 1.18–2.34), P=0.004] in unadjusted analysis, but (B) not when adjusted for sex, BMI, prior exacerbation, positive Pseudomonas status, and FEV1/FVC [HR 1.14 (95%CI 0.80–1.62), P=0.82].
Stability of PA:A
In the derivation cohort, all 74 patients included in the longitudinal analysis had a second CT scan performed at 1-year of follow-up. The PA diameter, A diameter, and the PA:A ratio did not significantly change from baseline (Table 4 and Figure S3). The baseline PA diameter was correlated to the follow-up PA diameter (r=0.68, p<0.001), and similar relationships were observed with the A diameter (r=0.81, p<0.001) and the PA:A ratio (r=0.53, p<0.001). The stability of the PA:A ratio was not affected by interval acute pulmonary exacerbations (data not shown).
Table 4.
12-month stability of pulmonary vessel measurements on CT.
| Baseline (n=74) | Follow-up (n=74) | Mean Change (SD) | p-value | |
|---|---|---|---|---|
| PA, cm | 2.55±0.30 | 2.53±0.31 | −0.02±0.24 | 0.40 |
| A, cm | 2.57±0.39 | 2.56±0.37 | −0.02±0.24 | 0.53 |
| PA:A ratio | 1.01±0.12 | 1.00±0.11 | −0.01±0.11 | 0.73 |
Data analyzed using paired sample t-test. Abbreviations: PA = pulmonary artery diameter, A = ascending aortic diameter, PA:A = pulmonary artery to ascending aortic diameter ratio
Discussion
We found that a PA:A>1is independently associated with the occurrence of acute pulmonary exacerbations of CF in two well-characterized populations with longitudinal follow-up. To our knowledge, this is the first study to investigate the PA:A ratio and PA enlargement as a risk factor for acute pulmonary exacerbations in CF. Moreover, PA:A>1 was associated with elevated sweat chloride, younger age, and lower FEV1 (or FEV1/FVC in the validation cohort) in cross-sectional analysis, and was independent of other demographic variables. These data indicate PA:A ratio is a useful means to stratify risk among CF adults for subsequent pulmonary exacerbations, an important contributor of respiratory decline3, and add to the predictive power of clinical variables alone, which are insufficient to identify those at greatest risk for deterioration. The PA:A ratio is easily measured on non-contrast enhanced CT scans, is reproducible between trained operators, and does not require IV contrast or special techniques10,11. Since many adult CF patients undergo CT scan for clinical indications, including testing for unexplained respiratory deterioration, evaluation of pulmonary complications such as hemoptysis or non-tuberculous mycobacterial infection, or in lung transplant evaluation, identifying PA enlargement on CT could be an important new tool to monitor adults with CF. Indeed, the relationship between PA:A>1 and pulmonary exacerbations is generalizable to an adult CF population given the confirmation of our results in the QPCH population.
Given the importance pulmonary exacerbations have on morbidity and mortality among patients with CF, there is a need to identify predictive markers for these events. Blood-based biomarkers17, CT metrics18, PET/CT19, and MRI techniques20 show promise for identifying physiologic changes that occur during exacerbations and track with response to therapy. The overall Brody score with its bronchiectasis and mucus-plugging component scores on high resolution CT are correlated with the rate of developing acute pulmonary exacerbations over a two-year period21. However, there is considerable variation among the scores in subjects who did not have pulmonary exacerbations, limiting the application of these measurements into clinical practice.
The PA:A ratio and PA enlargement correlate to invasive hemodynamic measurements and thus have been used primarily as non-invasive metrics for pulmonary hypertension. We believe the PA:A >1 likely represents underlying pulmonary hypertension as we and others have shown for COPD and other lung diseases22. However, the direct link between the PA:A and other means of detecting pulmonary hypertension has not yet been established in CF, either using echocardiography or right-sided heart catheterization. It is possible that in CF, PA enlargement may reflect other factors including vascular redistribution and centralization of blood flow due to parenchymal lung destruction, lung hyperinflation, or altered mechanical properties of the vascular matrix or smooth muscle – the latter being of great interest given the current findings linking CFTR function with PA enlargement. Nevertheless, our study highlights the importance of identifying PA enlargement in patients at increased risk for respiratory complications.
Pulmonary vascular disease and pulmonary hypertension are not well characterized in CF. Estimates of pulmonary hypertension in CF range between 26 and 63% of the population, however formal investigation of pulmonary hypertension in CF usually is limited to those with advanced lung disease in the course of lung transplant evaluation9. Our findings suggest that PA enlargement is highly prevalent – occurring in approximately 50% of adult CF patients with moderate airflow obstruction, and it is more common than the observed prevalence in COPD10. Given the implications of pulmonary hypertension for morbidity and mortality9, this finding highlights the need to be cognizant of pulmonary vascular disease at all degrees of airflow obstruction. Measuring the PA:A may be a viable non-invasive screening tool to aid in assessing CF patients for pulmonary hypertension, in addition to its utility as a prognostic indicator for acute respiratory events.
The effects of absent CFTR function have recently been recognized as protean, with CFTR activity recognized in non-epithelial tissues including leukocytes, osteobast, and bronchial smooth muscle23–25. The latter was associated with small airway sites in swine and human, suggesting clinically relevant smooth muscle constriction due to the absence of CFTR26. Interestingly, we found that increased sweat chloride, a marker of CFTR function, was also related to PA enlargement. Mechanistically, this relationship is intriguing for several reasons. First, CFTR channels exist on human lung microvascular cells27 and directly cause pulmonary vascular relaxation through CFTR channel activation28,29. Additionally, CFTR mediates vascular tone independent from its role as a chloride channel through other mechanisms including stimulating nitric oxide generation via ATP release30, altering lipid trafficking and vascular smooth muscle cell calcium handling31,32, and through increasing expression of vascular growth factors, including VEGF-A33. In addition, CFTR SNPs have previously been associated with altered flow mediated dilation and flow velocity in humans34. The implications of CFTR dysfunction being pathogenic for pulmonary vascular remodeling and pulmonary hypertension in CF may have prognostic and therapeutic implications. It is possible that novel therapies that restore CFTR function may not only improve lung function in CF, indirectly reducing pulmonary artery pressures, but also may have direct effects on the pulmonary vasculature.
It was notable that PA:A ratio remained stable over a 1-year period, independent of pulmonary exacerbations. This may be the result of inadequate time of follow-up, an uncoupling of change in PA size with further changes in PA hemodynamics35, or may be due to elastic properties intrinsic to the pulmonary vessels. While PA:A performs as a strong predictor of subsequent pulmonary exacerbations, these data suggest it may not be dynamic for monitoring disease progression.
Our study has several limitations. Given the observational nature of the study, we cannot conclude that either the PA:A ratio or pulmonary hypertension cause pulmonary exacerbations. We did not find associations between female sex, lower BMI and pulmonary exacerbations as has been previously reported, probably due to the small sample size3. However, acute pulmonary events in our study were related to lower lung function and Pseudomonas infection, as expected. Additionally, the method of reporting pulmonary exacerbations differed between our cohorts, though recapitulating the association between PA enlargement and the use of a registry-based definition of pulmonary exacerbations in a large CF population could be viewed as a strength. Further, we did not confirm a diagnosis of pulmonary hypertension based on echocardiographic or right-sided heart catheterization in these groups, although the PA:A is correlated to hemodynamic measurements in obstructive lung disease22. We posit there are similar findings in CF and are currently exploring these relationships as part of a separate study. Another limitation to our findings is the use of different CT protocols between our derivation and validation cohorts and the heterogeneous indications for CT in the validation cohort. However, the vascular dimensions and the PA:A were similar between the two study populations, suggesting the findings are valid and could be measured a priori in the clinical setting. Our study was also limited by the few numbers of sweat chloride tests available in the validation cohort. This cohort was adults and the vast majority of subjects were transitioned from pediatric care in the era before CFTR modulation therapies and routine eradication of Pseudomonas; often sweat electrolyte results were poorly documented and Pseudomonas infection rates were higher, whereas the derivation cohort all had sweat electrolytes performed during the RCT and lower Pseudomonas growth. We did observe a higher sweat chloride in patients with a PA:A>1, suggesting there could be a true relationship due to the role of CFTR in vascular smooth muscle, but this will need further mechanistic investigation for confirmation. The low prevalence of chronic Pseudomonas status in the derivation cohort may be due in-part to the entry criteria, multi-center design, and method for reporting chronic Pseudomonas infection in the parent clinical trial (including the use of a central lab, collection of sputum culture at enrollment, and reliance on patient history)12, may be a reflection of Pseudomonas eradication therapies in modern CF therapy, or may be the result of other factors including climate and geographical features that may also influence rates of chronic Pseudomonas infection, which could influence colonization in Australia36. Hence, differences between the two cohorts may not be unexpected. Finally, patients in both cohorts were not evaluated for cardiac disease, obstructive sleep apnea, or thromboembolic disease which could be a contributor to underlying pulmonary hypertension or PA enlargement; previous studies have shown low incidence of primary cardiovascular disease in adult CF patients, reducing the likelihood this is an important contributor7,37.
In conclusion, the PA:A ratio confers prognostic information about respiratory exacerbations in cystic fibrosis. Interestingly, changes observed on CT were also correlated to CFTR function as determined by sweat chloride analysis, suggesting a potential pathologic link. The PA:A ratio may be a useful predictive biomarker of pulmonary exacerbations in CF.
Supplementary Material
Research in context.
Evidence before this study
The development of pulmonary vascular disease is a critical event in the progression of cystic fibrosis and is associated with increased morbidity and mortality. We searched PubMed for articles published before January 1, 2016 using the terms “pulmonary vascular disease” OR “pulmonary hypertension” AND “cystic fibrosis” AND “exacerbation.” We did a second search with the terms “computed tomography” AND “cystic fibrosis” AND “pulmonary exacerbation.” We found 2 studies done in humans that examined the relationship between CF, imaging results, and acute pulmonary exacerbations. One study showed high resolution CT can identify changes in parenchymal appearance, peribronchial thickening, and mucus plugging during the course of an exacerbation in a small pediatric cohort. The other study linked CT imaging to regional airway inflammation during an exacerbation in a small pediatric cohort. We identified no published studies that examined pulmonary vascular disease on CT in the context of pulmonary exacerbations.
Added value of this study
We present data from relatively large derivation and validation cohorts of adult CF patients. We identified PA:A based on measurement on CT by expert operators blinded to clinical data. Pulmonary arterial enlargement is highly prevalent in adults with cystic fibrosis and is associated with developing acute pulmonary exacerbations, even when adjusted for other factors known to present increased exacerbation risk. We found a link between CFTR function measured by sweat chloride and PA enlargement in the derivation cohort. Measuring PA:A to estimate PA enlargement may be a useful predictive marker in CF.
Implications of all the available evidence
Our study demonstrates the link between pulmonary vascular abnormalities measured noninvasively and risk for acute pulmonary exacerbations of CF. PA enlargement may have important diagnostic implications in CF lung disease. The link between CFTR function and PA enlargement on CT and between PA enlargement and hemodynamics warrant further investigation.
Acknowledgments
Funding: NIH/NHLBI, Cystic Fibrosis Foundation, and UAB
Grant Support: UAB: Walter B. Frommeyer Jr. Fellowship in Investigational Medicine to JMW; the NIH: K08 HL123940 to JMW and the Cystic Fibrosis Foundation: SORSCH15R0 to JMW and P30 DK072482 ROWE14Y0 to SMR; Qld Health Fellowship to SCB.
We thank PTC Therapeutics for providing the data and blinded CT image files required for the creation and analysis of a derivation cohort. We thank both PTC and the original trial investigators for their efforts in putting together a successful clinical trial with robust CT imaging in cystic fibrosis. We also thank the patients who volunteered for the study. The authors thank Mrs. Andrea Beevers (Study Coordinator at the Adult CF Centre, TPCH), Mr. Paul Wallbridge (Medical Imaging at TPCH), and Mr. Geoff Sims (Manager of the Australian CF Data Registry) for their assistance in creating the validation cohort and Dr. Eric Sorscher for providing a number of helpful suggestions.
Footnotes
Authors’ Contributions:
Study conception and design: JMW, MTD, SCB, SMR
Sample acquisition and analysis: JMW, RFF, TAG, MEW, SCB, SMR
Data interpretation: JMW, MEW, SCB, SMR
Composition and revision of the manuscript: all authors
Accountability agreement: JMW and SMR are accountable for the accuracy and integrity of all parts of the work
Conflicts of Interest:
Dr. Wells reports grants from NIH/NHLBI, grants from Cystic Fibrosis Foundation, during the conduct of the study; other from AstraZeneca, other from GSK, other from AstraZeneca, other from Gilead, outside the submitted work.
Dr. Dransfield reports personal fees and other from AstraZeneca, personal fees and other from Boehringer Ingelheim, personal fees and other from Boston Scientific, personal fees and other from GlaxoSmithKline, personal fees from Ikaria, personal fees from Skyepharma, grants from NIH, grants from US Department of Defense, grants from American Heart Association, other from Aeris, other from Otsuka, other from Pearl, other from Pfizer, other from PneumRx, other from Pulmonx, other from Yungjin, outside the submitted work.
Ms. Wood reports non-financial support and other from Vertex Pharmaceuticals, non-financial support and other from PTC Pharmaceuticals, non-financial support and other from Galapagos NV, outside the submitted work.
Dr. Bell reports non-financial support and other from Vertex Pharmaceuticals, non-financial support and other from Gilead Pharmaceuticals, non-financial support and other from Novartis Pharmaceuticals, non-financial support and other from Raptor Pharmaceuticals, non-financial support and other from PTC Pharmaceuticals, outside the submitted work.
Dr. Rowe reports grants from NIH, grants from Cystic Fibrosis Foundation, during the conduct of the study; grants from Vertex Pharmaceuticals, grants from Novartis, grants from Nivalis Pharmaceuticals, grants from Bayer, grants from PTC Therapeutics, outside the submitted work.
All other authors declared no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352(19):1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- 2.Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med. 2003;168(8):918–51. doi: 10.1164/rccm.200304-505SO. [DOI] [PubMed] [Google Scholar]
- 3.de Boer K, Vandemheen KL, Tullis E, et al. Exacerbation frequency and clinical outcomes in adult patients with cystic fibrosis. Thorax. 2011;66(8):680–5. doi: 10.1136/thx.2011.161117. [DOI] [PubMed] [Google Scholar]
- 4.Zemanick ET, Harris JK, Conway S, et al. Measuring and improving respiratory outcomes in cystic fibrosis lung disease: opportunities and challenges to therapy. J Cyst Fibros. 2010;9(1):1–16. doi: 10.1016/j.jcf.2009.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rowe SM, Borowitz DS, Burns JL, et al. Progress in cystic fibrosis and the CF Therapeutics Development Network. Thorax. 2012;67(10):882–90. doi: 10.1136/thoraxjnl-2012-202550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minai OA, Chaouat A, Adnot S. Pulmonary hypertension in COPD: epidemiology, significance, and management: pulmonary vascular disease: the global perspective. Chest. 2010;137(6 Suppl):39S–51S. doi: 10.1378/chest.10-0087. [DOI] [PubMed] [Google Scholar]
- 7.Fraser KL, Tullis DE, Sasson Z, Hyland RH, Thornley KS, Hanly PJ. Pulmonary hypertension and cardiac function in adult cystic fibrosis: role of hypoxemia. Chest. 1999;115(5):1321–8. doi: 10.1378/chest.115.5.1321. [DOI] [PubMed] [Google Scholar]
- 8.Weitzenblum E, Hirth C, Ducolone A, Mirhom R, Rasaholinjanahary J, Ehrhart M. Prognostic value of pulmonary artery pressure in chronic obstructive pulmonary disease. Thorax. 1981;36(10):752–8. doi: 10.1136/thx.36.10.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hayes D, Jr, Tobias JD, Mansour HM, et al. Pulmonary hypertension in cystic fibrosis with advanced lung disease. Am J Respir Crit Care Med. 2014;190(8):898–905. doi: 10.1164/rccm.201407-1382OC. [DOI] [PubMed] [Google Scholar]
- 10.Wells JM, Washko GR, Han MK, et al. Pulmonary arterial enlargement and acute exacerbations of COPD. N Engl J Med. 2012;367(10):913–21. doi: 10.1056/NEJMoa1203830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iyer AS, Wells JM, Vishin S, Bhatt SP, Wille KM, Dransfield MT. CT scan-measured pulmonary artery to aorta ratio and echocardiography for detecting pulmonary hypertension in severe COPD. Chest. 2014;145(4):824–32. doi: 10.1378/chest.13-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kerem E, Konstan MW, De Boeck K, et al. Ataluren for the treatment of nonsense-mutation cystic fibrosis: a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Respir Med. 2014;2(7):539–47. doi: 10.1016/S2213-2600(14)70100-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee TW, Brownlee KG, Conway SP, Denton M, Littlewood JM. Evaluation of a new definition for chronic Pseudomonas aeruginosa infection in cystic fibrosis patients. J Cyst Fibros. 2003;2(1):29–34. doi: 10.1016/S1569-1993(02)00141-8. [DOI] [PubMed] [Google Scholar]
- 14.Fuchs HJ, Borowitz DS, Christiansen DH, et al. Effect of aerosolized recombinant human DNase on exacerbations of respiratory symptoms and on pulmonary function in patients with cystic fibrosis. The Pulmozyme Study Group. N Engl J Med. 1994;331(10):637–42. doi: 10.1056/NEJM199409083311003. [DOI] [PubMed] [Google Scholar]
- 15.Kidd TJ, Ramsay KA, Hu H, et al. Shared Pseudomonas aeruginosa genotypes are common in Australian cystic fibrosis centres. Eur Respir J. 2013;41(5):1091–100. doi: 10.1183/09031936.00060512. [DOI] [PubMed] [Google Scholar]
- 16.Smith DJ, Ramsay KA, Yerkovich ST, et al. Pseudomonas aeruginosa antibiotic resistance in Australian cystic fibrosis centres. Respirology. 2016;21(2):329–37. doi: 10.1111/resp.12714. [DOI] [PubMed] [Google Scholar]
- 17.Nick JA, Sanders LA, Ickes B, et al. Blood mRNA biomarkers for detection of treatment response in acute pulmonary exacerbations of cystic fibrosis. Thorax. 2013;68(10):929–37. doi: 10.1136/thoraxjnl-2012-202278. [DOI] [PubMed] [Google Scholar]
- 18.Horsley AR, Davies JC, Gray RD, et al. Changes in physiological, functional and structural markers of cystic fibrosis lung disease with treatment of a pulmonary exacerbation. Thorax. 2013;68(6):532–9. doi: 10.1136/thoraxjnl-2012-202538. [DOI] [PubMed] [Google Scholar]
- 19.Klein M, Cohen-Cymberknoh M, Armoni S, et al. 18F-fluorodeoxyglucose-PET/CT imaging of lungs in patients with cystic fibrosis. Chest. 2009;136(5):1220–8. doi: 10.1378/chest.09-0610. [DOI] [PubMed] [Google Scholar]
- 20.Wielputz MO, Puderbach M, Kopp-Schneider A, et al. Magnetic resonance imaging detects changes in structure and perfusion, and response to therapy in early cystic fibrosis lung disease. Am J Respir Crit Care Med. 2014;189(8):956–65. doi: 10.1164/rccm.201309-1659OC. [DOI] [PubMed] [Google Scholar]
- 21.Brody AS, Sucharew H, Campbell JD, et al. Computed tomography correlates with pulmonary exacerbations in children with cystic fibrosis. Am J Respir Crit Care Med. 2005;172(9):1128–32. doi: 10.1164/rccm.200407-989OC. [DOI] [PubMed] [Google Scholar]
- 22.Wells JM, Dransfield MT. Pathophysiology and clinical implications of pulmonary arterial enlargement in COPD. Int J Chron Obstruct Pulmon Dis. 2013;8:509–21. doi: 10.2147/COPD.S52204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sorio C, Buffelli M, Angiari C, et al. Defective CFTR expression and function are detectable in blood monocytes: development of a new blood test for cystic fibrosis. PLoS One. 2011;6(7):e22212. doi: 10.1371/journal.pone.0022212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stalvey MS, Clines KL, Havasi V, et al. Osteoblast CFTR inactivation reduces differentiation and osteoprotegerin expression in a mouse model of cystic fibrosis-related bone disease. PLoS One. 2013;8(11):e80098. doi: 10.1371/journal.pone.0080098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Norez C, Jayle C, Becq F, Vandebrouck C. Bronchorelaxation of the human bronchi by CFTR activators. Pulm Pharmacol Ther. 2014;27(1):38–43. doi: 10.1016/j.pupt.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 26.Meyerholz DK, Stoltz DA, Namati E, et al. Loss of cystic fibrosis transmembrane conductance regulator function produces abnormalities in tracheal development in neonatal pigs and young children. Am J Respir Crit Care Med. 2010;182(10):1251–61. doi: 10.1164/rccm.201004-0643OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tousson A, Van Tine BA, Naren AP, Shaw GM, Schwiebert LM. Characterization of CFTR expression and chloride channel activity in human endothelia. Am J Physiol. 1998;275(6 Pt 1):C1555–64. doi: 10.1152/ajpcell.1998.275.6.C1555. [DOI] [PubMed] [Google Scholar]
- 28.Robert R, Thoreau V, Norez C, et al. Regulation of the cystic fibrosis transmembrane conductance regulator channel by beta–adrenergic agonists and vasoactive intestinal peptide in rat smooth muscle cells and its role in vasorelaxation. J Biol Chem. 2004;279(20):21160–8. doi: 10.1074/jbc.M312199200. [DOI] [PubMed] [Google Scholar]
- 29.Robert R, Savineau JP, Norez C, Becq F, Guibert C. Expression and function of cystic fibrosis transmembrane conductance regulator in rat intrapulmonary arteries. Eur Respir J. 2007;30(5):857–64. doi: 10.1183/09031936.00060007. [DOI] [PubMed] [Google Scholar]
- 30.Liang G, Stephenson AH, Lonigro AJ, Sprague RS. Erythrocytes of humans with cystic fibrosis fail to stimulate nitric oxide synthesis in isolated rabbit lungs. Am J Physiol Heart Circ Physiol. 2005;288(4):H1580–5. doi: 10.1152/ajpheart.00807.2004. [DOI] [PubMed] [Google Scholar]
- 31.Tabeling C, Yu H, Wang L, et al. CFTR and sphingolipids mediate hypoxic pulmonary vasoconstriction. Proc Natl Acad Sci U S A. 2015;112(13):E1614–23. doi: 10.1073/pnas.1421190112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo JJ, Stoltz DA, Zhu V, et al. Genotype-specific alterations in vascular smooth muscle cell function in cystic fibrosis piglets. J Cyst Fibros. 2014;13(3):251–9. doi: 10.1016/j.jcf.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martin C, Coolen N, Wu Y, et al. CFTR dysfunction induces vascular endothelial growth factor synthesis in airway epithelium. Eur Respir J. 2013;42(6):1553–62. doi: 10.1183/09031936.00164212. [DOI] [PubMed] [Google Scholar]
- 34.Vasan RS, Larson MG, Aragam J, et al. Genome-wide association of echocardiographic dimensions, brachial artery endothelial function and treadmill exercise responses in the Framingham Heart Study. BMC Med Genet. 2007;8(Suppl 1):S2. doi: 10.1186/1471-2350-8-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boerrigter B, Mauritz GJ, Marcus JT, et al. Progressive dilatation of the main pulmonary artery is a characteristic of pulmonary arterial hypertension and is not related to changes in pressure. Chest. 2010;138(6):1395–401. doi: 10.1378/chest.10-0363. [DOI] [PubMed] [Google Scholar]
- 36.Collaco JM, McGready J, Green DM, et al. Effect of temperature on cystic fibrosis lung disease and infections: a replicated cohort study. PLoS One. 2011;6(11):e27784. doi: 10.1371/journal.pone.0027784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Panidis IP, Ren JF, Holsclaw DS, Kotler MN, Mintz GS, Ross J. Cardiac function in patients with cystic fibrosis: evaluation by two-dimensional and Doppler echocardiography. Journal of the American College of Cardiology. 1985;6(3):701–6. doi: 10.1016/s0735-1097(85)80134-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.