

Abstract
We systematically analyzed post-zygotic mutations (PZMs) in whole-exome sequences from the largest collection of trios (5,947) with autism spectrum disorder (ASD) available, including 282 unpublished trios, and performed re-sequencing using multiple independent technologies. We identified 7.5% of de novo mutations as PZMs, with 83.3% of these PZMs not discovered in previous studies. Damaging, non-synonymous PZMs within critical exons of prenatally-expressed genes were more common in ASD probands than controls (P<1×10-6), and genes carrying these PZMs were enriched for expression in the amygdala (P=5.4×10-3). Two genes (KLF16 and MSANTD2) were significantly enriched for PZMs genome-wide, and other PZMs involved genes (SCN2A, HNRNPU, SMARCA4) known to cause ASD or other neurodevelopmental disorders. PZMs constitute a significant proportion of de novo mutations and contribute importantly to ASD risk.
Introduction
Autism spectrum disorder (ASD) is a complex disorder with genetic and clinical heterogeneity. Beyond common variation1, previous studies focusing on germline mutations have demonstrated a significant contribution from de novo copy number variants (CNVs)2,3, and more recent whole-exome sequencing (WES) analyses have highlighted the role of de novo point mutations4,5. Although the number of exonic de novo mutations is similar between affected and unaffected individuals (≈1 de novo point mutation per exome), ASD probands harbor an excess of deleterious and loss-of-function (LoF) de novo mutations in exons compared to their unaffected siblings4,5. Collectively, 4-7% of probands have a de novo CNV and ∼7% of probands have a de novo point mutation that confers risk to ASD2. Additionally, WES studies have uncovered risk to ASD from rare autosomal recessive (3%) and X-linked variants (2%)6,7. However, a large portion of ASD risk cannot be explained by germline de novo, recessive and X-linked variants, and this warrants investigation of other genetic contributions to ASD risk.
Post-zygotic mutations (PZMs) result in distinct cell populations within the same individual, which can contribute to varying disease manifestations. These mutations are typically not transmitted to the offspring and it has been hypothesized that PZMs account for a significant proportion of genetic risk in sporadic disorders. There is increasing recent evidence that PZMs can contribute to brain malformations and epilepsy8,9, and that a fraction of clinically relevant PZMs can be detected in blood of affected individuals8,10. The role of PZMs in ASD risk is unknown and we therefore explored the contribution of this type of variation to ASD. PZMs are efficiently detected by candidate gene sequencing panels, given their deep sequencing coverage. However, PZMs present in greater than 25-30% of cells (or 15% alternate allele fraction (AAF)) can be detected with reasonable sensitivity using WES8. We re-called WES data from 5,947 trios, adding 282 newly sequenced trios, from the Autism Sequencing Consortium and Simons Simplex Collection, and using a custom pipeline, we resequenced PZMs detected from WES data using 3 resequencing technologies, providing a systematic evaluation of PZM's contribution to ASD risk.
Results
Excess of de novo mutations with low AAFs
We analyzed de novo mutations in WES data from 5,947 families, which included 4,032 ASD trios and 1,918 quads that also have unaffected siblings (Supplementary Tables 1-3)4,5. The vast majority of samples (96%) were derived from whole blood DNA, and a negligible fraction from lymphoblastoid cells (3%) and primary saliva (1%). We included all samples derived from various tissue types, but removed outlier samples with a large number of de novo or mosaic mutations from our analyses (see Methods). We increased specificity for likely pathogenic mutations by filtering out variants that are present in control exomes, resulting in modestly lower rates of de novo mutations than previously called (4,846 in total). Of these, a substantial portion (23%) showed low AAFs of ≤40% (Fig. 1A). The modal AAF was ≈50%, which is consistent with the expected AAF for a germline heterozygous mutation. We observed a 1.4-fold excess of mutations in the 40%-50% AAF category compared to the 50%-60% AAF category, suggesting a modest bias towards mutations with lower AAFs, possibly due to amplification, capture or sequencing biases for the alternate alleles. In contrast, we observed a robust (4.1-fold) excess of mutations with AAF≤40% (23.7% of all de novo mutations), compared to those with AAF≥60% (5.8% of all de novo mutations), suggesting that a significant proportion of mutations with AAF≤40% arose from a biological mechanism rather than a technical bias. In addition, there is an excess of de novo point mutations compared to inherited variants in the AAF≤40% category (odds ratio OR = 1.67), that was not seen in the AAF≥60% category (OR = 0.82). This suggests that a significant portion of de novo mutations is likely to have arisen post-zygotically rather than in the parental gametes.
Figure 1. De novo mutations in ASD show an excess of low alternate allele frequencies, consistent with post-zygotic mosaicism.
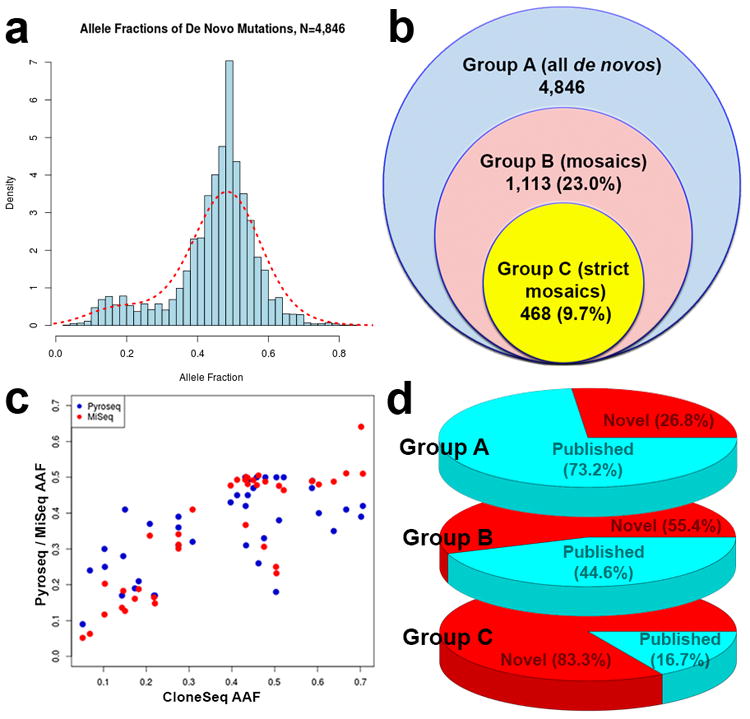
(a) There is an excess of variants with low AAFs among the de novo mutations, which are likely to be post-zygotic mutations. (b) Rates of mutations in the datasets for all de novos in Group A, as well as mosaics in Groups B and C. (c) Correlation of AAFs for PZMs across the AAF spectrum using multiple technologies (n=49 mutations for CloneSeq, n=46 mutations for Pyroseq, n=42 mutations for MiSeq), with higher correlations (Pearson's r2=0.85 for CloneSeq and MiSeq, r2=0.63 for CloneSeq and Pyroseq). (d) Percentages of identified de novo variants that were identified by previous analyses or novel from Groups A, B and C. The majority of high-confidence PZMs from Group C were not detected by previous calling algorithms.
Detection of PZMs from WES and secondary resequencing
Given our initial observations that some de novo mutations might be PZMs, we developed a pipeline to quantitatively categorize PZMs with high or low confidence in our cohort of 5,947 ASD families (see Methods). Of 4,846 total de novo mutations (which we define as Group A, Fig. 1B), 1,113 were candidate PZMs (23%, Group B), defined as having an AAF that was ≤80% of the modal AAFs, which ranged from 40-50%. Of these Group B mutations, 468 were interpreted as high-confidence PZMs (9.7%, Group C) because they showed statistically significant deviation from the modal AAFs.
We compared the 4,846 de novo mutations in Group A from our study with previous studies reporting these datasets4,5, and found that 1,297 of the de novo mutations (26.8%) we identified had not been previously reported in ASD. We enriched for de novo mutations that are most likely to be pathogenic and to have a large effect in ASD, by filtering away de novo mutations found in control individuals. As such, our reported rate of de novo mutations is conservative – on average, less than 1 de novo mutation per exome. However, we also re-called the exomes jointly using the latest GATK variant calling pipelines and best practices. This likely accounts for improved detection of previously unreported mutations.
To experimentally test the candidate PZMs, we applied independent resequencing methods in three phases. In Phase 1, we resequenced 50 mutations, based on sample availability, across the three groups (Table 1) using three independent technologies - pyrosequencing, subcloning with Sanger colony sequencing (CloneSeq), and targeted PCR followed by MiSeq resequencing (Supplementary Table 4), to test whether these mutations deviated from the expected AAF of 50%, and to compare these technologies. We found that 84.8-93.3% of the Group C mutations, predicted to be high-confidence PZMs, were indeed likely to arise post-zygotically with confirmed AAFs≤40% (Table 1). Of the less stringent candidate PZMs (in Group B but not in C), 25-38% were confirmed as post-zygotic with AAFs≤40%. In Phase 2, we resequenced another 181 mutations from all groups using targeted PCR and MiSeq, as well as pyrosequencing, and replicated the rates observed in Phase 1: 84.8-85.2% of high-confidence PZMs (Group C) and 13.5-25.6% of less stringent PZMs (Group B) showed AAFs≤40%. A small percentage (8.3%) of predicted germline de novo mutations (gDNMs) found only in Group A (and not identified as also being in Groups B or C) also showed AAFs≤40%. In Phase 3, we resequenced a larger number of 325 mutations using targeted PCR and MiSeq with DNA derived from blood samples, and found that 97% of high-confidence PZMs, 17.6% of less stringent PZMs, and 2.8% of predicted gDNMs have AAFs≤40%.
Table 1. Validation rates for mutations detected from WES.
Rates at which predicted PZMs from WES were also found to be de novo with unequal AAFs using three different technologies.
| Phase 1: Resequencing of initial 50 mutations to evaluate if AAFs≤40% | |||
|---|---|---|---|
| High-confidence PZMs from Group C | Less stringent PZMs found in Group B but not Group C | Potential germline de novos found in Group A but not Group B | |
| CloneSeq | 14 / 16 (87.5%) | 7 / 28 (25%) | 1 / 5 (20%) |
| Pyrosequencing | 13 / 15 (87%) | 10 / 26 (38%) | 2 / 5 (40%) |
| Targeted PCR + MiSeq | 14 / 15 (93.3%) | 6 / 24 (25%) | 0 / 3 (0%) |
| Phase 2: Resequencing of 181 mutations to evaluate if AAFs≤40% | |||
| Pyrosequencing | 28 / 33 (84.8%) | 20 / 78 (25.6%) | - |
| Targeted PCR + MiSeq | 52 / 61 (85.2%) | 10 / 73 (13.7%) | 1 / 12 (8.3%) |
| Phase 3: Resequencing of 325 mutations to evaluate if AAFs≤40% | |||
| Targeted PCR + MiSeq | 159 / 164 (97.0%) | 3 / 17 (17.6%) | 4 / 144 (2.8%) |
The Pearson's correlations between AAFs detected from WES compared to the 3 resequencing technologies ranged from 0.52 to 0.58, apparently reflecting mainly the relatively low coverage, and hence imprecise AAFs from WES. In contrast, AAFs determined using CloneSeq and targeted PCR with MiSeq were more highly correlated with one another, at 0.85 (Fig. 1C). Although CloneSeq is an excellent standard for measuring the AAF of PZMs, it is low-throughput and expensive. Our data suggest that targeted PCR with MiSeq is an acceptable alternative that is higher throughput. AAFs determined with pyrosequencing showed lower correlation with CloneSeq, at 0.63. In particular, pyrosequencing did not correlate well with CloneSeq at lower AAFs (Fig. 1C), e.g., AAFs≤40% (Pearson's correlation = 0.64), unlike targeted PCR with MiSeq (Pearson's correlation = 0.92), suggesting a larger variation in detecting lower AAFs using pyrosequencing.
We also tested 82 de novo mutations using Sanger sequencing, and found that 73 of them (or 89%) are confirmed as genuine de novo mutations, i.e., the mutations were not present at a detectable AAF in the parents' DNA samples. We confirmed this initial result using targeted PCR with MiSeq for another 327 de novo mutations and found that 84.1% of the PZMs from Group C were confirmed to arise de novo. Taken together, our data suggest that approximately 9.7% (the proportion of de novo mutations detected from WES that are high-confidence PZMs in Group C) × 0.84 (the average fraction of genuine de novo mutations in Group C) × 0.92 (the average fraction of genuine PZMs) = 7.5% of all detected de novo mutations are likely to be true PZMs detectable by WES, though the recovery of PZMs would be expected to be higher if the exomes had been sequenced at higher coverage.
It is possible that some potential PZMs might be falsely called as a result of copy number variants spanning across the region. As such, we performed TaqMan copy number assays on 36 PZMs in Group C to evaluate the rate of PZMs co-occurring with copy number variants, but did not detect any (Supplementary Table 5), suggesting that the rate at which copy number variants might overlap with PZMs is likely to be less than 3%.
PZMs were frequently missed with previous pipelines
Despite the lower overall rate of called de novo mutations using our approach compared to previous studies, we found that most PZMs in Group B had not been previously identified (617 out of 1,113 PZMs or 55.4%, Fig. 1D), and an even higher proportion of PZMs in Group C was not previously reported (390 out of 468 PZMs or 83.3%). This suggests that the previous pipelines were more likely to detect gDNMs found only in Group A, and confirms that our approach detects with high specificity many PZMs not previously identified, presumably because these PZMs might have been marked as variants with lower quality and were more likely to be flagged as falsely called variants, despite being readily confirmed by complementary technologies. Our data indicate that over 84.8% of the high-confidence PZMs in Group C were confirmed to be bona fide PZMs through the resequencing experiments, and that 83.3% of the high-confidence PZMs were not previously reported.
PZMs differ from gDNMs and cancer somatic mutations
Analysis of the mutational properties of PZMs reveal that they show several features that differ from gDNMs. PZMs are enriched on the anti-sense strand (relative to transcription) compared to gDNMs (OR = 1.30, 95% CI = [1.07, 1.58] for Group C, Supplementary Table 6). Anti-sense-strand bias typically reflects the inherent bias of transcription-coupled nucleotide excision repair, which has a higher fidelity on the sense strand. This results in a higher accumulation of mutations on the anti-sense strand11, and it is likely that PZMs arise at least in part from this mechanism, similar to previous reports for somatic mosaic mutations in cancers12.
The most common types of mutations among gDNMs and PZMs are C-T and G-A mutations. It has been reported that there is a strong preference for mutations from A to C or T to G in the nucleosome core13, and we observed a similar enrichment of A-C and T-G mutations in PZMs compared to gDNMs (OR = 2.23, 95% CI = [1.64, 2.99] for Group C, Supplementary Table 7). In particular, we found that the enrichment of A-C mutations was predominantly on the sense strand, whereas the enrichment of T-G mutations was predominantly on the anti-sense strand (Supplementary Table 8). This is a distinct mutational profile from the ones reported for somatic mosaic mutations in cancers12, but is suggestive that the enrichment of such mutations in the nucleosome core might affect chromatin remodeling, a process that has been previously found to be perturbed in ASD14.
Somatic mutations discovered in cancers have also been reported to be associated with late DNA replication12. We correlated PZMs against DNA replication timing during S phase15 and compared these against the gDNMs found only in Group A (Supplementary Table 9). We observed a similar trend for the PZMs with late replication timing (OR = 1.36, 95% CI = [0.83, 2.14] for Group C), but not with early replication timing (OR = 0.88, 95% CI = [0.72, 1.07] for Group C). However, the association of these PZMs with late replication timing was substantially less than that reported in cancers16 and was not statistically significant. Together, these results highlight some unique features of the PZMs. Our data suggest that the mechanisms generating PZMs and their mutational profile are distinct from those of gDNMs. Also, while PZMs detected in blood and somatic mosaic mutations in cancers accumulate preferentially on the anti-sense strand, they differ in the preference for nucleotide base substitutions.
It has been previously reported that germline de novo mutations are enriched on the paternal haplotype, and similarly, we observed a 1.69-fold excess of mutations in Group A on the paternal haplotype (1,321 paternal versus 781 maternal, binomial P=1.50×10-32, Supplementary Table 10). In contrast the high-confidence mosaic mutations in Group C did not show any significant excess of mutations on the paternal compared to maternal haplotypes (90 paternal versus 78 maternal, 1.15-fold, binomial P=0.2). This confirms that the mutations detected in Group C are likely to be enriched for true PZMs compared to the larger set of Group A mutations.
An excess of deleterious PZMs is found in brain-expressed critical exons in ASD probands
We next investigated whether PZMs might contribute to ASD risk. We first analyzed all de novo LoF mutations in Group A, and found the expected excess in probands compared to unaffected siblings, similar to previous reports4,5. However, the LoF PZMs from Groups B and C did not show an excess in probands versus siblings (Fig. 2A, Supplementary Table 11). When comparing missense PZMs predicted to be deleterious using three in-silico tools (PolyPhen217, SIFT18 and CADD19), we found more de novo missense mutations predicted to be deleterious in Groups A and B in probands compared to siblings, but no enrichment for PZMs in Group C (hypergeometric P = 0.024 for Group A, P = 0.041 for Group B, and P = 0.32 for Group C, Supplementary Table 12).
Figure 2. Post-zygotic mutations in ASD show excess deleterious mutations in critical exons of early developmental brain expressed genes.
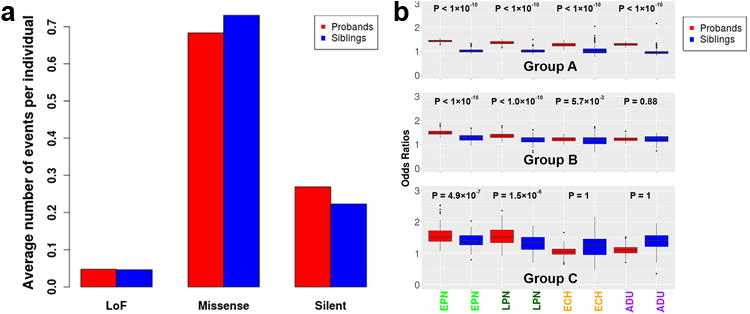
(a) There is no statistically significant global excess of Group C PZMs in the probands (red) compared to their unaffected siblings (blue), hypergeometric P=0.32 for fraction of LoF variants in probands compared to siblings. (b) As expected, there are highly significant excesses in overall gDNMs (Group A) for genes expressed in prenatal and adult brains. For Groups B and C, representing potential and high-confidence PZMs, there is a strong excess of LoF and missense mutations in critical exons that are expressed in EPN (early prenatal) and LPN (late prenatal), 1-tailed Wilcoxon rank sum test P<1×10-5, but not ECH (early childhood) or ADU (adult) post-mortem brain samples in the probands, 1-tailed Wilcoxon rank sum test P>1×10-5.
We wondered whether PZMs might contribute to ASD risk by selectively affecting genes expressed in the brain that are subjected to strong purifying selection. It has been previously shown that analysis of “critical exons” – i.e., those that are depleted for deleterious mutations in normal individuals - permits higher sensitivity in detecting differences in germline de novo mutations and shows an excess of deleterious PZMs in probands versus unaffected siblings in critical exons expressed in the brain20. In line with previous evidence, we observed an enrichment of LoF and missense mutations from Groups A and B found in critical exons in probands versus unaffected siblings (Fig. 2B). Importantly, we also observed an enrichment of high-confidence LoF and missense PZMs from Group C in the probands compared to siblings, further supporting the association of some of these PZMs with ASD.
Mutations in Group A that fall within critical exons are enriched in probands compared to their unaffected siblings in genes expressed across all developmental epochs - early (≤16 pcw) and late prenatal brains (>16pcw), early childhood (<15 years) and adulthood (≥15 years). Mutations in Groups B and C that fall within critical exons are enriched in probands for genes expressed in prenatal and early childhood brains, but not adult brains (Fig. 2B), suggesting a particular enrichment for these genes in processes that occur prenatally, including neurogenesis, neuronal migration, dendritogenesis and synaptogenesis. Assessment of PZMs in Group C that fall in critical exons across 16 brain regions during prenatal development pinpointed the amygdala as the top brain region where PZMs in critical exons are enriched in probands compared to unaffected siblings (Wilcoxon rank sum P=5.4×10-3, Fig. 3, Table 2). Our data suggest that further analyses of PZMs in ASD may begin to unveil brain regions important for the pathophysiology of the disorder.
Figure 3. Post-zygotic mutations implicate the prenatal amygdala in ASD.
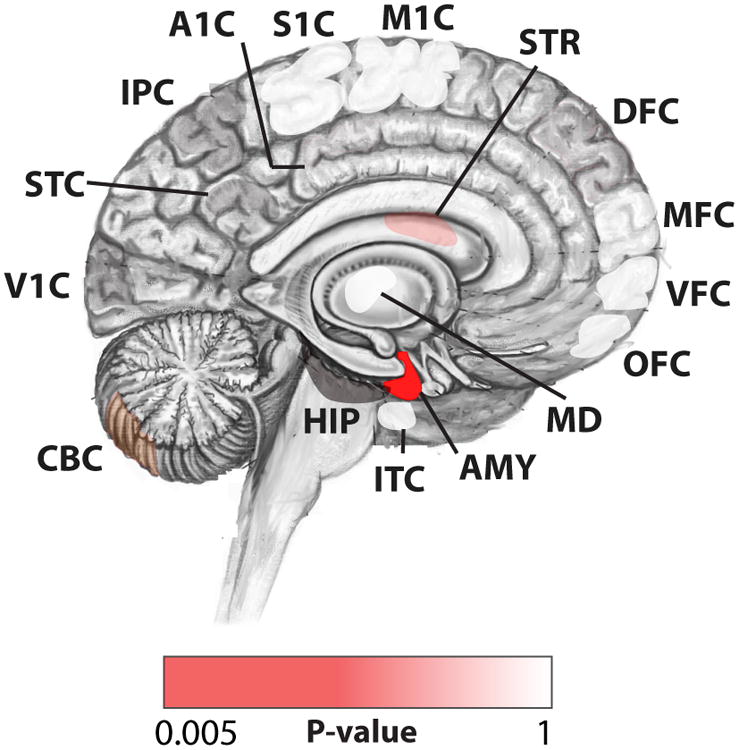
Spatial representation of the regions that are enriched for PZMs in Group C in the probands, and the 1-tailed Wilcoxon rank sum test P=5.4×10-3 for the top brain region (AMY – amygdala).
Table 2. Regions that are enriched for PZMs in Group C in the probands compared to their unaffected siblings.
The P-values reported are calculated using a 2-tailed Wilcoxon rank sum test.
| Brain Region | Group C Wilcoxon Test P |
|---|---|
| Amygdala (AMY) | 5.4×10-3 |
| Striatum (STR) | 0.065 |
| Cerebellar cortex (CBC) | 0.093 |
| Hippocampus (HIP) | 0.10 |
| Posteroinferior parietal cortex (IPC) | 0.27 |
| Primary visual cortex (V1C) | 0.43 |
| Primary auditory cortex (A1C) | 0.48 |
| Primary motor cortex (M1C) | 0.49 |
| Mediodorsal nucleus of thalamus (MD) | 0.59 |
| Posterior superior temporal cortex (STC) | 0.69 |
| Medial prefrontal cortex (MFC) | 0.71 |
| Ventrolateral prefrontal cortex (VFC) | 0.71 |
| Inferior temporal cortex (ITC) | 0.80 |
| Dorsolateral prefrontal cortex (DFC) | 0.96 |
| Orbital prefrontal cortex (OFC) | 0.96 |
| Primary somatosensory cortex (S1C) | 1 |
An excess of recurrent PZMs in genes found in probands implicate these genes in ASD
In probands, 27/735 genes (3.7%) showed recurrent non-synonymous PZMs, versus 2/322 genes (0.62%) with recurrent non-synonymous PZMs in siblings, representing a 6.1-fold excess of genes with recurrent non-synonymous PZMs in the probands (95% CI = [1.52,53.2], Fisher's Exact Test P=0.0035, permutation P = 0.0037). This strongly suggests that some of these genes with recurrent non-synonymous PZMs are relevant for ASD risk.
Given our finding that some genes with recurrent non-synonymous PZMs are likely to confer risk for ASD, we focused on these genes containing recurrent non-synonymous PZMs. We obtained a background set of 84,448 variants that are privately inherited (i.e., variants that are not found in our controls, consisting of parents and siblings, as well as in control databases such as the Exome Variant Server, but were inherited from a parent in an affected or unaffected offspring; see Methods and Supplementary Table 13). Amongst these, we selected a subset with an AAF of 80% or less from the expected modal AAF to obtain a background rate of PZMs in each gene. In addition, we filtered our genes in regions with segmental duplications as described previously10, allowing us to exclude genes with falsely called PZMs due to segmental duplications or common copy number variations.
We found 27 genes with recurrent non-synonymous PZMs in the probands, and amongst them, 2 genes (KLF16 and MSANTD2) harbored more PZMs than expected genome-wide based on their background rates (hypergeometric P<0.05/18,782 or 2.7×10-6, Table 3). Among the 27 genes, previous studies have reported an excess of germline de novo mutations in SCN2A found in ASD probands4,5, and de novo mutations in HNRNPU have been associated with epileptic encephalopathies21. Our approach detects genes with more recurrent, non-synonymous PZMs than expected from the number of falsely called mutations. There are several reasons why this might occur - for instance, some genes might be less likely to be repaired and thus may tend to accumulate PZMs. Nonetheless, multiple PZMs within well documented neurodevelopmental disease genes like SCN2A and HNRNPU provide strong evidence that at least some of the post-zygotic mosaic mutations can predispose to ASD.
Table 3. List of top 10 genes with recurrent non-synonymous PZMs from Group B.
Genes with recurrent non-synonymous PZMs from Group B found in the probands (observed), with the observed number of mosaics that are inherited (expected), as well as the hypergeometric test P-value. The genes that are expressed in the brain are highlighted in red.
| Expected | Observed | Hypergeometric P | |
|---|---|---|---|
| KLF16 | 0/84448 | 2/571 | <1×10-6 |
| MSANTD2 | 1/84448 | 2/571 | <1×10-6 |
| POLA2 | 2/84448 | 2/571 | 4.6×10-5 |
| SMARCA4 | 11/84448 | 3/572 | 4.9×10-5 |
| AZGP1 | 4/84448 | 2/571 | 2.7×10-4 |
| CNGB3 | 5/84448 | 2/571 | 4.5×10-4 |
| HNRNPU | 5/84448 | 2/571 | 4.5×10-4 |
| SCN2A | 5/84448 | 2/571 | 4.5×10-4 |
| EPPK1 | 58/84448 | 4/571 | 6.6×10-4 |
| CARD11 | 7/84448 | 2/571 | 9.4×10-4 |
Among the top genes with recurrent non-synonymous PZMs in probands, 8/10 were expressed in brain (Supplementary Table 14), whereas 2 of the bottom 10 genes with recurrent non-synonymous PZMs in probands showed brain expression. Although there are 2 genes with recurrent non-synonymous PZMs in unaffected siblings, neither of these genes was genome-wide significant (Supplementary Table 15). Germline de novo mutations in ASD probands have been reported to be found in genes that are more intolerant to mutation, defined by lower residual variation intolerance scores (RVIS)22. We found that genes with recurrent non-synonymous PZMs in probands that scored highest, i.e. had the lowest hypergeometric P-values, showed low RVIS scores, that is, are more intolerant to human variation (Supplementary Fig. 1). These data all further support a role for some of these PZMs in ASD risk.
It has been repeatedly reported that genes implicated in ASD based on de novo mutations are enriched for targets of the Fragile X Mental Retardation Protein (FMRP)5. We replicated this observation for de novo mutations in Group A (OR = 2.72 [2.35, 3.13], P<1×1-10). We also found a significant enrichment for PZMs in Groups B and C for FMRP target genes (OR = 2.65 [2.04, 3.41], P<1×1-10, and OR = 2.06 [1.30, 3.12], P=7.7×10-4 respectively).
PZMs in SMARCA4 down-regulates GRIN2B
One of the genes with recurrent non-synonymous PZMs is SMARCA4, which encodes BRG1, a critical component of the SWI/SNF chromatin-remodeling complex that regulates gene expression23. Germline and somatic LoF mutations in this gene have been implicated in a variety of cancers, including rhabdoid tumors and small cell carcinoma of the ovary of hypercalcemic type23 (Fig. 4C). On the other hand, germline heterozygous missense mutations in SMARCA4 have been associated with Coffin-Siris syndrome (OMIM #135900), characterized by intellectual disability. The absence of LoF mutations in SMARCA4 in Coffin-Siris syndrome suggests that the missense mutations act as gain-of-function or activating mutations, unlike the germline inactivating mutations in cancers24.
Figure 4. Recurrent non-synonymous post-zygotic mosaic mutations implicate novel genes with more mutations than expected false calls.
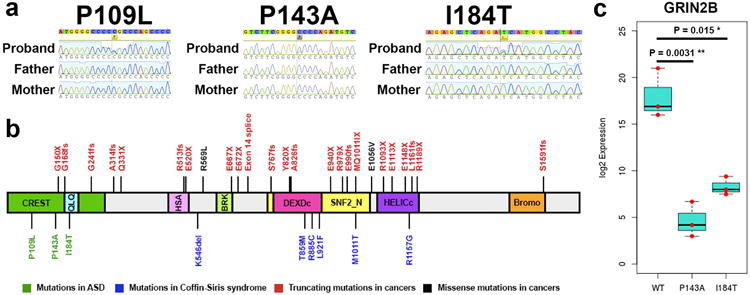
(a) Sanger sequencing traces for the 3 SMARCA4 mutations. (b) SMARCA4 mutations reported in cancers, Coffin-Siris syndrome and ASD. (c) qPCR results for GRIN2B after overexpression and selection of wildtype and mutant (p.P143A and p.I184T) human SMARCA4 in N2A cells, with the values for each replicate experiment (N=3 each for WT, P143A and I184T) in red dots (unpaired t-test P=0.0031 for P143A compared to wildtype and P=0.015 for I184T compared to wildtype).
We detected and confirmed the three missense mutations in SMARCA4 in the three probands with ASD (p.P143A with AAF 21%, p.I184T with AAF 33%, p.P109L with AAF 36%, Fig. 4A), all predicted to be deleterious using PolyPhen2 and SIFT17,18. The p.P143A mutation had a CADD score of 19.81, while the p.I184T and p.P109L mutations had CADD scores of ≥20 (26.4 and 34 respectively). The p.P109L mutation was previously reported as a somatic mutation in a lung carcinoma sample from the COSMIC database (COSM710132)25. CloneSeq on blood-derived DNA for these three individuals with the SMARCA4 mutations confirmed two of the mutations as likely PZMs (p.P143A: 45 alternate out of 164 total colonies, binomial P = 4.9×10-9, and p.I184T: 39 alternate out of 118 total colonies, binomial P = 1.5×10-4), while the p.P109L mutation is likely germline (p.P109L: 83 alternate out of 164 total colonies, binomial P = 0.59).
All three probands had IQs higher than 70 and were confirmed not to show the typical features of Coffin-Siris syndrome. Whereas most SMARCA4 mutations reported in cancers, such as medulloblastoma, fall within the helicase domains of the protein26, the PZMs in SMARCA4 in ASD probands fell in the N-terminal domain, in a region (between amino acids 1 and 282) that binds CREST27, encoded by the Synovial Sarcoma Translocation Gene On Chromosome 18-Like 1 (SS18L1) gene (Fig. 4B). The BRG1-CREST complex regulates the NR2B subunit of the ionotropic, N-methyl D-aspartate glutamate receptor27, encoded by the ASD risk gene GRIN2B4,5.
Therefore, we hypothesized that the PZMs in SMARCA4 might influence the BRG1-CREST interaction and thus the expression of the downstream target GRIN2B. To test the hypothesis, we overexpressed wild-type (WT), p.I184T or p.P143A SMARCA4 in mouse neuroblastoma (N2A) cells and measured the expression of GRIN2B by quantitative PCR. We found that overexpression of either SMARCA4 mutant led to significantly lower expression of GRIN2B compared to wildtype SMARCA4 (Fig. 4C).
Discussion
Our systematic analysis of WES from over 5,800 trios found that 7.5% of de novo mutations are PZMs, despite the limited sensitivity of WES to detect PZMs due to the relatively low coverage. We established a pipeline for detecting and analyzing PZMs, using 3 independent resequencing technologies, and showed that there is high specificity in our PZM detection. In particular, 84.8-93.3% of the high-confidence Group C PZMs are bona fide PZMs. We also discovered that ASD probands harbor more deleterious PZMs compared to their unaffected siblings in brain-expressed critical exons, supporting a role for some of these PZMs in ASD risk. Our estimate of 7.5% of de novo mutations being PZMs is similar to the 6.5% rate reported in an earlier cohort of 50 trios with intellectual disability28, as well as a recently reported estimate of 5.4% in 2,388 families with ASD29. Furthermore, the size of our dataset has allowed us to explore and confirm the role of PZMs in conferring risk to ASD, analyze the mutational characteristics of PZMs, and begin to use them to study the spatio-temporal distribution of PZMs in ASD. Our analysis also revealed striking enrichment of PZMs within genes that are clinically relevant to ASD, including the bona fide ASD risk gene SCN2A. The identification of recurrent non-synonymous PZMs in a small set of genes in ASD probands also provides strong evidence for the clinical importance of PZMs.
The finding that LoF and missense PZMs in critical exons in ASD probands showed enrichment in amygdala expression is intriguing since the amygdala plays key roles in emotional and social responses30, such as conditioned fear. Complete bilateral damage in the amygdala in humans results in impaired social judgement31, reaffirming the importance of the amygdala in regulating social conditioning and learning. An “amygdala theory” of autism32 has been supported by recent work that found impaired neuronal responses in the amygdala in individuals with ASD33. Sexual dimorphism has also been observed in response to testosterone in the amygdala34, which has been proposed to potentially account for some of the gender bias observed in ASD.
We have also identified two PZMs in SMARCA4, a gene that encodes a major chromatin factor implicated in cancer and Coffin-Siris syndrome. Both PZMs in SMARCA4 found in the ASD probands fall within the same N-terminal, CREST-binding domain, forming a complex that regulates the activity-dependent expression of key genes implicated in neuronal plasticity27. We discovered that overexpressing SMARCA4 mutants (with p.I184T and p.P143A) reduces the expression of GRIN2B, which encodes a key subunit of the NMDA glutamate receptor that has been previously implicated as an ASD risk gene based on de novo LoFs4,5. This suggests that the PZMs in SMARCA4 might impair the function of glutamatergic synapses35.
It has been reported that de novo CNVs and DNMs associated with ASD are more common in individuals with low non-verbal IQ scores5. To test the association of IQ with PZM carriers, we analyzed the 7 probands with recurrent PZMs in 9 of the genes (Fig. 4A) that had IQ scores available. Two of the 7 probands with PZMs (or 28.6%) had non-verbal IQs of at least 100, compared to two out of 65 probands (or 3.1%) with recurrent de novo LoF mutations having non-verbal IQs of at least 100, indicating a 9.3-fold excess of probands with higher non-verbal IQs harboring PZMs (hypergeometric test P=0.01). This preliminary observation would need replication in a larger number of individuals in the future to test the hypothesis that individuals harboring PZMs might be less severely affected than individuals harboring gDNMs in terms of cognitive abilities such as IQ, and if PZMs may be overrepresented in a subset of individuals with higher functioning forms of ASD.
Although the number of probands with IQ data is small, our data suggest that recurrent PZMs are found in individuals with higher IQs than previously reported gDNMs associated with ASD. This opens the intriguing possibility that some individuals with higher functioning forms of ASD might harbor PZMs that might distribute to and affect some but not all regions of the brain, such as the amygdala. This is also consistent with previous observations that high-functioning individuals such as unaffected parents might harbor low levels of parental mosaicism at low AAFs and can transmit these mosaic risk alleles to their affected offspring, which will present as germline mutations in the offspring36. A previous targeted resequencing experiment discovered a mosaic (AAF ∼10%) nonsense mutation in the ASD risk gene ADNP in an unaffected parent, providing further anecdotal evidence for this hypothesis37. It is also plausible that some PZMs could create mosaic clinical phenotypes where presence of the same mutant allele in the germline would be lethal, such as the AKT1 E17K mutation that causes Proteus syndrome38.
One limitation of our work is that we have not analyzed the potential role of post-zygotic copy number variants (CNV) in ASD. Given the strong association of de novo copy number variants with ASD2,39,40, it is possible that there might be mosaic CNV that are involved in ASD, and like the PZMs, mosaic CNV might be under-detected in previous large-scale genomics studies looking at primarily germline copy number variants in ASD. Another area worth pursuing in the future is the role of parental mosaicism in ASD. Such mutations, if present at low AAFs such as the ADNP example, might result in the parents appearing to be clinically unaffected, but can lead to an increased recurrent risk for disease in their offspring. It will be interesting to survey a large number of unaffected parents (or other control individuals) to understand the rates of mosaicism, and the distribution of AAFs in disease-associated genes, that do not result in a clinical presentation.
Multiple lines of evidence suggest that ASD-associated PZMs detectable in blood samples arose during early development, and are enriched in genes expressed in prenatal but not postnatal post-mortem brains. Many of the PZMs associated with ASD discovered in blood have relatively high AAFs and are thus likely to have arisen relatively early in development. Our previous studies have shown that functionally neutral PZMs with >5% AAF are likely to be found in multiple tissues8, suggesting that many of the PZMs discovered in blood are likely to be PZMs in brain tissue as well. Given that 83.3% of the high-confidence PZMs were missed using previous algorithms, it will be important in the future to perform a detailed reanalysis, as well as additional spatio-temporal analyses on PZMs in other neurodevelopmental and psychiatric disorders such as intellectual disability, epilepsy and schizophrenia, to understand the role and contribution of PZMs in these disorders.
Online Methods
Standard protocol approval and patient consent
Research performed on samples and data of human origin was conducted according to protocols approved by the institutional review boards of Boston Children's Hospital and Beth Israel Deaconess Medical Center.
Data processing and annotation
The Autism Sequencing Consortium (ASC) has performed joint calling of the variants in the 5,947 trios from the ASC and the Simons Simplex Collection (SSC) whose exome sequences have been previously published4,5. The variants were called using two versions of the GATK41 (the Unified Genotyper and the Haplotype Caller), and annotated using SnpEff versions 2.0.5 and 3.542. To remove exomes with inheritance errors, as well as potential artifactual mosaic mutations induced by cell passaging, we removed outlier exomes that had more than 2 PZMs or more than 5 de novo mutations from downstream analyses.
PZM detection pipeline applied on the ASC and SSC datasets
We first performed joint-calling of the raw files from the previously published and new exomes, in order to obtain standardized datasets for our analyses. Next, we developed a stringent pipeline to call autosomal de novo point mutations from our jointly-called exomes, i.e. mutations that are strictly present in the probands or siblings but are not found in both parents. We refer to all de novo mutations as Group A, whereas de novo point mutations with AAF equal to or less than 80% of the modal AAF for each cohort are defined as candidate PZMs called “Group B”. Mutations in Group B where the deviation from the modal AAF was statistically significant (binomial P≤1×10-4) formed “Group C”, the group most likely to be PZMs. The AAF was calculated using: number of alternate reads/(total number of reference + alternate reads).
For our initial analyses, we included all variants that passed a set of quality thresholds (genotype quality, GQ ≥ 20 and alternate read depth ≥ 7). All de novo variants that were observed only once in a proband and were not observed in 6,500 control individuals from the Exome Variant Server (http://evs.gs.washington.edu/EVS/) were included in Group A. In addition, to account for population-specific rare variation, we considered only de novo variants that were not observed in unaffected parents and siblings within each study. Given that there might be differences in capture and sequencing approaches across the various cohorts that can result in an over-calling of mosaic mutations, we defined PZMs as variants that deviated from the modal AAF (calculated from all de novo variants in Group A) for each cohort, instead of assuming that the modal AAF is 50%. In addition, to reduce false positives as a result of inaccurate realignment, we filtered away PZMs that were within 20bp of an inherited variant. For the final genes with recurrent non-synonymous PZMs, we lowered the quality thresholds to alternate DP ≥3 in order to screen for additional PZMs that might have been missed, and discovered only an additional non-synonymous PZM in SMARCA4 (I184T with alternate DP = 4).
Resequencing of PZMs
For both the ASC and SSC sequencing projects, DNA derived from mostly blood were used for exome sequencing. We resequenced the PZMs using DNA derived from mostly blood and some lymphoblastoid cells and saliva (from the ASC) or blood and lymphoblastoid cells (from the SSC). For our initial evaluation, we selected 50 de novo mutations where DNA samples were available (5 from Group A, 28 from Group B and 17 from Group C), and resequenced the mutations using subcloning and Sanger sequencing of the colonies (CloneSeq), targeted PCR followed by MiSeq and pyrosequencing (EpigenDx). Subcloning was performed using the standard protocol with the TA cloning kit (Life Technologies). For targeted PCR, we amplified the genomic regions around the mutations, performed PCR purification (Qiagen) and sheared the amplicons to ≈400bp fragments before library preparation and sequencing using MiSeq (paired-end 151bp).
To obtain an estimate of the rate of de novo mutations detected with our approach, we performed Sanger sequencing for 82 of the PZMs discovered (39 from Group B and 43 from Group C), using samples obtained from the trios and additional family members if available, to confirm the presence of the mutations, as well as the absence of the mutations in the family members, i.e. to confirm the de novo status of the mutations. Given that there is a limitation on detecting low AAFs from Sanger sequencing, we selected variants with AAF≥10% for the Sanger experiments, and confirmed 73/82 (89%) as de novo. In particular, 37/39 (94.9%) of the PZMs from Group B were confirmed to arise de novo, and 36/43 (83.7%) of the PZMs from Group C were confirmed to arise de novo. In addition, we performed targeted PCR with MiSeq for 327 de novo mutations where parental DNA was available, and found that 148/176 (84.1%) of the PZMs from Group C were confirmed to arise de novo, 0/18 (0%) of the PZMs from Group B were confirmed to arise de novo, and 131/133 (98.5%) of the gDNMs from Group A were confirmed to arise de novo.
Quantitative PCR for assaying copy number variants
For 36 PZMs in Group C where there are copy number variants in the Database of Genomic Variants (http://dgv.tcag.ca/dgv/app/home) that are within 2kb of the PZMs, we selected pre-designed primers from ThermoFisher that assay the copy number variants. The DNA samples used for these quantitative PCR assays were extracted from whole blood samples from the Simons Simplex Collection, and the quantitative PCR assays were performed by the Biopolymers core facility at Harvard Medical School. The reference assay used was AGO1.
DNA replication timing analyses
We used data that was previously published15, and mapped the genes from the human genome (hg19 assembly) to the regions with the reported replication timing. We defined early replicating genes as genes that fall within regions with replication timing Z≥1 and late replicating genes as genes that fall within regions with replication timing Z≤-1.
Phasing of de novo mutations
We ran the ReadBackedPhasing tool in GATK to phase the de novo mutations using a 100kb window around the mutation of interest. Out of the 4,846 de novo mutations in Group A, we phased 2,102 of these mutations (43.4%). Of the 1,113 mutations in Group B, we phased 464 of these (41.7%), and of the 468 mutations in Group C, we were able to phase 168 of these (35.9%).
In-silico prediction for missense mutations
We used three different tools (PolyPhen217, SIFT18 and CADD19) to obtain in-silico predictions for the missense mutations. We defined “deleterious mutations” as all mutations that were predicted by PolyPhen2 to be “probably damaging”, by SIFT to be “damaging”, and had CADD scores of ≥20. We further defined “benign mutations” as all mutations that were predicted by PolyPhen2 to be “possibly damaging” or “benign”, by SIFT to be “tolerated”, and had CADD scores of <20.
Critical exon analyses
We used whole-genome sequencing data from the 1000 Genomes Project43 to compute the burden of rare missense and loss-of-function mutations for each exon. Furthermore, exon level expression data from RNA sequencing was obtained for 524 brain tissues (prenatal and postnatal postmortem donors) from the BrainSpan project44. To classify critical exons, we computed the 75th percentile of brain expression and mutational burden for each exon as described in Uddin et al20. In short, a critical exon is defined as an exon where expression is high (>75th percentile) and the accumulation of deleterious mutation is low (<75th percentile). For each group (A, B and C) of mutations in the probands and siblings, we first computed the fraction of critical exons with non-synonymous and synonymous mutations for each brain tissue sample. Next, we computed the odds ratio for each tissue sample by normalizing the fraction of critical exons detected with the non-synonymous mutations by the fraction of critical exons detected with the synonymous mutations. Each data point corresponds to a ratio for each expression sample that was inferred from the non-synonymous/synonymous mutation counts in critical exons.
Inherited variant analyses
To obtain a background rate for comparing the non-synonymous post-zygotic mutations detected from the exomes beyond false calls, we obtained all the inherited variants that are ≤1% in the population, and selected all variants with AAFs≤80% of the modal AAF calculated from the de novo mutations. We used these inherited variants that deviated from the expected modal AAF for modeling the background rates of obtaining PZMs in each gene, in order to account for technical biases resulting from amplification, exome capture or sequencing. To evaluate the significance of observing recurrent PZMs in each gene beyond expected false calls, we calculated the hypergeometric test P-value by comparing the observed number of PZMs for each gene with the expected background gene-specific mutation rates (Supplementary Table 13). The genome-wide threshold was calculated as P<0.05/18,782 = 2.7×10-6 as there are 18,782 annotated genes in the data.
Spatial and temporal analyses
To evaluate the distributions of mutations found in genes that are expressed in post-mortem brains (prenatal and postnatal), as well as in specific regions of the brain, we downloaded the RNA sequencing data from the BrainSpan project44 (http://www.brainspan.org). For the spatial analyses, we focused on 16 brain regions (V1C: primary visual cortex; STC: posterior (caudal) superior temporal cortex; IPC: posterior inferior parietal cortex; A1C: primary auditory cortex; S1C: primary somatosensory cortex; M1C: primary motor cortex; DFC: dorsolateral prefrontal cortex; MFC: medial prefrontal cortex; VFC: ventrolateral prefrontal cortex; OFC: orbital frontal cortex; ITC: inferolateral temporal cortex; AMY: amygdaloid complex; CBC: cerebellar cortex; HIP: hippocampus; MD: mediodorsal nucleus of thalamus; and STR: striatum).
FMRP target dataset
To evaluate the enrichment of FMRP targets, we obtained a list of the transcripts published in Darnell JC et al.45 that were previously used to evaluate the de novos in ASD46 and schizophrenia47.
Residual Variation Intolerance Score (RVIS) analyses
We downloaded the RVIS gene scores based on variants reported in the ExAC database with allele frequencies up to 1% (http://genic-intolerance.org/data/RVIS_Unpublished_ExAC_May2015.txt, accessed: October 11th 2016).
Permutations for comparing proband PZMs to sibling PZMs
There are 786 PZMs in Group B found in the probands, resulting in 27/735 genes with recurrent non-synonymous PZMs. Conversely, there are 327 PZMs in Group B found in the unaffected siblings, resulting in 2/322 (0.62%) genes with recurrent non-synonymous PZMs. To evaluate the significance of the excess of recurrent PZMs found in probands compared to recurrent PZMs found in siblings, we randomly sampled 327 PZMs from the 786 PZMs discovered in the probands. 367/100,000 permutations resulted in the proportion of recurrent genes being less than or equal to 2/322.
Mutations in SMARCA4
We compiled a subset of germline and somatic mutations that were reported in cancers48,49, as well as Coffin-Siris syndrome50.
Mutagenesis of SMARCA4 plasmid
We used the human SMARCA4 transcript variant 3 that was cloned into a pCMV6-AC-GFP backbone (Origene cat no. RG219258). The primers used for the mutagenesis were designed using the Agilent QuikChange design tool, and mutagenesis was performed using the standard protocol with the Agilent QuikChange II XL kit. All mutants were confirmed using Sanger sequencing and plasmids were extracted using the endotoxin-free QIAGEN Plasmid Maxi Kit. We attempted mutagenesis for all 3 SMARCA4 mutants (c.326C>T or p.P109L, c.427C>G or p.P143A and c.551T>C or p.I184T), but only 2 of the mutagenesis experiments resulted in colonies (c.427C>G and c.551T>C). We repeated the mutagenesis experiments for the c.326C>T mutant using the Q5 Site-Directed Mutagenesis Kit (New England BioLabs), but did not get any colonies either.
The primers used for the p.P143A (c.427C>G) QuikChange mutagenesis experiment are:
Forward: 5′- gaagacatctgggcccccgaagacggg -3′
Reverse: 5′- cccgtcttcgggggcccagatgtcttc -3′
The primers used for the p.I184T (c.551T>C) QuikChange mutagenesis experiment are:
Forward: 5′- catcttgtaggccatggtctgagctctgagctg -3′
Reverse: 5′- cagctcagagctcagaccatggcctacaagatg -3′
Overexpression of SMARCA4 plasmids in N2A cells
P4 Mouse neuroblastoma (N2A) cells commercially available from ATCC were tested negative for mycoplasma, and were passaged in DMEM with L-Glutamine, 4.5g/L Glucose and Sodium Pyruvate (Thermo Fisher Scientific) with 10% Fetal Bovine Serum (Thermo Fisher Scientific) and 1% Penicillin Streptomycin (Thermo Fisher Scientific). 24μg of wildtype or mutant plasmids were transfected into 90% confluent N2A cells in 10cm tissue culture plates using Lipofectamine 2000 (Life Technologies). The transfections for each plasmid (wildtype and 2 mutants) were performed in triplicates. Selection was performed by adding 1000μg/ml of G418 antibiotic (Life Technologies) 24 hours after transfection to each plate for 10 days, changing fresh antibiotics every 3 days. 3 additional plates of wildtype N2A cells were grown without selection as controls.
RNA extraction and qPCR
The N2A cells were dissociated using 0.05% Trypsin-EDTA (Life Technologies) and washed with PBS (Life Technologies). RNA extraction was performed using the Ambion PureLink RNA Mini Kit (Life Technologies) and cDNA synthesis was performed using the SuperScript III First-Strand kit (Life Technologies). The KAPA SYBR FAST qPCR master mix was added to 1μg of cDNA and 1μl of each 10μM forward and reverse primers for the qPCR experiments.
The primers used for the mouse ACTB qPCR experiment are:
Forward: 5′- GGCTGTATTCCCCTCCATCG -3′
Reverse: 5′- CCAGTTGGTAACAATGCCATGT -3′
The primers used for the mouse GRIN2B qPCR experiment are:
Forward: 5′- CAGCAAAGCTCGTTCCCAAAA -3′
Reverse: 5′- GTCAGTCTCGTTCATGGCTAC -3′
To obtain the log2 expression levels for GRIN2B, we first calculated the ΔCt = CtGRIN2B - CtACTB for all the qPCR results obtained from the mutants, wildtypes and controls, and normalized the log2 expression levels by calculating ΔΔCt = ΔCtcontrol - ΔCt(mutant or wildtype).
Code availability
All analyses were performed using custom Perl and R scripts, which are available on request.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request. The codes and scripts have also been uploaded to https://pgpresearch.med.harvard.edu/mosaic/
BrainSpan Project: http://www.brainspan.org
Exome Variant Server: http://evs.gs.washington.edu/EVS/
RVIS: http://genic-intolerance.org/data/RVIS_Unpublished_ExAC_May2015.txt
Database of Genomic Variants: http://dgv.tcag.ca/dgv/app/home
Statistics
To compare the strand bias among mutations, we calculated the 2-tailed Fisher's Exact Test P-values for the numbers of mutations found on the sense and anti-sense strands in Groups B and C compared to the numbers of mutations in Group A (exact numbers are shown in Supplementary Table 6).
To compare the differences in mutational properties, we calculated the 2-tailed Fisher's Exact Test P-values for the numbers of A>C and T>G mutations in Groups B and C to the numbers of mutations in Group A (exact numbers are shown in Supplementary Table 7).
To compare the strand-specific differences in mutational properties, we calculated the 2-tailed Fisher's Exact Test P-values for the numbers of A>C and T>G mutations found on the sense and anti-sense strands in Groups B and C to the numbers of mutations in Group A (exact numbers are shown in Supplementary Table 8).
To compare the association of PZMs with replication timing, we calculated the 2-tailed Fisher's Exact Test P-values for the numbers of mutations with early or late replication times in Groups B and C compared to the numbers of mutations in Group A (exact numbers are shown in Supplementary Table 9).
To compare the enrichment of mutations on the paternal or maternal haplotypes, we calculated the binomial test P-values for the numbers of mutations in Groups A-C (exact numbers are shown in Supplementary Table 10).
To compare the functional distribution of mutations in probands versus unaffected siblings, we calculated the hypergeometric P-values for the numbers of mutations in Groups A-C for probands and siblings (exact numbers are shown in Supplementary Table 11).
To compare the rates of predicted deleterious missense mutations in probands compared to siblings, we calculated the 1-tailed Fisher's Exact Test P-values for Groups A-C (exact numbers are shown in Supplementary Table 12).
To prioritize the genes with recurrent non-synonymous PZMs found in the probands, we calculated the hypergeometric P-values for each gene (exact numbers are shown in Supplementary Tables 13-14). Similarly, we calculated the hypergeometric P-values for each gene with recurrent non-synonymous PZMs found in the unaffected siblings and the exact numbers are shown in Supplementary Table 15.
No statistical methods were used to pre-determine sample sizes but our sample sizes are similar to those reported in previous publications4,5. Data collection and analysis were not performed blind to the conditions of the experiments.
Supplementary Material
Acknowledgments
We are grateful to all the families who participated in the research, including the Simons Foundation Autism Research Initiative (SFARI) Simplex Collection (SSC), the Autism Sequencing Consortium (ASC) and Autism Speaks. We acknowledge the clinicians and organizations that contributed to samples used in this study, including the ASC and SSC principal investigators, the coordinators and staff at the ASC and SSC sites for the recruitment and comprehensive assessment of simplex families, and the ASC, SFARI and NDAR staff for facilitating access to the datasets. We also thank the 3 anonymous reviewers for their critical suggestions, which have helped to improve the work significantly. This work was supported by a grant from the Simons Foundation (178093, C.A.W.), grants from the National Institutes of Health (NIH) R01MH083565, RC2MH089952, U01MH106883 (C.A.W.), as well as grants R01MH097849, U01MH100233, U01MH100209, U01MH100229, U01MH100239 and U01MH111661-01 to the Autism Sequencing Consortium, grants from the Centre for Applied Genomics, the University of Toronto McLaughlin Centre, Genome Canada and Autism Speaks (S.W.S.), SRPBS and Brain/MINDS grants from AMED (I.K., B.A., N.O.), grants from the Spanish Ministry of Economy and Competitiveness (M.P.), Instituto de Salud Carlos III (M.P.), PI10/02989 (M.P.), CIBERSAM (M.P.) and ERA-NET NEURON (M.P., C.M.F.), Network of European Funding for Neuroscience Research (M.P.), and Fundación María José Jove and The Institute of Health Carlos III-Fondo de Investigaciones Sanitarias grant project PI13/01136 (A.C.). We thank A. Hossain and N. Hatem for their help with sample preparation, F. Zhao and C. Stevens for their help with reprocessing the BAM files, as well as M. Daly, S. McCarroll, G. Genovese and J. Hirschhorn for helpful comments and suggestions. Research reported in this paper was supported by the Office of Research Infrastructure of the National Institutes of Health under award number S10OD018522. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This work was supported in part through the computational resources and staff expertise provided by the Department of Scientific Computing at the Icahn School of Medicine at Mount Sinai. Additional computing support was provided by the Harvard Medical School's Orchestra High-Performance Computing Group, which is partially supported by NIH grant NCRR 1S10RR028832-01. The NHLBI GO Exome Sequencing Project and its ongoing studies produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926) and the Heart GO Sequencing Project (HL-103010). C.A.W. is an Investigator of the Howard Hughes Medical Institute. S.W.S is the GlaxoSmithKline-Canadian Institutes of Heath Research Chair in Genome Sciences at the Hospital for Sick Children and University of Toronto. M.U. is a Banting postdoctoral fellow. A.M.D. is supported by the NIGMS (T32GM007753) and NRSA (5T32 GM007226-39). S.D.R. is a Seaver fellow, supported by the Seaver Foundation
Footnotes
Accession codes: The raw WES data has been in part deposited into NDAR (#2337), and we will ensure that the new data will be deposited upon publication.
Competing Financial Interests: Drs. Scherer and Uddin and the Hospital for Sick Children hold intellectual property used in this analysis, which is also licensed by Lineagen, Inc. Dr Parellada has received educational honoraria from Otsuka, research grants from Fundación Alicia Koplowitz and Mutua Madrileña and travel grants from Otsuka and Janssen. Dr Arango has been a consultant to or has received honoraria or grants from Abbot, Amgen, AstraZeneca, Bristol-Myers-Squibb, Caja Navarra, CIBERSAM, Fundación Alicia Koplowitz, Instituto de Salud Carlos III, Janssen Cilag, Lundbeck, Merck, Ministerio de Ciencia e Innovación, Ministerio de Sanidad, Ministerio de Economía y Competitividad, Mutua Madrileña, Otsuka, Pfizer, Roche, Servier, Shire, Takeda, and Schering-Plough.
References
- 1.Gaugler T, et al. Most genetic risk for autism resides with common variation. Nature genetics. 2014;46:881–885. doi: 10.1038/ng.3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanders SJ, et al. Insights into Autism Spectrum Disorder Genomic Architecture and Biology from 71 Risk Loci. Neuron. 2015;87:1215–1233. doi: 10.1016/j.neuron.2015.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pinto D, et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. American journal of human genetics. 2014;94:677–694. doi: 10.1016/j.ajhg.2014.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.De Rubeis S, et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature. 2014;515:209–215. doi: 10.1038/nature13772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iossifov I, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature. 2014;515:216–221. doi: 10.1038/nature13908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yu TW, et al. Using whole-exome sequencing to identify inherited causes of autism. Neuron. 2013;77:259–273. doi: 10.1016/j.neuron.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lim ET, et al. Rare complete knockouts in humans: population distribution and significant role in autism spectrum disorders. Neuron. 2013;77:235–242. doi: 10.1016/j.neuron.2012.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jamuar SS, et al. Somatic mutations in cerebral cortical malformations. The New England journal of medicine. 2014;371:733–743. doi: 10.1056/NEJMoa1314432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poduri A, Evrony GD, Cai X, Walsh CA. Somatic mutation, genomic variation, and neurological disease. Science. 2013;341:1237758. doi: 10.1126/science.1237758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Genovese G, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. The New England journal of medicine. 2014;371:2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pleasance ED, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Polak P, et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature. 2015;518:360–364. doi: 10.1038/nature14221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prendergast JG, Semple CA. Widespread signatures of recent selection linked to nucleosome positioning in the human lineage. Genome research. 2011;21:1777–1787. doi: 10.1101/gr.122275.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cotney J, et al. The autism-associated chromatin modifier CHD8 regulates other autism risk genes during human neurodevelopment. Nature communications. 2015;6:6404. doi: 10.1038/ncomms7404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koren A, et al. Differential relationship of DNA replication timing to different forms of human mutation and variation. American journal of human genetics. 2012;91:1033–1040. doi: 10.1016/j.ajhg.2012.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Woo YH, Li WH. DNA replication timing and selection shape the landscape of nucleotide variation in cancer genomes. Nature communications. 2012;3:1004. doi: 10.1038/ncomms1982. [DOI] [PubMed] [Google Scholar]
- 17.Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Current protocols in human genetics / editorial board, Jonathan L. Haines … [et al.] 2013;Chapter 7 doi: 10.1002/0471142905.hg0720s76. Unit7 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nature protocols. 2009;4:1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 19.Kircher M, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nature genetics. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Uddin M, et al. Brain-expressed exons under purifying selection are enriched for de novo mutations in autism spectrum disorder. Nature genetics. 2014;46:742–747. doi: 10.1038/ng.2980. [DOI] [PubMed] [Google Scholar]
- 21.Carvill GL, et al. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nature genetics. 2013;45:825–830. doi: 10.1038/ng.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Petrovski S, Wang Q, Heinzen EL, Allen AS, Goldstein DB. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS genetics. 2013;9:e1003709. doi: 10.1371/journal.pgen.1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jelinic P, et al. Recurrent SMARCA4 mutations in small cell carcinoma of the ovary. Nature genetics. 2014;46:424–426. doi: 10.1038/ng.2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kosho T, Okamoto N Coffin-Siris Syndrome International, C. Genotype-phenotype correlation of Coffin-Siris syndrome caused by mutations in SMARCB1, SMARCA4, SMARCE1, and ARID1A. American journal of medical genetics Part C, Seminars in medical genetics. 2014;166C:262–275. doi: 10.1002/ajmg.c.31407. [DOI] [PubMed] [Google Scholar]
- 25.Forbes SA, et al. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic acids research. 2015;43:D805–811. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pugh TJ, et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488:106–110. doi: 10.1038/nature11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiu Z, Ghosh A. A calcium-dependent switch in a CREST-BRG1 complex regulates activity-dependent gene expression. Neuron. 2008;60:775–787. doi: 10.1016/j.neuron.2008.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Acuna-Hidalgo R, et al. Post-zygotic Point Mutations Are an Underrecognized Source of De Novo Genomic Variation. American journal of human genetics. 2015;97:67–74. doi: 10.1016/j.ajhg.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Freed D, Pevsner J. The Contribution of Mosaic Variants to Autism Spectrum Disorder. PLoS genetics. 2016;12:e1006245. doi: 10.1371/journal.pgen.1006245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Morris JS, Ohman A, Dolan RJ. Conscious and unconscious emotional learning in the human amygdala. Nature. 1998;393:467–470. doi: 10.1038/30976. [DOI] [PubMed] [Google Scholar]
- 31.Adolphs R, Tranel D, Damasio AR. The human amygdala in social judgment. Nature. 1998;393:470–474. doi: 10.1038/30982. [DOI] [PubMed] [Google Scholar]
- 32.Baron-Cohen S, et al. The amygdala theory of autism. Neuroscience and biobehavioral reviews. 2000;24:355–364. doi: 10.1016/s0149-7634(00)00011-7. [DOI] [PubMed] [Google Scholar]
- 33.Rutishauser U, et al. Single-neuron correlates of atypical face processing in autism. Neuron. 2013;80:887–899. doi: 10.1016/j.neuron.2013.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu X, et al. Modular genetic control of sexually dimorphic behaviors. Cell. 2012;148:596–607. doi: 10.1016/j.cell.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gkogkas CG, et al. Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature. 2013;493:371–377. doi: 10.1038/nature11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Campbell IM, et al. Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. American journal of human genetics. 2014;95:173–182. doi: 10.1016/j.ajhg.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Roak BJ, et al. Recurrent de novo mutations implicate novel genes underlying simplex autism risk. Nature communications. 2014;5:5595. doi: 10.1038/ncomms6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lindhurst MJ, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. The New England journal of medicine. 2011;365:611–619. doi: 10.1056/NEJMoa1104017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weiss LA, et al. Association between microdeletion and microduplication at 16p11.2 and autism. The New England journal of medicine. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- 40.Sebat J, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DePristo MA, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature genetics. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cingolani P, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Genomes Project C, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kang HJ, et al. Spatio-temporal transcriptome of the human brain. Nature. 2011;478:483–489. doi: 10.1038/nature10523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Darnell JC, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iossifov I, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012;74:285–299. doi: 10.1016/j.neuron.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fromer M, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014;506:179–184. doi: 10.1038/nature12929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ramos P, et al. Small cell carcinoma of the ovary, hypercalcemic type, displays frequent inactivating germline and somatic mutations in SMARCA4. Nature genetics. 2014;46:427–429. doi: 10.1038/ng.2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Le Loarer F, et al. SMARCA4 inactivation defines a group of undifferentiated thoracic malignancies transcriptionally related to BAF-deficient sarcomas. Nature genetics. 2015;47:1200–1205. doi: 10.1038/ng.3399. [DOI] [PubMed] [Google Scholar]
- 50.Tsurusaki Y, et al. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nature genetics. 2012;44:376–378. doi: 10.1038/ng.2219. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request. The codes and scripts have also been uploaded to https://pgpresearch.med.harvard.edu/mosaic/
BrainSpan Project: http://www.brainspan.org
Exome Variant Server: http://evs.gs.washington.edu/EVS/
RVIS: http://genic-intolerance.org/data/RVIS_Unpublished_ExAC_May2015.txt
Database of Genomic Variants: http://dgv.tcag.ca/dgv/app/home