

SUMMARY
Emerging evidence suggests that microbes resident in the human intestine represent a key environmental factor contributing to obesity-associated disorders. Here we demonstrate that the gut microbiota-initiated trimethylamine-N-oxide (TMAO)-generating pathway is linked to obesity and energy metabolism. In multiple clinical cohorts, systemic levels of TMAO were observed to strongly associate with type 2 diabetes. In addition, circulating TMAO levels were associated with obesity traits in the different inbred strains represented in the Hybrid Mouse Diversity Panel. Further, antisense oligonucleotide-mediated knockdown or genetic deletion of the TMAO-producing enzyme, flavin-containing monooxygenase 3 (FMO3), conferred protection against obesity in mice. Complimentary mouse and human studies indicate a negative regulatory role for FMO3 in the beiging of white adipose tissue. Collectively, our studies reveal a link between the TMAO-producing enzyme FMO3 and obesity and the beiging of white adipose tissue.
Keywords: microbiota, nutrition, obesity, diabetes, adipose
Graphical abstract
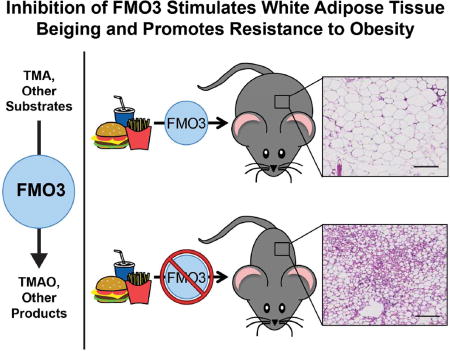
INTRODUCTION
There is strong evidence that microbes resident in the human intestine represent a key environmental factor contributing to obesity and associated insulin resistance (Bäckhed et al., 2004; Ley et al., 2005; Turnbaugh and Gordon, 2009; Cox et al., 2014). However, molecular mechanisms by which gut microbiota promote obesity and insulin resistance in humans are incompletely understood. Recently, several independent groups have identified the gut microbiota-initiated TMA/FMO3/TMAO pathway as a potential modulator of cardiometabolic phenotypes in the host (Wang et al., 2011; Warrier et al., 2015; Miao et al., 2015; Shih et al., 2015), although our mechanistic understanding of this meta-organismal pathway is still incomplete. The TMA/FMO3/TMAO pathway is a microbe-to-host endocrine axis by which gut microbial metabolism of nutrients common in Western diets (phosphatidylcholine, choline, and L-carnitine) results in the production of the metabolite trimethylamine (TMA), which is exclusively generated by certain communities of gut microbiota (Wang et al., 2011; Koeth et al., 2013; Gregory et al., 2015; Romano et al., 2015). Then, the host hepatic enzyme flavin-containing monooxygenase 3 (FMO3) further metabolizes gut microbe-derived TMA to produce trimethylamine-N-oxide (TMAO) (Wang et al., 2011; Bennett et al., 2013). Importantly, the end product of this meta-organismal nutrient metabolism pathway, TMAO, is both a prognostic biomarker and is mechanistically linked to cardiovascular disease (CVD) pathogenesis in humans (Wang et al., 2011; Koeth et al., 2013; Tang et al., 2013; Wang et al., 2014; Koeth et al., 2014; Tang et al., 2014; Suzuki et al., 2016; Missailidis et al., 2016; Mafune et al., 2016; Trøseid et al., 2015; Wang et al., 2015; Zhu et al., 2016). Given the strong link between gut microbiota and both obesity and obesity-related disease in humans, and the links between the TMAO pathway and cardiometabolic diseases, we hypothesized that the TMAO pathway may be mechanistically linked to the pathogenesis of obesity. Here we show that both antisense oligonucleotide-mediated knockdown and genetic deletion of the TMAO-producing enzyme FMO3 protects mice against high fat diet-induced obesity, in part by stimulating the beiging of white adipose tissue, which may reduce the adverse effects of increased adiposity and improve overall metabolic health (Bartelt and Heeren, 2014). Collectively, our studies have uncover a link between the gut microbe-driven TMA/FMO3/TMAO pathway and adipose tissue function.
RESULTS
Elevated Systemic Levels of TMAO are Associated with Type 2 Diabetes in Humans
To first establish clinical relevance, we investigated the relationship of fasting plasma levels of choline or TMAO with type 2 diabetes mellitus (T2DM) risk in two independent cohorts of s- subjects undergoing elective cardiac risk factor evaluation and recommendations in our preventative cardiology clinic (n=187) or evaluation for suspected non-alcoholic fatty liver disease in our hepatology clinic (n=248). Patient demographics, laboratory values, and clinical characteristics for both cohorts (n=435 combined) are provided in Tables S1–S3. Plasma concentrations of TMAO were significantly higher in subjects with T2DM in each of the individual cohorts, as well as when the cohorts were combined (Figure 1A). Fasting choline levels were significantly higher only in T2DM subjects from the hepatology cohort (Figure 1B). Similarly, we observed a dose-dependent association between higher TMAO concentrations and the presence of T2DM (Figure 1C), while the association between choline and T2DM was seen only in the hepatology cohort (Figure 1D). After adjustments for multiple comorbidities, prevalent CVD and CVD risk factors, medications, and renal function, TMAO remained a strong predictor of T2DM risk in both cohorts analyzed alone, as well as when the cohorts were combined (Figure S1). Collectively, these data suggest that circulating levels of the meta-organismal metabolite TMAO are closely correlated with T2DM risk in humans.
Figure 1. Elevated Circulating Levels of TMAO are Associated with Type 2 Diabetes Mellitus in Humans.

We recruited two separate cohorts of stable subjects in preventative cardiology (n=187) or hepatology clinics (n=248) to evaluate associations between fasting circulating choline or trimethylamine-N-oxide (TMAO) levels with prevalent type 2 diabetes (T2DM). The total number of subjects recruited in both studies was n=435. Patient demographics, laboratory values, and clinical characteristics are provided in Tables S1–S3 and Figure S1.
(A) Relationship of fasting plasma TMAO concentrations and prevalent T2DM. Boxes represent the 25th, 50th and 75th percentiles of plasma TMAO concentration, and whiskers represent the 10th and 90th percentiles.
(B) Relationship of fasting plasma choline concentrations and prevalent T2DM. Boxes represent the 25th, 50th and 75th percentiles of plasma choline concentration, and whiskers represent the 10th and 90th percentiles.
(C) Forest plots of the odds ratio of prevalent T2DM and quartiles of TMAO; bars represent 95% confidence intervals.
(D) Forest plots of the odds ratio of prevalent T2DM and quartile of choline; bars represent 95% confidence intervals.
Plasma TMAO Levels in Mice and FMO3 mRNA Expression in Men Demonstrate Positive Correlations with Obesity
First, using a systems genetics approach in mice, we examined various obesity-related traits and circulating TMAO levels in mice from the Hybrid Mouse Diversity Panel (HMDP) fed an obesogenic high fat and high sucrose diet (Parks et al., 2013). Across the different inbred strains represented in the HMDP, circulating levels of TMAO were positively associated with body weight, fat mass, mesenteric adiposity, and subcutaneous adiposity (Figure 2A–D). Given the observed associations between TMAO and obesity across the diverse inbred strains of mice, we next set out to determine whether expression of FMO3, which encodes the TMAO-producing enzyme, was differentially expressed in the adipose tissue of overweight or obese humans. To do this we first examined a random sampling (n=770) of a large population-based study of Finnish men known as the METSIM study (Stancakova et al., 2009). This study performed dense phenotypic characterization of subjects for characteristics related to adiposity and insulin sensitivity, including adipose biopsies and microarray expression analysis (Stancakova et al., 2009; Civelek et al., 2017). When we examined the correlation between expression levels of all members of the flavin-containing monooxygenase (FMO) family in adipose tissue with metabolic traits in this population, we found that FMO3 was positively correlated with body mass index (BMI) and waist-to-hip ratio, and negatively correlated with the Matsuda Index (Matsuda and DeFronzo, 1999), which is a measure of insulin sensitivity (Figure 2E). Interestingly, FMO3 mRNA expression levels in human adipose tissue were significantly negatively correlated with several genes that represent selective markers of beige or brown adipocytes that have recently been reported (Wu et al., 2012; Ussar S et al., 2014) (Figure 2E). These data suggest that FMO3 expression is negatively associated with beiging signatures in human subcutaneous white adipose tissue.
Figure 2. Plasma TMAO Levels in Mice and FMO3 mRNA Expression in Men Demonstrate Positive Correlations with Obesity.

(Panels A–D) Correlation of plasma trimethylamine-N-oxide (TMAO) levels with obesity-related traits in 180 male mice from 92 inbred strains within the hybrid mouse diversity panel (HMDP) after 8 week feeding of a high fat and high sucrose diet. Correlation coefficient (r) and p value (p) are indicated for each obesity trait.
(A) Correlation between plasma TMAO and body weight
(B) Correlation between plasma TMAO and fat mass
(C) Correlation between plasma TMAO and mesenteric fat weight
(D) Correlation between plasma TMAO and subcutaneous fat weight
(E) Correlations between human white adipose tissue flavin monooxygenase 3 (FMO3) mRNA expression and metabolic traits or brown/beige adipocyte marker gene expression (n=770). The gene name and probeset ID is provided for each of the brown/beige adipocyte marker genes.
Given that the METSIM study only includes Finnish men, we set out to validate microarray expression data in several distinct cohorts spanning both men and women of European American and African American ethnicity. Importantly, these validation cohorts were also chosen for their gender, ethnic and racial diversity, and because each has been extensively characterized for obesity and cardiometabolic phenotypes (Das et al., 2015; Sharma et al., 2016). The first cohort included n=99 non-Hispanic Caucasian Americans, comprised of 42 males and 57 females (Das et al., 2015). The second cohort included n=260 African Americans, comprised of 139 males and 121 females (Sharma et al., 2016). In agreement with findings from the METSIM study (Figure 2E), we found that FMO3 was positively correlated with BMI and adiposity, and negatively correlated with insulin sensitivity in both European American and African American men and women (Figure S2). Also, the primary transcript variant of FMO3 was negatively correlated with the beige/brown marker genes uncoupling protein 1 (UCP1) and PR domain-containing 16 (PRDM16) (Figure S2).
In separate studies we sought to examine whether similar associations were observed between FMO3 expression in human liver and metabolic traits using liver biopsies from obese patients undergoing bariatric surgery and normal weight controls. In contrast to our findings in human adipose tissue (Figure 2E and Figure S2), we did not find significant correlations between liver FMO3 protein levels and metabolic traits in this cohort (Figure S3). Of note, the protein expression of FMO3 in human liver does not significantly differ between males and females in the cohort under study (n=15 males, n=35 females; p=0.79) (Figure S3).
Knockdown of FMO3 Protects Mice from High Fat Diet-Induced Obesity by Stimulating the Beiging of White Adipose Tissue
To further examine the potential of the meta-organismal TMAO pathway to impact obesity, we utilized a second-generation antisense oligonucleotide (ASO) to inhibit the expression of Fmo3 in mice challenged with a high fat diet (HFD) (Figure 3). Fmo3 ASO treatment resulted in >90% knockdown of Fmo3 mRNA levels in mouse liver in both chow-fed and HFD-fed mice (Figure 3A). In line with this, FMO3 knockdown caused accumulation of the FMO3 substrate, TMA, in mice on both diets, while reductions in its product, TMAO, were seen only in chow-fed mice (Figure 3B, 3C). Indeed, the overall lower TMAO levels in the HFD groups may be due to less overall choline substrate in the HFD (Table S4). Despite documenting comparable food intake on HFD (Figure S4D), FMO3 knockdown resulted in significantly decreased body weight gain (Figure 3D). The attenuation in body weight gain observed appeared to be largely attributed to decreased white adipose tissue weight (Figure 3E), with magnetic resonance imaging (Figure 3F) demonstrating that both peritoneal (Figure 3G) and subcutaneous (Figure 3H) adipose tissue mass were markedly reduced with Fmo3 ASO-treatment. Collectively, Fmo3 ASO treatment altered body composition, reducing the overall percentage of fat mass (Figure 3I), while increasing the percent of lean mass (Figure 3J) in the HFD-fed cohort. In additional studies, gonadal white adipose tissue Fmo3 mRNA levels were observed to be approximately 1000-fold lower than hepatic Fmo3 mRNA levels (Figure S4A). Importantly, Fmo3 ASO treatment protected against HFD-induced obesity in both female (Figure 3) and male (Figure S4B) mice, despite sexual dimorphism in total FMO activity in liver and gonadal white adipose tissue (Figure S4C).
Figure 3. FMO3 Knockdown Protects Mice from High Fat Diet-Induced Obesity by Stimulating the Beiging of White Adipose Tissue.

At 6–8 weeks of age, female C57BL/6 mice were treated with either a non-targeting control ASO or or Fmo3 ASO in conjunction with either standard rodent chow or high fat diet (HFD) feeding for the indicated times.
(A) Hepatic Fmo3 mRNA expression was quantified by qPCR after 10 weeks
(B) Plasma levels of TMA after 6 weeks
(C) Plasma levels of TMAO after 6 weeks
(D) Body weight changes over 12 weeks
(E) Gonadal white adipose tissue (WAT) weight at necropsy
Panels (F–J) MRI images and subsequent quantification of adiposity in control and Fmo3 ASO-treated mice maintained on diets for 14 weeks.
(F) MRI images of control and Fmo3 ASO-treated mice
(G) Peritoneal adipose tissue mass
(H) Subcutaneous adipose tissue mass
(I) % Fat mass
(J) % Lean mass
All data represent the mean ± S.E.M. for n=5–10 mice per group; *, p≤0.05, **, p≤0.01, ***, p≤0.001, ****, p≤0.0001 vs. control ASO-treated mice fed the same diet. +, p≤0.05, ++, p≤0.01, +++, p≤0.001, ++++, p≤0.0001 vs. chow-fed mice treated with the same ASO.
Given the fact that FMO3 expression was negatively correlated with brown and beige adipocyte gene markers in human adipose tissue (Figure 2E), we next examined the effect of FMO3 knockdown on thermogenic reprogramming in adipose tissue (Figure 4). An initial clue that FMO3 knockdown may alter thermogenic programs was that gonadal adipose depots had morphologic (lipid droplet multi-locularity) and gene expression profiles that are consistent with the appearance of beige adipocytes (Wu et al., 2012) in a classically white adipose tissue depot (Figure 4A, 4B). In the gonadal white adipose tissue depots, FMO3 knockdown elicited a 5-fold upregulation of the β1 adrenergic receptor (Adrb1), a 150-fold increase of uncoupling protein 1 (Ucp1), and a 3-fold increase in transmembrane protein 26 (Tmem26) (Figure 4B). In parallel, plasma membrane-associated β1-AR abundance was increased 4.7-fold in Fmo3 ASO-treated mice (Figure 4C), which was associated with a 3-fold increase in cyclic AMP levels in gonadal adipose tissue (Figure 4D). To examine the role of FMO3 in cold-induced transcriptional reprogramming, we housed mice in either room temperature (22°C) or cold (4°C) conditions. Interestingly, the normal cold-induced upregulation of the thermogenic transcriptional regulator peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Ppargc1a) (Puigserver et al., 1998) was much higher in mice treated with Fmo3 ASO in multiple adipose depots (Figure 4E).
Figure 4. FMO3 Knockdown Stimulates the Beiging of White Adipose Tissue.

Panels (A–E) At 6–8 weeks of age, female C57BL/6 mice were treated with either a non-targeting control ASO or Fmo3 ASO in conjunction with high fat diet feeding (HFD) for 8–12 weeks.
(A) Microscopic examination of hematoxylin and eosin stained gonadal white adipose tissue; scale bar equals 200µm.
(B) Gonadal white adipose tissue (WAT) mRNA expression of β1-adrenergic receptor (Adrb1), uncoupling protein 1 (Ucp1), and transmembrane protein 26 (Tmem26) quantified by qPCR.
(C) Surface density of β1-adrenergic receptor (β1-AR) in gonadal white adipose tissue (WAT) quantified by radio-ligand binding assay.
(D) Cyclic AMP (cAMP) levels in gonadal white adipose tissue (WAT).
(E) Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Ppargc1a) mRNA expression was quantified by qPCR in gonadal (gWAT), inguinal (iWAT) and brown (BAT) adipose tissue.
Panels (F–H) Mice were housed in metabolic cages for indirect calorimetry measurements. Gray background denotes dark cycle.
(F) Oxygen consumption (VO2)
(G) Heat production
(H) Respiratory exchange ratio (RER)
Panels (I–K) At 6–8 weeks of age, female C57BL/6 mice were treated with either a non-targeting control ASO or Fmo3 ASO in conjunction with feeding of HFD or HFD supplemented with 0.02% w/w TMAO for 12 weeks. See also Figure S4.
(I) Body weight changes over 12 weeks
(J) Gonadal white adipose tissue (WAT) expression of uncoupling protein 1 (Ucp1)
(K) Gonadal white adipose tissue (WAT) expression of peroxisome proliferator-activated receptor gamma coactivator 1-alpha (Ppargc1a), PR domain-containing 16 (Prdm16), T-box transcription factor (Tbx1), and transmembrane protein 26 (Tmem26)
All data represent the mean ± S.E.M. for n=5–10 mice per group; *, p≤0.05, **, p≤0.01, ***, p≤0.001, ****, p≤0.0001 vs. control ASO-treated mice fed the same diet. +++, p≤0.001 vs. chow-fed mice treated with the same ASO. &&&, p≤0.001 vs. Fmo3 ASO-treated mice fed HFD.
We next investigated the physiological role of FMO3 in cold-induced thermogenic reprogramming by performing indirect calorimetry in HFD-fed Fmo3 ASO-treated mice housed at thermoneutrality (30°C), room temperature (22°C), or under cold stress (4°C). FMO3 knockdown increased oxygen consumption (VO2) and heat production under all temperature conditions (Figure 4F, 4G). Interestingly, the normal cold-induced increase in VO2 and heat seen in control mice was significantly enhanced in the Fmo3 ASO-treated mice housed at 4°C (Figure 4F, 4G). It is well documented that the preferred macronutrient fuel source (carbohydrates vs. fats) differs under fasted and fed states as well as during cold stress, and dysregulation of this metabolic flexibility is thought to be a key component of the metabolic syndrome (Muoio, 2014). HFD-fed Fmo3 ASO-treated mice have a marked improvement in metabolic flexibility in response to feeding (during the dark cycle) and cold (Figure 4H). In fact, Fmo3 ASO-treated mice have a slightly larger increase in glucose oxidation (indicated by increased respiratory exchange ratio) during the dark cycle at thermoneutrality, and this enhanced metabolic flexibility becomes much more striking at 22°C and 4°C (Figure 4H). These data suggest that FMO3 inhibition facilitates feeding-induced fuel switching (from fats to carbohydrates) especially under conditions of cold stress. Collectively, these data suggest that knockdown of FMO3 increases energy expenditure and enhances metabolic flexibility under conditions of cold stress.
Given that TMAO levels were linked to adiposity across multiple strains of mice (Figure 2), we hypothesized that TMAO itself may be directly involved in regulating adiposity. Therefore, we set out to determine the involvement of TMAO in the adipose tissue phenotype seen with FMO3 knockdown. To achieve this, we provided TMAO as a dietary supplement as previously described (Warrier et al., 2015) to control and Fmo3 ASO-treated mice maintained on HFD. Dietary provision of TMAO effectively raised levels of TMAO in the circulation, liver, and white adipose tissue in control and Fmo3 ASO-treated mice (Figure S4E, S4F). Interestingly, despite commensurate food consumption (Figure S4D), Fmo3 ASO-treated mice had significantly lower plasma TMAO levels compared to control ASO-treated mice following dietary supplementation (Figure S4E). Importantly, dietary provision of TMAO did not reverse the ability of Fmo3 ASO treatment to attenuate HFD-induced body weight gain (Figure 4I). Likewise, dietary TMAO provision did not alter the ability of Fmo3 ASO treatment to elevate Ucp1 expression in gonadal white adipose tissue (Figure 4J) However, Fmo3 ASO-driven increases in gonadal white adipose tissue genes involved in energy metabolism and the development of brown adipose tissue including Ppargc1a, Prdm16, and T-box transcription factor, Tbx1, were reversed by dietary TMAO provision (Figure 4K). Collectively, these data demonstrate that provision of dietary TMAO can reverse a portion, but not all, of the transcriptional reorganization observed in white adipose tissue driven by FMO3 knockdown, and suggest that future studies should investigate the role of additional FMO3 products or substrates in energy metabolism.
Genetic Deletion of the TMAO-Producing Enzyme FMO3 Protects Mice from Obesity
To further examine the role of FMO3 in obesity, we examined global FMO3 knockout (Fmo3−/−) mice generated using CRISPR-Cas9-mediated gene editing (Shih, et al., in preparation) (Figure 5, Table S5). Hepatic FMO3 protein was undetectable by Western blot in Fmo3−/− mice (Figure 5A). In initial studies, we maintained Fmo3−/− mice on a C57BL/6 background and fed a choline-supplemented chow-based diet. Under these non-obesogenic conditions, Fmo3−/− mice accumulate plasma TMA and have diminished TMAO as predicted, and while there were no differences in food intake or body weight, they do exhibit significantly reduced adiposity compared to Fmo3+/+ mice (Figure 5B). To examine effects of FMO3 knockout on adiposity under obesogenic conditions we crossed Fmo3−/− mice to the low-density lipoprotein knockout (Ldlr−/−) background and maintained mice on a Western diet (Figure 5C–5H). Western diet-fed Ldlr−/−;Fmo3−/− mice had markedly reduced hepatic FMO activity (Figure 5C) and circulating TMAO levels (Figure 5D), when compared to Ldlr−/−;Fmo3+/+ control mice. In agreement with our studies in HFD-fed Fmo3 ASO-treated mice (Figures 3 and 4), Western diet-fed Ldlr−/−;Fmo3−/− mice were protected against diet-induced obesity (Figure 5E–G), and had increased expression of brown/beige adipocyte marker genes in the subcutaneous fat depots compared to Ldlr−/− ;Fmo3+/+ control mice (Figure 5H). Collectively, these data provide genetic evidence that FMO3 is a negative regulator of beiging programs in white adipose tissue.
Figure 5. Genetic Deletion of FMO3 Protects Mice From Diet-Induced Obesity.

(A) Top panel: CRISPR-Cas9 strategy for generating Fmo3−/− mice. The sequence of exon 2 of the murine Fmo3 coding sequence is shown. The target sequence (underlined) used for construction of the guide RNA is shown with arrows indicating predicted cleavage sites by Cas9. Bottom panel: immunoblotting analysis of FMO3 protein levels in the livers of wild-type (WT) and Fmo3−/− (KO) mice.
(B) Decreased adiposity in Fmo3−/− mice. Fmo3+/+ (n=9), Fmo3+/− (n=6), and Fmo3−/− (n=11) mice were fed a 1.3% choline chloride (w/w) diet for 12 weeks before tissue collection. Plasma TMAO and TMA levels (top left), food intake (top right), body weight (bottom left) and 4 fat pads/body weight (%; bottom right) are shown. The four fat pads included in the 4 fat pads/body weight measurement were gonadal, mesentery, perirenal, and subcutaneous. *: p≤0.05 between Fmo3+/+ and Fmo3−/− groups. &: p≤0.05 between Fmo3+/− and Fmo3−/− genotype groups.
Panels (C–H) Ldlr−/−; Fmo3−/− mice are more resistant to obesity than Ldlr−/− littermates when fed a Western diet for 12 weeks.
(C) Liver FMO activity
(D) Plasma TMAO levels
(E) Body weight changes over 12 weeks
(F) Fat mass/body weight (%)
(G) 4 fat pads weight/body weight (%); the 4 fat pads measured were gonadal, mesentery, perirenal, and subcutaneous
(H) Gene expression analysis of subcutaneous fat pads of Ldlr−/− (n=17) and Ldlr−/−; Fmo3−/− (n=9) mice. Cell death-inducing DFFA-like effector A (Cidea), Cytochrome C oxidase subunit 8b (Cox8b), Elongation of very long chain fatty acids protein 3 (Elovl3), Uncoupling protein 1 (Ucp1), Diglyceride acyltransferase 1 (Dgat1), Leptin (Lep), Stearoyl CoA desaturase-1 (Scd1), Transducin-like enhancer of split 3 (Tle3)*, p≤0.05, **, p≤0.01, and ***, p≤0.0001 between the two genotype groups.
DISCUSSION
Obesity, insulin resistance, and atherosclerotic cardiovascular disease (CVD) are closely linked diseases that can be heavily impacted by the quantity and quality of dietary inputs. In a time where genetic and genomic approaches dominate clinical investigation, we are still constantly reminded that environmental factors such as diet can play a major role in disease pathogenesis. The meta-organismal TMAO pathway was initially discovered using untargeted metabolomics approaches to identify small molecules in plasma associated with CVD risk (Wang et al., 2011). In multiple follow-up studies, it has been shown that feeding atherosclerosis-prone mice diets enriched in either distinct nutrient precursors to TMA synthesis or TMAO itself enhances atherosclerotic CVD and thrombosis potential by altering host cholesterol metabolism and platelet hyper-reactivity (Koeth et al., 2013; Koeth et al., 2014; Zhu et al., 2016). In parallel, ASO-mediated inhibition of the TMAO producing enzyme FMO3 protects mice from atherosclerosis (Shih et al., 2015; Miao et al., 2015), possibly in part by stimulating an intestinal pathway of reverse cholesterol transport called transintestinal cholesterol excretion (TICE) and altering tissue sterol metabolism (Warrier et al., 2015; Shih et al. 2015). More recently, inhibition of the microbial enzymes responsible for generating TMA from choline, thereby reducing TMAO levels, was shown to similarly inhibit atherosclerosis in mice (Wang et al., 2015). Therefore, while significant mechanistic and clinical data indicates that the meta-organismal TMA/FMO3/TMAO pathway is closely linked to the pathogenesis of atherosclerosis in mice and humans (Brown and Hazen, 2015), here we provide evidence that the pathway can also impact the beiging of white adipose tissue. The key findings of the current study are: (1) circulating levels of the gut microbe-derived metabolite TMAO are associated with enhanced risk of T2DM in humans; (2) TMAO levels are associated with adiposity traits across mouse strains within the Hybrid Mouse Diversity Panel; (3) adipose tissue expression of FMO3 is positively associated with obesity in humans; (4) FMO3 mRNA expression is negatively associated with brown/beige adipocyte gene expression in white adipose tissue in humans; (5) FMO3 knockdown or genetic deletion protects mice against high fat diet-induced obesity; and (6) FMO3 knockdown or genetic deletion is associated with the beiging of white adipose tissue in mice.
Although the vast majority of studies have focused on the TMA/FMO3/TMAO pathway in the context of CVD, several recent studies have linked this meta-organismal pathway to diabetes. In agreement with our findings here, several independent groups have found an association between circulating TMAO levels and both T2DM and the heightened adverse CVD outcomes in diabetics (Lever et al., 2014; Dambrova et al., 2016; Tang et al., 2017). Using a metabolomics platform, Miao and colleagues recently showed that mice with selective hepatic insulin resistance (liver insulin receptor knockout mice; LIRKO) have elevated levels of circulating TMAO, and a profound upregulation of the TMAO-producing enzyme FMO3 in the liver (Miao et al., 2015). This study also demonstrated that the hepatic expression of FMO3 is suppressed by the postprandial hormone insulin, yet is reciprocally stimulated by the fasting hormone glucagon (Miao et al., 2015). In addition to these findings in LIRKO mice, dietary supplementation with the FMO3 product TMAO is reported to exacerbate glucose intolerance in high fat diet-fed mice (Gao et al., 2014). Collectively, a growing body of evidence suggests that the gut microbial TMAO pathway may be an attractive drug target for subjects with T2DM. Of note, while TMAO has been causally-linked to atherosclerosis and thrombosis (Wang et al., 2011; Koeth et al., 2013; Zhu et al., 2016), and associated with obesity here, there is also literature that shows diets high in the TMAO source nutrient choline are associated with beneficial effects on fetal development and cognitive function in adults (Jiang et al., 2012; Shaw et al., 2009; Poly et al., 2011).
As drug discovery forges ahead, identifying the mechanisms driving the links between TMA, FMO3, TMAO, and human health and disease will be needed to understand where to therapeutically intervene. It is important to note that FMO3 is a promiscuous xenobiotic metabolizing enzyme with many substrates and products (Cashman J, 2006). In fact, FMO3 knockdown-associated effects on metabolic disease may be driven by factors other than TMA or TMAO. Although provision of supplemental TMAO reversed a subset of the Fmo3 ASO-induced effects on adipose gene expression, it also makes clear that not all of the phenotypes observed by FMO3 knockdown are mediated by either TMA or TMAO. Additional work is needed to understand the most therapeutically tractable targets in the entire TMA/FMO3/TMAO meta-organismal pathway. Based on the present studies, it appears that the phenotypic effects of FMO3 knockdown or deletion could be driven by a combination of factors including: (1) chronic increases in the levels of TMA, (2) chronic decreases in the levels of TMAO, and/or (3) effects driven by other FMO3 substrates or products. It is interesting to note that dietary provision of TMAO reversed Fmo3 ASO-driven increases in Ppargc1a, but did not rescue Fmo3 ASO-driven reductions in body weight or expression of Ucp1 in white adipose tissue (Figure 5I–5K). Such findings support a model where TMAO may be involved in specific transcriptional reprogramming in adipocytes, but how TMAO is being sensed is still an unanswered question. TMA is known to activate the G protein-coupled receptor trace amine-associated receptor 5 (TAAR5); however, TAAR5 does not recognize TMAO (Li et al., 2013). It will be important in future studies to determine whether TMA-driven activation of TAAR5 couples metabolic reprogramming in the host, and to identify potential host TMAO receptor(s). In conclusion, this work highlights a role for the TMA/FMO3/TMAO meta-organismal pathway in the progression of obesity-related disorders. Given the numerous strong associations of the gut microbie-driven TMAO pathway with human disease, this work has broad implications for drug discovery efforts targeting gut microbes themselves instead of the human host in which they reside.
EXPERIMENTAL PROCEDURES
Human Studies
To examine whether circulating choline and TMAO levels were associated with T2DM risk, we recruited two unique cohorts including both men and women with diverse cardiometabolic risk profiles in cardiology and hepatology clinics at the Cleveland Clinic. These studies were approved by the Cleveland Clinic Institutional Review Board and every subject provided written informed consent. Extended patient demographics, laboratory values, and clinical characteristics are available in Supplemental Experimental Procedures. To examine the relationship between FMO3 expression and metabolic traits, we took advantage of adipose biopsy microarray data (n=770) within the Metabolic Syndrome in Men (METSIM) study, which has been previously described in detail (Stancakova et al., 2009). The study was approved by the ethics committee of the University of Eastern Finland and Kuopio University Hospital and was conducted in accordance with the Helsinki Declaration. All study participants gave written informed consent. To validate human adipose microarray findings from the METSIM cohort, we analyzed additional microarray data from two distinct cohorts spanning both men and women of European American and African American ethnicity, which have previously been described (Das et al., 2015; Sharma et al., 2016). These studies were approved by the University of Arkansas for Medical Sciences and the Institutional Review Board of Wake Forest School of Medicine. All study participants gave written informed consent. Finally, we examined the protein expression of FMO3 in human liver from normal BMI or bariatric surgery patients. This study was approved by the Institutional Review Board of Wake Forest School of Medicine and all study participants gave written informed consent. Detailed information for all human studies is provided in the Supplemental Experimental Procedures.
Animal Studies
To study the role of the TMAO-producing enzyme FMO3 in diet-induced obesity, we initially employed an in vivo antisense oligonucleotide (ASO)-mediated knockdown approach as previously described (Warrier et al., 2015; Shih et al., 2015). Because of the known sexual dimorphism of hepatic FMO3 expression in mice, all studies were conducted in adult female mice unless otherwise noted. Mice were maintained on either standard rodent chow (2918 Teklad Global 18% Protein Rodent Diet) or a custom high fat diet comprised of 45% kcal derived from fat (Brown et al., 2010). ASO-treated mice were then subjected to cold tolerance and indirect calorimetry studies using methods previously described (Thomas et al., 2013). To establish FMO3 knockout mice, we used CRISPR-Cas9 gene editing as described in the Supplemental Experimental Procedures. Plasma TMA and TMAO quantification and FMO activity measurements were measured using stable isotope dilution mass spectrometry-based assays as previously described (Wang et al., 2014; Warrier et al., 2015) on a Shimadzu 8050 triple quadrupole mass spectrometer. All mice studies were approved by Institutional Animal Care and Use Committees of the Cleveland Clinic, Case Western Reserve University, or University of California – Los Angeles. Detailed information for all mouse studies and biochemical workup of mouse tissue is provided in the Supplemental Experimental Procedures.
Statistical Analysis
To examine the association between circulating choline and TMAO with T2DM, Wilcoxon rank-sum tests were used for continuous variables and χ2 tests were used for categorical variables. Multilogistic regression models were used to estimate odds ratio and 95% confidence interval for diabetes. All analyses were performed using R 3.1.0 (Vienna, Austria) and p≤0.05 was considered statistically significant. All mouse data were analyzed using either one-way or two-way analysis of variance (ANOVA), where appropriate, followed by post hoc analysis. Differences were considered significant at p≤0.05. All mouse data analyses were performed using JMP Pro 10 (SAS Institute; Cary, NC) or GraphPad Prism 6 (La Jolla, CA) software.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health and Office of Dietary Supplements grants R00 HL096166 (J.M.B.), R01 HL122283 (J.M.B.), P50 AA024333 (J.M.B.), R01 HL103866 (S.L.H.), R01 HL126827 (S.L.H. and W.H.W.T), R01 DK106000 (S.L.H. and W.H.W.T), R01 HL130819 (Z.W.), R01 DK090111 (S.K.D.), R00 HL12172 (M.C.), P01 HL028481 (A.J.L.), P01 HL030568-31A1 (A.J.L. and D.M.S.), P01 HL49373 (L.L.R.), the Deutsche Forschungsgemeinschaft in the framework of SFB841 (J.H.), the Academy of Finland and the Finnish Cardiovascular Research Foundation (M.L.), and the American Heart Association (Postdoctoral Fellowship 15POST2535000 to R.C.S, Postdoctoral Fellowship 14POST18700001 to M.W., and Postdoctoral Fellowship 17POST3285000 to R.N.H.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
R.C.S., D.M.S., M.W., and J.M.B. planned the project, designed experiments, analyzed data, and wrote the manuscript; J.M.B., S.L.H., S.V.N.P., W.H.W.T., A.J.L., M.C., and M.L. designed experiments and provided useful discussion directing the project; R.C.S., D.M.S., M.W., R.N.H., A.B., D.F., A.L.B., A.D.G., M.H., A.C., L.L., X.S.L., Z.W., B.W., Y.M., H.K., N.C., C.P., R.E.M., C.D.L., S.K.D., L.L.R., N.Z., A.J.M., S.D., M.L., M.C., and J.H. either recruited human subjects or conducted mouse experiments, performed biochemical workup of mouse tissues, analyzed data, and aided in manuscript preparation; B.O.E and C.A.F. performed imagining studies in mice; R.G.L., R.M.C., and M.J.G. provided antisense oligonucleotides; All authors were involved in the editing of the final manuscript.
CONFLICTS OF INTEREST
R.C.S., D.M.S., M.W. R.N.H, A.B., D.F., A.L.B., A.D.G., M.H., A.C., L.L., X.S.L., B.W., Y.H.M, H.K., N.C., C.P., R.E.M., C.D.L., S.K.D., L.L.R., N.Z., A.J.M., S.D., W.H.W.T., B.O.E, C.A.F, M.L., M.C., S.V.N.P., J.H., A.J.L, and J.M.B. have no conflicts of interest to declare. S.L.H. and Z.W. are named as co-inventors on pending and issued patents held by the Cleveland Clinic relating to cardiovascular diagnostics and therapeutics. S.L.H. reports he has been paid as a consultant by the following companies: Esperion, and Procter & Gamble. S.L.H. also reports he has received research funds from Astra Zeneca, Procter & Gamble, Roche, and Takeda. S.L.H. has the rights to receive royalty payments for inventions or discoveries related to cardiovascular diagnostics from Cleveland Heart Lab Inc., Frantz Biomarkers, and Siemens Healthcare. R.G.L., R.M.C., and M.J.G. are employees at Ionis Pharmaceuticals, Inc. (Carlsbad, CA).
References
- Bäckhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. 2004;101:15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartelt A, Heeren J. Adipose tissue browning and metabolic health. Nat Rev Endocrinol. 2014;10:24–36. doi: 10.1038/nrendo.2013.204. [DOI] [PubMed] [Google Scholar]
- Bennett BJ, de Aguiar Vallim TQ, Wang Z, Shih DM, Meng Y, Gregory J, Allayee H, Lee R, Graham M, Crooke R, et al. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013;17:49–60. doi: 10.1016/j.cmet.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JM, Hazen SL. The gut microbial endocrine organ: bacterially derived signals driving cardiometabolic diseases. Annu. Rev. Med. 2015;66:343–359. doi: 10.1146/annurev-med-060513-093205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cashman JR, Zhang J. Human flavin-containing monooxygenases. Annu. Rev. Pharmacol. Toxicol. 2006;46:65–100. doi: 10.1146/annurev.pharmtox.46.120604.141043. [DOI] [PubMed] [Google Scholar]
- Civelek M, Wu Y, Pan C, Raulerson CK, Ko A, He A, Tilford C, Saleem NK, Stancakova A, Scott LJ, et al. Genetic regulation of adipose gene expression and cardio-metabolic traits. Am. J. Human Genet. 2017;100:428–443. doi: 10.1016/j.ajhg.2017.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox LM, Yamanishi S, Sohn J, Alekseyenko AV, Leung JM, Choi I, Kim SG, Li H, Gao Z, Mahana D, et al. Altering the intestinal microbiota during a critical development window has lasting metabolic consequences. Cell. 2014;158:705–721. doi: 10.1016/j.cell.2014.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dambrova M, Latkovskis G, Kuka J, Strele I, Konrade I, Grinberga S, Hartmane D, Pugovics O, Erglis A, Liepinsh E. Diabetes is associated with higher trimethylamine N-oxide plasma levels. Exp. Clin. Endocrinol. Diabetes. 2016;124:251–256. doi: 10.1055/s-0035-1569330. [DOI] [PubMed] [Google Scholar]
- Das SK, Sharma NK, Zhang B. Integrative network analysis reveals different pathophysiological mechanisms of insulin resistance among Caucasians and African Americans. BMC Med. Genomics. 2015;8:4. doi: 10.1186/s12920-015-0078-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Liu X, Xu J, Xue C, Xue Y, Wang Y. Dietary trimethylamine-N-oxide exacerbates impaired glucose tolerance in mice fed a high fat diet. J. Biosci. Bioeng. 2014;118:476–81. doi: 10.1016/j.jbiosc.2014.03.001. [DOI] [PubMed] [Google Scholar]
- Gregory JC, Buffa JA, Org E, Wang Z, Levison BS, Zhu W, Wagner MA, Bennett BJ, Li L, DiDonato JA, et al. Transmission of Atherosclerosis Susceptibility with Gut Microbial Transplantation. J Biol Chem. 2015;290:5647–5660. doi: 10.1074/jbc.M114.618249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Yan J, West AA, Perry CA, Malysheva OV, Devapatia S, Pressman E, Vermeylen F, Caudill MA. Maternal choline intake alters the epigenetic state of fetal cortisol-regulating genes in humans. FASEB J. 2012;26:3563–3574. doi: 10.1096/fj.12-207894. [DOI] [PubMed] [Google Scholar]
- Koeth RA, Wang Z, Levison BS, Buffa JA, Org E, Sheehy BT, Britt EB, Fu X, Wu Y, Li L, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeth RA, Levison BS, Culley MK, Buffa JA, Wang Z, Gregory JC, Org E, Wu Y, Li L, Smith JD, et al. γ-Butyrobetaine is a proatherogenic intermediate in gut microbial metabolism of L-carnitine to TMAO. Cell Metab. 2014;20:799–812. doi: 10.1016/j.cmet.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA. 2005;102:11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lever M, George PM, Slow S, Bellamy D, Young JM, Ho M, McEntyre CJ, Elmslie JL, Atkinson W, Molyneux SL, et al. Betaine and trimethylamine-N-oxide as predictors of cardiovascular outcomes show different patterns in diabetes mellitus: an observational study. PLoS One. 2014;9:e114969. doi: 10.1371/journal.pone.0114969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Korzan WJ, Ferrero DM, Chang RB, Roy DS, Buchi M, Lemon JK, Kaur AW, Stowers L, Fendt M, et al. Synchronous evolution of an odor biosynthesis pathway and behavioral response. Curr. Biol. 2013;23:11–20. doi: 10.1016/j.cub.2012.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mafune A, Iwamoto T, Tsutsumi Y, Nakashima A, Yamamoto I, Yokoyama K, Yokoo T, Urashima M. Associations among serum trimethylamine-N-oxide (TMAO) levels, kidney function and infarcted coronary artery number in patients undergoing cardiovascular surgery: a cross-sectional study. Clin Exp Nephrol. 2016;20:731–739. doi: 10.1007/s10157-015-1207-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda M, DeFronzo R. Insulin sensitivity indices obtained from oral glucose tolerance testing: comparison with the euglycemic insulin clamp. Diabetes Care. 1999;22:1462–1470. doi: 10.2337/diacare.22.9.1462. [DOI] [PubMed] [Google Scholar]
- Missailidis C, Hällqvist J, Qureshi AR, Barany P, Heimbürger O, Lindholm B, Stenvinkel P, Bergman P. Serum Trimethylamine-N-Oxide Is Strongly Related to Renal Function and Predicts Outcome in Chronic Kidney Disease. PLoS One. 2016;11:e0141738. doi: 10.1371/journal.pone.0141738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao J, Ling AV, Manthena PV, Gearing ME, Graham MJ, Crooke RM, Croce KJ, Esquejo RM, Clish CB Morbid Obesity Study Group. Flavin-containing monooxygenase 3 as a potential player in diabetes-associated atherosclerosis. Nat Commun. 2015;6:6498. doi: 10.1038/ncomms7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muoio DM. Metabolic inflexibility: when mitochondrial indecision leads to metabolic gridlock. Cell. 2014;159:1253–1262. doi: 10.1016/j.cell.2014.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks BW, Nam E, Org E, Kostem E, Norheim F, Hui ST, Pan C, Civelek M, Rau CD, Bennett BJ, et al. Genetic control of obesity and gut microbiota composition in response to high-fat, high-sucrose diet in mice. Cell Metab. 2013;17:141–152. doi: 10.1016/j.cmet.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poly C, Massaro JM, Seshardri S, Wolf PA, Cho E, Krall E, Jacques PF, Au R. The relation of dietary choline to cognitive performance and white-matter hyperintesity in the Framingham Offspring Cohort. Am. J. Clin. Nutr. 2011;94:1584–1591. doi: 10.3945/ajcn.110.008938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- Romano KA, Vivas EI, Amador-Noguez D, Rey FE. Intestinal microbiota composition modulates choline bioavailability from diet and accumulation of the proatherogenic metabolite trimethylamine-N-oxide. mBio. 2015;6:e02481–14. doi: 10.1128/mBio.02481-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma NK, Sajuthi SP, Chou JW, Calles-Escandon J, Demons J, Rogers S, Ma L, Palmer ND, McWilliams D, Beal J, et al. Tissue-specific and genetic regulation of insulin sensitivity-associated transcripts in African Americans. J. Clin. Endocrinol. Metab. 2016;101:1455–1468. doi: 10.1210/jc.2015-3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw GM, Finnell RH, Blom HJ, Carmichael SL, Vollset SE, Yang W, Ueland PM. Choline and risk of neural tube defects in a folate-fortified population. Epidemiology. 2009;20:714–719. doi: 10.1097/EDE.0b013e3181ac9fe7. [DOI] [PubMed] [Google Scholar]
- Shih DM, Wang Z, Lee R, Meng Y, Che N, Charugundla S, Qi H, Wu J, Pan C, Brown JM, et al. Flavin containing monooxygenase 3 exerts broad effects on glucose and lipid metabolism and atherosclerosis. J Lipid Res. 2015;56:22–37. doi: 10.1194/jlr.M051680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stancakova A, Kuulasmaa T, Paananen J, Jackson AU, Bonnycastle LL, Collins FS, Boehnke M, Muusisto J, Laakso M. Association of 18 confirmed susceptibility loci for type 2 diabetes with indices of insulin release, proinsulin conversion, and insulin sensitivity in 5,327 nondiabetic Finnish men. Diabetes. 2009;58:2129–2136. doi: 10.2337/db09-0117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Heaney LM, Bhandari SS, Jones DJ, Ng LL. Trimethylamine N-oxide and prognosis in acute heart failure. Heart. 2016;102:841–848. doi: 10.1136/heartjnl-2015-308826. [DOI] [PubMed] [Google Scholar]
- Tang WH, Wang Z, Levison BS, Koeth RA, Britt EB, Fu X, Wu Y, Hazen SL. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med. 2013;368:1575–1584. doi: 10.1056/NEJMoa1109400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang WH, Wang Z, Fan Y, Levison B, Hazen JE, Donahue LM, Wu Y, Hazen SL. Prognostic value of elevated levels of intestinal microbe-generated metabolite trimethylamine-N-oxide in patients with heart failure: refining the gut hypothesis. J Am Coll Cardiol. 2014;64:1908–1914. doi: 10.1016/j.jacc.2014.02.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang WH, Wang Z, Li XS, Fan Y, Li DS, Wu Y, Hazen SL. Increased trimethylamine N-oxide portends high mortality risk independent of glycemic control in patients with type 2 diabetes mellitus. Clin. Chem. 2017;63:297–306. doi: 10.1373/clinchem.2016.263640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas G, Better JL, Lord CC, Brown AL, Marshall S, Ferguson D, Sawyer J, Davis MA, Melchior JT, Blume LC, et al. The serine hydrolase ABHD6 is a critical regulator of the metabolic syndrome. Cell Rep. 2013;5:508–520. doi: 10.1016/j.celrep.2013.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trøseid M, Ueland T, Hov JR, Svardal A, Gregersen I, Dahl CP, Aakhus S, Gude E, Bjørndal B, Halvorsen B, et al. Microbiota-dependent metabolite trimethylamine-N-oxide is associated with disease severity and survival of patients with chronic heart failure. J Intern Med. 2015;277:717–726. doi: 10.1111/joim.12328. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Gordon JI. The core gut microbiome, energy balance and obesity. J. Physiol. 2009;587:4153–4158. doi: 10.1113/jphysiol.2009.174136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ussar S, Lee KY, Dankel SN, Boucher J, Haering MF, Kleinridders A, Thomou T, Xue R, Macotela Y, Cypress AM, et al. ASC-1, PAT2, and P2RX5 are cell surface markers for white, beige, and brown adipocytes. Sci. Transl. Med. 2014;6:247re103. doi: 10.1126/scitranslmed.3008490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, Feldstein AE, Britt EB, Fu X, Chung YM, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Tang WH, Buffa JA, Fu X, Britt EB, Koeth RA, Levison BS, Fan Y, Wu Y, Hazen SL. Prognostic value of choline and betaine depends on intestinal microbiota-generated metabolite trimethylamine-N-oxide. Eur Heart J. 2014;35:904–910. doi: 10.1093/eurheartj/ehu002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Roberts AB, Buffa JA, Levison BS, Zhu W, Org E, Gu X, Huang Y, Zamanian-Daryoush M, Culley MK, et al. Non-lethal inhibition of gut microbial trimethylamine production for the treatment of atherosclerosis. Cell. 2015;163:1585–1595. doi: 10.1016/j.cell.2015.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrier M, Shih DM, Burrows AC, Ferguson D, Gromovsky AD, Brown AL, Marshall S, McDaniel A, Schugar RC, Wang Z, et al. The TMAO-Generating Enzyme Flavin Monooxygenase 3 Is a Central Regulator of Cholesterol Balance. Cell Rep. 2015;10:1–13. doi: 10.1016/j.celrep.2014.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Bostrom P, Sparks LM, Ye L, Choi JH, Giang AH, Khandekar M, Virtanen KA, Nuutila P, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. 2012;150:366–376. doi: 10.1016/j.cell.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Gregory JC, Org E, Buffa JA, Gupta N, Wang Z, Li L, Fu X, Wu Y, Mehrabian M, et al. Gut microbial metabolite TMAO enhances platelet hyperreactivity and thrombosis risk. Cell. 2016;165:111–124. doi: 10.1016/j.cell.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.