

Abstract
Objective
To evaluate the safety and tolerability of repeated intratympanic administration of the gel-formulated NMDA receptor antagonist AM-101 in acute patients with inner ear tinnitus.
Study Design
Prospective, double-blind, randomized, placebo-controlled study.
Setting
Sixty-nine secondary and tertiary sites in North America, Europe, and Asia.
Subjects and Methods
In total, 343 subjects with persistent acute tinnitus after traumatic cochlear injury or otitis media were randomized to receive 3 intratympanic doses of either AM-101 0.87 mg/mL or placebo over 3 to 5 days. They were followed for 84 days. The primary safety end point was the incidence of a clinically meaningful hearing deterioration from baseline to study day 35. Further safety assessments included tympanic membrane closure rates, analysis of adverse events, hematology, blood chemistry, and vital signs. In addition, data were collected on applied anesthetics and injection techniques.
Results
The treatment was well tolerated, with no intervention-related serious adverse events. The incidence of clinically meaningful hearing deterioration was low, comparable between treatment groups (P = .82 for the primary safety end point) and not different between treated and untreated ears in unilaterally treated subjects. The rate of treatment and procedure-related adverse events was similar among treatment groups. The tympanic membrane was closed in 92% of subjects within 1 week and in all subjects by study day 84. Blood values and vital signs were inconspicuous.
Conclusion
Repeated intratympanic injections of AM-101 over a 3- to 5-day period appear to be safe and well tolerated, demonstrating the ability to potentially use this delivery approach over longer time periods.
Keywords: tinnitus, hearing loss, traumatic, otitis media, cochlea, intratympanic, treatment, safety, local tolerance, AM-101, anesthetic
In recent years, there has been growing interest in intratympanic therapeutics for treating inner ear disorders. Intratympanic drug delivery allows for a highly targeted delivery of medication to the cochlea with minimal systemic exposure and is generally considered as safe by otolaryngologists.1,2 While tympanic membrane perforation, pain, otitis media, vertigo, and hearing loss have been reported as potential complications of intratympanic therapy,3 so far only limited data have been available on actual risk.
The phase 3 trial TACTT2 (Efficacy and Safety of AM-101 in the Treatment of Acute Peripheral Tinnitus 2) was designed to assess the safety and efficacy of AM-101 (esketamine otic gel; Auris Medical AG, Basel, Switzerland) in acute peripheral tinnitus. AM-101 had previously been tested in 1 phase 1/24 and 2 phase 2 trials5,6 involving >900 injections. Administered in single or triple doses at concentrations of up to 0.81 mg/mL, the study drug had been well tolerated in these previous trials, and only small traces of the active substance and its primary metabolite had been detected in plasma, confirming the minimal systemic exposure afforded by the intratympanic approach.
The TACTT2 protocol was agreed by the US Food and Drug Administration (FDA) under a Special Protocol Assessment procedure. Only minor changes that could affect safety were made compared with the previous AM-101 trials. The esketamine concentration was slightly increased from 0.81 to 0.87 mg/mL to achieve precisely a 1-mg/mL concentration in hydrochloride salt, the study drug was provided in prefilled syringes instead of vials, and the 3 doses could be administered over 3 to 5 days instead of 3 straight days to allow for more scheduling flexibility.
Methods
TACTT2 was designed as a multicenter, double-blind, randomized, placebo-controlled phase 3 trial involving 69 sites in the United States, Canada, Czech Republic, Turkey, Israel, and South Korea (ENT departments and private practices). The trial was registered at ClinicalTrials.gov (NCT 01803646) and conducted in compliance with the Declaration of Helsinki and the relevant guidelines of the International Conference on Harmonisation and Good Clinical Practice. The study was approved by the central institutional review board (IRB; Quorum Review IRB, Seattle, Washington) and local IRBs/ethics committees as well as by the FDA and corresponding non-US health authorities.
Eligible participants were aged 18 years or older, had persistent tinnitus following a documented incident of traumatic cochlear injury (acute acoustic trauma, blast trauma, middle ear surgery, inner ear barotrauma, tympanic membrane trauma) or acute otitis media with onset up to 3 months prior. Exclusion criteria included fluctuating or intermittent tinnitus, tinnitus resulting from traumatic head or neck injury, Ménière’s disease or endolymphatic hydrops, history of repeated idiopathic sudden sensorineural hearing loss or acoustic neuroma, fluctuating hearing, hearing threshold ≥75 dB in any audiometric test frequency, ongoing acute or chronic otitis media or externa, any ongoing therapy known to induce tinnitus, concomitant use of any other N-methyl-D-aspartate (NMDA) receptor antagonist or other tinnitus treatments, or diagnosed psychiatric disorders requiring drug treatment. The use of any antidepressant or antianxiety medication was not allowed, unless it was taken in a low dose, maintained throughout the duration of the study, and not used for the treatment of tinnitus. Breastfeeding and pregnant women or women of childbearing potential who declared being unwilling or unable to practice an effective method of contraception were excluded. Written informed consent was obtained from each subject prior to the performance of any study-specific procedures.
Randomization and Masking
Subjects were randomized to receive AM-101 0.87 mg/mL or placebo (vehicle only) at a 3:2 ratio. The likelihood of receiving active treatment was decreased from 2 in 3 in phase 2 to 3 in 5 to counter potential upward drift in the placebo response size that has been observed with psychiatry treatments in phase 3 trials.7 AM-101 and the placebo had the same appearance and viscosity, and the formulation revealed no differences during or following administration. The study drug was provided to study sites in numbered but otherwise identical kits, each containing one single-dose syringe for the treatment of 1 ear. Subjects were randomized using an interactive web response system and based on a dynamic allocation method8 with stratification regarding study country, tinnitus etiology (traumatic or otitis media), and treatment laterality (unilateral or bilateral). Patients and investigators remained blinded to the study drug allocation throughout the study.
Procedures
For each subject, the study consisted of a screening visit, a 2-week screening period, a baseline assessment on the first treatment visit (study day 0), 2 additional treatment visits between day 2 and day 4, and 3 follow-up visits (FUVs), which were scheduled on days 10 (FUV1), 35 (FUV2), and 84 (FUV3). The duration of follow-up was in line with the phase 2 program, where it had sufficiently covered the safety signals and the gradual changes in tinnitus metrics following the (short) treatment phase. Safety-relevant screening assessments included a general physical examination, vital signs, hematology and blood chemistry tests, otoscopy, tympanometry, and pure-tone audiometry (air and bone conducted, using the ascending method of limits). At baseline, vital signs, otoscopy, tympanometry, and pure-tone audiometry were repeated; in addition, a urine drug test was performed, women of childbearing age did a urine pregnancy test, and spontaneous nystagmus was evaluated with Frenzel goggles in the dark, eyes open, and the number of beats, if any, and their direction recorded. If beats were >10/30 seconds, bithermal caloric video- or electronystagmography was performed.
The study drug was administered by intratympanic injection after the tympanic membrane had been locally anesthetized in accordance with the site’s standard practice. The use of phenol was not recommended based on prior study experience.6 Prior to study drug administration, any remaining anesthetic had to be suctioned off. The injection was to be performed in the posterior-inferior quadrant or, alternatively, in the anterior-inferior quadrant. After puncturing the tympanic membrane with a needle (tympanopunction) or performing a small incision (tympanotomy), approximately 0.25 mL of the study drug was gently injected into the middle ear. If the subject had a tympanostomy tube placed prior to enrollment, the study medication could be injected through it. The second and third injections were to be performed in the same quadrant and at the same injection site to minimize the risk of damage to the eardrum.
During the study drug administration, subjects were in a reclined or supine position with their head tilted about 45° toward the untreated ear. Patients remained in this position for approximately 30 minutes to allow for diffusion of the active substance into the cochlea. In case of bilateral tinnitus, the second ear was treated no later than 90 minutes after the first ear.
Vital signs, otoscopy, nystagmometry, tympanometry, and pure-tone audiometry were performed as safety measures at all follow-up visits. Presence of an opening in the tympanic membrane was considered medically normal for up to 7 days after the administration procedure and hence not considered an adverse event. If the tympanic membrane was still open at study day 10, an additional follow-up visit was scheduled and treatment measures were to be taken as deemed medically indicated.
Objectives and End Points
The primary safety end point was the frequency of clinically relevant hearing deterioration in treated ears, defined as hearing deterioration ≥15 dB from baseline to FUV2 at the average of any 2 contiguous test frequencies (based on air conduction thresholds, repeated with bone conduction values). Any permanent threshold shift induced by the treatment or the procedure could be expected to be fully revealed at that point.9 Secondary safety end points included the frequency of clinically relevant hearing deterioration at FUV1 and FUV3, the difference in such frequency between the treated and untreated contralateral ear (unilaterally treated subjects), and the frequency and severity of adverse events (AEs) and serious adverse events (SAEs). The causal relationship of the AE to the study drug (“treatment related”) or to an administration procedure, assessment, or blood draw (“procedure related”), if any, was assessed by the investigator, following relevant guidelines.10,11 The changes in hematology, blood chemistry, and vital sign values were evaluated as exploratory safety end points.
Statistical Methods
The sample size of 330 subjects was based on the planned efficacy analysis with the change in the Tinnitus Functional Index (TFI) and subjective tinnitus loudness as co-primary end points; powering assumptions and test methods for efficacy outcomes will be reported elsewhere. Safety analyses were performed on all subjects who had received study drug at least once (safety analysis set). The percentage of subjects experiencing a clinically relevant hearing deterioration (in at least one of the treated ears in the case of bilateral tinnitus) was compared between treatment groups with Fisher’s exact test. For the comparison of occurrence in treated and untreated ears in unilaterally treated subjects, the McNemar test for symmetry was used with 2 × 2 contingency tables.
Results
Patient Demographics and Characteristics
The trial profile in accordance with the CONSORT statement9 is shown in Figure 1 . A total of 478 subjects were screened, of whom 343 were randomized and 336 treated. In total, 316 of the treated subjects (94%) completed the study. Baseline demographics and patient characteristics for the study population are presented in Table 1 . Similar to the previous AM-101 trials,5,6 subjects were in the majority male (77%), had a mean age of 44 years, and were enrolled on average 65 days after tinnitus onset. In contrast, in TACTT2, unilateral cases represented only a slight majority over bilateral cases, and the share of traumatic cochlear injury as a tinnitus trigger was higher relative to that of otitis media (84% vs 16%). Of the study subjects, 69% were enrolled in North America, 23% in Europe, and 8% in Asia. The TFI score was 51.6 points (ie, tinnitus on average was a moderate problem at baseline);10 the mean hearing threshold at 4 to 8 kHz was 28 dB. Baseline values for hearing and tinnitus variables are presented in Table 2 . Overall, subject demographics and characteristics were similar for the treatment groups.
Figure 1.

Patient flowchart for the TACTT2 trial.
Table 1.
Patient Demographics.
| Characteristic | AM-101 (n = 201) | Placebo (n = 135) | Total (N = 336) |
|---|---|---|---|
| Sex, No. (%) | |||
| Male | 159 (86) | 99 (73) | 60 (77) |
| Female | 42 (21) | 36 (27) | 24 (23) |
| Age, y | |||
| Mean (SD) | 43.4 (14.6) | 44.2 (15.2) | 43.7 (14.8) |
| Range | 18 to 74 | 20 to 73 | 18 to 74 |
| Region of recruitment, No. (%) | |||
| North America | 139 (69) | 93 (69) | 232 (69) |
| Europe | 47 (23) | 31 (23) | 78 (23) |
| Asia | 15 (7) | 11 (8) | 26 (8) |
| Race, No. (%) | |||
| White | 174 (87) | 114 (84) | 288 (86) |
| Asian | 18 (9) | 12 (9) | 30 (9) |
| Black or African American | 6 (3) | 1 (1) | 7 (2) |
| American Indian or Alaska Native | 0 (0) | 1 (1) | 1 (0) |
| Other | 3 (1) | 7 (5) | 10 (3) |
Abbreviation: SD, standard deviation.
Table 2.
Baseline Values for Hearing and Tinnitus Variables.
| Characteristic | AM-101 (n = 201) | Placebo (n = 135) | Total (N = 336) |
|---|---|---|---|
| Days since tinnitus onset | |||
| Mean (SD) | 64.8 (19.9) | 64.6 (24.1) | 64.7 (21.6) |
| Range | 20 to 104 | 18 to 225 | 18 to 225 |
| Tinnitus laterality, No. (%) | |||
| Unilateral | 105 (52) | 64 (47) | 169 (50) |
| Bilateral | 96 (48) | 71 (53) | 167 (50) |
| Tinnitus etiology, No. (%) | |||
| Traumatic | 167 (83) | 116 (86) | 283 (84) |
| Otitis media | 34 (17) | 18 (13) | 52 (16) |
| Other | 0 (0) | 1 (0) | 1 (0) |
| Tinnitus Functional Index, points | n = 187 | n = 125 | n = 312 |
| Mean (SD) | 52.6 (19.5) | 50.2 (19.6) | 51.6 (19.6) |
| Range | 11.6 to 95.6 | 7.6 to 96.0 | 7.6 to 96.0 |
| Hearing threshold (4, 6, 8 kHz), dB | n = 199 | n = 134 | n = 333 |
| Mean (SD) | 27.4 (17.8) | 28.7 (16.9) | 28.0 (17.4) |
| Range | –0.8 to 75.0 | –1.7 to 66.7 | –1.7 to 75.0 |
Abbreviation: SD, standard deviation.
Procedures
In total, TACTT2 subjects received 1392 treatment administrations. Compliance was high, with 95% of subjects taking the complete course of 3 injections. Six subjects received the treatment only twice, 8 subjects received it only once, and 3 subjects with bilateral tinnitus received a complete treatment course only on one ear and an incomplete one on the other. Half the subjects were treated over 3 consecutive days, whereas the other half had treatments spread out over more days. As shown in Figure 2 , most procedures were performed by puncturing the tympanic membrane (86%) and in the posterior-inferior quadrant (84%). More investigators used spinal needles (87%) than microsuction tubes (13%). As shown in Figure 3 , lidocaine was used most often for local anesthesia (37%), followed by EMLA cream (31%) and tetracaine (10%). Phenol was used in 7% of cases, and no anesthetic was applied in 11% of cases.
Figure 2.
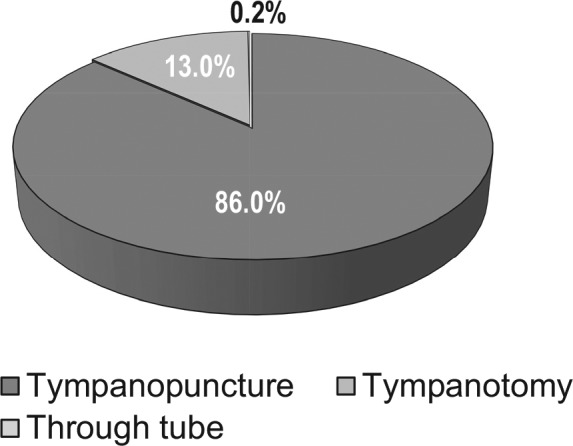
Frequency of use of tympanopunction (puncturing of eardrum), tympanotomy, or application through a preexisting ventilating tube (1392 administrations in total).
Figure 3.
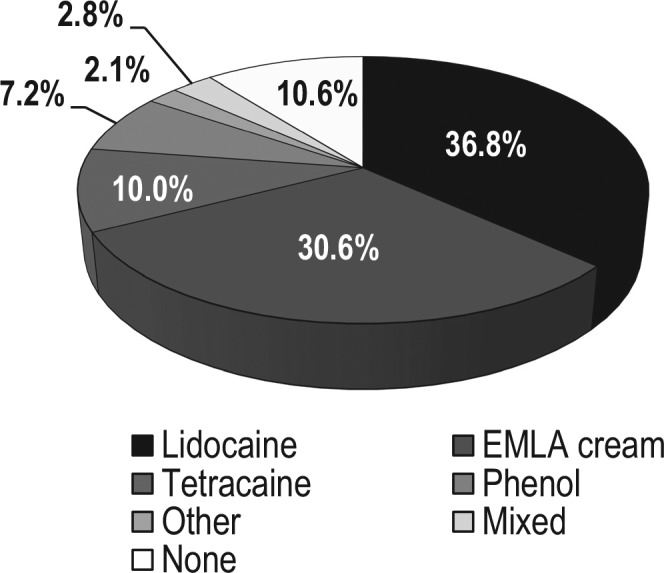
Choice of local anesthetic for numbing of eardrum prior to intratympanic drug delivery (1392 administrations in total).
Clinically Relevant Hearing Deterioration
The primary safety end point for the study was met as there was no difference in the incidence of clinically relevant hearing loss between treatment groups at FUV2 (6.2% vs 6.9% in the AM-101 and placebo groups, respectively; P = .821). As shown in Figure 4a , the incidence was less than 7% based on air conduction threshold shifts. In addition, no significant differences were found at FUV1 or FUV3 (10.9% vs 16.2%, P = .18 and 4.3% vs 7.8%, P = .223). The incidence at FUV2 dropped to 2% when using bone conduction data; again, there was no difference between treatment groups at that point (2.1% vs 2.3%, P = 1.000) or at FUV1 or FUV3 (3.1% vs 3.7%, P = .554 and 2.2% vs 4.7%, P = .328; Figure 4b ).
Figure 4.

Frequency of clinically relevant hearing loss in treated ear(s) (threshold shift ≥15 dB in any 2 contiguous test frequencies from baseline). (a) Air conduction hearing thresholds and (b) bone conduction hearing thresholds. n = 323. FUV, follow-up visit.
Comparison of the incidence of clinically relevant hearing deterioration in treated vs untreated ears, in the case of unilateral treatment, showed no significant differences with the exception of FUV1. In placebo-treated subjects, the treated ear showed a significantly higher incidence than the untreated ear at FUV1 (15.3% vs 1.4%, P = .003; Figure 5a ), whereas active-treated ears showed only a trend toward higher incidence in the treated ear (9.0% vs 3.6%, P = .090; Figure 5b ). With bone conduction thresholds, there were no statistically significant differences between treated and untreated ears ( Figure 6a , b ). The mean 4- to 8-kHz hearing threshold essentially remained unchanged to day 84 (+0.6 and +0.2 dB on average for AM-101 and placebo groups; median unchanged).
Figure 5.

Frequency of clinically relevant hearing loss in treated and untreated ears in unilaterally treated subjects (≥15 dB air conduction in any 2 contiguous test frequencies from baseline). (a) AM-101 group (n = 111); (b) placebo group (n = 72). FUV, follow-up visit.
Figure 6.

Frequency of clinically relevant hearing loss in treated and untreated ears in unilaterally treated subjects (≥15 dB bone conduction in any 2 contiguous test frequencies from baseline). (a) AM-101 group (n = 109); (b) placebo group (n = 72). FUV, follow-up visit.
Adverse Events
Treatment-emergent AEs were observed for similar proportions of patients across the treatment groups with no clinically meaningful differences in frequency or intensity ( Table 3 ). Among the AEs considered as related, more than 70% were attributed to the procedure. For 20 subjects (5.9%), treatment-related AEs were reported; 15 were considered mild and the remaining 5 moderate in intensity. AEs related to the procedure were reported for 56 subjects (16.7%); 47 were considered mild, 7 moderate, and 2 severe. The most frequent procedure-related AEs were ear pain (21 subjects, 6.2%) and ear discomfort (15 subjects, 4.4%; Table 4 ); all except 3 cases were rated as mild, and all were fully resolved, except for 1 mild case of intermittent nature. Vertigo was rarely observed (AM-101, 2 cases; placebo, 4 cases) and always considered mild.
Table 3.
Treatment-Emergent Adverse Events.a
| Adverse Event | AM-101 (n = 201), No. (%) | Placebo (n = 135), No. (%) | Total (N = 336), No. (%) |
|---|---|---|---|
| Any adverse event | 90 (45) | 46 (34) | 136 (41) |
| Adverse events, treatment related | 14 (7) | 6 (4) | 20 (6) |
| Mild | 11 (5) | 4 (3) | 15 (4) |
| Moderate | 3 (1) | 2 (1) | 5 (1) |
| Severe | 0 (0) | 0 (0) | 0 (0) |
| Adverse events, procedure related | 34 (17) | 22 (16) | 56 (17) |
| Mild | 26 (13) | 21 (16) | 47 (14) |
| Moderate | 6 (3) | 1 (1) | 7 (2) |
| Severe | 2 (1) | 0 (0) | 2 (1) |
| All severe adverse events (including unrelated cases) | 3 (1) | 1 (1) | 4 (1) |
| Serious adverse events | 5 (2) | 1 (1) | 6 (2) |
| Adverse events leading to | |||
| Study drug withdrawal/interruption | 5 (2) | 3 (2) | 8 (2) |
| Premature study termination | 1 (0) | 1 (1) | 2 (1) |
Frequencies and percentages of patients with adverse events; occurrence of an adverse event in the same patient was counted only once.
Table 4.
Most Common Treatment-Emergent Adverse Events and Their Causality.a
| AM-101 (n = 201) |
Placebo (n = 135) |
Total (N = 336) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Characteristic | All | Rx | Proc | All | Rx | Proc | All | Rx | Proc |
| Ear related | |||||||||
| Ear discomfort | 14 (7) | 4 (2) | 10 (5) | 8 (6) | 3 (2) | 5 (4) | 22 (6) | 7 (2) | 15 (4) |
| Ear pain | 16 (8) | 2 (1) | 12 (6) | 10 (7) | 1 (1) | 9 (7) | 26 (8) | 1 (1) | 21 (6) |
| Tinnitus | 8 (4) | 1 (0) | 2 (1) | 5 (4) | 2 (1) | 1 (1) | 13 (4) | 3 (1) | 3 (1) |
| Hypoacusis | 8 (4) | 3 (1) | 1 (0) | 4 (3) | 1 (1) | 3 (2) | 12 (4) | 4 (1) | 4 (1) |
| Tympanic membrane perforation | 5 (2) | 0 (0) | 3 (1) | 4 (3) | 0 (0) | 4 (3) | 9 (3) | 0 (0) | 7 (2) |
| General | |||||||||
| Headache | 11 (5) | 0 (0) | 3 (1) | 6 (4) | 1 (1) | 0 (0) | 17 (5) | 1 (0) | 3 (1) |
| Dizziness | 10 (5) | 2 (1) | 4 (2) | 1 (1) | 0 (0) | 0 (0) | 11 (3) | 2 (1) | 4 (1) |
| Nasopharyngitis | 6 (3) | 0 (0) | 0 (0) | 4 (3) | 0 (0) | 0 (0) | 10 (3) | 0 (0) | 0 (0) |
Numbers and percentages of subjects with adverse events (>3% overall or in any of the treatment groups) for all causalities (All), treatment related (Rx), or procedure related (Proc); any difference between All and the sum of Rx and Proc represents unrelated adverse events. Occurrence of an adverse event in the same patient was counted only once. Preferred term is based on the MedDRA coding dictionary, version 19.0.
Six subjects experienced nonfatal, SAEs; none of the SAEs concerned the ear or were considered related ( Table 3 ). Four subjects experienced a total of 6 SAEs (5 in 3 AM-101-treated subjects); all were considered not related and fully resolved. Study drug administration was prematurely terminated or interrupted due to AEs in 8 cases: 5 in the AM-101 group (subjective worsening of hearing [1], otitis externa/inflammation of ear canal [2], otitis media [1], and nausea [1]) and 3 in the placebo group (ear blockage and discomfort [1], clogged ear and postprocedural headache [1], and hearing loss and new tinnitus [1]). All of these cases resolved. In each treatment group, there was 1 AE leading to study discontinuation (AM-101: subjective worsening of hearing; placebo: ear blockage and discomfort).
Tympanic Membrane Closure
At the time of FUV1 (ie, about 5-7 days after the last administration), 92% of tympanic membranes were closed ( Table 5 ). For the 20 subjects whose eardrums were still open at that point, the extra study visit at day 21 revealed closure in all cases but 3. At FUV2, 99% of tympanic membranes were closed, and at FUV3 the rate was 100%.
Table 5.
Tympanic Membrane Closure.a
| Characteristic | AM-101 (n = 201), No. (%) | Placebo (n = 135), No. (%) | Total (N = 336), No. (%) |
|---|---|---|---|
| Treatment visit 2 (days 2-4) | 36 (18) | 28 (21) | 64 (19) |
| Treatment visit 3 (days 3-5) | 32 (16) | 23 (17) | 55 (17) |
| Follow-up visit 1 (day 10) | 16 (8) | 9 (7) | 25 (8) |
| Follow-up visit 2 (day 35) | 2 (1) | 1 (1) | 3 (1) |
| Follow-up visit 3 (day 84) | 0 (0) | 0 (0) | 0 (0) |
Values are numbers and percentages of subjects with open tympanic membrane in the treated ear(s).
Other Safety Outcomes
Hematology and blood chemistry values did not show any meaningful differences between treatment groups or relevant changes from baseline that could be attributable to the treatment. Apart from a slight transient increase in mean blood pressure and pulse around the treatment phase, vital signs did not show any peculiarities. The number of cases of clinically relevant spontaneous nystagmus was very small, and there was no difference between treatment groups.
Discussion
The TACTT2 study was the fourth randomized and placebo-controlled trial in the AM-101 clinical development program. The previous trials had shown good tolerance of the treatment with no impact on hearing; adverse events that were predominantly local, ear related, and transient; and minimal systemic exposure.5,6 Based on almost 1400 intratympanic injections, the present study confirmed the favorable safety profile.
Importantly, the TACTT2 study showed only a few cases of clinically relevant hearing deterioration and no drug-related effect. These results confirm previous results obtained in preclinical animal models—esketamine, the active substance of AM-101, had no effects on auditory thresholds even at much higher concentrations than the clinically tested 0.87 mg/mL. A procedure-related effect on hearing was seen in <7% of active-treated subjects at FUV1 (ie, shortly after the treatment course) based on air conduction thresholds. No such effect was observed when analyzing bone conduction thresholds. It appears likely that in some subjects, sound conduction was affected at that point by residues of the gel in the middle ear and/or an eardrum that had not fully closed. Using untreated contralateral ears as within-subject controls in unilaterally treated subjects confirmed a transient procedure-related effect at FUV1 based on air conduction and only a slight but statistically not significant preponderance of deteriorations in the treated ear based on bone conduction. This suggests that apart from the transient procedure-related effect, observed hearing deterioration represented normal variation.
The rapid closure of the tympanic membrane in the present study—92% closed at FUV1—vindicates the recommendation for using other local anesthetics rather than phenol. Whereas in the first phase 2 study with AM-101, a 93% closure rate had been achieved at FUV1 with the use of a 10% lidocaine pump spray,5 the protocol for the second phase 2 study had not specified any particular anesthetic, and the closure rate was only <80%.6 Here phenol and, in rare cases, Bonain’s solution (a mixture of phenol, cocaine, and menthol) were applied to 43% of subjects, and these 2 products accounted for a disproportionate 87% of cases with open eardrums at FUV1. Apart from phenol’s potential retarding effect on tympanic membrane closure, it should also be considered that it is a highly caustic chemical and can cause severe burns.11 Although some studies have shown or suggested that intratympanic administration without anesthesia may result in similar pain perception as with anesthesia,12,13 it appears advisable to use a local anesthetic for patient acceptance and comfort and as the experience of pain is highly individual.14
To our knowledge, TACTT2 was the first study to prospectively collect AE data differentiated by relatedness to the procedure or to the treatment. For the further adoption of intratympanic administration, it will be important to dispose of more detailed, differentiated, and evidence-based information on the nature and rate of potential complications. For example, it appears from a review of some of the largest trials with intratympanic administration of recent years that the rate of persistent tympanic membrane perforation 2 to 4 months following intratympanic corticosteroid administration ranged between 1.6% and 15.5% ( Table 6 ). Since no such outcome was observed with intratympanic IGF-115 and only in 1 of more than 2000 cases over the entire AM-101 program so far, this points rather to an effect of the drug, specifically corticosteroids’ well-known retarding effects on wound healing, than to the procedure itself. Furthermore, when high incidences of AEs for ear pain or injection pain are reported—54.3% and 27.1% in 1 well-conducted trial16—the number of injections (4 in that case) and, more importantly, the tolerability of the drug formulation have to be taken into account (methylprednisolone solution). As pointed out by previous authors,17 none of the drug compositions that are commonly used for local application to the middle ear have been formulated specifically for good middle ear tolerance and may contain preservatives or have a pH or osmolarity that can cause irritation or pain.
Table 6.
Reported Rates of Persistent Tympanic Membrane Perforation following Intratympanic Administration.
| Study | Indication | Treatment | Anesthetic | No. | Follow-up, d | Persistent TM Perforation, % |
|---|---|---|---|---|---|---|
| Rauch et al, 201118 | ISSNHL | 4 × methylprednisolone 40 mg/mL of over 2 weeks | Phenol | 129 | 60 | 3.9 |
| Lambert et al, 201219 | Ménière’s disease | 1 × 3 or 12 mg dexamethasone in poloxamer gel | Phenol | 30 | 84 | 3.3 |
| Nakagawa et al, 201420 | SSHL | 4 × 3.8 mg/mL dexamethasone over 4-7 days | Lidocaine | 58 | 112 | 15.5 |
| 1 × IGF-1 10 mg/mL in gelatin hydrogel | 62 | 0.0 | ||||
| Lambert et al, 201621 | Ménière’s disease | 1 × 12 mg dexamethasone in poloxamer gel | Unknown | 77 | 120 | 2.7 |
| Topf et al, 201722 | Ménière’s disease or ISSHNL | ≥ 1 × dexamethasone 10 mg/mL | Phenol | 192 | 90 | 1.6 |
Abbreviations: IGF-1, insulin-like growth factor 1; ISSNHL, idiopathic sudden sensorineural hearing loss; SSHL, sudden sensorineural hearing loss; TM, tympanic membrane.
In conclusion, repeated intratympanic injections of AM-101 over a 3- to 5-day period appear to be safe and well tolerated. No drug-related side effects were observed, while procedure-related effects were essentially limited to a transient increase in hearing thresholds (<7% of subjects) and ear discomfort or ear pain in 4% to 6% of cases, which in the vast majority was mild in intensity. The study outcomes support the use of repeated-dose intratympanic administration over longer time periods, especially when using medication adapted for intratympanic use as well as appropriate local anesthetics.
Author Contributions
Hinrich Staecker, substantial contribution to conception and design, principal investigator in the study, contribution to analysis and interpretation of data, contribution to drafting the work, final approval of work, accountable for all aspects therein; Michael Morelock, principal investigator in the study, contribution to critical review, final approval of work, accountable for all aspects therein; Timothy Kramer, principal investigator in the study, contribution to critical review, final approval of work, accountable for all aspects therein; Pavel Chrbolka, principal investigator in the study, contribution to critical review, final approval of work, accountable for all aspects therein; Joong Ho Ahn, principal investigator in the study, contribution to critical review, final approval of work, accountable for all aspects therein; Thomas Meyer, substantial contribution to conception and design, directing the sponsor of the study, participating in the analysis and interpretation of data, contribution to drafting the work, final approval of work, accountable for all aspects therein.
Disclosures
Competing interests: Hinrich Staecker, recipient of speaker honoraria for participating in symposia organized by sponsor at AAO annual meetings and advisor in FDA meeting with the sponsor. Thomas Meyer, shareholder of sponsor and office bearer (chairman of the board, chief executive officer).
Sponsorships: Auris Medical AG: design and conduct of study, collection, analysis, and interpretation of the data. The sponsor reviewed and approved the final version of the submitted manuscript.
Funding source: Auris Medical AG, Basel, Switzerland.
Acknowledgments
We thank Dr Manfred Wargenau and Dr Marcos Marin-Galiano, MARCO Institute, Dusseldorf, Germany, for their biostatistical contributions and Cindy McGee for assistance with coordinating the manuscript submission. Sincere gratitude is also expressed to all participating patients with tinnitus who made this study possible and to all investigators and their staff.
Footnotes
Sponsorships or competing interests that may be relevant to content are disclosed at the end of this article.
This article was presented at the 2016 AAO-HNSF Annual Meeting and OTO EXPO; September 18-21, 2016; San Diego, California.
References
- 1. Lustig LR. The history of intratympanic drug therapy in otology. Otolaryngol Clin North Am. 2004;37:1001-1017. [DOI] [PubMed] [Google Scholar]
- 2. Meyer T. Intratympanic treatment for tinnitus: a review. Noise Health 2013;15:83-90. [DOI] [PubMed] [Google Scholar]
- 3. Haynes DS, O’Malley M, Cohen S, Watford K, Labadie RF. Intratympanic dexamethasone for sudden sensorineural hearing loss after failure of systemic therapy. Laryngoscope 2007;117:3-15. [DOI] [PubMed] [Google Scholar]
- 4. Muehlmeier G, Biesinger E, Maier H. Safety of intratympanic injection of AM-101 in patients with acute inner ear tinnitus. Audiol Neurotol. 2011;16:388-397. [DOI] [PubMed] [Google Scholar]
- 5. Van de Heyning P, Muehlmeier G, Cox T, et al. Efficacy and safety of AM-101 in the treatment of acute inner ear tinnitus—a double-blind, randomized, placebo-controlled phase II study. Otol Neurotol. 2014;35:589-597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Staecker H, Maxwell KS, Morris JR, et al. Selecting appropriate dose regimens for AM-101 in the intratympanic treatment of acute inner ear tinnitus. Audiol Neurotol. 2015;20:172-182. [DOI] [PubMed] [Google Scholar]
- 7. Rutherford BR, Roose SP. A model of placebo response in antidepressant clinical trials. Am J Psychiatry. 2013;170:723-733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pocock SJ, Simon R. Sequential treatment assignment with balancing for prognostic factors in the controlled clinical trial. Biometrics. 1975;31:103-115. [PubMed] [Google Scholar]
- 9. Ryan AF, Kujawa SG, Hammill T, Le Prell C, Kil J. Temporary and permanent noise-induced threshold shifts: a review of basic and clinical observations. Otol Neurotol. 2016;37:271-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. US Food and Drug Administration. Safety Reporting Requirements for INDs and BA/BE Studies: Guidance for Industry and Investigators. Washington, DC: US Food and Drug Administration; December 2012. [Google Scholar]
- 11. European Medicines Agency. Guideline on Good Pharmacovigilance Practices (GVP), Module VI—Management and Reporting of Adverse Reactions to Medicinal Products (Rev 1). London, England: European Medicines Agency and Heads of Medicines Agencies; September 2014. [Google Scholar]
- 12. Moher D, Schulz KF, Altman D. The CONSORT statement: revised recommendations for improving the quality of reports of parallel-group randomized trials. JAMA 2001;285:1987-1991. [DOI] [PubMed] [Google Scholar]
- 13. Henry JA, Griest S, Thielman E, McMillan G, Kaelin C, Carlson KF. Tinnitus Functional Index: development, validation, outcomes research, and clinical application. Hear Res. 2016;334:58-64. [DOI] [PubMed] [Google Scholar]
- 14. Santa Maria PL, Corrales CE, Sevy AB, Jackler RK. Iatrogenic phenol injury causing facial paralysis with tympanic membrane and ossicular necrosis. Otol Neurotol. 2016;37:385-387. [DOI] [PubMed] [Google Scholar]
- 15. Fillingim RB. Individual differences in pain responses. Curr Rheumatol Rep. 2005;7:342-347. [DOI] [PubMed] [Google Scholar]
- 16. Ata N, Erdur O, Görgülü MH, Yilmaz E. Effects of two different local anaesthetic methods vs no anaesthesia on pain scores for intratympanic injections. Laryngol Otol. 2016;130:1153-1157. [DOI] [PubMed] [Google Scholar]
- 17. Mikulec AA, Hartsock JJ, Salt AN. Permeability of the round window membrane is influenced by the composition of applied drug solutions and by common surgical procedures. Otol Neurotol. 2008;29:1020-1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rauch SD, Halpin CF, Antonelli PJ, et al. Oral vs intratympanic corticosteroid therapy for idiopathic sudden sensorineural hearing loss: a randomized trial. JAMA. 2011;305:2071-2079. [DOI] [PubMed] [Google Scholar]
- 19. Lambert PR, Nguyen S, Maxwell KS, et al. A randomized, double-blind, placebo-controlled clinical study to assess safety and clinical activity of OTO-104 given as a single intratympanic injection in patients with unilateral Ménière’s disease. Otol Neurotol. 2012;33:1257-1265. [DOI] [PubMed] [Google Scholar]
- 20. Nakagawa T, Kumakawa K, Usami S, et al. A randomized controlled clinical trial of topical insulin-like growth factor-1 therapy for sudden deafness refractory to systemic corticosteroid treatment. BMC Med. 2014;12:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lambert PR, Carey J, Mikulec AA, LeBel C; Otonomy Ménière’s Study Group. Intratympanic sustained-exposure dexamethasone thermosensitive gel for symptoms of Ménière’s disease: randomized phase 2b safety and efficacy trial. Otol Neurotol. 2016;37:1669-1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Topf MC, Hsu DW, Adams DR, et al. Rate of tympanic membrane perforation after intratympanic steroid injection. Am J Otolaryngol. 2017;38:21-25. [DOI] [PubMed] [Google Scholar]