

Abstract
Genetics-based studies of women with polycystic ovary syndrome (PCOS) implicate >20 PCOS risk genes that collectively account for <10% of PCOS. Clinicians now consider that either rare alleles or non-genetic, potentially epigenetic, developmental origins may contribute key pathogenic components to >90% of PCOS cases. Animal models convincingly demonstrate excess fetal testosterone exposure in females as a reliable, epigenetic, developmental origin for PCOS-like traits. In particular, nonhuman primates (NHPs) provide the most faithful emulation of PCOS-like pathophysiology, likely because of close similarities to humans in genomic, developmental, reproductive and metabolic characteristics, as well as aging. Recent appreciation of potential molecular mechanisms contributing to enhanced LH action in both PCOS women (GWAS-based) and PCOS-like monkeys (DNA methylation-based) suggest commonality in pathogenic origins. This review examines the translational relevance of NHP studies to PCOS, identifying characteristics of newborn females at risk for PCOS-like traits and potential prepubertal treatment interventions to ameliorate PCOS onset.
Keywords: Androgen excess, developmental origins, fetal programming, Barker hypothesis, gestational hyperglycemia, lipotoxicity, animal models
BACKGROUND
Polycystic ovary syndrome (PCOS), while prevalent (~15%), highly familial, and with an onset in adolescence, has no known cause [1]. Women with PCOS exhibit at least two of the following three diagnostic criteria: clinical or biochemical hyperandrogenism, intermittent or absent menstrual cycles and/or polycystic ovaries. Establishing the diagnosis of PCOS in adolescence, however, is contentious because of considerable overlap between true PCOS and normal, transient adolescent androgen excess, intermittent menstrual cycles and metabolic fluctuations [2, 3]. Accompanying PCOS sequelae include luteinizing hormone (LH) hypersecretion, antimullerian hormone (AMH) overproduction, insulin resistance and obesity, together with increased risks for metabolic syndrome, type 2 diabetes, gestational diabetes and endometrial cancer [1]. PCOS pathophysiology thus extends well beyond ovarian dysfunction to include hypothalamic dysregulation and, perhaps more concerning, increased risks for metabolic disease and cancer associated with long-term morbidity and mortality [4, 5].
Contemporary understanding of PCOS considers its etiology as polygenic, with developmental origins likely preceding puberty [1, 6, 7]. At least 21 replicated candidate genes have been identified, regulating gonadotropin secretion and action, extracellular matrix development and a variety of common cellular functions [6, 8]. Each, however, accounts for only a small percentage of the estimated 70% heritability of PCOS, implying a considerable epigenetic contribution to the phenotypic expression of PCOS [9, 10]. The most comprehensive epigenetic phenotypes that mimic PCOS arise from animal models that employ experimentally-induced fetal testosterone(T) excess to permanently induce (‘program’) PCOS-like reproductive and metabolic traits in female rodents [11–13], sheep [14, 15] and nonhuman primates(NHPs) [16, 17]. The absence of a naturally occurring PCOS animal model, however, has hindered progress towards understanding pathogenic mechanisms that may bestow both genetic and epigenetic contributions to the etiology of PCOS.
Macaque monkeys, including rhesus (Macaca mulatta) and cynomolgus (M. fascicularis), share over 90% of their genome with humans, and provide close parallels throughout fetal, infant and juvenile development in regards to reproduction, metabolism and aging [17]. Experimental induction of T excess in fetal female rhesus macaques during early-to-mid gestation provides the most faithful emulation of PCOS in women (Table 1) [17–19]. Apart from humans, moreover, female macaques are unique in exhibiting naturally-occurring, PCOS-like phenotypes [20, 21], as well as demonstrating concomitant external genital biomarkers indicative of fetal T excess [21–23]. Gestational T excess, whether experimentally-induced or naturally-occurring, may thus provide an early developmental origin for life-long epigenetic changes to the female macaque genome that closely mimic both genetic and epigenetic components of PCOS women.
Table 1.
Fetal, infant and adult phenotypic characteristics of PCOS-like monkeys, PCOS and High T Women.
| PA Monkeysa | Prepubertal Tb | High T monkeysc | PCOSd | High Te | |||
|---|---|---|---|---|---|---|---|
| Trait | Fetus | Infant | Adult | Adult | Adult | Women | Women |
| Ovarian | |||||||
| High T | + | + | + | − | +e | +e | + |
| High AMH | NA | − | − | − | + | + | + |
| Polycystic ovaries | ? | − | + | + | ? | + | − |
| Intermittent/absent menstrual cycles | NA | NA | +f | − | −e | +e | + |
| Hypothalamic-Pituitary | |||||||
| High LH | + | + | + | + (transient) | + | + | + |
| Increased frequency LH release | ? | ? | + | + (transient) | ? | + | ? |
| Decreased E2/P4 negative feedback | ? | ? | + | − | ? | + | ? |
| Positive feedback (LH surge) | NA | NA | + | + | + | + | + |
| Metabolic | |||||||
| Increased body weight or BMI | +/−g | + | − | + | − | + | − |
| Increased visceral fat | NA | ? | + | − | ? | +/−h | ? |
| Increased type 2 diabetes | NA | − | + | − | − | + | − |
| Pancreatic defect | ? | + | + | − | ? | + | ? |
| Lipid abnormalities | + | − | + | − | ? | + | ? |
| Maternal gestational hyperglycemia | + | + | ? | NA | ? | + | ? |
This review will consider the developmental origins of PCOS-like traits in T-exposed female fetal macaques by first exploring a newborn female phenotype preceding adult PCOS-like pathophysiology, and then discussing how such findings may forge translational approaches to basic science and clinical medicine in the field of PCOS. We will focus on T exposure during early-to-mid gestation, and not late gestation or starting before puberty, since the former best emulates PCOS in the NHP model [17]. We will include preliminary studies of naturally-occurring high T adult female monkeys as an additional NHP model that combines genetic/epigenetic components in the developmental origin for PCOS and its accompanying squeals in PCOS women.
HYPERANDROGENIC GESTATIONAL CONTRIBUTIONS TO PCOS-LIKE TRAITS
Neuroendocrine-Related
From rodents [11–13] and sheep [14, 15] to NHPs [16–18], animal studies overwhelmingly demonstrate how fetal T excess, and likely accompanying gestational hyperglycemia and hyperinsulinemia [24–26], provide developmental origins for PCOS-like phenotypes. Experimental induction of T exposure in NHPs is achieved by daily injection of macaque dams with 10–15 mg T propionate (TP) during early-to-mid gestation. Such maternal treatment overwhelms substantial placental capacity to aromatize and inactivate T, and raises circulating T levels in fetal females to those normally found in comparably aged fetal males [27]. This discrete androgen excess distinguishes the fetal NHP model from those in non-primates, such as sheep [26], because fetal estradiol in NHPs remains at normal female levels and does not increase [27], due in part to estrogen conjugation.
Following cessation of TP injections at mid-gestation, circulating fetal levels of pituitary LH and follicle stimulating hormone (FSH) dramatically increase, likely due to escape from T-imposed negative feedback and consequent increased release of hypothalamic gonadotropin-releasing hormone (GnRH), illustrating a possible male-like outcome of T programming on negative feedback at the fetal female hypothalamus-pituitary level [27]. T-exposed fetal females thus resemble fetal males in precocious development of negative feedback regulation of GnRH/LH [28], likely mediated through de-sensitized estradiol and/or progesterone mediated mechanisms [13, 18, 29, 30]. Such PCOS-like, de-sensitized negative feedback regulation of GnRH/LH is also pronounced in T-exposed female rodents [31] and sheep [32], but unlike those non-primate species, positive feedback regulation of the ovulation-inducing LH surgeremains functional in T-exposed and high T adult female monkeys, as it does in PCOS and high T women (Table 1). Adult T-exposed female NHPs, in contrast to fetal counterparts, do not exhibit elevated circulating FSH levels probably due to matured ovarian negative feedback, and emulating PCOS women in whom FSH levels are either normal or low [33].
In a recent revival of the importance of this neuroendocrine-related PCOS pathophysiology, genetic studies in women implicate dysregulated LH release [10, 34, 35, 36] or action [6, 9, 37–40] in the PCOS pathogenic mechanism. In the T-exposed NHP model, altered gene expression involving LH signaling is also implicated from gene network analysis of DNA methylation changes in T-exposed infant and adult monkeys [41]. In the T-exposed female sheep model, deficient LH signaling also has been recently implicated in abnormal ovarian follicle development [42]. Taken together, these findings suggest that dysfunctional LH signaling may promote ovarian hyperandrogenism [40], which impairs steroid negative feedback to further exaggerate LH hypersecretion [43], implicating excessive LH release as a primary characteristic of women who ultimately manifest PCOS. This feed-forward amplification hypothesis is illustrated in Fig. 2. Figure 3 additionally illustrates the overlap between recently hypothesized molecular dysfunction of LH signaling in PCOS [40] and related LH signaling abnormalities identified through DNA methylation analyses of T-exposed female monkeys [41].
Fig. (2).
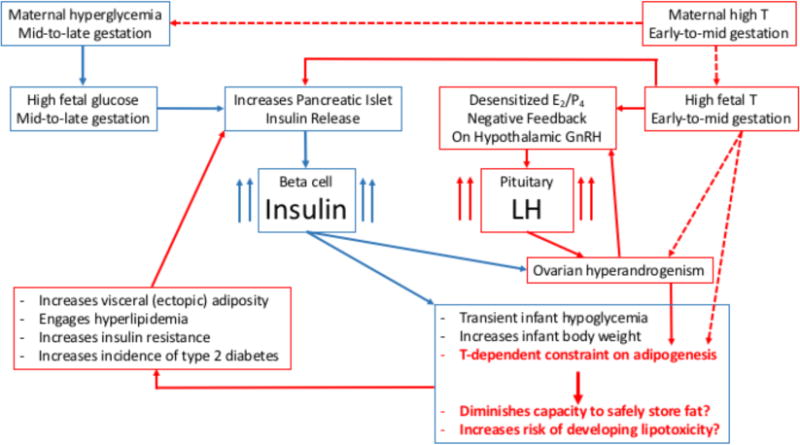
Diagrammatic representation of hypothesized contributions from fetal testosterone (T) excess and gestational hyperglycemia to subsequent LH and insulin hypersecretion and postnatal PCOS-like metabolic and reproductive pathophysiology in NHP models for PCOS. Dashed arrows indicate hypothesized fetal programming that has not yet been demonstrated in sheep or monkeys.
Fig. (3).
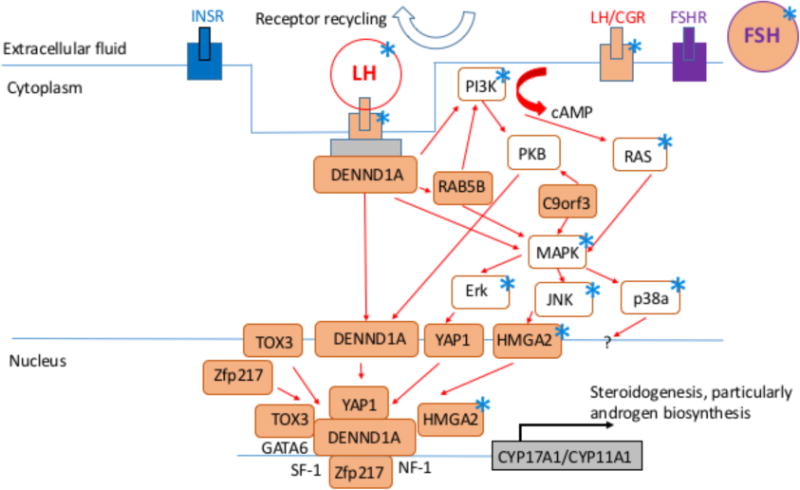
Diagrammatic representation of hypothesized molecular mechanism of LH signaling dysfunction in PCOS women modified from [40]. Each asterisk indicates a molecule also identified in the most significant signaling pathway or gene network from DNA methylation analyses of visceral adipose from infant and adult T-exposed female monkeys [41].
Ovarian-Related
No endogenous fetal hyperandrogenism becomes apparent in T-exposed fetal female monkeys following exogenous induction of T excess [27]. Postnatal evidence of their exogenous T exposure presents in the form of lengthened anogenital distance (a biomarker of fetal T exposure) that is positively correlated with their duration of exogenous T treatment [23]. Interestingly in this regard, naturally occurring high T females may also experience fetal hyperandrogenism. They exhibit positive correlations between circulating androgen levels and anogenital distance, as well as between clitoral volume and postnatal age. These associations strongly suggest that naturally occurring high T females experience endogenous T excess from at least mid-gestation [21–23]. Recently, lengthened newborn anogenital distance [44], and mid-gestation amniotic fluid T excess [45], have been shown in daughters of women with PCOS, strongly implicating mid-gestational T excess during human female fetal development with the likely acquisition of PCOS in later life. Certainly, the fetal human mid-gestational ovary has developed androgen biosynthetic capacity and androgen receptors [46, 47], in analogous ovarian developmental progression to the monkey [48].
As another biomarker of fetal T exposure, adult second-to-fourth (2D: 4D) finger length ratio correlates with anogenital distance and duration of exogenous T in female NHPs [23]. Consistent with a fetal T origin, women with PCOS also exhibit lengthened 2D: 4D finger length ratios [49, 50], although another study found a more masculinized finger length ratio in PCOS women [51]. To illustrate PCOS-like dysfunction programmed by gestational exposure to excess T in female monkeys, Fig. 2 employs a fetal origins of disease approach [52, 53], with an emphasis on LH and T excess as causal factors that develop into a feed-forward, amplification loop. As illustrated in Fig. 2, early onset elevation of circulating insulin levels (as discussed below) may contribute additional metabolic dysfunction to such a feed-forward process.
Following birth, infant T-exposed female monkeys are hyper-androgenic [27]. Their endogenous hyperandrogenism may originate from ovarian production, potentially driven by high circulating LH levels acting on LH receptors in the ovary [54]. The early onset of this hyperandrogenic function in young female, T-exposed monkeys emulates increased 5 alpha-reductase activity found in 1–3 year old daughters of women with PCOS [7], suggesting comparably developmental onsets of PCOS-like androgenic activity in female monkeys and humans. The ovaries of infant T-exposed monkeys, however, are not polycystic and contain age-appropriate numbers of follicles, but exhibit diminished follicle commitment to growth (unpublished results) suggestive of disrupted follicle development. By adolescence, prior fetal T exposure delays menarche and promotes luteal phase insufficiency in initial menstrual cycles compared to controls [55], suggesting a perturbed hypothalamic-pituitary-ovarian axis at the onset of reproductive maturity in PCOS-like animals.
In contrast to female infant monkeys that experienced T excess during early-to-mid gestation, 40% of similarly T-exposed females when adult exhibit large, polycystic ovaries [18] with accompanying ovarian [56] and adrenal [57] hyperandrogenism combined with intermittent or absent menstrual cycles [18]. Increased body mass index (BMI) exaggerates absence of menstrual cycles [58]. The adult T-exposed monkeys thus show all the diagnostic hallmarks of women with PCOS, including the diversity of phenotypes defined by NIH [59], “Rotterdam” [60] or Androgen Excess-PCOS Society criteria [61]. When monkey equivalents of the PCOS diagnostic criteria are applied to individual T-exposed adult female macaques [62], 42% exhibit classic PCOS (combining the two PCOS phenotypes: hyperandrogenism + intermittent/absent cycles + polycystic ovaries; and hyperandrogenism + intermittent/absent cycles) and 25% show the milder two forms (hyperandrogenism + polycystic ovaries; and intermittent/absent cycles + polycystic ovaries). Surprisingly, phenotyping these monkeys as PCOS-like accounts for only 67% of T-exposed adult females. Neithermild variations in duration of T exposure [23, 62], and elevation of circulating fetal T levels [27], nor reliable responses of androgen sensitive fetal tissues [23], explain the absence of PCOS-like phenotypes in 33% of T-exposed females or differences in PCOS-like phenotypic expression. Different maternal responses to T-exposure (see Metabolic-related section, below) and/or differences in fetal female genome may be key modifiers of endogenous or exogenous fetal T exposure. Such phenotypic diversity from a discrete, homogenous gestational elevation of T may reflect multiple “hits” contributing to the development of adult disease: namely, genetic variation upon which epigenetic reprogramming from gestational T or glucose excess (see below) promotes susceptibility to PCOS-like phenotypic expression in the presence of increased postnatal weight gain. Animal and human studies are increasingly implicating such complex developmental origins.
Metabolic-Related
In addition to gestational T exposure, fetal T-exposed female monkeys experience transient, mid-gestational hyperglycemia from their dams’ diminished ability to regulate glucose [24], a trait emulated by T-treated pregnant ewes that deliver T-exposed female lambs [25]. As might be expected from such gestational hyperglycemia, fetal female size increases, along with a ~5% enlarged ultrasonography-assessed head diameter, elevated late gestation circulating insulin levels (exceeding the control range in 33–55% of cases), and abnormal fetal lipids levels [24]. Gestational T exposure per se, however, may also induce fetal hyperinsulinemia, since direct injections of T into fetal female sheep induce hyperinsulinemic responses to glucose [63], and beta cells of such T-exposed fetal females express androgen receptors [63]. These findings raise the possibility of combined contributions from fetal T excess and gestational hyperglycemia on subsequent development of female offspring hyperinsulinemia, as illustrated in Fig. 2.
Consistent with these findings in T-exposed female monkeys, pre- and peripubertal daughters of women with PCOS exhibit hyperinsulinemic responses to glucose when compared to peers born to women without PCOS [64–66]. Pancreatic decompensation [67, 68] and insulin resistance become obvious in PCOS daughters later in puberty [68, 69], but may have been undetected at a younger age due to the absence of dynamic testing.
As expected in hyperglycemic pregnancies, 55% of newborn T-exposed infants are transiently hypoglycemic, exhibit relative hyperinsulinemia [24], and demonstrate a greater proportion of beta-to-alpha cells in their pancreatic islets, the latter positively correlated with infant insulin levels [70]. Taken together, these results suggest onset of abnormal pancreatic islet beta cell function and morphology in infancy that precedes PCOS-like insulin-related defects in adulthood [71]. Not surprisingly, relative hyperinsulinemia in hyperandrogenic T-exposed infants that are insulin sensitive accelerates the increase in body weight over time [24].
At menarche, T-exposed females are older and heavier than contemporary controls [55]. In adulthood, T-exposed adult female monkeys exhibit preferential accumulation of visceral fat without differences in BMI [72, 73], DNA methylation changes in visceral fat gene promoter sites [41], and a defect in subcutaneous abdominal adipogenesis [74]. Taken together, these results suggest a diminished ability to safely store lipid in subcutaneous adipose depots, resulting in dyslipidemia from lipotoxicity with insulin resistance, pancreatic beta cell and islet morphology defects, as well as type 2 diabetes mellitus [66–68, 70]. Such adult female metabolic dysfunction may result from a combination of gestational hyperglycemia and T excess promoting postnatal insulin-driven fat storage, in parallel with T-constrained subcutaneous adipogenesis, leading to lipotoxocity and metabolic dysfunction. Fig. 2 illustrates this metabolically-related programming in our experimentally induced, T-exposed monkeys, with an emphasis on hyperinsulinemia and T excess as causal factors. The development of hyperinsulinemia from insulin resistance with reduced hypothalamic steroid negative feedback leading to LH hypersecretion would constrain through androgen excess the capacity of normal adipose to safely store fat, leading to lipotoxicity, metabolic dysfunction and reproductive PCOS-like traits, as has also been proposed for girls who will likely manifest PCOS [66, 68]. The combination of reproductive and metabolic developmental origins may thus comprise a more complete fetal origin for PCOS, since gestational diabetes, common among pregnant women with PCOS [77, 78], increases the incidence of obesity-related insulin resistance in offspring [79, 80], but not the full PCOS phenotype. Interestingly, recent exploration of molecular pathogenesis underlying LH signaling dysfunction in PCOS also implicates dysfunctional insulin signaling, as illustrated in Fig. 3 modified from [40], suggesting that molecular bases for PCOS-like phenotypes among women, and possibly monkeys, may have common elements. Weight gain linked to dyslipidemia is certainly part of a hyperandrogenic adolescent progression into PCOS in women [82].
Pre-Pubertal Exposure to Exogenous High T
One NHP model, employing a pre-pubertal onset of exogenous high T treatment, induces transient LH hypersecretion and increased body weight, but without accompanying ovarian hyperandrogenism, menstrual cycle dysfunction and altered glucose regulation [83]. Subsequent addition of a high fat diet, increases body weight in T-exposed and control females, but increases ovarian follicle recruitment in T-exposed females alone [19]. Such polyfollicular ovarian presentation, however, occurs without ovarian hypersecretion of AMH [83], in contrast to elevated AMH levels found in PCOS women and their daughters [84, 85]. At present, it is unclear whether initiating such exposure to high T preceding puberty produces mild aspects of PCOS-like traits or merely induces anabolic hormone-like disruption of female physiology.
Translation Relevance of NHP High T Female Models to PCOS
One major translational issue exists, however, between NHP models and curative therapies in humans. At present, we cannot safely obtain blood samples from human fetuses during mid-gestation in order to confirm high circulating T levels in girls who will develop PCOS as adults [71]. So our ability to link fetal T exposure with development of PCOS in women remains unattainable, and constrains development of a preventive health-care strategy. Therefore, since current clinical practice has yet to develop safe and routine methods for evaluation of the human fetal hormonal milieu, investigation of developmental origins of PCOS in humans has relied on indirect assessments or postnatal outcomes considered as biomarkers of fetal T excess. In this regard, anogenital distance and lengthened 2D: 4D finger length ratio, biomarkers of mid-gestation fetal T exposure characteristic of T-exposed female monkeys [23], are associated with PCOS [44, 49–50]. Naturally occurring high T female monkeys also exhibit an association between circulating T levels and anogenital distance [22]. These reports increasingly provide circumstantial evidence for mid-gestation androgen excess in PCOS pregnancies.
In addition, since pregnant PCOS women have elevated circulating T levels [86, 87], subtle reductions in placental aromatase [88] may expose female offspring to elevated T during gestation. Interestingly, elevated mid-gestation serum T levels in PCOS mothers predict in their adolescent daughters elevated levels of AMH, a transforming growth factor-β (TGF-β) superfamily protein normally produced by granulosa cells of preantral and small antral ovarian follicles [89]. As elevated AMH levels are characteristic of adolescents and women with PCOS [84, 90] and newborn daughters of PCOS women [85], such high AMH levels may represent a cross-generational outcome of hyperandrogenism on the development of PCOS in daughters. Mid-gestational human female fetuses may generate their own T excess (Fig. 1) as their ovaries become capable of producing [46] and responding [47] to androgens, as evidenced by elevated mid-gestational amniotic fluid levels of T in fetal daughters of women with PCOS [45].
Fig. (1).
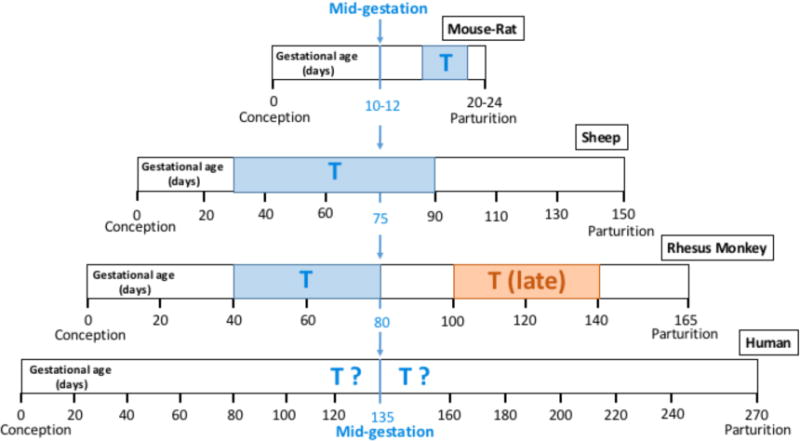
Diagrammatic representation of gestational ages at fetal T exposure resulting in PCOS-like traits (blue boxes with T) in female mice and rats [11–13], sheep [14, 15] and rhesus monkeys [16–18]. All gestational periods are aligned at mid-gestation. Effective T exposure drifts progressively earlier in gestation when when comparing rodents to sheep to monkeys, reflecting relatively earlier onset of organ differentiation with respect to both mid-gestation and parturition in sheep and monkeys. In monkeys, however, late gestation T exposure (brown box with “T (late)”) produces a sufficient degree of PCOS-like phenotype to suggest that, at least in primates, the gestational developmental window for T programming of PCOS-like traits may span either side of mid-gestation [17, 18]. The hypothesized mid-gestational ages during which girls may be vulnerable to fetal T exposure contributing to PCOS development (blue “T?”) avoid the potential for obvious genital virilization in early gestation (ambiguous genitalia are rare in newborn daughters of women with PCOS), are consistent with occurrence of high T levels in PCOS daughters during mid-gestation [45] and with the ability of the human fetal ovary to synthesize and respond to T [46, 47], and take into account the continuing vulnerability for T exposure inducing PCOS-like attributes beyond mid-gestation [17].
Perinatal studies vary in their support of gestational T exposure as a fetal programming origin for PCOS, possibly because onset of labor variably reduces T levels in umbilical cord blood [91]. In studies of umbilical cord venous blood levels in newborn daughters born to women with PCOS, one study shows elevated T levels [92], whereas two studies show reduced androstenedione levels [88, 93] and one shows normal T levels [87]. In a fourth study involving adolescent girls diagnosed with PCOS, and including an inherently high prevalence (~28%) of diagnosis at this young age, umbilical cord blood shows no elevation in T levels [94]. With the fetal ovary as a key site for gestational T excess during mid-gestational target tissue differentiation [44, 45], studies of infants at birth are likely to be too late to detect any remaining hormonal differences [95]. Quantification of androgens in the scalp hair of newborn, however, holds promise for discerning prevailing fetal androgen levels during the third trimester [96] and a more relevant measure of fetal T levels.
Overall, the current evidence from NHP and non-primate models, together with human studies, implicates the gestational environment in the ontogeny of PCOS. Such a hypothesis supports a long-standing developmental origins basis for PCOS [58, 97, 98] that may provide key opportunities to implement early intervention.
Therapeutic Translation from Animal Models to Treatment of PCOS Women
To date, no NHP studies have attempted prenatal or pre-pubertal therapies to prevent or ameliorate PCOS-like outcomes. Therapeutic treatment of adult T-exposed females, however, has been conducted using six months of daily insulin sensitizer (PPAR-gamma agonist, pioglitazone). Such treatment of T-exposed females with insulin sensitizer restores ovulatory menstrual cycles, diminishes ovarian androgenic responses, improves insulin-mediated glucose regulation without weight gain, but does not correct LH hypersecretion [99]. Impaired insulin signaling thus underlies both impaired glucose regulation and ovarian dysfunction, as found in PCOS women [100] and T-exposed ewes [101]. Notably, hypothalamic dysregulation of LH release may not involve a component of impaired insulin signaling in T-exposed monkeys, and pioglitazone is inconsistent in its ameliorating effects on LH hypersecretionin PCOS women [102, 103]. This potential separation of dysfunctional components of PCOS pathogenesis is reflected in the hypothetical mechanism illustrated in Fig. 2.
Prenatal therapies, however, have been attempted in sheep, employing anti-androgen (androgen receptor antagonist, flutamide) and insulin sensitizer (PPARgamma agonist, rosiglitazone) administration to ewe dams receiving T injections during early-to-mid gestation. In T-exposed female lambs, prenatal anti-androgen normalizes anogenital distance, prevents early onset of puberty, normalizes LH surge responses to estrous synchronization in the first breeding season, but does not ameliorate metabolic abnormalities [15, 25]. In comparison, insulin sensitizer prenatal treatment prevents insulin resistance and normalizes onset of puberty. As both prenatal treatments normalize puberty onset, perturbed androgenic and metabolic aspects of the fetal environment may regulate hypothalamic-mediated onset of reproductive maturity.
Proposing to treat pregnant PCOS women with anti-androgen or insulin sensitizers, however, are not attractive options. Anti-androgens compromise male fetal development and development of normal female behavior [104]. Insulin sensitizers, such as pioglitazone and rosiglitazone, are potential teratogens [105]. The insulin sensitizer, metformin, while a class B drug categorized as safe for pregnant women, has raised concerns regarding increased infant weight gain and elevated basal glucose levels in pre-adolescent daughters of PCOS women [106, 107], although high AMH and estradiol levels are normalized in infant daughters of PCOS women who received metformin throughout pregnancy [85]. Evaluating efficacy of prenatal treatments in ameliorating PCOS-like outcomes in NHP models could provide timely translatable findings (in ~5–8 years) without risk to future human generations.
A safer approach involves pre-pubertal intervention to ameliorate PCOS onset. In T-exposed sheep, anti-androgen or metformin treatments commencing before puberty ameliorate aspects of reproductive dysfunction, including precocious puberty [53]. Not surprisingly, insulin sensitizer treatment alone or combined with anti-androgen, has proved efficacious in ameliorating reproductive and metabolic dysfunction in adolescent girls suspected of having PCOS or demonstrating hyperandrogenic anovulation [108, 109]. Early clinical intervention in adolescents suspected of having PCOS, however, needs to be delayed until at least 2 years after menarche due to the transient changes in ovarian function that accompany the onset of puberty [1, 110]. Early intervention, however, may be the key to efficacious prevention of PCOS pathogenesis illustrated in Fig. 2. In this respect, beneficial outcomes following therapeutic reversal of adiposity and insulin resistance in adolescent girls with hyperinsulinemic hyperandrogenism (attributes that commonly precede PCOS) are encouraging. One year of combined anti-androgen and insulin sensitizer treatment enabled at least 9–12 months (study ongoing) of normal, ovulatory menstrual cycles following cessation of treatment. In contrast, onset of anovulation followed cessation of an identical duration of oral contraceptive therapy in a comparable hyperinsulinemic and hyperandrogenic peer group [111].
CONCLUSION
NHP, sheep and many rodent models, identify T excess during fetal life as a reliable developmental origin for PCOS-like traits. Recent genetic and morphological studies of PCOS women and their daughters have also implicated developmental origins, fetal T excess and potentially epigenetic changes in the etiology of PCOS. Future studies employing individual genotyping of NHPs with naturally occurring high T and PCOS-like traits may shed novel insight on molecular mechanisms contributing to the pathogenesis of PCOS.
Acknowledgments
Funded, in part, by NIH grants P50 HD044405 (PI: Dunaif), P50 HD028934 (PI: Marshall), P50 HD071836 (PI: Stouffer) and a competitive supplement to P51 OD011106 (PI: Mallick).
Biography

D.H. Abbott
Footnotes
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
DISCLAIMER: The above article has been published in Epub (ahead of print) on the basis of the materials provided by the author. The Editorial Department reserves the right to make minor modifications for further improvement of the manuscript.
References
- 1.Dumesic DA, Oberfield SE, Stener-Victorin E, Marshall JC, Laven JS, Legro RS. Scientific statement on the diagnostic criteria, epidemiology, pathophysiology, and molecular genetics of polycystic ovary syndrome. Endocr Rev. 2015;36:487–525. doi: 10.1210/er.2015-1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carmina E, Oberfield SE, Lobo RA. The diagnosis of polycystic ovary syndromein adolescents. Am J Obstet Gynecol. 2010;203:201.e1–5. doi: 10.1016/j.ajog.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 3.Witchel SF, Oberfield S, Rosenfield RL, et al. The diagnosis of polycystic ovary syndrome during adolescence. Horm Res Paediatr. 2015 Apr 1; doi: 10.1159/000375530. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 4.Wild RA, Carmina E, Diamanti-Kandarakis E, et al. Assessment of cardiovascular risk and prevention of cardiovascular disease in women with the polycystic ovary syndrome: a consensus statement by the Androgen Excess and Polycystic Ovary Syndrome (AE-PCOS) Society. J Clin Endocrinol Metab. 2010;95:2038–49. doi: 10.1210/jc.2009-2724. [DOI] [PubMed] [Google Scholar]
- 5.Dumesic DA, Lobo RA. Cancer risk and PCOS. Steroids. 2013;78:782–5. doi: 10.1016/j.steroids.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 6.Dunaif A. Perspectives in Polycystic Ovary Syndrome: From Hair to Eternity. J Clin Endocrinol Metab. 2016;101:759–68. doi: 10.1210/jc.2015-3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Torchen LC, Idkowiak J, Fogel NR, et al. Evidence for increased 5α-reductase activity during early childhood in daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2016 Mar 18;:jc20153926. doi: 10.1210/jc.2015-3926. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Azziz R. PCOS in 2015: New insights into the genetics of polycystic ovary syndrome. Nat Rev Endocrinol. 2016;12:74–5. doi: 10.1038/nrendo.2015.230. [DOI] [PubMed] [Google Scholar]
- 9.Hayes MG, Urbanek M, Ehrmann DA, et al. Genome-widea ssociation of polycystic ovary syndrome implicates alterations in gonadotropin secretion in European ancestry populations. Nat Commun. 2015;6:7502. doi: 10.1038/ncomms8502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones MR, Brower MA, Xu N, et al. Systems genetics reveals the functional context of PCOS Loci and Identifies genetic and molecular mechanisms of disease heterogeneity. PLoS Genet. 2015;11:e1005455. doi: 10.1371/journal.pgen.1005455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walters KA. Androgens in polycystic ovary syndrome: lessons from experimental models. Curr Opin Endocrinol Diabetes Obes. 2016 Feb 10; doi: 10.1097/MED.0000000000000245. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 12.Caldwell AS, Eid S, Kay CR, et al. Haplosufficient genomic androgen receptor signaling is adequate to protect female mice from induction of polycystic ovary syndrome features by prenatal hyperandrogenization. Endocrinology. 2015;156:1441–52. doi: 10.1210/en.2014-1887. [DOI] [PubMed] [Google Scholar]
- 13.Foecking EM, McDevitt MA, Acosta-Martínez M, Horton TH, Levine JE. Neuroendocrine consequences of androgen excess in female rodents. Horm Behav. 2008;53:673–92. doi: 10.1016/j.yhbeh.2007.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puttabyatappa M, Cardoso RC, Padmanabhan V. Effect of maternal PCOS and PCOS-like phenotype on the offspring’s health. Mol Cell Endocrinol. 2015 Nov 27; doi: 10.1016/j.mce.2015.11.030. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cardoso RC, Puttabyatappa M, Padmanabhan V. Steroidogenic versus metabolic programming of reproductive neuroendocrine, ovarian and metabolic dysfunctions. Neuroendocrinology. 2015;102:226–37. doi: 10.1159/000381830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dumesic DA, Goodarzi MO, Chazenbalk GD, Abbott DH. Intrauterine environment and polycystic ovary syndrome. Semin Reprod Med. 2014;32:159–65. doi: 10.1055/s-0034-1371087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abbott DH, Nicol LE, Levine JE, Xu N, Goodarzi MO, Dumesic DA. Nonhuman primate models of polycystic ovary syndrome. Mol Cell Endocrinol. 2013;373:21–8. doi: 10.1016/j.mce.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abbott DH, Barnett DK, Bruns CM, Dumesic DA. Androgen excess fetal programming of female reproduction: a developmental aetiology for polycystic ovary syndrome? Hum Reprod Update. 2005;11:357–74. doi: 10.1093/humupd/dmi013. [DOI] [PubMed] [Google Scholar]
- 19.McGee WK, Bishop CV, Pohl CR, et al. Effects of hyperandrogenemia and increased adiposity on reproductive and metabolic parameters in young adult female monkeys. Am J Physiol Endocrinol Metab. 2014;306:E1292–304. doi: 10.1152/ajpendo.00310.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arifin E, Shively CA, Register TC, Cline JM. Polycystic ovary syndrome with endometrial hyperplasia in a cynomolgus monkey (Macaca fascicularis) Vet Pathol. 2008;45:512–5. doi: 10.1354/vp.45-4-512. [DOI] [PubMed] [Google Scholar]
- 21.Abbott DH, Dumesic DA, Rayome BH, et al. Clustering of PCOS-like traits in female macaques. Endocr Rev. 2016;37(2, Suppl):SUN-161. [Google Scholar]
- 22.Abbott DH, Dumesic DA, Lewis KC, et al. Naturally occurring hyperandrogenism and intermittent menstrual cycles in female rhesus monkeys. Endocr Rev. 2012;33(3, Suppl):MON-16. [Google Scholar]
- 23.Abbott AD, Colman RJ, Tiefenthaler R, Dumesic DA, Abbott DH. Early-to-midgestation fetal testosterone increases right hand 2D: 4D finger length ratio in polycystic ovary syndrome-like monkeys. PLoS One. 2012;7:e42372. doi: 10.1371/journal.pone.0042372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abbott DH, Bruns CR, Barnett DK, et al. Experimentally induced gestational androgen excess disrupts glucoregulationin rhesus monkey dams and their female offspring. Am J Physiol Endocrinol Metab. 2010;299:E741–51. doi: 10.1152/ajpendo.00058.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cardoso RC, Veiga-Lopez A, Moeller J, et al. developmental programming: impact of gestational steroid and metabolic milieus on adiposity and insulin sensitivity in prenatal testosterone-treated female sheep. Endocrinology. 2016;157:522–35. doi: 10.1210/en.2015-1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Veiga-Lopez A, Steckler TL, Abbott DH, et al. Developmental programming: impact of excess prenatal testosterone on intrauterine fetal endocrine milieu and growth in sheep. Biol Reprod. 2011;84:87–96. doi: 10.1095/biolreprod.110.086686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abbott DH, Barnett DK, Levine JE, et al. Endocrine antecedents of polycystic ovary syndrome in fetal and infant prenatally androgenized female rhesus monkeys. Biol Reprod. 2008;79:154–63. doi: 10.1095/biolreprod.108.067702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ellinwood WE, Baughman WL, Resko JA. The effects of gonadectomy and testosterone treatment on luteinizing hormone secretion in fetal rhesus monkeys. Endocrinology. 1982;110:183–9. doi: 10.1210/endo-110-1-183. [DOI] [PubMed] [Google Scholar]
- 29.Steiner RA, Clifton DK, Spies HG, Resko JA. Sexual differentiation and feedback control of luteinizing hormone secretion in the rhesus monkey. Biol Reprod. 1976;15:206–12. doi: 10.1095/biolreprod15.2.206. [DOI] [PubMed] [Google Scholar]
- 30.Dumesic DA, Schramm RD, Peterson E, Paprocki AM, Zhou R, Abbott DH. Impaired developmental competence of oocytes in adult prenatally androgenized female rhesus monkeys undergoing gonadotropin stimulation for in vitro fertilization. J Clin Endocrinol Metab. 2002;87:1111–9. doi: 10.1210/jcem.87.3.8287. [DOI] [PubMed] [Google Scholar]
- 31.Moore AM, Prescott M, Marshall CJ, Yip SH, Campbell RE. Enhancement of arobust arcuate GABAergic input to gonadotropin-releasing hormone neurons in amodel of polycystic ovarian syndrome. Proc Natl Acad Sci USA. 2015;112:596–601. doi: 10.1073/pnas.1415038112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sarma HN, Manikkam M, Herkimer C, et al. Fetal programming: excess prenatal testosterone reduces postnatal luteinizing hormone, but not follicle-stimulating hormone responsiveness, toestradiol negative feedback in the female. Endocrinology. 2005;146:4281–91. doi: 10.1210/en.2005-0322. [DOI] [PubMed] [Google Scholar]
- 33.Catteau-Jonard S, Dewailly D. Pathophysiology of polycystic ovary syndrome: the role of hyperandrogenism. Front Horm Res. 2013;40:22–7. doi: 10.1159/000341679. [DOI] [PubMed] [Google Scholar]
- 34.Hayes MG, Urbanek M, Ehrmann DA, et al. Genome-wide association of polycystic ovary syndrome implicates alterations in gonadotropin secretion in European ancestry populations. Nat Commun. 2015;6:7502. doi: 10.1038/ncomms8502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cui L, Zhao H, Zhang B, et al. Genotype-phenotype correlations of PCOS susceptibility SNP sidentified by GWAS in a large cohort of Han Chinese women. Hum Reprod. 2013;28:538–44. doi: 10.1093/humrep/des424. [DOI] [PubMed] [Google Scholar]
- 36.Tian Y, Zhao H, Chen H, et al. Variants in FSHB are associated with polycystic ovary syndrome and luteinizing hormonelevel in Han Chinese women. J Clin Endocrinol Metab. 2016 Mar 3; doi: 10.1210/jc.2015-3776. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 37.Chen ZJ, Zhao H, He L, et al. Genome-wide association study identifies susceptibility loci for polycystic ovary syndrome on chromosome 2p16.3, 2p21 and 9q33.3. Nat Genet. 2011;43:55–9. doi: 10.1038/ng.732. [DOI] [PubMed] [Google Scholar]
- 38.Mutharasan P, Galdones E, Peñalver Bernabé B, et al. Evidence for chromosome 2p16.3polycystic ovary syndrome susceptibility locus in affected women of Europeanancestry. J Clin Endocrinol Metab. 2013;98:E185–90. doi: 10.1210/jc.2012-2471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McAllister JM, Modi B, Miller BA, et al. Overexpression of a DENND1A isoform produces a polycystic ovary syndrometheca phenotype. Proc Natl Acad Sci USA. 2014;111:E1519–27. doi: 10.1073/pnas.1400574111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McAllister JM, Legro RS, Modi BP, Strauss JF., 3rd Functional genomics of PCOS: from GWAS to molecular mechanisms. Trends Endocrinol Metab. 2015;26:118–24. doi: 10.1016/j.tem.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu N, Kwon S, Abbott DH, et al. Epigenetic mechanism underlying the development of polycystic ovary syndrome(PCOS)-like phenotypes in prenatally androgenized rhesus monkeys. PLoS One. 2011;6:e27286. doi: 10.1371/journal.pone.0027286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Veiga-Lopez A, Moeller J, Abbott DH, Padmanabhan V. Developmental programming: Rescuing disruption in preovulatory follicle growth and steroidogenesis from prenatal testosterone disruption. J Ovarian Res. doi: 10.1186/s13048-016-0250-y. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blank SK, McCartney CR, Marshall JC. The origins and sequelae of abnormal neuroendocrine function in polycystic ovary syndrome. Hum Reprod Update. 2006;12:351–61. doi: 10.1093/humupd/dml017. [DOI] [PubMed] [Google Scholar]
- 44.Barrett E, Hoeger K. Anogenital distance, A biomarker of prenatal androgen exposure, is longer among newborn daughters of women with polycystic ovary syndrome (PCOS) Endocr Rev. 2016;37(2, Suppl):SAT 182. [Google Scholar]
- 45.Palomba S, Marotta R, Di Cello A, et al. Pervasive developmental disorders in children of hyperandrogenic women with polycystic ovary syndrome: a longitudinal case-controlstudy. Clin Endocrinol (Oxf) 2012;77:898–904. doi: 10.1111/j.1365-2265.2012.04443.x. [DOI] [PubMed] [Google Scholar]
- 46.Cole B, Hensinger K, Maciel GA, Chang RJ, Erickson GF. Human fetal ovary development involves the spatiotemporal expression of p450c17 protein. J Clin Endocrinol Metab. 2006;91:3654–61. doi: 10.1210/jc.2006-0641. [DOI] [PubMed] [Google Scholar]
- 47.Fowler PA, Anderson RA, Saunders PT, et al. Development of steroid signaling pathways during primordial follicle formation in the human fetal ovary. J Clin Endocrinol Metab. 2011;96:1754–62. doi: 10.1210/jc.2010-2618. [DOI] [PubMed] [Google Scholar]
- 48.Ellinwood WE, McClellan MC, Brenner RM, Resko JA. Estradiol synthesis by fetal monkey ovaries correlates with antral follicle formation. Biol Reprod. 1983;28:505–16. doi: 10.1095/biolreprod28.2.505. [DOI] [PubMed] [Google Scholar]
- 49.Lujan ME, Bloski TG, Chizen DR, Lehotay DC, Pierson RA. Digit ratios do not serve as anatomical evidence of prenatal androgen exposure in clinical phenotypes of polycystic ovary syndrome. Hum Reprod. 2010;25:204–11. doi: 10.1093/humrep/dep363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lujan ME, Podolski AJ, Chizen DR, Lehotay DC, Pierson RA. Digit ratios by computer-assisted analysis confirm lack of anatomical evidence of prenatal androgen exposure in clinical phenotypes of polycystic ovary syndrome. Reprod Biol Endocrinol. 2010;8:156. doi: 10.1186/1477-7827-8-156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cattrall FR, Vollenhoven BJ, Weston GC. Anatomical evidence for in utero androgen exposure in women with polycystic ovary syndrome. Fertil Steril. 2005;84:1689–92. doi: 10.1016/j.fertnstert.2005.05.061. [DOI] [PubMed] [Google Scholar]
- 52.Barker DJ. The origins of the developmental origins theory. J Intern Med. 2007;261:412–7. doi: 10.1111/j.1365-2796.2007.01809.x. [DOI] [PubMed] [Google Scholar]
- 53.Padmanabhan V, Cardoso RC, Puttabyatappa M. Developmental programming, a pathway to disease. Endocrinology. 2016 Feb 9; doi: 10.1210/en.2016-1003. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Comim FV, Teerds K, Hardy K, Franks S. Increased protein expression of LHCG receptor and 17α-hydroxylase/17–20-lyase in human polycystic ovaries. Hum Reprod. 2013;28:3086–92. doi: 10.1093/humrep/det352. [DOI] [PubMed] [Google Scholar]
- 55.Goy RW, Robinson JA. Prenatal exposure of rhesus monkeys to patent androgens: morphological, behavioral, and physiological consequences. In: Hunt VR, Smith MK, Worth D, editors. Banbury Report II: Environmental factors in human growth and development. Cold Spring Harbor: Cold Spring Harbor Laboratory; 1982. pp. 355–78. [Google Scholar]
- 56.Eisner JR, Barnett MA, Dumesic DA, Abbott DH. Ovarian hyperandrogenism inadult female rhesus monkeys exposed to prenatal androgen excess. Fertil Steril. 2002;77:167–72. doi: 10.1016/s0015-0282(01)02947-8. [DOI] [PubMed] [Google Scholar]
- 57.Zhou R, Bird IM, Dumesic DA, Abbott DH. Adrenal hyperandrogenism is induced by fetal androgen excess in a rhesus monkey model of polycystic ovary syndrome. J Clin Endocrinol Metab. 2005;90:6630–7. doi: 10.1210/jc.2005-0691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Abbott DH, Dumesic DA, Eisner JR, Colman RJ, Kemnitz JW. Insights into the development of polycystic ovary syndrome (PCOS) from studies of prenatally androgenized female rhesus monkeys. Trends Endocrinol Metab. 1998;9:62–7. doi: 10.1016/s1043-2760(98)00019-8. [DOI] [PubMed] [Google Scholar]
- 59.Zawadski JK, Dunaif A. Diagnostic criteria for polycystic ovary syndrome: towards a rational approach. In: Dunaif A, Givens JR, Haseltine FP, Merriam GR, editors. Polycystic Ovary Syndrome. Boston: Blackwell Scientific Publications; 1992. pp. 377–84. [Google Scholar]
- 60.Fauser BC, Tarlatzis BC, Rebar RW, et al. Consensus on women’s health aspects of polycystic ovary syndrome (PCOS): the Amsterdam ESHRE/ASRM-Sponsored 3rd PCOS Consensus Workshop Group. Fertil Steril. 2012;97:28–38.e25. doi: 10.1016/j.fertnstert.2011.09.024. [DOI] [PubMed] [Google Scholar]
- 61.Azziz R, Carmina E, Dewailly D, et al. Task Force on the Phenotype of the Polycystic Ovary Syndrome of The Androgen Excess and PCOS Society. The Androgen Excess and PCOS Society criteria for the polycystic ovary syndrome: the complete task force report Fertil Steril. 2009;91:456–88. doi: 10.1016/j.fertnstert.2008.06.035. [DOI] [PubMed] [Google Scholar]
- 62.Abbott DH, Tarantal AF, Dumesic DA. Fetal, infant, adolescent and adult phenotypes of polycystic ovary syndrome in prenatally androgenized female rhesus monkeys. Am J Primatol. 2009;71:776–84. doi: 10.1002/ajp.20679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rae M, Grace C, Hogg K, et al. The pancreas is altered by in utero androgen exposure: implications for clinical conditions such as polycystic ovary syndrome (PCOS) PLoS One. 2013;8:e56263. doi: 10.1371/journal.pone.0056263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sir-Petermann T, Codner E, Pérez V, et al. Metabolic and reproductive features before and during puberty in daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2009;94:1923–30. doi: 10.1210/jc.2008-2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Maliqueo M, Galgani JE, Pérez-Bravo F, et al. Relationship of serum adipocyte-derived proteins with insulin sensitivity and reproductive features in pre-pubertal and pubertal daughters of polycystic ovary syndrome women. Eur J Obstet Gynecol Reprod Biol. 2012;161:56–61. doi: 10.1016/j.ejogrb.2011.12.012. [DOI] [PubMed] [Google Scholar]
- 66.Kent SC, Gnatuk CL, Kunselman AR, Demers LM, Lee PA, Legro RS. Hyperandrogenism and hyperinsulinism in children of women with polycystic ovary syndrome: a controlled study. J Clin Endocrinol Metab. 2008;93:1662–9. doi: 10.1210/jc.2007-1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Torchen LC, Fogel NR, Brickman WJ, Paparodis R, Dunaif A. Persistent apparent pancreatic β-cell defects in premenarchal PCOS relatives. J Clin Endocrinol Metab. 2014;99:3855–62. doi: 10.1210/jc.2014-1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Trottier A, Battista MC, Geller DH, et al. Adipose tissue insulin resistance in peripubertal girls with first-degree family history of polycystic ovary syndrome. Fertil Steril. 2012;98:1627–34. doi: 10.1016/j.fertnstert.2012.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Raissouni N, Kolesnikov A, Purushothaman R, et al. Altered glucose disposition and insulin sensitivity in peripubertal first-degreerelatives of women with polycystic ovary syndrome. Int J Pediatr Endocrinol. 2012;2012:14. doi: 10.1186/1687-9856-2012-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nicol LE, O’Brien TD, Dumesic DA, Grogan T, Tarantal AF, Abbott DH. Abnormal infant islet morphology precedes insulin resistance in PCOS-like monkeys. PLoS One. 2014;9:e106527. doi: 10.1371/journal.pone.0106527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Abbott DH, Bacha F. Ontogeny of polycystic ovary syndrome and insulin resistance in utero and early childhood. Fertil Steril. 2013;100:2–11. doi: 10.1016/j.fertnstert.2013.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Eisner JR, Dumesic DA, Kemnitz JW, Colman RJ, Abbott DH. Increased adiposityin female rhesus monkeys exposed to androgen excess during early gestation. Obes Res. 2003;11:279–86. doi: 10.1038/oby.2003.42. [DOI] [PubMed] [Google Scholar]
- 73.Bruns CM, Baum ST, Colman RJ, et al. Prenatal androgen excess negatively impacts body fat distribution in anonhuman primate model of polycystic ovary syndrome. Int J Obes (Lond) 2007;31:1579–85. doi: 10.1038/sj.ijo.0803638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Keller E, Chazenbalk GD, Aguilera P, et al. Impaired preadipocyte differentiation into adipocytes in subcutaneous abdominal adipose of PCOS-like female rhesus monkeys. Endocrinology. 2014;155:2696–703. doi: 10.1210/en.2014-1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Virtue S, Vidal-Puig A. Adipose tissue expandability, lipotoxicity and the Metabolic Syndrome–an allostatic perspective. Biochim Biophys Acta. 2010;1801:338–49. doi: 10.1016/j.bbalip.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 76.Ibáñez L, Ong KK, López-Bermejo A, Dunger DB, de Zegher F. Hyperinsulinaemic androgen excess in adolescent girls. Nat Rev Endocrinol. 2014;10:499–508. doi: 10.1038/nrendo.2014.58. [DOI] [PubMed] [Google Scholar]
- 77.Palomba S, de Wilde MA, Falbo A, Koster MP, La Sala GB, Fauser BC. Pregnancy complications in women with polycystic ovary syndrome. Hum Reprod Update. 2015;21:575–92. doi: 10.1093/humupd/dmv029. [DOI] [PubMed] [Google Scholar]
- 78.Joham AE, Palomba S, Hart R. Polycystic Ovary Syndrome, Obesity, and Pregnancy. Semin Reprod Med. 2016;34:93–101. doi: 10.1055/s-0035-1571195. [DOI] [PubMed] [Google Scholar]
- 79.Spaight C, Gross J, Horsch A, Puder JJ. Gestational Diabetes Mellitus. Endocr Dev. 2016;31:163–78. doi: 10.1159/000439413. [DOI] [PubMed] [Google Scholar]
- 80.Nicholas LM, Morrison JL, Rattanatray L, Zhang S, Ozanne SE, McMillen IC. The early origins of obesity and insulin resistance: timing, programming and mechanisms. Int J Obes (Lond) 2016;40:229–38. doi: 10.1038/ijo.2015.178. [DOI] [PubMed] [Google Scholar]
- 81.McGee WK, Bishop CV, Bahar A, et al. Elevated androgens during puberty in female rhesus monkeys lead to increased neuronal drive to the reproductive axis: a possible component of polycystic ovary syndrome. Hum Reprod. 2012;27:531–40. doi: 10.1093/humrep/der393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ollila MM, Piltonen T, Puukka K, et al. Weight gain and dyslipidemia in early adulthood associate with polycystic ovary syndrome: prospective cohort study. J Clin Endocrinol Metab. 2016;101:739–47. doi: 10.1210/jc.2015-3543. [DOI] [PubMed] [Google Scholar]
- 83.Xu J, McGee WK, Bishop CV, et al. Exposure of female macaques to Western-style diet with or without chronic T in vivo alters secondary follicle function during encapsulated 3-dimensional culture. Endocrinology. 2015;156:1133–42. doi: 10.1210/en.2014-1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pigny P, Merlen E, Robert Y, et al. Elevated serum level of anti-mullerian hormone in patients with polycysticovary syndrome: relationship to the ovarian follicle excess and to the follicular arrest. J Clin Endocrinol Metab. 2003;88:5957–62. doi: 10.1210/jc.2003-030727. [DOI] [PubMed] [Google Scholar]
- 85.Crisosto N, Echiburú B, Maliqueo M, et al. Improvement of hyperandrogenism and hyperinsulinemia during pregnancy in women with polycystic ovary syndrome: possible effect in the ovarian follicular mass of their daughters. Fertil Steril. 2012;97:218–24. doi: 10.1016/j.fertnstert.2011.10.002. [DOI] [PubMed] [Google Scholar]
- 86.Sir-Petermann T, Maliqueo M, Angel B, Lara HE, Pérez-Bravo F, Recabarren SE. Maternal serum androgens in pregnant women with polycystic ovarian syndrome: possible implications in prenatal androgenization. Hum Reprod. 2002;17:2573–9. doi: 10.1093/humrep/17.10.2573. [DOI] [PubMed] [Google Scholar]
- 87.Caanen MR, Kuijper EA, Hompes PG, et al. Mass spectrometry methods measured androgen and estrogen concentrations during pregnancy and in newborns of mothers with polycystic ovary syndrome. Eur J Endocrinol. 2016;174:25–32. doi: 10.1530/EJE-15-0699. [DOI] [PubMed] [Google Scholar]
- 88.Maliqueo M, Lara HE, Sánchez F, Echiburú B, Crisosto N, Sir-Petermann T. Placental steroidogenesis in pregnant women with polycystic ovary syndrome. Eur J Obstet Gynecol Reprod Biol. 2013;166:151–5. doi: 10.1016/j.ejogrb.2012.10.015. [DOI] [PubMed] [Google Scholar]
- 89.Hart R, Sloboda DM, Doherty DA, et al. Circulating maternal testosterone concentrations at 18 weeks of gestation predict circulating levels of antimüllerian hormone in adolescence: aprospective cohort study. Fertil Steril. 2010;94:1544–7. doi: 10.1016/j.fertnstert.2009.12.060. [DOI] [PubMed] [Google Scholar]
- 90.Sopher AB, Grigoriev G, Laura D, et al. Anti-Mullerian hormone may be a useful adjunct in the diagnosis of polycystic ovary syndrome in nonobese adolescents. J Pediatr Endocrinol Metab. 2014;27:1175–9. doi: 10.1515/jpem-2014-0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Keelan JA, Mattes E, Tan H, et al. Androgen concentrations in umbilical cord blood and their association with maternal, fetal and obstetric factors. PLoS One. 2012;7:e42827. doi: 10.1371/journal.pone.0042827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Barry JA, Kay AR, Navaratnarajah R, et al. Umbilical vein testosterone in female infants born to mothers with polycystic ovary syndrome is elevated to male levels. J Obstet Gynaecol. 2010;30:444–6. doi: 10.3109/01443615.2010.485254. [DOI] [PubMed] [Google Scholar]
- 93.Anderson H, Fogel N, Grebe SK, Singh RJ, Taylor RL, Dunaif A. Infants of women with polycystic ovary syndrome have lower cord blood androstenedione andestradiol levels. J Clin Endocrinol Metab. 2010;95:2180–6. doi: 10.1210/jc.2009-2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hickey M, Sloboda DM, Atkinson HC, et al. The relationship between maternal and umbilical cord androgen levels and polycystic ovary syndrome in adolescence: a prospective cohort study. J Clin Endocrinol Metab. 2009;94:3714–20. doi: 10.1210/jc.2009-0544. [DOI] [PubMed] [Google Scholar]
- 95.Beck-Peccoz P, Padmanabhan V, Baggiani AM, et al. Maturation of hypothalamic-pituitary-gonadal function in normal human fetuses: circulating levels of gonadotropins, their common alpha-subunit and free testosterone, and discrepancy between immunologicaland biological activities of circulating follicle-stimulating hormone. J Clin Endocrinol Metab. 1991;73:525–32. doi: 10.1210/jcem-73-3-525. [DOI] [PubMed] [Google Scholar]
- 96.Kapoor A, Lubach G, Hedman C, Ziegler TE, Coe CL. Hormones in infant rhesus monkeys’ (Macaca mulatta) hair at birth provide a window into the fetal environment. Pediatr Res. 2014;75:476–81. doi: 10.1038/pr.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Barnes RB, Rosenfield RL, Ehrmann DA, et al. Ovarian hyperandrogynism as a result of congenital adrenal virilizing disorders: evidence for perinatal masculinization of neuroendocrine function in women. J Clin Endocrinol Metab. 1994;79:1328–33. doi: 10.1210/jcem.79.5.7962325. [DOI] [PubMed] [Google Scholar]
- 98.Abbott DH, Dumesic DA, Franks S. Developmental origin of polycystic ovary syndrome - a hypothesis. J Endocrinol. 2002;174:1–5. doi: 10.1677/joe.0.1740001. [DOI] [PubMed] [Google Scholar]
- 99.Zhou R, Bruns CM, Bird IM, et al. Pioglitazone improves insulin action and normalizes menstrual cycles in a majority of prenatally androgenized female rhesus monkeys. Reprod Toxicol. 2007;23:438–48. doi: 10.1016/j.reprotox.2006.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rice S, Christoforidis N, Gadd C, et al. Impaired insulin-dependent glucose metabolism ingranulosa-lutein cells from anovulatory women with polycystic ovaries. Hum Reprod. 2005;20:373–81. doi: 10.1093/humrep/deh609. [DOI] [PubMed] [Google Scholar]
- 101.Veiga-Lopez A, Lee JS, Padmanabhan V. Developmental programming: insulin sensitizer treatment improves reproductive function in prenatal testosterone-treated female sheep. Endocrinology. 2010;151:4007–17. doi: 10.1210/en.2010-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Glintborg D, Hermann AP, Andersen M, et al. Effect of pioglitazone on glucose metabolism and luteinizing hormone secretion in women with polycystic ovary syndrome. Fertil Steril. 2006;86:385–97. doi: 10.1016/j.fertnstert.2005.12.067. [DOI] [PubMed] [Google Scholar]
- 103.Stabile G, Borrielli I, Artenisio AC, et al. Effects of the insulin sensitizer pioglitazone on menstrual irregularity, insulin resistance and hyperandrogenism in young women with polycystic ovary syndrome. J Pediatr Adolesc Gynecol. 2014;27:177–82. doi: 10.1016/j.jpag.2013.09.015. [DOI] [PubMed] [Google Scholar]
- 104.Wallen K. Hormonal influences on sexually differentiated behavior in nonhuman primates. Front Neuroendocrinol. 2005;26:7–26. doi: 10.1016/j.yfrne.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 105.Valsamakis G, Lois K, Kumar S, Mastorakos G. Metabolic and other effects of pioglitazone as an add-on therapy to metformin in the treatment of polycystic ovary syndrome (PCOS) Hormones (Athens) 2013;12:363–78. doi: 10.1007/BF03401302. [DOI] [PubMed] [Google Scholar]
- 106.Helseth R, Vanky E, Stridsklev S, Vogt C, Carlsen SM. Maternal and fetal insulin levels at birth in women with polycystic ovary syndrome: data from a randomized controlled study on metformin. Eur J Endocrinol. 2014;170:769–75. doi: 10.1530/EJE-13-0859. [DOI] [PubMed] [Google Scholar]
- 107.Carlsen SM, Martinussen MP, Vanky E. Metformin’s effect on first-year weightgain: a follow-up study. Pediatrics. 2012;130:e1222–6. doi: 10.1542/peds.2012-0346. [DOI] [PubMed] [Google Scholar]
- 108.Ibáñez L, Díaz M, Sebastiani G, Marcos MV, López-Bermejo A, de Zegher F. Oral contraception vs insulin sensitization for 18 months in nonobese adolescents with androgen excess: posttreatment differences in C-reactive protein, intima-media thickness, visceral adiposity, insulin sensitivity, and menstrual regularity. J Clin Endocrinol Metab. 2013;98:E902–7. doi: 10.1210/jc.2013-1041. [DOI] [PubMed] [Google Scholar]
- 109.Ibáñez L, López-Bermejo A, del Rio L, Enríquez G, Valls C, de Zegher F. Combined low-dose pioglitazone, flutamide, and metformin for women with androgen excess. J Clin Endocrinol Metab. 2007;92:1710–4. doi: 10.1210/jc.2006-2684. [DOI] [PubMed] [Google Scholar]
- 110.Witchel SF, Oberfield S, Rosenfield RL, et al. The Diagnosis of Polycystic Ovary Syndrome during Adolescence. Horm Res Paediatr. 2015 Apr 1; doi: 10.1159/000375530. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 111.Ibanez L, Diaz M, Sebastiani G, Lopez-Bermejo A, de Zegher E. Early Normalization of Ovulation in the Polycystic Ovary Syndrome. Endocr Rev. 2016;37(2, Suppl):OR21–2. [Google Scholar]
- 112.Sjaarda LA, Mumford SL, Kissell K, et al. Increased androgen, anti-Müllerian hormone, and sporadic anovulation in healthy, eumenorrheic women: a mild PCOS-like phenotype? J Clin Endocrinol Metab. 2014;99:2208–16. doi: 10.1210/jc.2013-3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Barber TM, Golding SJ, Alvey C, et al. Global adiposity rather than abnormal regional fat distribution characterizes women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2008;93:999–1004. doi: 10.1210/jc.2007-2117. [DOI] [PubMed] [Google Scholar]
- 114.Androulakis II, Kandaraki E, Christakou C, et al. Visceral adiposity index (VAI) is related to the severity of anovulation and other clinical features in women with polycystic ovary syndrome. Clin Endocrinol (Oxf) 2014;81:426–31. doi: 10.1111/cen.12447. [DOI] [PubMed] [Google Scholar]