

Abstract
Our laboratory has contributed to the areas of B cell receptor (BCR) and pre-BCR gene identification and transcription and has focused on the problem of the aged immune system in mice and humans for the last 15 years. We have found biomarkers for the decrease in B cell function in aged mice and humans. These include decreases in immunoglobulin (Ig) class switch (e.g., IgM to IgG), decreases in the enzyme AID (activation-induced cytidine deaminase) and decreases in the transcription factor E47. The E47 mRNA stability is decreased in old B cells due to decreased phospho-MAPKinase and phospho-TTP (tristetraprolin). Inflammation, e.g., TNF-α, which increases with age, impacts B cells directly by increasing their TNF-α and NF-κB and leads to the above decreased pathway. Both class switch and affinity maturation are decreased in elderly responses to the influenza vaccine and biomarkers we have found (numbers and percentages of switched memory B cells and AID in stimulated B cells in culture) can predict a beneficial or decreased immune response to the vaccine. Current and future avenues to improve the humoral immune response in the elderly are discussed.
Keywords: Aging, B cells, Transcription factors, Antibody production, Vaccines
Introduction
The impaired humoral immune responses seen with age in mice and humans lead to increased frequency and severity of infectious diseases and are responsible for the reduced vaccine responses observed in elderly individuals [1–5]. The decreased ability of aged individuals to produce high affinity protective antibody/immunoglobulin (Ig) against infectious agents is due to combined defects in T cell signaling to B cells [4, 6, 7], decreases in somatic hypermutation (SHM) and class switch recombination (CSR) in germinal center B cells [8–10], as well as intrinsic VH repertoire shifts [11, 12]. We have characterized intrinsic B cell defects occurring in aging mice [13–17] and humans [18–21] and have identified and characterized several human B cell biomarkers that are reduced by aging. These are activation-induced cytidine deaminase (AID), the enzyme of CSR and SHM, both required for the generation of protective memory B cell responses and a transcription factor E47 which transcriptionally activates aicda, the gene for AID.
Class switch recombination requires chromatin opening of the switch (S) region, upstream (5′) of all heavy chain constant region gene segments except delta, via T cell cytokine-induced germ line transcription. The class switch process requires AID [22, 23], which deaminates cytidine residues in S regions, thus creating uracils, and the resulting mismatches are recognized by specific enzymes and excised, leading to DNA double-strand breaks [22, 24]. In humans, mutations in the AID gene are associated with the absence of secondary antibodies (IgG, IgA, IgE) and SHM and lead to a hyper-IgM syndrome [25], a disease associated with increased vulnerability to infections [26]. AID can also induce point mutations in oncogenes, such as Bcl-6 [27], and DNA double-strand breaks in the Ig heavy chain loci that are recognized as substrates for chromosome translocations [28].
The AID gene, aicda, is transcriptionally regulated by E proteins [29, 30] among others, which are class I basic helix loop helix (bHLH) proteins, first identified based on their ability to bind with relatively high affinity to the palindromic DNA sequence CANNTG, referred to as an E-box site [31–33]. E-boxes have been found in the promoter and enhancer regions of many B lineage-specific genes and regulate a large number of processes involved in B cell commitment and differentiation [34–37]. We have found that E47 and Pax-5 are crucial transcription factors regulating aicda which are down-regulated during aging in mice [17, 38] (Landin et al. manuscript in preparation) and human [21] B cells.
CSR in mice and humans
Mouse studies
In senescent mice, we have shown that splenic B cells, stimulated in vitro with anti-CD40/IL-4 [17], BAFF/IL-4 [14] or lipopolysaccharide (LPS) [39], are deficient in CSR and production of class switched secondary isotypes. This occurs concomitant with decreased induction of E47 and AID and is not a consequence of defective B cell proliferation [40]. Although it is known that there are defects in T as well as B cells during aging, our studies indicate that an intrinsic B cell defect may not be able to be rescued by modifying/enhancing T cell activity alone in aged individuals. Both DNA-binding (measured by EMSA) and expression (measured by Western blot) of E47 are decreased in stimulated splenic B cells from old mice. We have also shown [41] that basal/unstimulated E47 DNA-binding is low, and importantly, twofold lower than that in unstimulated young spleen cells in the majority of aged mice individually tested. Activation of B cells up-regulates E47 DNA-binding in young and to a significantly lower (fivefold) extent in old mice. Therefore, both basal and activated levels of E47 are decreased in splenic B cells in aged mice. These findings suggest that the down-regulation of this transcriptional regulator may help to explain not only decreased CSR in activated splenic B cells from old mice, but also age-related changes in affinity maturation and SHM affecting the quality of the antibody response. Other results from our laboratory showing that CSR is perturbed in E2A heterozygous knockout mice, and even more as they are aged (20 months or more), further support the important role of this transcription factor in the generation of antibodies with different isotypes [17, 42].
In order to determine a mechanism for the age-related decrease in the amounts of E47 protein in nuclear extracts, we found that E47 mRNA levels were decreased in stimulated splenic B cells from old as compared to young mice. RNA stability assays showed that the rate of E47 mRNA decay was accelerated in stimulated splenic B cells from old mice, but E47 protein degradation rates were comparable in young versus aged B cells, indicating that the regulation of E47 expression in activated splenic B cells occurs primarily by mRNA stability [16]. In contrast to splenic-activated B cells, E47 mRNA expression is comparable in bone marrow-derived IL-7-expanded pro-B/early pre-B cells from young and old mice [43], in which the reduced expression and DNA-binding of the E47 transcription factor with age is due to reduced protein stability [43, 44] and is mediated by the ubiquitin–proteasome pathway [45, 46]. This instability is largely due to PEST (proline, glutamic acid, serine, threonine) residues common to degradation domains [47].
A general mechanism for the decreased stability of labile mRNA is accomplished by signal transduction cascades, where the final product of the cascade phosphorylates a protein which interacts with adenylate-/uridylate-rich elements (ARE) in the 3′ untranslated region (UTR) of mRNA and modifies its stability [48, 49]. ARE sequences have been found in the 3′-UTR of many mRNAs, including E47, which contains one AUUUA sequence and multiple AU/U-rich regions. At least part of the decreased stability of E47 mRNA seen in aged B cells is mediated by proteins. We have found that tristetraprolin (TTP), a physiological regulator of mRNA stability, is involved in the degradation of the E47 mRNA. TTP is a zinc finger protein, also known as Nup475, TIS11 and ZFP36. It is a prototypical member of a small family of mammalian proteins with tandem CCCH (CX8CX5CX3H) zinc finger motifs separated by 18-aa residues. It has originally been identified on the basis of its rapid induction in response to a variety of stimuli, such as serum, insulin, platelet-derived growth factor and PMA [50, 51]. TTP KO mice develop a severe inflammatory syndrome, which is largely due to the increased stability of mRNA for TNF-α and the resulting enhanced secretion of the pro-inflammatory cytokine [52, 53]. Our published results [13] show that TTP mRNA and protein levels are higher in stimulated splenic B cells from old as compared to young mice, and these results are consistent with our other results where stimulated aged B cells do make less TNF-α (please see below). TTP is directly phosphorylated by p38 MAPK in macrophages [54, 55]. We have shown that inhibition of the p38 MAPK signaling pathway significantly reduces TTP protein expression in B cells. Old B cells in response to LPS make less phospho-p38 MAPK [13] and therefore, as would be expected, make less phospho-TTP. This leads to an increase in the amount of TTP bound to the 3′-UTRs, and therefore decreased mRNA stability (of E47) in old B cells. Our published studies were the first demonstration that TTP is regulated in activated B cells during aging and that TTP is involved in the degradation of the E47 mRNA, suggesting a molecular mechanism for the decreased expression of E47, AID and CSR in aged B cells [13].
Protein phosphatase 2A (PP2A) is a serine/threonine protein phosphatase that plays an important role in the regulation of a number of signaling pathways. PP2A is a ubiquitously expressed serine/threonine phosphatase composed of a 36-kDa catalytic C unit, a 64-kDa scaffolding A subunit and multiple regulatory B subunits which influence enzyme activity, substrate specificity and subcellular localization [56, 57]. We have shown that not only the amount but also the activity of PP2A is increased in old B cells [15]. As a consequence of this higher phosphatase activity in old B cells, p38 MAPK and TTP (either directly or indirectly by PP2A) are less phosphorylated as compared to young B cells. PP2A dephosphorylation of p38 MAPK and/or TTP likely generates more binding of the hypophosphorylated TTP to the E47 mRNA, inducing its degradation. This mechanism may be at least in part responsible for the age-related decrease in class switch.
More recently, we have shown that it is possible to rescue at least in vitro intrinsic B cell defects in senescence [58]. We first used a retroviral construct containing the DsRED reporter and the 3′-UTR of E47 to confirm that the 3′-UTR drives mRNA degradation, and particularly in B cells from old mice, providing the first demonstration that the E47 3′-UTR directly regulates its degradation. We also showed that young and old primary B cells over-expressing a stable E47 mRNA (without the 3′-UTR) were able to up-regulate E47, AID and CSR and improve B cell immune responses to young levels in senescent murine B cells.
Work in progress in the laboratory is aiming at evaluating the role of another transcription factor, Pax-5, in age-related defects of CSR. We have shown (Landin et al. manuscript in preparation) that over-expression of E47, alone or together with Pax-5, rescues AID levels in old murine B cells to the levels observed in young B cells. Furthermore, lentiviral transduction of B cells with E47 or Pax-5 siRNA leads to decreased AID. We are also in the process of measuring the effects of small molecules from the LOPAC 1280 and other libraries in up-regulation of E47, Pax-5 and AID in cultures of stimulated primary human B cells.
There is a connection, albeit not clearly defined, between deficits in the immune system and increased inflammation with age, which also negatively impacts other biological systems (for example, the neurological and cardiovascular systems). Our recent published novel findings have shown that TNF-α produced by unstimulated aged murine B cells is at least in part responsible for their decreased function and that this can be attenuated/reversed by treatments in vivo and in vitro to decrease TNF-α [39]. Incubation of B cells with TNF-α before LPS stimulation decreased both young and old B cell markers (E47, AID, CSR) and B cell responses. Importantly, B cell function is restored by adding an anti-TNF-α antibody to cultured B cells. An anti-TNF-α antibody given in vivo is also able to increase B cell function in old, but not in young B cells. To address a molecular mechanism, we found that incubation of B cells with TNF-α before LPS stimulation induced TTP.
Human studies
Human peripheral B cells have been shown to decrease in percentages and absolute numbers with age; however, understanding how age affects B cell subsets is complicated by heterogeneity among individuals as well as by the variety of phenotyping approaches employed [21, 59–62]. For example, using CD19, CD27 and IgD antibodies, it is possible to identify four major circulating B cell subsets: naïve (IgD+CD27−), IgM or unswitched memory (IgD+CD27+), switched memory (IgD−CD27+) and exhausted memory or “double negative” (IgD−CD27−). Our results have shown that with age, naïve and exhausted memory B cells increase in percentage but not in numbers, IgM memory B cells do not change and switched memory B cells decrease in both percentages and numbers. Using CD19, CD24 and CD38 antibodies, it is possible to identify the population of transitional B cells (CD24bright CD38bright), on which the effects of age have been controversial [63, 64], as they were reported to be decreased [64] or not [63] by aging, as we have also found (Frasca et al. unpublished).
We have extended our mouse studies to investigate whether aging also affects E47, AID and CSR in B cells isolated from the peripheral blood of human subjects (20–80 years). Our initial studies published a few years ago have shown that the expression of E47, AID and Igγ1 transcripts progressively decreases with age [21]. We wanted to measure whether these markers were important for generating an in vivo response and with that aim we have evaluated E47, AID and CSR in adults and elderly responding to the influenza vaccine. Aging significantly decreases the influenza vaccine-specific response as we [18–20] and others [65–67] have previously shown. We have found [21, 61, 68] that age-related intrinsic defects in B cell function occur in humans as well as in mice and we have identified AID, CSR and switched memory B cells (CD19+CD27+IgD−) as human B cell biomarkers that are reduced by aging. AID and switched memory B cells can help not only to monitor but also to predict quantitatively and qualitatively the in vivo response of an individual to the influenza vaccine and we would suggest also for other antigens and vaccines [19, 20]. Specifically, we have measured the antibody response to influenza vaccination in vivo and associated this with the purified B cell response to the vaccine in vitro. Our results have shown that the specific responses of B cells to vaccination in vivo (measured in serum by hemagglutination inhibition assay) and in vitro (measured by AID in cultures of restimulated B cells) are both decreased with age and are significantly correlated, and thus, we argue that the in vitro AID response recapitulates what has occurred in vivo in the germinal center in the generation of memory B cells. We realize that aged alterations in T (and other) cells would also contribute to the declines seen in vivo but assert that the autonomous B cell deficiencies we have described in aged mice and humans would be critical to correct in order to generate optimal humoral immune responses in the elderly.
The role of AID in CSR has been well characterized, but its role in SHM and polyclonal antibody affinity maturation has not within a vaccine response. In collaboration with Hana Golding’s laboratory, we have shown that AID correlates with higher affinity antibodies to the vaccine [69]. Antibody affinity maturation is a key aspect of an effective response to vaccination, which also has a significant impact on clinical outcomes following exposure to infectious agents. Briefly, we have investigated whether AID induction in human B cells following pandemic (p)H1N1 vaccination correlated with in vivo antibody affinity maturation against hemagglutinin (HA) domains in plasma of young and elderly individuals. Polyclonal antibody affinity in human plasma for the HA1 and HA2 domains of the pH1N1 HA was measured by antibody–antigen complex dissociation rates using real-time kinetics in surface plasmon resonance. Results showed an age-related decrease in AID induction in B cells following vaccination, as we have previously shown [20]. Levels of AID mRNA before vaccination and fold-increase in AID mRNA after vaccination directly correlated with increase in polyclonal antibody affinity to the HA1 globular domain, but not to the HA2 stalk domain, which is highly conserved among multiple influenza subtypes and strains and thus already has generated an optimal memory response. In the younger individuals, significant affinity maturation to the HA1 globular domain was observed, which associated with initial levels of AID and fold-increase in AID after vaccination. In most of the older individuals (>65 yr), higher affinity to the HA1 domain was observed before vaccination which resulted in minimal change in antibody affinity and correlated with low AID induction in this age group. These findings have demonstrated for the first time a strong correlation between AID induction and in vivo antibody affinity maturation in humans.
We have also evaluated in vivo and in vitro B cell responses to the pH1N1 or to the seasonal influenza vaccine in autoimmune disease patients and in Type-2 Diabetes (T2D) patients, respectively. In the first study, we evaluated the response to the pH1N1 vaccine in patients with inflammatory bowel disease (IBD) undergoing therapy with anti-TNF-α, alone or combined with immunosuppressants (IS). We found a suboptimal in vivo [70] and in vitro [71] response to pH1N1 vaccine in IBD patients on therapy with anti-TNF-α and IS compared to those on anti-TNF-α monotherapy and healthy controls, similarly to what we have previously shown for elderly individuals. In the second study, we evaluated the immune response to the seasonal influenza vaccine in young and elderly T2D patients. We demonstrated that both in vivo and in vitro responses decrease by age in healthy individuals but not in T2D patients, despite high levels of B cell-intrinsic inflammation (TNF-α) in T2D patients [19], which was surprising as we had previously demonstrated this negatively impacts B cell function. One explanation may be due to the fact that the innate immune system of T2D patients is beneficially hyperactivated, as we found elevated serum levels of bacterial LPS and soluble (s)CD14, and we believe that these may not only counteract the negative effects of inflammation from increased TNF-α, IL-6 and CRP, but also to induce a direct stimulation of B cells. Another explanation we are currently investigating is that the T2D patients above were all taking anti-inflammatory agents, such as metformin, which blocks TNF-α signaling in all cells, including B cells.
Based on our previous data in aged mice, we have hypothesized that the inflammatory status of the individual and of B cells themselves would impact B cell function. We have recently shown (Frasca et al. submitted) that the ability to generate a vaccine-specific antibody response in humans is negatively correlated with levels of serum TNF-α. Moreover, human unstimulated B cells from elderly make higher levels of TNF-α than those from young individuals, and this positively correlates with serum TNF-α levels. These all negatively correlate with B cell function, measured by AID from in vitro stimulated B cells. Although it has recently been shown that human B cells from young donors can be directly activated via Toll-like receptors or anti-CD40 and BCR cross-linking to secrete TNF-α [72], our results are the first to show that aging increases TNF-α production by B cells before stimulation in vivo or with in vitro stimulation and we refer to these here as “unstimulated” B cells. Our results support our hypothesis that initial pre-stimulation levels of endogenous TNF-α negatively impact the ability of these unstimulated B cells to generate optimal function, that this is elevated in aged B cells, and thus aged B cells are refractory to further stimulation.
Our collaborative work in the area of breast cancer and psychosocial intervention with Drs. M. Antoni and S. Lechner here at the University of Miami has also concluded that these interventions to accomplish stress reduction and less depression have improved the immune system such as decreasing inflammatory mediators (IL-6, IL1) and improving Th1 responses.
Conclusions and further directions
Our model for autonomous deficiencies in aged B lymphocytes is given in Fig. 1 and highlights that elevations in TNF-α with age predispose the B cell to respond less well to further stimulation (and are also associated with increases in basal levels of NF-κB). The molecular pathways that are affected upon antigen stimulation of the aged B cells are lower MAPK activation (phosphorylation), less TTP phosphorylation and hence more active E47 mRNA degradation, less E47 leading to less Pax-5 and both leading to less AID, which leads to less CSR and SHM and therefore less affinity maturation and less effective antibody.
Fig. 1.
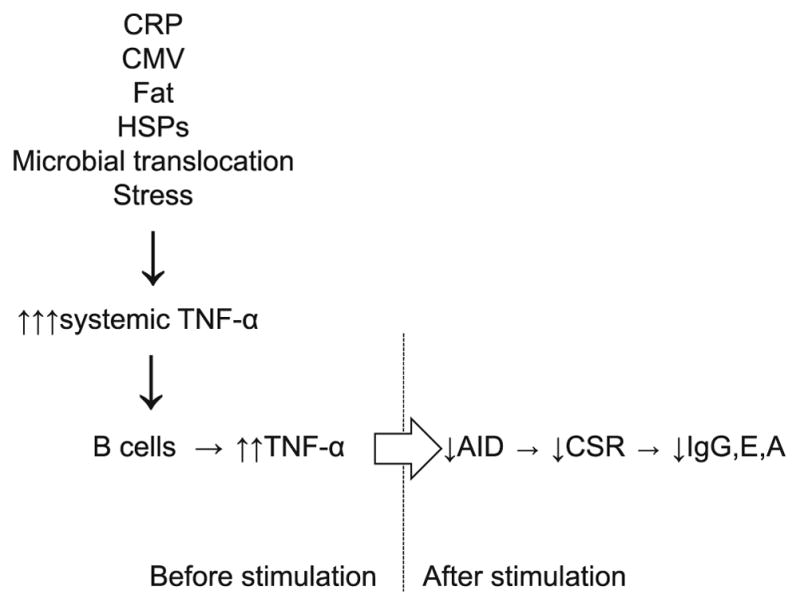
Model. Systemic inflammation (TNF-α) increases with age. This occurs concomitantly or is a consequence of the increase in other markers of inflammation [CRP, CMV, heat shock proteins (HSPs)] and increase in microbial translocation from the intestine and in fat. Stress can also induce high systemic TNF-α. Systemic TNF-α induces TNF-α production by B cells. The levels of TNF-α in B cells before stimulation regulate their capacity to be optimally stimulated by antigens/mitogens to up-regulate AID, undergo CSR and produce secondary switched antibodies will include studies in breast cancer patients to measure the quality of their B cell function and response to vaccines as a function of psychosocial intervention along with the markers and pathways above.
Further directions include a more complete analysis of B cell pathways involved in the aged reduction in B cell function, expansion to other increased comorbidities/diseases of aging including rheumatoid arthritis and T2D with study of interventions which decrease inflammation, such as metformin, with a better analysis of inflammatory markers and also possible involvement of microbiota, and discovery of small molecules which improve the B cell biomarkers we have discovered. Other future directions
Acknowledgments
This study was supported by the following NIH grants: AG-17618 and AG-28586 (to BBB); AI096446-01A1 and AG042826-01A1 (to BBB and DF).
References
- 1.Hodes RJ. Aging and the immune system. Immunol Rev. 1997;160:5–8. doi: 10.1111/j.1600-065x.1997.tb01022.x. [DOI] [PubMed] [Google Scholar]
- 2.LeMaoult J, Szabo P, Weksler ME. Effect of age on humoral immunity, selection of the B-cell repertoire and B-cell development. Immunol Rev. 1997;160:115–26. doi: 10.1111/j.1600-065x.1997.tb01032.x. [DOI] [PubMed] [Google Scholar]
- 3.Linton PJ, Dorshkind K. Age-related changes in lymphocyte development and function. Nat Immunol. 2004;5(2):133–9. doi: 10.1038/ni1033. [DOI] [PubMed] [Google Scholar]
- 4.Pawelec G, Barnett Y, Forsey R, Frasca D, Globerson A, McLeod J, et al. T cells and aging, January 2002 update. Front Biosci J virtual Library. 2002;7:d1056–183. doi: 10.2741/a831. [DOI] [PubMed] [Google Scholar]
- 5.Sadighi Akha AA, Miller RA. Signal transduction in the aging immune system. Curr Opin Immunol. 2005;17(5):486–91. doi: 10.1016/j.coi.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 6.Haynes L, Eaton SM, Burns EM, Randall TD, Swain SL. CD4 T cell memory derived from young naive cells functions well into old age, but memory generated from aged naive cells functions poorly. Proc Natl Acad Sci USA. 2003;100(25):15053–8. doi: 10.1073/pnas.2433717100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pawelec G, Derhovanessian E. Role of CMV in immune senescence. Virus Res. 2011;157(2):175–9. doi: 10.1016/j.virusres.2010.09.010. [DOI] [PubMed] [Google Scholar]
- 8.Kosco MH, Burton GF, Kapasi ZF, Szakal AK, Tew JG. Antibody-forming cell induction during an early phase of germinal centre development and its delay with ageing. Immunology. 1989;68(3):312–8. [PMC free article] [PubMed] [Google Scholar]
- 9.van Dijk-Hard I, Soderstrom I, Feld S, Holmberg D, Lundkvist I. Age-related impaired affinity maturation and differential D-JH gene usage in human VH6-expressing B lymphocytes from healthy individuals. Eur J Immunol. 1997;27(6):1381–6. doi: 10.1002/eji.1830270613. [DOI] [PubMed] [Google Scholar]
- 10.Zheng B, Han S, Takahashi Y, Kelsoe G. Immunosenescence and germinal center reaction. Immunol Rev. 1997;160:63–77. doi: 10.1111/j.1600-065x.1997.tb01028.x. [DOI] [PubMed] [Google Scholar]
- 11.Wang X, Stollar BD. Immunoglobulin VH gene expression in human aging. Clin Immunol. 1999;93(2):132–42. doi: 10.1006/clim1999.4781. [DOI] [PubMed] [Google Scholar]
- 12.Yang X, Stedra J, Cerny J. Repertoire diversity of antibody response to bacterial antigens in aged mice. IV. Study of VH and VL gene utilization in splenic antibody foci by in situ hybridization. J Immunol. 1994;152(5):2214–21. [PubMed] [Google Scholar]
- 13.Frasca D, Landin AM, Alvarez JP, Blackshear PJ, Riley RL, Blomberg BB. Tristetraprolin, a negative regulator of mRNA stability, is increased in old B cells and is involved in the degradation of E47 mRNA. J Immunol. 2007;179(2):918–27. doi: 10.4049/jimmunol.179.2.918. [DOI] [PubMed] [Google Scholar]
- 14.Frasca D, Riley RL, Blomberg BB. Aging murine B cells have decreased class switch induced by anti-CD40 or BAFF. Exp Gerontol. 2007;42(3):192–203. doi: 10.1016/j.exger.2006.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frasca D, Romero M, Landin AM, Diaz A, Riley RL, Blomberg BB. Protein phosphatase 2A (PP2A) is increased in old murine B cells and mediates p38 MAPK/tristetraprolin dephosphorylation and E47 mRNA instability. Mech Ageing Dev. 2010;131(5):306–14. doi: 10.1016/j.mad.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frasca D, Van der Put E, Landin AM, Gong D, Riley RL, Blomberg BB. RNA stability of the E2A-encoded transcription factor E47 is lower in splenic activated B cells from aged mice. J Immunol. 2005;175(10):6633–44. doi: 10.4049/jimmunol.175.10.6633. [DOI] [PubMed] [Google Scholar]
- 17.Frasca D, Van der Put E, Riley RL, Blomberg BB. Reduced Ig class switch in aged mice correlates with decreased E47 and activation-induced cytidine deaminase. J Immunol. 2004;172(4):2155–62. doi: 10.4049/jimmunol.172.4.2155. [DOI] [PubMed] [Google Scholar]
- 18.Frasca D, Diaz A, Romero M, Landin AM, Phillips M, Lechner SC, et al. Intrinsic defects in B cell response to seasonal influenza vaccination in elderly humans. Vaccine. 2010;28(51):8077–84. doi: 10.1016/j.vaccine.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Frasca D, Diaz A, Romero M, Mendez NV, Landin AM, Ryan JG, et al. Young and elderly patients with type 2 diabetes have optimal B cell responses to the seasonal influenza vaccine. Vaccine. 2013 doi: 10.1016/j.vaccine.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frasca D, Diaz A, Romero M, Phillips M, Mendez NV, Landin AM, et al. Unique biomarkers for B-cell function predict the serum response to pandemic H1N1 influenza vaccine. Int Immunol. 2012;24(3):175–82. doi: 10.1093/intimm/dxr123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frasca D, Landin AM, Lechner SC, Ryan JG, Schwartz R, Riley RL, et al. Aging down-regulates the transcription factor E2A, activation-induced cytidine deaminase, and Ig class switch in human B cells. J Immunol. 2008;180(8):5283–90. doi: 10.4049/jimmunol.180.8.5283. [DOI] [PubMed] [Google Scholar]
- 22.Nussenzweig MC, Alt FW. Antibody diversity: one enzyme to rule them all. Nat Med. 2004;10(12):1304–5. doi: 10.1038/nm1204-1304. [DOI] [PubMed] [Google Scholar]
- 23.Okazaki IM, Kinoshita K, Muramatsu M, Yoshikawa K, Honjo T. The AID enzyme induces class switch recombination in fibroblasts. Nature. 2002;416(6878):340–5. doi: 10.1038/nature727. [DOI] [PubMed] [Google Scholar]
- 24.Rada C, Williams GT, Nilsen H, Barnes DE, Lindahl T, Neuberger MS. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr Biol: CB. 2002;12(20):1748–55. doi: 10.1016/s0960-9822(02)01215-0. [DOI] [PubMed] [Google Scholar]
- 25.Durandy A. Hyper-IgM syndromes: a model for studying the regulation of class switch recombination and somatic hypermutation generation. Biochem Soc Trans. 2002;30(4):815–8. doi: 10.1042/bst0300815. doi:10.1042/ [DOI] [PubMed] [Google Scholar]
- 26.Notarangelo LD, Lanzi G, Peron S, Durandy A. Defects of class-switch recombination. J Allergy Clin Immunol. 2006;117(4):855–64. doi: 10.1016/j.jaci.2006.01.043. [DOI] [PubMed] [Google Scholar]
- 27.Pasqualucci L, Migliazza A, Fracchiolla N, William C, Neri A, Baldini L, et al. BCL-6 mutations in normal germinal center B cells: evidence of somatic hypermutation acting outside Ig loci. Proc Natl Acad Sci USA. 1998;95(20):11816–21. doi: 10.1073/pnas.95.20.11816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dorsett Y, Robbiani DF, Jankovic M, Reina-San-Martin B, Eisenreich TR, Nussenzweig MC. A role for AID in chromosome translocations between c-myc and the IgH variable region. J Exp Med. 2007;204(9):2225–32. doi: 10.1084/jem.20070884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sayegh CE, Quong MW, Agata Y, Murre C. E-proteins directly regulate expression of activation-induced deaminase in mature B cells. Nat Immunol. 2003;4(6):586–93. doi: 10.1038/ni923. [DOI] [PubMed] [Google Scholar]
- 30.Tran TH, Nakata M, Suzuki K, Begum NA, Shinkura R, Fagarasan S, et al. B cell-specific and stimulation-responsive enhancers derepress Aicda by overcoming the effects of silencers. Nat Immunol. 2010;11(2):148–54. doi: 10.1038/ni.1829. [DOI] [PubMed] [Google Scholar]
- 31.Ephrussi A, Church GM, Tonegawa S, Gilbert W. B lineage–specific interactions of an immunoglobulin enhancer with cellular factors in vivo. Science. 1985;227(4683):134–40. doi: 10.1126/science.3917574. [DOI] [PubMed] [Google Scholar]
- 32.Henthorn P, Kiledjian M, Kadesch T. Two distinct transcription factors that bind the immunoglobulin enhancer microE5/kappa 2 motif. Science. 1990;247(4941):467–70. doi: 10.1126/science.2105528. [DOI] [PubMed] [Google Scholar]
- 33.Quong MW, Romanow WJ, Murre C. E protein function in lymphocyte development. Annu Rev Immunol. 2002;20:301–22. doi: 10.1146/annurev.immunol.20.092501.162048. [DOI] [PubMed] [Google Scholar]
- 34.Massari ME, Murre C. Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol. 2000;20(2):429–40. doi: 10.1128/mcb.20.2.429-440.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murre C, McCaw PS, Baltimore D. A new DNA binding and dimerization motif in immunoglobulin enhancer binding, daughterless, MyoD, and myc proteins. Cell. 1989;56(5):777–83. doi: 10.1016/0092-8674(89)90682-x. [DOI] [PubMed] [Google Scholar]
- 36.Schlissel M, Voronova A, Baltimore D. Helix-loop-helix transcription factor E47 activates germ-line immunoglobulin heavy-chain gene transcription and rearrangement in a pre-T-cell line. Genes Dev. 1991;5(8):1367–76. doi: 10.1101/gad.5.8.1367. [DOI] [PubMed] [Google Scholar]
- 37.Sigvardsson M, O’Riordan M, Grosschedl R. EBF and E47 collaborate to induce expression of the endogenous immunoglobulin surrogate light chain genes. Immunity. 1997;7(1):25–36. doi: 10.1016/s1074-7613(00)80507-5. [DOI] [PubMed] [Google Scholar]
- 38.Anspach J, Poulsen G, Kaattari I, Pollock R, Zwollo P. Reduction in DNA binding activity of the transcription factor Pax-5a in B lymphocytes of aged mice. J Immunol. 2001;166(4):2617–26. doi: 10.4049/jimmunol.166.4.2617. [DOI] [PubMed] [Google Scholar]
- 39.Frasca D, Romero M, Diaz A, Alter-Wolf S, Ratliff M, Landin AM, et al. A molecular mechanism for TNF-alpha-mediated downregulation of B cell responses. J Immunol. 2012;188(1):279–86. doi: 10.4049/jimmunol.1003964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Frasca D, Nguyen D, Riley RL, Blomberg BB. Effects of aging on proliferation and E47 transcription factor activity induced by different stimuli in murine splenic B cells. Mech Ageing Dev. 2003;124(4):361–9. doi: 10.1016/s0047-6374(03)00009-5. [DOI] [PubMed] [Google Scholar]
- 41.Frasca D, Nguyen D, Riley RL, Blomberg BB. Decreased E12 and/or E47 transcription factor activity in the bone marrow as well as in the spleen of aged mice. J Immunol. 2003;170(2):719–26. doi: 10.4049/jimmunol.170.2.719. [DOI] [PubMed] [Google Scholar]
- 42.Quong MW, Harris DP, Swain SL, Murre C. E2A activity is induced during B-cell activation to promote immunoglobulin class switch recombination. EMBO J. 1999;18(22):6307–18. doi: 10.1093/emboj/18.22.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van der Put E, Frasca D, King AM, Blomberg BB, Riley RL. Decreased E47 in senescent B cell precursors is stage specific and regulated posttranslationally by protein turnover. J Immunol. 2004;173(2):818–27. doi: 10.4049/jimmunol.173.2.818. [DOI] [PubMed] [Google Scholar]
- 44.King AM, Van der Put E, Blomberg BB, Riley RL. Accelerated Notch-dependent degradation of E47 proteins in aged B cell precursors is associated with increased ERK MAPK activation. J Immunol. 2007;178(6):3521–9. doi: 10.4049/jimmunol.178.6.3521. [DOI] [PubMed] [Google Scholar]
- 45.Huggins GS, Chin MT, Sibinga NE, Lee SL, Haber E, Lee ME. Characterization of the mUBC9-binding sites required for E2A protein degradation. J Biol Chem. 1999;274(40):28690–6. doi: 10.1074/jbc.274.40.28690. [DOI] [PubMed] [Google Scholar]
- 46.Kho CJ, Huggins GS, Endege WO, Hsieh CM, Lee ME, Haber E. Degradation of E2A proteins through a ubiquitin-conjugating enzyme, UbcE2A. J Biol Chem. 1997;272(6):3845–51. doi: 10.1074/jbc.272.6.3845. [DOI] [PubMed] [Google Scholar]
- 47.Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1 alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci USA. 1998;95(14):7987–92. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bevilacqua A, Ceriani MC, Capaccioli S, Nicolin A. Post-transcriptional regulation of gene expression by degradation of messenger RNAs. J Cell Physiol. 2003;195(3):356–72. doi: 10.1002/jcp.10272. [DOI] [PubMed] [Google Scholar]
- 49.Chen CY, Shyu AB. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem Sci. 1995;20(11):465–70. doi: 10.1016/s0968-0004(00)89102-1. [DOI] [PubMed] [Google Scholar]
- 50.DuBois RN, McLane MW, Ryder K, Lau LF, Nathans D. A growth factor-inducible nuclear protein with a novel cysteine/histidine repetitive sequence. J Biol Chem. 1990;265(31):19185–91. [PubMed] [Google Scholar]
- 51.Lai WS, Stumpo DJ, Blackshear PJ. Rapid insulin-stimulated accumulation of an mRNA encoding a proline-rich protein. J Biol Chem. 1990;265(27):16556–63. [PubMed] [Google Scholar]
- 52.Carballo E, Lai WS, Blackshear PJ. Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science. 1998;281(5379):1001–5. doi: 10.1126/science.281.5379.1001. [DOI] [PubMed] [Google Scholar]
- 53.Taylor GA, Carballo E, Lee DM, Lai WS, Thompson MJ, Patel DD, et al. A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity. 1996;4(5):445–54. doi: 10.1016/s1074-7613(00)80411-2. [DOI] [PubMed] [Google Scholar]
- 54.Carballo E, Cao H, Lai WS, Kennington EA, Campbell D, Blackshear PJ. Decreased sensitivity of tristetraprolin-deficient cells to p38 inhibitors suggests the involvement of tristetraprolin in the p38 signaling pathway. J Biol Chem. 2001;276(45):42580–7. doi: 10.1074/jbc.M104953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chrestensen CA, Schroeder MJ, Shabanowitz J, Hunt DF, Pelo JW, Worthington MT, et al. MAPKAP kinase 2 phosphorylates tristetraprolin on in vivo sites including Ser178, a site required for 14-3-3 binding. J Biol Chem. 2004;279(11):10176–84. doi: 10.1074/jbc.M310486200. [DOI] [PubMed] [Google Scholar]
- 56.Eichhorn PJ, Creyghton MP, Bernards R. Protein phosphatase 2A regulatory subunits and cancer. Biochim Biophys Acta. 2009;1795(1):1–15. doi: 10.1016/j.bbcan.2008.05.005. [DOI] [PubMed] [Google Scholar]
- 57.Millward TA, Zolnierowicz S, Hemmings BA. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem Sci. 1999;24(5):186–91. doi: 10.1016/s0968-0004(99)01375-4. [DOI] [PubMed] [Google Scholar]
- 58.Landin AM, Frasca D, Harrison P, Scallan M, Riley RL, Blomberg BB. E47 retroviral rescue of intrinsic B-cell defects in senescent mice. Aging Cell. 2011;10(2):327–37. doi: 10.1111/j.1474-9726.2011.00673.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ademokun A, Wu YC, Dunn-Walters D. The ageing B cell population: composition and function. Biogerontology. 2010;11(2):125–37. doi: 10.1007/s10522-009-9256-9. [DOI] [PubMed] [Google Scholar]
- 60.Colonna-Romano G, Bulati M, Aquino A, Pellicano M, Vitello S, Lio D, et al. A double-negative (IgD−CD27−) B cell population is increased in the peripheral blood of elderly people. Mech Ageing Dev. 2009;130(10):681–90. doi: 10.1016/j.mad.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 61.Frasca D, Diaz A, Romero M, Landin AM, Blomberg BB. Age effects on B cells and humoral immunity in humans. Ageing Res Rev. 2011;10(3):330–5. doi: 10.1016/j.arr.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shi Y, Yamazaki T, Okubo Y, Uehara Y, Sugane K, Agematsu K. Regulation of aged humoral immune defense against pneumococcal bacteria by IgM memory B cell. J Immunol. 2005;175(5):3262–7. doi: 10.4049/jimmunol.175.5.3262. [DOI] [PubMed] [Google Scholar]
- 63.Buffa S, Pellicano M, Bulati M, Martorana A, Goldeck D, Caruso C, et al. A novel B cell population revealed by a CD38/CD24 gating strategy: CD38(−)CD24 (−) B cells in centenarian offspring and elderly people. Age (Dordr) 2013;35(5):2009–24. doi: 10.1007/s11357-012-9488-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Duggal NA, Upton J, Phillips AC, Sapey E, Lord JM. An age-related numerical and functional deficit in CD19(+) CD24(hi) CD38(hi) B cells is associated with an increase in systemic autoimmunity. Aging Cell. 2013;12(5):873–81. doi: 10.1111/acel.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Derhovanessian E, Theeten H, Hahnel K, Van Damme P, Cools N, Pawelec G. Cytomegalovirus-associated accumulation of late-differentiated CD4 T-cells correlates with poor humoral response to influenza vaccination. Vaccine. 2013;31(4):685–90. doi: 10.1016/j.vaccine.2012.11.041. [DOI] [PubMed] [Google Scholar]
- 66.McElhaney JE. Influenza vaccine responses in older adults. Ageing Res Rev. 2011;10(3):379–88. doi: 10.1016/j.arr.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Murasko DM, Bernstein ED, Gardner EM, Gross P, Munk G, Dran S, et al. Role of humoral and cell-mediated immunity in protection from influenza disease after immunization of healthy elderly. Exp Gerontol. 2002;37(2–3):427–39. doi: 10.1016/s0531-5565(01)00210-8. [DOI] [PubMed] [Google Scholar]
- 68.Frasca D, Blomberg BB. Aging affects human B cell responses. J Clin Immunol. 2011;31(3):430–5. doi: 10.1007/s10875-010-9501-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Khurana S, Frasca D, Blomberg B, Golding H. AID activity in B cells strongly correlates with polyclonal antibody affinity maturation in vivo following pandemic 2009-H1N1 vaccination in humans. PLoS Pathog. 2012;8(9):e1002920. doi: 10.1371/journal.ppat.1002920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Andrisani G, Frasca D, Romero M, Armuzzi A, Felice C, Marzo M, et al. Immune response to influenza A/H1N1 vaccine in inflammatory bowel disease patients treated with anti TNF-alpha agents: effects of combined therapy with immunosuppressants. J Crohns Colitis. 2013;7(4):301–7. doi: 10.1016/j.crohns.2012.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Frasca D, Andrisani G, Diaz A, Felice C, Guidi L, Blomberg BB. AID in aging and autoimmune diseases. Autoimmunity. 2013;46(2):168–75. doi: 10.3109/08916934.2012.750300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Agrawal S, Gupta S. TLR1/2, TLR7, and TLR9 signals directly activate human peripheral blood naive and memory B cell subsets to produce cytokines, chemokines, and hematopoietic growth factors. J Clin Immunol. 2011;31(1):89–98. doi: 10.1007/s10875-010-9456-8. [DOI] [PMC free article] [PubMed] [Google Scholar]