

Abstract
Immunotherapies targeting immune checkpoints have proven efficacious in reducing the burden of lung cancer in patients; however, the antigenic targets of these re-invigorated T cells remain poorly defined. Lung cancer tumors contain tumor-associated macrophages (TAM) and neutrophils, which release the serine proteases neutrophil elastase (NE) and proteinase 3 (P3) into the tumor microenvironment. NE and P3 shape the antitumor adaptive immune response in breast cancer and melanoma. In this report, we demonstrate that lung cancer cells cross-presented the tumor-associated antigen PR1, derived from NE and P3. Additionally, NE and P3 enhanced the expression of human leukocyte antigen (HLA) class I molecules on lung cancer cells and induced unique, endogenous peptides in the immunopeptidome, as detected with mass spectrometry sequencing. Lung cancer patient tissues with high intratumoral TAM were enriched for MHC class I genes and T-cell markers, and patients with high TAM and cytotoxic T lymphocyte (CTL) infiltration had improved overall survival. We confirmed the immunogenicity of unique, endogenous peptides with cytotoxicity assays against lung cancer cell lines, using CTL from healthy donors that had been expanded against select peptides. Finally, CTL specific for serine proteases–induced endogenous peptides were detected in lung cancer patients using peptide/HLA-A2 tetramers and were elevated in tumor-infiltrating lymphocytes. Thus, serine proteases in the tumor microenvironment of lung cancers promote the presentation of HLA class I immunogenic peptides that are expressed by lung cancer cells, thereby increasing the antigen repertoire that can be targeted in lung cancer.
Keywords: Tumor antigens, tumor microenvironment, lung cancer, cancer immunology, immune recognition
INTRODUCTION
Cytotoxic T lymphocytes (CTL) recognize and lyse cancer cells presenting neoantigens in the context of HLA class I molecules (1,2). Reported mechanisms of lung cancer attenuation of the antitumor immune response include expression of T-cell inhibitory molecules such as PD-L1 by lung cancer cells to evade CTL activity (1) and recruitment of regulatory T cells (Tregs) by tumor-associated neutrophils (TAN) (3). This inhibition can be overcome by immune-checkpoint blockade therapies targeting PD-1 and CTLA-4 on the T cells, thus permitting recognition of tumor neoantigens presented on HLA class I molecules (1,2).
The HLA-A2-restricted, leukemia-associated antigen PR1 (VLQELNVTV), derived from neutrophil elastase (NE) and proteinase 3 (P3) has proven to be an effective leukemia target in preclinical animal models as well as clinical trials (4–9). PR1 serves as an exogenous antigen on the surface of solid tumors, but has not been reported for lung cancer (6–8,10). In breast cancer, NE cleaves the endogenous antigen–encoded protein cyclin E, generating an HLA-A2–restricted, immunogenic antigen, ILLDWLMEV (11). NE also increases expression of surface HLA-A,B,C on breast cancer cell lines, H2023 lung cancer cells, and MEL526 melanoma cells (12). In the solid tumor setting, NE and P3, along with other serine proteases, are delivered to cancer cells by TAN and tumor-associated macrophages (TAM) (10). In early-stage lung cancer, TAN, which can comprise up to 25% of cells of the tumor, can activate T cells (13).
In view of (i) the demonstrated capacity of NE to elicit immunogenic peptide presentation from exogenous and endogenous proteins on solid tumors (10,11), (ii) previous studies reporting cell-extrinsic perturbations to the immunopeptidome (14), and (iii) improved outcomes in lung cancer patients that correlated with neoantigen presentation on the tumor cell surface (1), we hypothesized that serine proteases alter the immunopeptidome by inducing immunogenic presentation, including but not limited to PR1, on the lung cancer cell surface. These peptides may elicit CTL recognition, and therefore could serve as targets for existing and novel immunotherapies.
In this study, we demonstrated that lung cancer cells cross-presented the leukemia-associated antigen PR1 and were lysed by PR1-targeted immunotherapy. Increased surface expression of HLA class I molecules on lung cancer cells was induced by NE, which led to enhanced presentation of novel self-antigens and mutant peptides. We validated the immunogenicity of novel self-antigens using in vitro assays and found CTL specific for these novel, endogenous self-antigens in lung cancer patients. Additionally, we provided evidence that TAM could alter the antitumor adaptive immune response.
MATERIALS AND METHODS
Cells, cell culture, and reagents
Healthy-donor peripheral blood mononuclear cells (PBMC) and polymorphonuclear leukocytes (PMN) were isolated from apheresis obtained from the MD Anderson Blood Bank by single or double Ficoll gradient, respectively, using Histopaque®-1077 and Histopaque®-1119 (Sigma-Aldrich). Four-digit, molecular HLA typing was performed for lung cancer cell lines: H2023 (A*02:01, A*02:01, B*07:02, B*08:01, C*07:01, C*07:02), H441 (A*02:01, A*03:01, B*38:01, B*44:03, C*12:03, C*16:02), DFCI024 (A*24:02, A*33:01, B*14:02, B*35:02, C*08:02, C*04:01), DFCI032 (A*02:01, A*02:01, B*44:02, B*51:01, C*05:01, C*05:01), H1299 (A*24:02, A*32:01, B*40:01, B*40:02, C*03:03, C*02:02), and HCC2935 (A*02:01, A*26:01, B*37:01, B*49:01, C*06:02, C*07:01) at the HLA Typing Laboratory at MD Anderson. The H441, U937 (histiocytic leukemia), and T2 cell lines were obtained from American Type Culture Collection in 2012, 2014, and 2015, respectively. Cell lines were cultured continuously for 6 months or less. The remaining lung cancer lines were a generous gift Dr. Adi Gazdar (University of Texas Southwestern) in 2009. Cell lines were maintained in RPMI 1640 supplemented with 100 U/ml penicillin, 100 μg/ml streptavidin (Invitrogen), and 10% fetal bovine serum that had been heat-shocked at 56°C for 30 min. All cells were cultured at 5% CO2 at 37°C. Cell lines were validated at the MD Anderson Sequencing and Microarray Facility using short tandem repeat DNA fingerprinting and routinely checked for mycoplasma by PCR (PromoKine).
Purified neutrophil elastase (NE) and proteinase 3 (P3) were purchased from Athens Research. Recombinant human cytokines were purchased from R&D Systems. All primers were ordered from Sigma-Aldrich: ELANE (forward 5′-CACGGAGGGGCAGAGACC-3′; reverse 5′-TATTGTGCCAGATGCTGGAG-3′) and PRTN3 (forward 5′-5′-GACCCCACCATGGCTCAC-3′; reverse 5′-ATGGGAAGGACAGACAGGAG-3′). GAPDH (forward 5′-TAGACGGGAAGCTCACTGGC-3′; reverse 5′-AGGTCCACCACCCTGTTGCT-3′) oligonucleotides served as loading controls. Real-time PCR amplifications were performed using an iCycler iQ thermal cycler (Bio-Rad Laboratories). Peptides of >95% purity were purchased from Bio-Synthesis Inc.
Flow cytometry, serotyping, and immunofluorescence
For flow cytometry, fluorochrome-conjugated antibodies to the following proteins were used: PR1/HLA-A2 complex clone 8F4-AlexaFluor-647 (in house), pan-HLA-A,B,C clone W6/32-FITC or phycoerythrin (PE), HLA-A2 clone BB7.2-FITC (BD Bioscience). Mouse anti-NE (Santa Cruz) and rabbit anti-P3 (NeoMarkers) were directly conjugated to fluorchromes (in house). Ghost Dye Violet 510 (Tonbo Biosciences) was used to select viable cells for all flow studies. Healthy donor PBMCs were serotyped with the anti-HLA-A2 (clone BB7.2). Peptide/HLA-A2 tetramers were generated by the MHC Tetramer Production Laboratory at Baylor College of Medicine and were used to determine CTL target specificity, as previously described (10,11,15). Briefly, patient samples were stained with CD8-PE/Cy7 (Tonbo), CD3-FITC (BioLegend), peptide/HLA-A2-tetramer-allophycocyanin (APC) or PE-conjugated, and the following pacific blue-conjugated lineage Abs: CD4 (BD Biosciences), CD14 (BD Biosciences), CD16 (BD Biosciences), and CD19 (BioLegend). Healthy donor and patient samples were fixed with 4% paraformaldehyde. CTL frequency was determined as the percentage of live cells that were lineage−CD3+CD8+, and peptide/HA-A2 tetramer+. All staining was performed at 4°C for 30–45 min. Flow cytometry events were collected on a BD LSRFortessa, analyzed using FlowJo software (Tree Star) and graphed using GraphPad Prism software. Antibody-binding capacity was determined using Quantum™ Simply Cellular® anti-Mouse IgG (Bangs Laboratories, Inc.). Intracellular staining was performed with BD Perm/Wash™ (BD Biosciences). Confocal images were captured on a Leica Microsystems SP2 SE confocal microscope with a 63×/1.4 oil objective at the Optical Microscopy Core Laboratory (MD Anderson). Leica LCS software (version 2.61) was used for the image analysis.
Surface MHC class I expression following serine protease and cytokine incubation
Lung cancer cells were incubated for 24 h with purified NE or P3 (10 μg/mL), or lysates from healthy-donor PMN at a 2:1 PMN:target cell ratio. Interferon-gamma (100 U/mL IFNγ) served as a positive control. The HLA-A2-binding peptide PR1 served as a negative control. The synergistic effects of NE and P3 with IFNγ was evaluated by co-incubation. Surface MHC class I was evaluated by flow cytometry (described above).
Peptide extraction, mass spectrometry identification, and analysis
Peptides presented by HLA class I molecules were identified by mass spectrometry following isolation by immunoprecipitation. Briefly, HLA class I molecules were captured from 1 × 108 lysed cells per treatment, using anti-HLA-A,B,C (30 μg; clone W6/32 supernatant grown in house and purified by the Monoclonal Antibody Core Facility at MD Anderson) crosslinked to 100 μL of 50% w/v slurry of Protein A/G resin. Peptides were eluted in 0.1 N acetic acid and filtered using a 3-kDa Amicon Ultra column (Millipore). Peptides were fractioned using an off-line 1100 series HPLC system and were run on a nanoLC-MS/MS using LTQ-Orbitrap Elite. Sequest HT from Proteome discoverer was used for peptide identification. HLA class I–bound peptides were further inspected for mass accuracy and MS/MS spectra were validated manually. All identified HLA class I peptides were blasted against non-redundant NCBInr human entries to identify their corresponding source proteins (Gene ID). Predicted binding of discovered peptides was performed in silico using HLArestrictor v 1.2 (16) and NetHLApan version 3.0 (17,18) under the following restraints: matched 4-digit HLA-A and HLA-B allele typing, length restricted to match discovered peptide, strong (50 nM affinity or 0.5% rank) and weak (500 nM or 2% rank) binding criteria.
For mutant peptide identification, a lookup table was generated from reported mutations with predicted binding to endogenous HLA molecules (19,20). Briefly, reported missense mutations encoded within a peptide predicted to bind endogenous HLA class I alleles were accessed from the TRON cell line portal to create a lookup table for mutant peptides for mass spectrometry discovery. Predicted binding to endogenously-encoded HLA class I alleles of the mutant neoantigens and wild-type counterparts for each cell line were examined. Peptides were assigned as mutant, wild-type or potential, with the lattermost indicating the single amino-acid substitution was not resolved in that particular event. Peptides were quantified by PSM and the fraction of peptides containing potential mutagenic peptides was compared to the potential mutagenic peptide fraction following coculture of lung cancer cells with NE or P3.
CTL expansions and cytotoxicity assays
CTL specific for peptide/HLA-A2 complexes were generated as previously described (7,8,21). Briefly, T2 cells were loaded with peptide (30 μg) individually for 2 h in 10% RPMI, pooled, irradiated and washed four times to remove unbound peptide. Healthy donors were serotyped for HLA-A2 using fluorochorome-conjugated BB7.2 antibody. PBMC were cultured in RPMI supplemented with 10% human AB serum (Gemini Bio-Products) and stimulated weekly with peptide-loaded T2 cells for 2.5 weeks; after one week in culture, interleukin-2 (20 IU/mL) was added to the culture medium at the time of peptide stimulation (weekly).
Cytotoxicity was determined against T2 loaded with individual peptides. HIV GAG nonomer (BioSynthesis) served as an irrelevant peptide control (22). Target cells were fluorescently labeled with 5 μg of Calcein AM (BD Biosciences) for 15 min, excess Calcein AM was removed by repeat washes and 10,000 cells were seeded into a 96-well plate. CTL and target cells were incubated for 4 h at 37°C in a humidified incubator at the indicated effector-to-target ratios. Trypan blue (Sigma Life Sciences) was used as a fluorescence quencher, and fluorescence of live target cells was determined using a CytoFluor II plate reader (PerSeptive Biosystems). Percent specific cytotoxicity was calculated according to the following equation:
Tumor-infiltrating lymphocytes (TIL) were expanded from tumor fragments ex vivo. Briefly, fresh tumor was minced into 3–5 mm2 fragments and cultured in media containing high concentration of IL2. Cells were divided into fresh culture media with expansion over 3–5 weeks. Subsequently, TILs were rapidly expanded per the Rapid Expansion Protocol using anti-CD3, irradiated feeder cells, and high dose IL2. TIL were expanded to ≥ 1000-fold in a two-week period, recapitulating the product that would be infused into a patient in the context of adoptive TIL therapy.
T-cell proliferation
PBMC from healthy donors were measured for proliferation using the carboxyfluorescein succinimidyl ester (CFSE, Life Technologies). Briefly, PBMC were isolated by Ficoll gradient density 1.077 g/mL and labeled with CFSE (Invitrogen). T cells were cocultured with the indicated lung cancer cell line (that had or had not been pretreated with purified human NE (10 μg/mL; Athens Research) and washed with PBS) at a 1:5 PBMC:lung cancer cell ratio and/or stimulated with the addition of mAbs to CD3 and CD28 (3μg/mL; BD Biosciences). After four days in culture, cells were stained with PE-conjugated anti-CD4 and APC-conjugated anti-CD8 (BioLegend) and Ghost Dye Violet 510 viability dye and analyzed by flow cytometry.
Computational analysis and statistics
Bar and line graphs display the mean with error bars denoting the standard deviation, unless otherwise indicated. As appropriate, two-way ANOVA two-tailed and Mann-Whitney unpaired t-test was used for statistical analysis, with the Dunnett’s correction applied to the former as needed; the criterion for significance in all studies was P = 0.05.
Provisional lung adenocarcinoma patient data from TCGA Research Network (http://cancergenome.nih.gov/) was retrieved from the online portal www.cbioportal.org. Patients were grouped as “high” or “low” based on quartile expression of the macrophage marker CD68 or the CTL marker CD8 (CD8A and CD8B). Gene enrichment analysis was performed on mRNA expression (RNA Seq V2 RSEM) in the TCGA web interface; P values from the two-tailed, Fisher’s Exact Test are reported. Patient survival was analyzed in Prism v 7.0b; the log-rank (Mantel-Cox) test P value is reported. Hierarchal clustering of immunoproteasome core components in lung cancer cell lines was performed in RStudio version 3.3.0 (2016-05-03) using the heatmap.2 function from the gplots package version 3.0.1.
Human Subjects
All investigations involving humans were performed after approval by MD Anderson Cancer Center Institutional Review Board. Lung adenocarcinoma PBMCs and TILs were obtained from genotyped HLA-A2 patients enrolled in the immunogenomic profiling of non-small cell lung cancer (ICON) project after informed consent was obtained from each patient or patient’s guardian. Patients had no evidence of metastasis and were diagnosed with adenocarcinoma (n = 4) or squamous cell carcinoma (n = 2).
RESULTS
Lung cancer cells internalize NE and P3 from multiple sources
Uptake of NE and P3 by lung cancer cell lines has been previously reported (23,24). We first confirmed that the full-length transcripts of NE and P3 were not endogenously expressed in a panel of lung cancer cell lines (Fig. 1A), in agreement with publically available data of cell lines from the Cancer Cell Line Encyclopedia (Supplementary Fig. S1) (25). However, lung cancer cells can contain NE and P3 protein after co-incubation with exogenous sources; the H2023 cell line internalized NE and P3 from soluble, purified proteins (Fig. 1B) and healthy donor-PMN (Fig. 1C). We extended these findings in a panel of lung cancer cell lines with purified NE (Fig. 1D). These data demonstrate that lung cancer cells lack endogenous NE and P3 but can take up NE and P3 from exogenous sources.
Figure 1. Exogenous serine proteases are internalized by lung cancer cells.
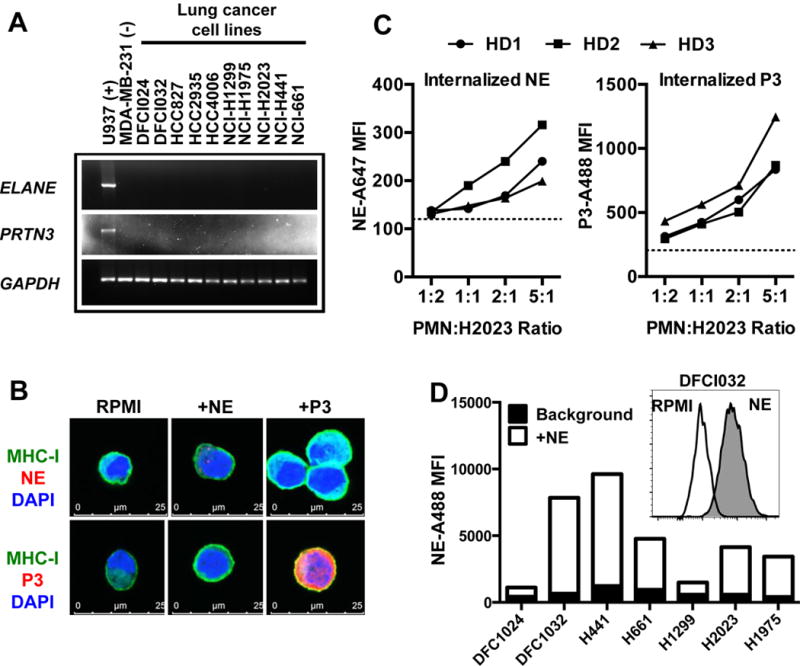
(A) Lung cancer cells do not express full-length NE (ELANE) or P3 (PRTN3) transcripts, as determined by RT-PCR. (B) Confocal imaging of internalized proteins containing the PR1 epitope, NE and P3 in H2023 lung cancer cells. (C) Flow cytometry of internalized NE and P3 from three separate healthy donor (HD) peripheral mononuclear (PMN) lysates by H2023; dashed line indicates staining of unpulsed cells. (D) Uptake of purified NE detected by flow cytometry in a panel of lung cancer cells with autofluorescence removed; inset shows DFCI032 as a representative histogram with NE (gray) and without (white, RPMI).
Serine proteases enhance antigen presentation via elevated HLA class I surface expression
The serine proteases NE and P3 contain the immunogenic, HLA-A2–restricted leukemia-associated antigen PR1, which was shown to be cross-presented by breast cancer and other solid tumors. Although NE was shown to be internalized by lung cancer (24), there are no reports of lung cancer cells presenting the exogenous antigen PR1. Using the PR1/HLA-A2–specific monoclonal antibody 8F4 (5,26), we detected cross-presentation of PR1 from NE by HLA-A2+ lung cancer cells via flow cytometry (Fig. 2A), as previously shown in antigen-presenting cells, breast cancer, and melanoma (10,27). Moreover, lung cancer cells exposed to NE had increased susceptibility to destruction by CTL bearing the PR1-specific 8F4–chimeric antigen-receptor (CAR) (28) (Fig. 2B). Thus, lung cancer cells could cross-present PR1 in the presence of exogenous NE and were susceptible to PR1-targeted immunotherapies.
Figure 2. NE and P3 enhance antigen presentation.
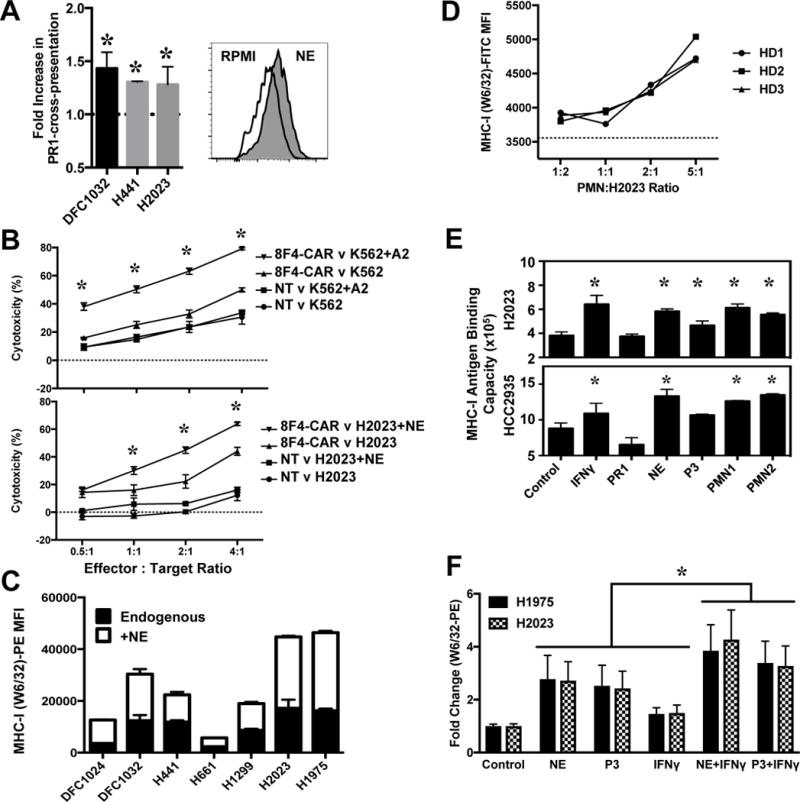
(A) Increase of the immunogenic PR1 peptide on HLA-A2+ lung cancer cells after 24 h co-incubation with NE, displayed as the fold-increase in PR1 cross-presentation over baseline, determined with flow cytometry by binding of 8F4 antibody (anti-PR1/HLA-A2) directly conjugated to AlexaFluor647. Inset is a representative histogram of DFCI032 with NE (gray) and without (white, RPMI). (B) CAR-T cells recognizing PR1/HLA-A2 from healthy donors lyse NE-exposed H2023 lung cancer cells. The NE- and P3-endogenously expressing K562 cell line transduced with HLA-A2 (to allow PR1 presentation) and nontransduced T cells (NT) were used as effector controls. Graph displays the mean of 3 technical replicates representative of 2 independent experiments; asterisks indicates significant difference between target cell populations of 8F4-CAR–transduced effectors by the two-way ANOVA with P = 0.05 as the criterion for significance. (C) Purified NE, and (D) NE supplied by PMNs from healthy donors, enhanced total HLA class I surface expression, as detected by flow cytometry with W6/32, the antibody to-folded, HLA-A,B,C. (E) NE contributes to enhanced surface HLA class I. Lung cancer cells were incubated for 24 h with IFNγ (100 U/mL, positive control), PR1 peptide (10 μg/mL, negative control), purified NE or P3 (10 μg/mL), or lysates from healthy-donor PMN at a 2:1 PMN:target cell ratio. Asterisks denote significant elevation over baseline HLA class I antigen binding capacity via two-way ANOVA with P = 0.05 as the criterion for significance. (F) Serine proteases NE and P3 synergize with IFNγ to upregulate surface MHC-I. Cells treated as in (E); the mean and standard deviation from 5 independent experiments performed in triplicate is shown. Asterisk indicates significant increase in surface MHC-I of combined treatments compared to respective single treatments alone by the two-way ANOVA with P = 0.05 as the criterion for significance.
We have demonstrated an increase in surface HLA class I molecules on breast cancer cell lines and one lung cancer cell line after coculture with NE (12). We expanded these findings using an antibody specific for folded, pan-HLA-A,B,C (mAb W6/32) to detect expression of HLA class I molecules, which increased after exposure to NE, on all lung cancer cell lines examined (Fig. 2C). In the context of the lung cancer tumor microenvironment, NE is packaged in azurophilic granules and delivered to the tumor via TAN and TAM, which release the granule-contained serine proteases upon activation. The elevation in surface HLA class I expression was validated by a dose-dependent increase in surface HLA class I expression as the ratios of PMN to tumor cells was increased (Fig. 2D). Furthermore, our studies demonstrate that in lung cancer, NE contributes to a greater upregulation of HLA class I surface expression than P3 (Fig. 2E), and synergizes with the proinflammatory cytokine IFNγ to increase MHC class I on the surface of lung cancer cells (Fig. 2F).
Next, we validated these findings in tissue from primary lung cancer patients, by dividing TCGA lung adenocarcinoma provisional dataset into “high” and “low” TAM infiltration, based on quartile expression of the TAM marker CD68 (Fig. 3). Significantly higher expression of MHC class I heavy chain (HLA-A,B,C) and light chain (B2M) genes was present in CD68HI patient tissue (Fig. 3A), consistent with our findings in lung cancer cell lines that PMN contribute to enhanced MHC class I surface presentation on cancer cells. In order for TAM-enhanced MHC class I molecules to contribute to antitumor immunity, T cells must be present to recognize peptide/MHC class I molecule complexes. CD68HI patient tissues were enriched for CTL marker CD8 (CD8A and CD8B) in addition to the T helper marker CD4 (Fig. 3B). Lung cancer cell lines demonstrated a suppressive effect on healthy donor CTL proliferation (Supplementary Fig. S2). However, we determined that prior exposure to NE by lung cancer cells did not augment suppression of CTL proliferation; in some instances, a slight increase in CTL proliferation was observed when incubated with lung cancer cell lines previously exposed to NE (Supplementary Fig. S2). CD68HI lung cancer patient tissue was then stratified into CTLHI and CTLLO based on quartile expression of CD8A and CD8B. A significant improvement in overall survival was observed in patients with greater CTL gene expression (Fig. 3C). Independently, the upper quartile for TAM or CTL gene expression did not select patients with significantly improved overall survival over that of patients within the respective lower quartile (data not show). Thus, higher co-expression of both the TAM marker CD68 and CTL marker CD8 selected patients with an improved overall survival, in agreement with our hypothesis that innate immune cells, including PMN and macrophages, contribute to CTL antitumor immunity. Thus, the observed in vitro enrichment of MHC class I genes and T-cell markers in lung cancer tumors with higher TAM gene marker expression, in addition to improved overall survival of CD68HICTLHI patients, supports our studies in cell lines that TAN and TAM contributed to antitumor immunity via enhanced MHC class I surface expression of immunogenic antigens on lung cancer cells.
Figure 3. Enriched expression of T-cell markers and MHC class I genes with CD68 expression in lung adenocarcinoma patient tissues.
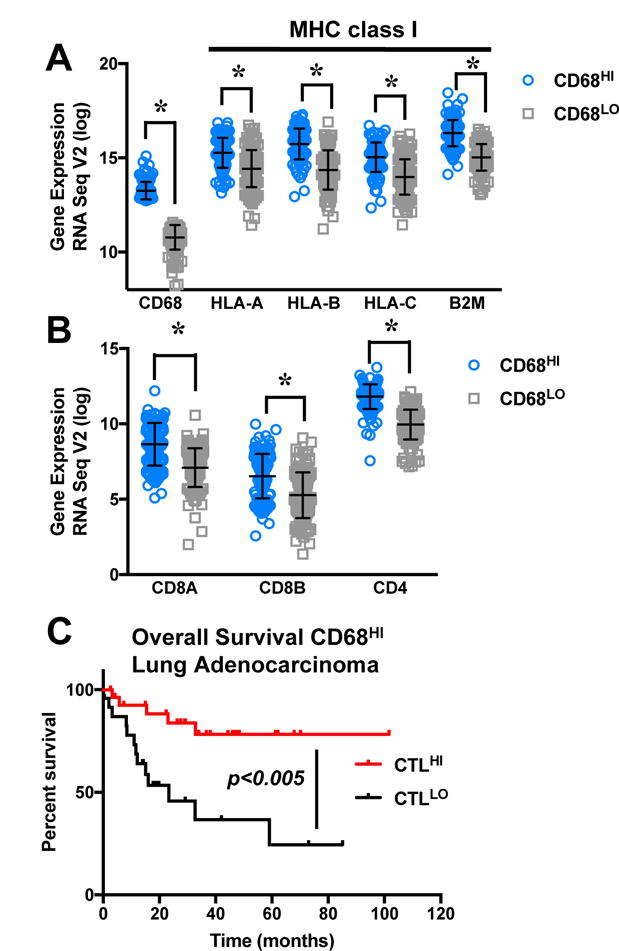
(A, B) TCGA lung adenocarcinoma provisional dataset was divided into CD68HI and CD68LO based on upper and lower quartiles. (A) Patient tissue with high CD68 expression had higher expression of the MHC class I heavy chains (HLA-A,B,C) and light chain (B2M) genes. (B) Enriched expression of CTL marker CD8 (CD8A and CD8B) as well as the helper T-cell marker CD4 was observed in CD68HI patient tissue. Asterisks indicate significance derived from the Fisher’s Exact Test. (C) CD68HI patients were divided into two groups based on quartile expression of the CTL genes CD8A and CD8B. CTLHI patients had significantly improved overall survival, compared to CTLLO patients (P = 0.005).
NE and P3 induce unique, endogenous peptides that are novel antigens
We then sought to identify if NE and P3 affected the antigenic landscape and used mass spectrometry to identify unique, endogenous antigens presented on lung cancer cells after exposure to NE or P3. Two HLA-A2+ lung cancer cell lines were selected for investigation of the immunopeptidome repertoire (Fig. 4A). Briefly, lung cancer cells were incubated in serum-free RPMI with or without NE and P3 for 24 h. Immunoprecipitation was performed to capture HLA class I molecules, and peptides were detected by mass spectrometry after size exclusion filtration and reversed-phase HPLC fractionation. By quantifying detected eluted peptides by peptide spectrum matches (PSM), we determined that the examined serine proteases increased peptides with 25 PSM or fewer (H2023: 81% NE, 80% P3; H441: 86% NE, 90% P3) to a far greater extent than peptides with more than 25 PSM (H2023: 52% NE, 66% P3; H441: 55% NE, 50% P3) (Fig. 4B). Additionally, a slight enrichment of mutant neoantigens was observed after NE or P3 exposure (Supplementary Fig. S3) (19). Thus, NE and P3 had the greatest impact on the lung cancer immunopeptidome by increasing low-abundance, endogenous peptides, with “low” indicating 25 or fewer events detected by MS.
Figure 4. Strategy and profiling of HLA class I-bound antigens on lung cancer cells after NE or P3.
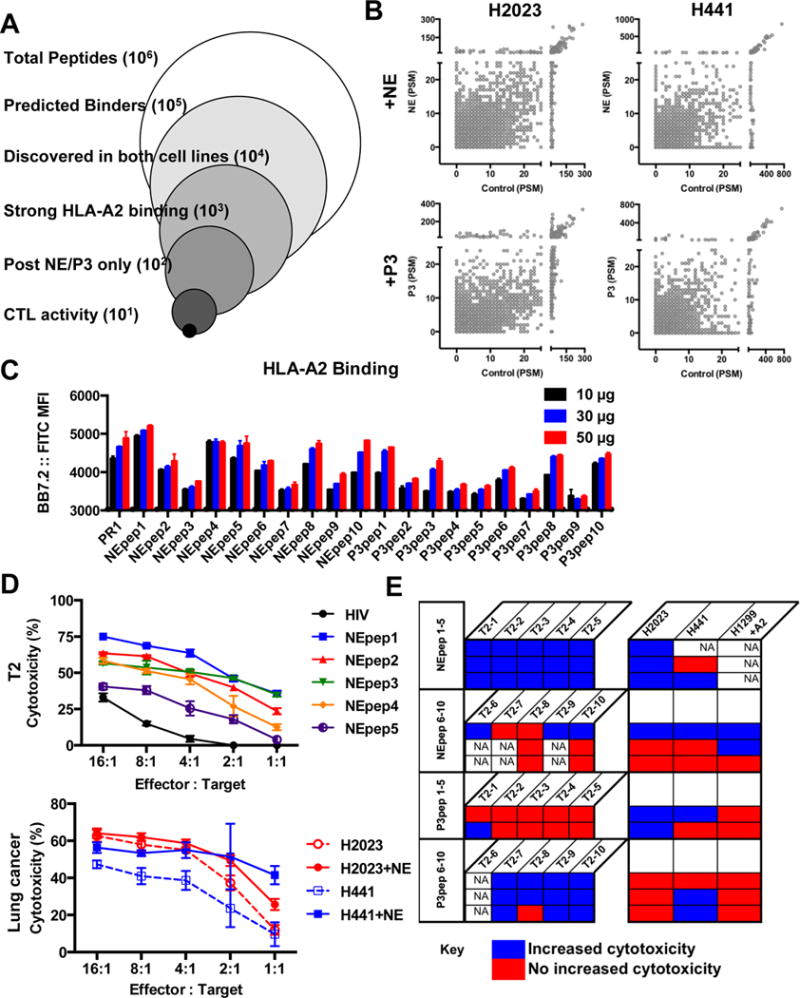
Two HLA-A2+ lung cancer cell lines, H2023 and H441, were incubated for 24 h with NE or P3. (A) HLA class I molecules were captured from lysed cells by the anti-HLA-A,B,C antibody clone W6/32, peptides were eluted and separated with a 3 kDa ultraspin filter and discovered by mass spectrometry. HLARestrictor 1.2 was used to determine in silico binding to HLA-A and HLA-B. Peptides predicted to bind to HLA-A2 (the only conserved HLA allele) were then analyzed, and peptides discovered within untreated cells were removed to retain unique peptides only. (B) Scatter plot of eluted peptides from control (x-axis) and NE/P3-treated lung cancer cells (y-axis) quantified by peptide spectrum matches (PSM). A shift towards the upper-left indicates upregulated or novel peptides. (C-E) Ten peptides per treatment were selected for immunogenicity testing with CTL expansion. (C) Predicted HLA-A2 binding was validated by the T2 binding assay after 2 h; stabilized surface HLA class I was detected by flow cytometry with the anti-HLA-A2 antibody BB7.2. (D-E) CTL from healthy donors were expanded against pools of five peptides for 2.5 weeks, then examined for cytotoxic capacity against T2 cells loaded with individual peptide. An HLA-A2–binding peptide from HIV served as a negative control. Expanded CTL demonstrated enhanced cytotoxicity against lung cancer cell lines incubated with serine protease (compare solid and dash lines of similar color). Representative cytotoxicity assays are shown in (D). (E) Table summarizing cytotoxicity results from 11 different HLA-A2+ healthy donor CTL expansions, denoted by each row. Blue boxes denote expansion against specific peptide/HLA-A2 complexes and statistically improved CTL cytotoxicity against peptide-loaded T2 cells compared to HIV or unpulsed T2 cells and greater CTL lysis of serine protease-treated lung cancer cells compared to unexposed cells. Red indicates statistically enhanced CTL killing of targets was not achieved. Peptide loading was confirmed by flow cytometry for each T2 target cell set; not assessed (NA) denotes no peptide loading for T2 controls or the lung cancer cell line was not assayed. H2023 and H441 were discovery cell lines, and the HLA-A2-transduced H1299 cell served as a validation cell line.
We then interrogated the immunogenicity of unique, endogenous peptides detected after coculture with NE or P3. The strongest, predicted HLA-A2 binders based on affinity were selected (Table 1), because greater binding to HLA class I correlates with immunogenicity (29,30). Many of the eluted peptides were encoded by genes that were implicated in biological processes altered by serine proteases such as cell adhesion (PCDH20), proliferation (24,31,32) (ALK), transcription (33,34) (INSM2, ZFHX3), signal transduction (ABCA1) and protein transport (CLPB, SLC28A1, SLC32A1, SLC41A3, SNX14).
Table 1.
Peptides in lung cancer immunopeptidome
| NE-Novel | ||||
|---|---|---|---|---|
| Name | Peptide | Affinity (nM) | Gene Name | Description |
| NEpepl | LLSAVLPSV | 3 | MCMBP | Mini-chromosome maintenance complex-binding protein |
| NEpep2 | ALAGLSPV | 4 | ZFHX3 | Zinc finger homeobox protein 3 |
| NEpep3 | ALIGGPPV | 5 | ABCA1 | ATP-binding cassette sub-family A member 1 |
| NEpep4 | LLLELAGVTHV | 7 | SIRPD | Signal-regulatory protein delta |
| NEpep5 | SLLGLALLAV | 13 | VSIG10L | V-set and immunoglobulin domain-containing protein 10-like |
| NEpep6 | LLAGPPGV | l5 | SPATA5L1 | Spermatogenesis-associated protein 5-like protein 1 |
| NEpep7 | ALAAFLGL | 20 | SLC28A1 | Sodium/nucleoside cotransporter 1 |
| NEpep8 | LLGPAPGV | 2l | ALK | ALK tyrosine kinase receptor |
| NEpep9 | LLLRSPAGV | 24 | GLS | Glutaminase kidney isoform, mitochondrial |
| NEpeplO | GLGPVAAV | 26 | CELF6 | CUGBP Elav-like family member 6 |
| P3-Novel | ||||
|---|---|---|---|---|
| Name | Peptide | Affinity (nM) | Gene Name | Description |
| P3pepl | LLLPALAGA | 9 | INSM2 | Insulinoma-associated protein 2 |
| P3pep2 | ALAAALVV | l2 | CLPB | Caseinolytic peptidase B protein homolog |
| P3pep3 | LLWRKLLWHQV | l2 | SLC32A1 | Vesicular inhibitory amino acid transporter |
| P3pep4 | ALLGIPKV | l3 | BBS10 | Bardet-Biedl syndrome 10 protein |
| P3pep5 | GLLAIVKV | 16 | FAM222B | Protein FAM222B |
| P3pep6 | LLFPYILPPKA | l7 | SNX14 | Sorting nexin-14 |
| P3pep7 | ALVSIIKV | 18 | PCDH20 | Protocadherin-20 |
| P3pep8 | LLAAVAALL | 18 | SLC41A3 | Solute carrier family 41 member 3 |
| P3pep9 | ALISKNPV | 19 | AMZ2 | Archaemetzincin-2 |
| P3peplO | LLGCPVPLGV | 19 | NLRP5 | NACHT, LRR and PYD domains-containing protein 5 |
We experimentally confirmed HLA-A2–binding of selected, unique, endogenous peptides in Table 1 using the T2 binding assay (Fig. 4C). In order to interrogate immunogenicity, peptides were pooled into groups of five, and healthy donor CTL were expanded. After expansion, T2 cells were loaded with individual peptides to confirm the presence of successfully expanded CTL (Fig. 4D, top). We detected CTL with cytotoxic activity against unique peptide/HLA-A2 complexes in 10 out of 11 different HLA-A2+ healthy donors post expansion (represented by row in Fig. 4E). Lung cancer cell lines were then tested as targets (Fig. 4D, bottom); enhanced CTL lysis of lung cancer cells with serine protease co-incubation was observed in 9 out of 10 confirmed expansions (Fig. 4E). Thus demonstrating that the identified unique, endogenous, self-antigens are indeed valid immunogenic tumor antigens.
Enrichment of CTLs reactive to induced endogenous peptides and PR1 in lung cancer patients
Having identified that CTLs from healthy donors were present and functional against serine protease–induced endogenous peptides in the immunopeptidome of lung cancer cells, we then expanded our studies into lung cancer patients via peptide/HLA-A2 tetramer staining of patient circulating PBMCs as well as TIL. Building upon our findings from healthy donors, peptides were selected based on their ability to form peptide/HLA-A2 complexes in vitro (used for the tetramer staining), and their capacity to elicit potent T-cell expansions with the greatest cytotoxic activity against lung cancer cells (Fig. 4), as well as correlation of the encoding gene with T cells, neutrophils, and/or macrophage gene markers in the non-small cell lung adenocarcinoma provisional dataset from TCGA. Endogenous peptides folded into HLA-A2 tetramers were NEpep1 (MCMBP), NEpep3 (ABCA1), and NEpep4 (SIPRD). Attempts to generate peptide/HLA-A2 complexes for P3pep7 (PCDH20) and P3pep9 (AMZ2) were unsuccessful. The exogenous antigen PR1 was also included in our studies.
Circulating, peptide/HLA-A2 tetramer+ PBMC were detected in both healthy donor (white bars) and lung cancer patients (gray bars) at comparable frequencies (Fig. 5). The CTL populations specific for the three endogenous NE-induced peptides (NEpep1, NEpep3, and NEpep4) were enriched in the TIL populations. PR1/HLA-A2–specific CTL were also detected in TIL populations. Therefore, CTLs specific for endogenous peptides induced by NE, in addition to the exogenous antigen PR1, are present in lung cancer patients and are enriched in their TIL populations.
Figure 5. CTL specific for endogenous and exogenous antigens induced by and from serine proteases presented by HLA-A2 are enriched in lung cancer patients.
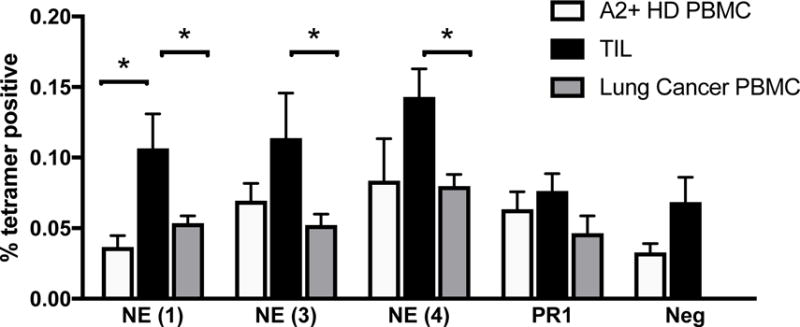
The frequency of CTL reactive to peptide/HLA-A2 complexes was determined by tetramer staining of healthy donor PBMC (white bars, n = 3), circulating PBMC from lung cancer patients (gray bars, n=5) and tumor-infiltrating lymphocytes (TIL, black bars, n = 5). The following peptides were used: NE (1) peptide (LLSAVLPSV); NE (3) peptide (ALIGGPPV); NE (4) peptide (LLLELAGVTHV); PR1 (VLQELNVTV); negative peptide (ALIAPVHAV). Bar graph displays the mean frequency; error bars denote the standard error of the mean. Asterisks indicate statistical significance by the two-tailed, unpaired Mann-Whitney t test, with a P = 0.05 as the criterion for significance.
DISCUSSION
In these studies, we demonstrated that components of the tumor microenvironment, the serine proteases NE and P3, increased antigen presentation of immunogenic peptides by lung cancer cells. We found that healthy donor–derived PMN enhanced surface HLA class I expression and showed the upregulation of surface HLA class I molecules by serine proteases in a panel of lung cancer cell lines that encompassed a spectrum of the epithelial-to-mesenchymal phenotype (35), placing an emphasis on peptide discovery in an epithelial-like (H441) and mesenchymal-like (H2023) cell line.
Lung cancer cells contain a high mutational burden, second only to melanoma (30,36). Peptides resulting from mutations are ideal targets for T cell–based therapy, as they are highly immunogenic, due to lack of tolerance against these antigens in the TCR repertoire, and are sourced from the tumor cells specifically (1). Additionally, immunogenic mutations are associated with favorable patient survival (1,37). Our detection here of predicted mutant antigens (19) and immunogenic endogenous antigens after exposure to NE and P3, and the presence of endogenous antigen/HLA-A2–specific CTL in lung cancer patients, supports previous findings that neoantigen presentation may account for some of the benefits that have been observed in some patients who receive immune-checkpoint blockade (1), which enables T-cell activation in the presence of stimulating peptide/HLA class I complexes.
Specifically, we demonstrated that serine proteases delivered to the tumor by TAN and TAM enhanced immunogenic antigen presentation by lung cancer cells. Tumor tissues from lung cancer patients with high expression of the TAM marker CD68 were found to be enriched for MHC class I genes and T-cell markers. As evidence that TAN and TAM support T-cell recognition of tumor antigens in patients, patients with CD68HI tumor tissues, and concurrent high expression of the genes encoding the CTL marker CD8, had significantly improved survival compared to patients with high CD68 expression in tumor tissues, but low CTL infiltration. When TAM and CTL markers were evaluated independently, high expression did not confer a survival benefit (data not shown), suggestive that these two cell populations synergize to improve host antitumor immunity. In murine pancreatic cancer models, reduction of alternatively-activated myeloid cell population (i.e. type II TAM or M2) improved antigen presentation, antitumor T-cell activity, and response to immune-checkpoint blockade therapy (38). Thus, in view of our data here and our previous reports (10–12), lung cancer patients with relatively large N1 TAN or or M1 TAM populations may have a better prognosis and response to immunotherapies that activate T-cell responses, specifically immune-checkpoint blockade.
Antigen presentation impacts the survival of non-small cell lung cancer patients. Deficiency of immunoproteasome components correlating with poor patient outcomes was identified in mesenchymal-like cells (21), and cells undergoing a epithelial-mesenchymal transition are associated with an inflammatory microenvironment and increased immune-checkpoint molecules (39). Thus mesenchymal-like lung cancer cells are specifically poised to escape CTL destruction. Our analysis here of 186 lung cancer cell lines available through the Cancer Cell Line Encyclopedia reaffirmed a diversity in expression of IFNγ-inducible immunoproteasome subunits within the core components PSMB9, PSMB8, and PSMB10, and ubiquitous expression of proteasome core subunits (Supplementary Fig. S4A–B). We tested whether NE and P3 altered the antigen repertoire via induction of the immunoproteasome; however, we did not find evidence to support this hypothesis (Supplementary Fig. S4C). Because NE-induced surface HLA class I enhancement on breast cancer cells results from stabilized surface molecules and is dependent upon serine protease activity (12), neoantigens are likely generated as a direct result of serine protease cleavage, as in the case of the E75 cyclin E antigen (11). This mechanism agrees with our increased surface HLA class I expression and neoantigens on epithelial- and mesenchymal-like lung cancer cells. Thus, the increase in antigen presentation on lung cancer cells induced by serine protease is not restricted by epithelial-mesenchymal phenotype.
NE is internalized via a receptor-dependent mechanism (11,23,24), but P3 displays receptor-independent internalization despite reported receptor associations (40–42). Platelet-derived growth factor receptor (PDGFR) has been implicated in the proliferative effects of NE on lung cancer cells; however, the receptor responsible for directing internalized NE towards PR1 cross-presentation has yet to be reported. Elucidation of a receptor that modulates cross-presentation is beyond the scope of these studies, but underscores the foundation to understanding the tumor context in which cross-presentation occurs.
To summarize, we demonstrated that PMN/macrophage-associated NE enhanced antigen presentation by lung cancer cells, including the exogenous antigen PR1 and induced endogenous antigens, and that lung cancer cells were susceptible to lysis by CTL that target these peptide/HLA-A2. We also showed that CTL specific for NE-induced, endogenous antigens are present in lung cancer patients, and specifically in the TIL populations. Thus, lung cancer patients, particularly those with abundant TAN and TAM, could greatly benefit from immune-checkpoint blockade and PR1-based immunotherapies, such as CAR-T cells (28,43), monoclonal antibodies (5,26), and vaccines (9). Finally, PR1-based immunotherapies could synergize with immune-checkpoint blockade to achieve profound and long-term benefits for lung cancer patients.
Supplementary Material
Acknowledgments
This research was supported by Leukemia & Lymphoma Society Translational Research Program Grant (6030-12), and NIH SPORE (P50 CA100632) and Research Program Project (P01 CA049639 and P01 CA148600) Grants (to JJM); an NIH Training Program in Cancer Immunobiology T32 CA009598 (to HLP and CK); a Leukemia & Lymphoma Society Translational Research Program (6477-15) Grant (to GA); and a Lung Cancer SPORE (2P50 CA070907) grant (to DLG). The work was also supported by the generous philanthropic contributions to The University of Texas MD Anderson Lung Cancer Moon Shots Program. The MD Anderson Flow Cytometry and Cellular Imaging Core at South Campus is supported by NCI Cancer Center Support Grant P30CA16672. The authors thank the following from MD Anderson: Dr. Amin Momin for data retrieval, Dr. Anne V. Philips for thoughtful and technical contributions, Amanda Cernosek Herrmann for the H1299+A2 cell line, Dr. Anna Sergeeva for technical expertise and reagents, and Joy Nichols for coordination and communication.
Footnotes
The authors have no conflicts of interest to disclose.
References
- 1.McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351(6280):1463–9. doi: 10.1126/science.aaf1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. The New England journal of medicine. 2015;373(17):1627–39. doi: 10.1056/NEJMoa1507643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mishalian I, Bayuh R, Eruslanov E, Michaeli J, Levy L, Zolotarov L, et al. Neutrophils recruit regulatory T-cells into tumors via secretion of CCL17–a new mechanism of impaired antitumor immunity. Int J Cancer. 2014;135(5):1178–86. doi: 10.1002/ijc.28770. [DOI] [PubMed] [Google Scholar]
- 4.Rezvani K, Yong AS, Mielke S, Savani BN, Musse L, Superata J, et al. Leukemia-associated antigen-specific T-cell responses following combined PR1 and WT1 peptide vaccination in patients with myeloid malignancies. Blood. 2008;111(1):236–42. doi: 10.1182/blood-2007-08-108241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sergeeva A, He H, Ruisaard K, St John L, Alatrash G, Clise-Dwyer K, et al. Activity of 8F4, a T-cell receptor-like anti-R1/HLA-A2 antibody, against primary human AML in vivo. Leukemia. 2016 doi: 10.1038/leu.2016.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Molldrem J, Dermime S, Parker K, Jiang YZ, Mavroudis D, Hensel N, et al. Targeted T-cell therapy for human leukemia: cytotoxic T lymphocytes specific for a peptide derived from proteinase 3 preferentially lyse human myeloid leukemia cells. Blood. 1996;88(7):2450–7. [PubMed] [Google Scholar]
- 7.Molldrem JJ, Lee PP, Wang C, Champlin RE, Davis MM. A PR1-human leukocyte antigen-A2 tetramer can be used to isolate low-frequency cytotoxic T lymphocytes from healthy donors that selectively lyse chronic myelogenous leukemia. Cancer Res. 1999;59(11):2675–81. [PubMed] [Google Scholar]
- 8.Molldrem JJ, Lee PP, Wang C, Felio K, Kantarjian HM, Champlin RE, et al. Evidence that specific T lymphocytes may participate in the elimination of chronic myelogenous leukemia. Nat Med. 2000;6(9):1018–23. doi: 10.1038/79526. [DOI] [PubMed] [Google Scholar]
- 9.Qazilbash MH, Wieder E, Thall PF, Wang X, Rios R, Lu S, et al. R1 peptide vaccine induces specific immunity with clinical responses in myeloid malignancies. Leukemia. 2016 doi: 10.1038/leu.2016.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alatrash G, Mittendorf EA, Sergeeva A, Sukhumalchandra P, Qiao N, Zhang M, et al. Broad cross-presentation of the hematopoietically derived PR1 antigen on solid tumors leads to susceptibility to PR1-targeted immunotherapy. Journal of immunology. 2012;189(11):5476–84. doi: 10.4049/jimmunol.1201221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mittendorf EA, Alatrash G, Qiao N, Wu Y, Sukhumalchandra P, St John LS, et al. Breast cancer cell uptake of the inflammatory mediator neutrophil elastase triggers an anticancer adaptive immune response. Cancer Res. 2012;72(13):3153–62. doi: 10.1158/0008-5472.CAN-11-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chawla A, Alatrash G, Philips AV, Qiao N, Sukhumalchandra P, Kerros C, et al. Neutrophil elastase enhances antigen presentation by upregulating human leukocyte antigen class I expression on tumor cells. Cancer Immunol Immunother. 2016 doi: 10.1007/s00262-0161841-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eruslanov EB, Bhojnagarwala PS, Quatromoni JG, Stephen TL, Ranganathan A, Deshpande C, et al. Tumor-associated neutrophils stimulate T cell responses in early-stage human lung cancer. The Journal of clinical investigation. 2014;124(12):5466–80. doi: 10.1172/JCI77053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caron E, Vincent K, Fortier MH, Laverdure JP, Bramoulle A, Hardy MP, et al. The MHC I immunopeptidome conveys to the cell surface an integrative view of cellular regulation. Mol Syst Biol. 2011;7:533. doi: 10.1038/msb.2011.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.St John LS, Wan L, He H, Garber HR, Clise-Dwyer K, Alatrash G, et al. PR1-specific cytotoxic T lymphocytes are relatively frequent in umbilical cord blood and can be effectively expanded to target myeloid leukemia. Cytotherapy. 2016;18(8):995–1001. doi: 10.1016/j.jcyt.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Erup Larsen M, Kloverpris H, Stryhn A, Koofhethile CK, Sims S, Ndung’u T, et al. HLArestrictor-a tool for patient-specific predictions of HLA restriction elements and optimal epitopes within peptides. Immunogenetics. 2011;63(1):43–55. doi: 10.1007/s00251-010-0493-5. [DOI] [PubMed] [Google Scholar]
- 17.Nielsen M, Andreatta M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med. 2016;8(1):33. doi: 10.1186/s13073-016-0288-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoof I, Peters B, Sidney J, Pedersen LE, Sette A, Lund O, et al. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics. 2009;61(1):1–13. doi: 10.1007/s00251-008-0341-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scholtalbers J, Boegel S, Bukur T, Byl M, Goerges S, Sorn P, et al. TCLP: an online cancer cell line catalogue integrating HLA type, predicted neo-epitopes, virus and gene expression. Genome Med. 2015;7:118. doi: 10.1186/s13073-015-0240-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature. 2014;515(7528):572–6. doi: 10.1038/nature14001. [DOI] [PubMed] [Google Scholar]
- 21.Tripathi SC, Peters HL, Taguchi A, Katayama H, Wang H, Momin A, et al. Immunoproteasome deficiency is a feature of non-small cell lung cancer with a mesenchymal phenotype and is associated with a poor outcome. Proc Natl Acad Sci U S A. 2016 doi: 10.1073/pnas.1521812113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kan-Mitchell J, Bisikirska B, Wong-Staal F, Schaubert KL, Bajcz M, Bereta M. The HIV-1 HLA-A2-SLYNTVATL is a help-independent CTL epitope. Journal of immunology. 2004;172(9):5249–61. doi: 10.4049/jimmunol.172.9.5249. [DOI] [PubMed] [Google Scholar]
- 23.Gregory AD, Hale P, Perlmutter DH, Houghton AM. Clathrin pit-mediated endocytosis of neutrophil elastase and cathepsin G by cancer cells. J Biol Chem. 2012;287(42):35341–50. doi: 10.1074/jbc.M112.385617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Houghton AM, Rzymkiewicz DM, Ji H, Gregory AD, Egea EE, Metz HE, et al. Neutrophil elastase-mediated degradation of IRS-1 accelerates lung tumor growth. Nat Med. 2010;16(2):219–23. doi: 10.1038/nm.2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–7. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sergeeva A, Alatrash G, He H, Ruisaard K, Lu S, Wygant J, et al. An anti-PR1/HLA-A2 T-cell receptor-like antibody mediates complement-dependent cytotoxicity against acute myeloid leukemia progenitor cells. Blood. 2011;117(16):4262–72. doi: 10.1182/blood-2010-07-299248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alatrash G, Ono Y, Sergeeva A, Sukhumalchandra P, Zhang M, St John LS, et al. The role of antigen cross-presentation from leukemia blasts on immunity to the leukemia-associated antigen PR1. Journal of immunotherapy. 2012;35(4):309–20. doi: 10.1097/CJI.0b013e31824b3b14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma Q, Garber HR, Lu S, He H, Tallis E, Ding X, et al. A novel TCR-like CAR with specificity for R1/HLA-A2 effectively targets myeloid leukemia in vitro when expressed in human adult peripheral blood and cord blood T cells. Cytotherapy. 2016 doi: 10.1016/j.jcyt.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paul S, Weiskopf D, Angelo MA, Sidney J, Peters B, Sette A. HLA class I alleles are associated with peptide-binding repertoires of different size, affinity, and immunogenicity. Journal of immunology. 2013;191(12):5831–9. doi: 10.4049/jimmunol.1302101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348(6230):69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 31.Relle M, Mayet WJ, Strand D, Brenner W, Galle PR, Schwarting A. Proteinase 3/myeloblastin as a growth factor in human kidney cells. J Nephrol. 2003;16(6):831–40. [PubMed] [Google Scholar]
- 32.Bories D, Raynal MC, Solomon DH, Darzynkiewicz Z, Cayre YE. Down-regulation of a serine protease, myeloblastin, causes growth arrest and differentiation of promyelocytic leukemia cells. Cell. 1989;59(6):959–68. doi: 10.1016/0092-8674(89)90752-6. [DOI] [PubMed] [Google Scholar]
- 33.Kawata J, Yamaguchi R, Yamamoto T, Ishimaru Y, Sakamoto A, Aoki M, et al. Human Neutrophil Elastase Induce Interleukin-10 Expression in Peripheral Blood Mononuclear Cells through Protein Kinase C Theta/Delta and Phospholipase Pathways. Cell J. 2016;17(4):692–700. doi: 10.22074/cellj.2016.3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duan Z, Li FQ, Wechsler J, Meade-White K, Williams K, Benson KF, et al. A novel notch protein, N2N, targeted by neutrophil elastase and implicated in hereditary neutropenia. Mol Cell Biol. 2004;24(1):58–70. doi: 10.1128/MCB.24.1.58-70.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schliekelman MJ, Taguchi A, Zhu J, Dai X, Rodriguez J, Celiktas M, et al. Molecular portraits of epithelial, mesenchymal, and hybrid States in lung adenocarcinoma and their relevance to survival. Cancer Res. 2015;75(9):1789–800. doi: 10.1158/0008-5472.CAN-14-2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214–8. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, et al. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014;24(5):743–50. doi: 10.1101/gr.165985.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74(18):5057–69. doi: 10.1158/0008-5472.CAN-13-3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lou Y, Diao L, Parra Cuentas ER, Denning WL, Chen L, Fan YH, et al. Epithelial-mesenchymal transition is associated with a distinct tumor microenvironment including elevation of inflammatory signals and multiple immune checkpoints in lung adenocarcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2016 doi: 10.1158/1078-0432.CCR-15-1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gabillet J, Millet A, Pederzoli-Ribeil M, Tacnet-Delorme P, Guillevin L, Mouthon L, et al. Proteinase 3, the autoantigen in granulomatosis with polyangiitis, associates with calreticulin on apoptotic neutrophils, impairs macrophage phagocytosis, and promotes inflammation. Journal of immunology. 2012;189(5):2574–83. doi: 10.4049/jimmunol.1200600. [DOI] [PubMed] [Google Scholar]
- 41.Kurosawa S, Esmon CT, Stearns-Kurosawa DJ. The soluble endothelial protein C receptor binds to activated neutrophils: involvement of proteinase-3 and CD11b/CD18. Journal of immunology. 2000;165(8):4697–703. doi: 10.4049/jimmunol.165.8.4697. [DOI] [PubMed] [Google Scholar]
- 42.Ramachandran R, Mihara K, Chung H, Renaux B, Lau CS, Muruve DA, et al. Neutrophil elastase acts as a biased agonist for proteinase-activated receptor-2 (PAR2) J Biol Chem. 2011;286(28):24638–48. doi: 10.1074/jbc.M110.201988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bensinger WI, Maloney D, Storb R. Allogeneic hematopoietic cell transplantation for multiple myeloma. Seminars in hematology. 2001;38(3):243–9. doi: 10.1016/s0037-1963(01)90016-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.