

Abstract
The selective activation of unreactive hydrocarbons by biosynthetic enzymes has inspired new synthetic methods in C–H bond activation. Herein, we report the unprecedented two-step biosynthetic conversion of thiotetromycin to thiotetroamide C involving the tandem oxidation and amidation of an unreactive ethyl group. We detail the genetic and biochemical basis for the terminal amidation in thiotetroamide C biosynthesis, which involves a uniquely adapted cytochrome P450-amidotransferase enzyme pair and highlights the first oxidation-amidation enzymatic cascade reaction leading to the selective formation of a primary amide group from a chemically inert alkyl group. Motivated by the ten-fold increase in antibiotic potency of thiotetroamide C ascribed to the acetamide group and the unusual enzymology involved, we enzymatically interrogated diverse thiolactomycin analogues and prepared an unnatural thiotetroamide C analogue with potentiated bioactivity compared to the parent molecule.
Keywords: biosynthesis, selective C–H activation, cytochrome P450, amidotransferase, unnatural thiotetronates, potentiated bioactivity
Table-of-Content Graphics
Enzymatic oxidation-amidation cascade: Uncovering the biochemical genesis of the terminal amide group of thiotetroamide C illuminated a uniquely adapted cytochrome P450-amidotransferase enzyme pair that selectively and sequentially oxidizes and amidates an unreactive ethyl group, leading to a significant increase in antibiotic potency. Substrate promiscuity of this enzyme pair provides an opportunity in chemoenzymatic synthesis.
The selective transformation of ubiquitous but inert C–H bonds to other functional groups is fundamentally important to life and immensely useful in chemical industry.[1] The discovery and development of new selective (bio)synthetic transformations is a goal that has long been pursued but still remains significantly challenging.[2] To address this challenge, organic chemists have elaborated new chemical strategies, such as the design of novel directing groups or templates[3] and the development of new catalysts or catalyst-controlled methods without pre-installation of a directing group.[2, 4] On the other hand, it is well appreciated that a myriad of naturally occurring metalloenzymes selectively activate C–H bonds through the docking of a substrate in a specific orientation.[1a, 5] In addition, enzyme engineering as well as hybridization of enzymes and synthetic organometallic systems hold promise for controlling site selectivity of reactions conducted in abiological systems.[6] Thus, development of “biomimetic” catalysts have been extensively studied[1a, 7] and have led to a variety of biologically inspired C–H bond functionalization reactions, e.g., hydroxylation,[8] amination,[9] and cyclization.[10] Among these catalysts, some have achieved predictable selectivities on a broad range of substrates.[11] In turn, the success of biomimetic catalysis has prompted the discovery of new biosynthetic reactions for potential inspiration or utilization in organic synthesis and medicinal chemistry.[1a]
The interplay of chemical and enzymatic C–H bond activation is further illuminated by the realization that many pharmaceutically significant natural compounds only gain biological activity upon C–H bond functionalization by dedicated tailoring enzymes, as represented by the remarkable series of stereospecific and regiospecific hydroxylation reactions at eight reduced carbon sites during the maturation of paclitaxel from taxadiene.[12] Thus, the merits of unreactive hydrocarbon functionalization in both chemistry and biological activity motivated our study on the unprecedented C–H bond amidation of an unactivated ethyl group in the biosynthetic conversion of thiotetromycin (5) to thiotetroamide C (TTM C, also known as Tü 3010[13]; 1) that leads to a ten-fold increase in antibiotic potency (Figure 1). Herein, we report the genetic and biochemical characterization of the first oxidation-amidation enzymatic cascade to functionalize an inert terminal C–H bond and achieve the chemoenzymatic synthesis of natural and unnatural thiotetroamides with enhanced bioactivity.
Figure 1.

Representative thiotetronate antibiotics biosynthetic gene clusters and metabolites. a) Gene organization of the thiotetroamide (ttm) and thiolactomycin (tlm) gene clusters from Streptomyces afghaniensis NRRL 5621 and Salinispora pacifica CNS 863, respectively.[18] b) Chemical structures of the major products of the ttm (thiotetroamide C, 1) and tlm (thiolactomycin, 2, and its analogues 3–5) pathways.
TTM C (1) and thiotetromycin (5) belong to a class of rare and structurally unique thiotetronate antibiotics that selectively inhibits type II fatty acid synthase (FAS-II) in both Gram-positive and Gram-negative bacteria.[14] The selectivity at FAS-II makes thiotetronate antibiotics prominent due to the lack of this target in humans. Moreover, this class of antibiotics has emerged as an attractive lead for developing new anti-tuberculosis,[15] anti-malarial,[16] anti-trypanosomal,[16a] and anti-toxoplasmosis drugs.[17] Among the thiotetronate antibiotics (e.g., 1–5), TTM C (1) bears an acetamide moiety (Figure 1b) and consequently exhibits 10-fold enhancement of potency compared to other members.[14c] The acetamide group in 1, in comparison to methyl and ethyl substituents in the other thiotetronate antibiotics (e.g., 2–5), was strongly suggestive of an unprecedented C–H bond amide functionalization that selectively occurs at one of the two side chain ethyl groups of thiotetromycin 5. Thus, motivated by the anticipated selective C–H bond activation in TTM C biosynthesis that links to bioactivity enhancement, we set out to elucidate the biosynthetic logic of how the acetamide group in TTM C is constructed at both the genetic and biochemical levels.
We first performed a comparative bioinformatics analysis. We recently discovered, captured, and heterologously expressed thiotetroamide (ttm) and thiolactomycin (tlm) gene clusters in Streptomyces coelicolor M1152, leading to the isolation of 1 as the major compound from the Streptomyces afghaniensis NRRL 5621 ttm gene cluster and four TLMs (2–5) from the Salinispora pacifica CNS-863 tlm pathway (Figure 1).[18] These two gene clusters, and related homologs independently reported by Leadlay and co-workers,[19] contain four core genes (ttm/tlmF-I) that encode hybrid polyketide synthase-nonribosomal peptide synthetase proteins for the construction of the parent thiolactone skeleton of this family of antibiotics.[20] Different from the tlm cluster, the ttm locus harbors three additional genes (ttmN-P) which were predicted from sequence to code for an ATP-dependent amidotransferase (TtmN), a cytochrome P450 (CYP450, TtmP), and its electron shuttle ferredoxin (TtmO) (Figure 1a). Based on this comparative bioinformatics analysis and the structural differences between 1 and 5 featuring an acetamide moiety in 1, we[18] and Leadlay[19] previously proposed the involvement of TtmN-P in the terminal amide formation of 1.
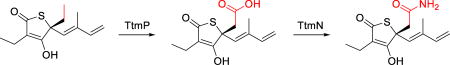
To explore this biosynthetic proposal, we sequentially deleted the genes by λ-Red-mediated recombination[21] (Supporting Materials and Methods; Tables S1 and S2) and analyzed the chemical extracts of the resulting mutants by LC-MS. In-frame deletion of the annotated CYP450 gene, ttmP, completely abolished the production of 1 with concomitant accumulation of 5 as the major product (Figure 2a). While disruption of ttmN encoding an amidotransferase also eliminated the production of 1, the mutant instead produced prethiotetroamide C (8; Figure 2a), which we earlier identified as a minor metabolite in the ttm heterologous expression host.[18] Based on these findings, we envision an unprecedented oxidation-amidation two-step process for the construction of the acetamide group in 1. First, the CYP450 (TtmP) selectively activates the terminal C–H bond of the C-4 ethyl group of 5 via a triple oxidation[22] to form the carboxyl group in 8 (Scheme 1). Second, a transfer of ammonia[23] to this carboxylate intermediate (8) is catalyzed by the amidotransferase (TtmN) in an ATP dependent manner to complete the terminal amidation process and generate 1 (Scheme 1).
Figure 2.

In vivo and in vitro characterization of the C–H bond oxidation-amidation in the biosynthesis of thiotetroamide C. a) HPLC chromatograms (239 nm) of organic extracts of the heterologous host harboring ttm gene cluster, and ΔttmP and ΔttmN mutants. b) HR-LCMS analysis of the in vitro assays showing three sequential oxidation reactions catalyzed by the CYP450 TtmP to ultimately form the carboxylic acid product 8, which was further amidated by the Mg2+/ATP-dependent amidotransferase TtmN with l-glutamine as the preferred amino donor to generate the final amide product 1. All traces are extracted ion chromatograms (EICs) for m/z 268.1002, 239.1100, 255.1049, 253.0893, and 269.0842 that correspond to [M+H]+ of 1 and 5–8, respectively.
Scheme 1.

Biosynthetic conversion of thiotetromycin (5) to thiotetroamide C (1) by the oxidation-amidation cascade catalyzed by the TtmP P450 and the TtmN amidotransferase. The in vitro chemoenzymatic synthesis of 1 was achieved in two sequential steps through intermediates 6–8 (reactions a and b) and in a single, one-pot dual reaction (reaction c) using an NADH regeneration assay with phosphite dehydrogenase (PtdH). Reaction details are provided in the Supporting Information document. Abbreviations: AMP = adenosine monophosphate; ATP = adenosine triphosphate; NADH/NAD+ = reduced/oxidized nicotinamide adenine dinucleotide; PPi = inorganic pyrophosphate.
To rigorously validate the functions of TtmP and TtmN, we next biochemically characterized these two enzymes using 5 and 8 as substrates that we isolated from the corresponding mutants (Supporting Materials and Methods; Tables S3 and S4; Figures S1–S4, S8, and S36–S44). TtmP and TtmO were each cloned, expressed, and purified as soluble recombinant N-hexahistadyl-tagged proteins from Escherichia coli, while TtmN was cloned as a C-hexahistadyl-tagged recombinant protein and produced solubly in S. lividans TK23 (Supporting Materials and Methods; Tables S1 and S2; Figure S9). As a gene encoding a ferredoxin reductase, needed as an ancillary electron transport protein for the CYP450 TtmP, was not identified within or adjacent to the ttm gene cluster, we used the ferredoxin reductase Bmp10 previously characterized from Pseudoalteromonas luteoviolacea 2ta16[24] for the TtmP assay. Thus, upon incubation of TtmP, TtmO, and Bmp10 with 100-fold molar excess of 5, we observed by LC-MS the initial formation of an alcohol intermediate, 11-hydroxythiotetromycin (6), followed by the complete conversion to the fully oxidized carboxylate product 8 (Figure 2b). The structure of 6 was proposed based on its high-resolution MS (Figure S8), characteristic thiotetronate UV profile (Figure S2), and reaction mechanism, and the identity of 8 was initially confirmed by LC-MS comparison to an authentic standard (Figures S8 and S10). An anticipated aldehyde intermediate (7) was only detected by LC-MS at a trace level (Figure 2b; Figure S8), potentially due to its rapid conversion to 8 or its relative instability to the extraction and workup procedures.
We next investigated the function of the amidotransferase TtmN using compound 8 as a substrate. As expected, we observed that TtmN catalyzed a Mg2+/ATP-dependent conversion of 8 to the amide product 1 using l-glutamine as the preferred nitrogen donor (Figure 2b; Supporting Materials and Methods; Table S3; Figures S2, S3, S8, S11, S12, and S18–S23), which was consistent with TtmN’s anticipated function as a glutamine amidotransferase homologue.[23] In the absence of Mg2+ or ATP, no production of 1 was detected (Figure S12a). During the conversion of 8 to 1, we observed consumption of l-glutamine with concomitant production of l-glutamic acid (Figure S12b), consistent with the side chain of l-glutamine as the nitrogen source for TtmN. While using one equivalent of l-glutamine to the substrate led to an approximately 90% conversion of 8 to 1 after 16 h incubation (30 °C) at high concentration of TtmN (1:5 molar ratio of TtmN to 8), increasing l-glutamine to 30 equivalents facilitated the full conversion within 2 hours with significantly less amount of TtmN used (1:100 molar ratio of TtmN to 8). In contrast to the full conversion using l-glutamine as the nitrogen donor, a marginal production of 1 was detected when replacing l-glutamine with l-asparagine (Figure S12a). Following the functional investigation of TtmN, we next showed that the one-pot coupled assay with TtmP and TtmN successfully demonstrated the dual oxidation-amidation conversion of 5 to 1 (Figure 2b).
Although the chemical synthesis of unamidated thiotetronate members (e.g., 2–5) has been previously reported, the total synthesis of TTM C (1) has yet to be achieved.[16, 25] Thus, we were motivated to chemoenzymatically synthesize 1 at semipreparative scale (Scheme 1 and Supporting Materials and Methods). We performed the synthesis using TtmP and TtmN successively or together in a one-pot reaction, with 3.6 mg of 5 added as starting material for each reaction. To allow for efficient regeneration of NADH,[26] we included sodium phosphite and phosphite dehydrogenase in the reaction involving TtmP. Given that the substrate and product were previously observed unstable, we used a 50:1 molar ratio of substrate to enzyme to facilitate the full conversion of each reaction within 2 hours, as monitored by LC-MS. The product of the TtmP reaction, 8, as well as the final amide product (1) generated from both reactions, were isolated and characterized by NMR (Figures S18 and S40). Our utilization of this new enzyme pair thus enabled for the first time the chemoenzymatic synthesis of 1 from 8 in high isolated yield of 67% (TtmP and TtmN used successively) or 80% (TtmP and TtmN combined for a one-pot synthesis).
With the functional characterization of the acetamide forming biosynthetic enzymes TtmP-TtmN completed, we explored their potential as biocatalysts for the synthesis of unnatural TTM analogues in a preliminary set of experiments. We assembled a focused chemical library of thiotetronate analogues (2–4 and 9–18; Figures 1b, 3a, and 3b; See Supporting Materials and Methods as well as Figure S1 for isolation process) from native strains and heterologous expression hosts cultured under assorted media conditions. Compounds 2–4, 10, 12, 13, 17, and 18, as representatives of each structural subcategory, were fully characterized (see Supporting Materials and Methods for structure elucidation; Tables S4–S7; Figures S2, S4–S8, S13, S14, S24–S35, and S45–S66) and evaluated as enzyme substrates. Altogether these molecules feature a broad range of structural differences compared to 5, including the size of the thiolactone ring, the length of the C-2 and C-4 alkyl side chains, and the oxidation state of the C-4 butadienyl moiety (Figures 3a and 3b). We observed a conversion of all γ-thiolactone analogues with a C-4 ethyl group (3, 10, 12, 17, and 18) to their corresponding carboxylic acids (19, 21, 23, 25, and 27) by TtmP in approximately 60–80% yields, as estimated by LC-MS, followed by a complete amidation of these acid intermediates to form amide products 20, 22, 24, 26, and 28, respectively, by TtmN (Figures 3a, S8, and S15). Conversely, we did not observe oxidation at C-4 methyl groups nor C-2 methyl/ethyl substituents of all the analogues tested (Figures 3a and 3b). In the case of the δ-thiolactone series 13–16, we tested them collectively as a single group and as 13 alone, and in all cases no product was observed (Figure 3b). This small substrate series highlights the inherent selectivity and specificity of the TtmP-TtmN acetamide forming enzymes and, as a consequence, provides future opportunities for biocatalytic C–H amide functionalization reactions in organic synthesis.
Figure 3.

Chemoenzymatic production of unnatural thiotetroamide analogues and evaluation of antimicrobial activity. Compounds in panel (a) were recognized as enzymatic substrates of TtmP and TtmN and converted to carboxylic acid and amidated products, respectively, while those in panel (b) were not substrate analogues reacted upon by TtmP-TtmN. c) Disk diffusion assay with S. coelicolor M1152 showing the increased potency of the unnatural amidated product 20 compared to the substrate 3, which is comparable to that observed between the natural thiotetroamide C (1) and thiotetromycin (5).
With the previous observation that 1 exhibited significantly enhanced FAS-II inhibitory activity over its non-oxidized homologue 5,[14c] we next explored the impact of the terminal amidation on the bioactivity of the thiotetronate analogues shown to be substrates of the TtmP-TtmN enzyme pair. As a representative example, compound 3 was used for a semipreparative-scale in vitro enzymatic conversion to the corresponding amide product 20 in ~75% isolated yield (Figure 3a; Figure S16). This unnatural TTM analogue, assigned the name thiotetroamide D, was structurally characterized by HRMS, comparative MS2 fragmentation, and 1H NMR analyses (Figures S8, S17, and S67). As anticipated, 20 displayed potentiated antimicrobial activity against S. coelicolor M1152 in a disk diffusion assay (Figure 3c). We plan to report the bioactivity evaluation of this and other new TTM analogues against a broad range of microbes in a separate publication.
Terminal amide functionality is prevalent in Nature, as many mammalian and arthropodic biologically active peptide hormones, neurotransmitters, and signaling lipids terminate in a primary amide group (Figure S68),[27] which contributes to both the biological activity and chemical stability.[28] Incorporation of a terminal amide group is also frequently utilized in medicinal chemistry for lead optimization, and this functionality is present in a fair number of marketed drugs (e.g., Briviact® and Adlyxin® among the total 14 non-antibody drugs approved by U.S. FDA in 2016[29]). Accordingly, the biosynthesis of terminal amides have been actively studied. In mammals, this process involves in situ generation of ammonia and subsequent transamidation (Scheme S1 A)[23] or oxidative cleavage of C-terminal glycine by a peptidylglycine α-amidating monooxygenase (PAM; Scheme S1 B).[27b, 27d, 28b] Although less well-studied in microbes,[30] the terminal amidation in myxothiazol A biosynthesis has been hypothesized to be catalyzed by an integrated unusual monooxygenase domain that appears to act similarly to PAM, despite the absence of known PAM homologues in bacteria.[31] Additionally, two divergent approaches have been recently discovered to perform terminal amidation in the biosynthesis of thiopeptide family of antibiotics via an enamide dealkylation (Scheme S1 C)[32] or a sequential deesterification-amidation mechanism (Scheme S1 D).[33] Thus, the present study reports an unprecedented oxidation-amidation cascade for activation of an inert C–H bond and expands the molecular understanding of terminal amidation biosynthetic chemistry.
In summary, we describe here the first genetic and biochemical characterization of a oxidation-amidation enzymatic cascade reaction leading to the efficient and selective formation of a terminal amide group from a chemically inert alkyl group in thiotetroamide C (1). This enzymatic functionalization reaction, catalyzed by a uniquely adapted CYP450-amidotransferase enzyme pair, expands current strategies for selective C–H bond activation, oxidation, and subsequent amidation. Our in vitro chemoenzymatic transformation using this enzyme pair has completed the first reported synthesis of thiotetroamide C as well as the preparation of the unnatural thiotetroamide D with potentiated bioactivity. Given the prevalence and significance of primary amides in pharmaceutical application, the C–H functionalization enzymes TtmP-TtmN described here may further have utility as biocatalysts in synthetic biology and drug discovery programs, especially when coupled with the possibility for further protein engineering to expand their substrate scope.
Supplementary Material
Acknowledgments
We thank our UCSD colleagues Ellis O’Neill, Jan Wohlfarth, Abrahim El Gamal, Shaun McKinnie, Vinayak Agarwal, Marlene Rothe, Yuta Kudo, Hanna Luhavaya, Ethan Older, and Brendan M. Duggan for discussions and help with experiments. The authors thank Professor Michihiko Kobayashi (University of Tsukuba, Japan) for providing the vector pHSA81, Professor Satish K. Nair (University of Illinois at Urbana-Champaign, USA) for providing the PtdH clone, and Professor Mervyn Bibb (John Innes Centre, UK) for providing S. coelicolor M1152 strain. Funding was generously provided by NIH grants R01-AI047818 and R01-AI117712 to B.S.M. and by the Uehara Memorial Foundation through a postdoctoral fellowship to T.A.
Contributor Information
Dr. Jie Li, Center of Marine Biotechnology and Biomedicine, Scripps Institution of Oceanography, University of California at San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0204 (USA).
Dr. Xiaoyu Tang, Center of Marine Biotechnology and Biomedicine, Scripps Institution of Oceanography, University of California at San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0204 (USA).
Dr. Takayoshi Awakawa, Center of Marine Biotechnology and Biomedicine, Scripps Institution of Oceanography, University of California at San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0204 (USA) Graduate School of Pharmaceutical Sciences, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-0033 (Japan).
Dr. Bradley S. Moore, Center of Marine Biotechnology and Biomedicine, Scripps Institution of Oceanography, University of California at San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0204 (USA); Skaggs School of Pharmacy and Pharmaceutical Sciences, University of California at San Diego, 9500 Gilman Drive, La Jolla, CA 92093 (USA).
References
- 1.a) Que L, Jr, Tolman WB. Nature. 2008;455:333–340. doi: 10.1038/nature07371. [DOI] [PubMed] [Google Scholar]; b) Labinger JA, Bercaw JE. Nature. 2002;417:507–514. doi: 10.1038/417507a. [DOI] [PubMed] [Google Scholar]
- 2.Hartwig JF, Larsen MA. ACS Cent. Sci. 2016;2:281–292. doi: 10.1021/acscentsci.6b00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Colby DA, Bergman RG, Ellman JA. Chem. Rev. 2010;110:624–655. doi: 10.1021/cr900005n. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang Z, Tanaka K, Yu JQ. Nature. 2017;543:538–542. doi: 10.1038/nature21418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Cook AK, Schimler SD, Matzger AJ, Sanford MS. Science. 2016;351:1421–1424. doi: 10.1126/science.aad9289. [DOI] [PubMed] [Google Scholar]; b) Liao K, Negretti S, Musaev DG, Bacsa J, Davies HM. Nature. 2016;533:230–234. doi: 10.1038/nature17651. [DOI] [PubMed] [Google Scholar]; c) Obligacion JV, Bezdek MJ, Chirik PJ. J. Am. Chem. Soc. 2017;139:2825–2832. doi: 10.1021/jacs.6b13346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Narayan ARH, et al. Nat. Chem. 2015;7:653–660. doi: 10.1038/nchem.2285. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Walsh CT. Nat. Chem. Biol. 2005;1:122–124. doi: 10.1038/nchembio0805-122. [DOI] [PubMed] [Google Scholar]
- 6.Hoarau M, Hureau C, Gras E, Faller P. Coordin. Chem. Rev. 2016;308:445–459. [Google Scholar]
- 7.a) Bordeaux M, Galarneau A, Drone J. Angew. Chem. Int. Ed. 2012;51:10712–10723. doi: 10.1002/anie.201203280. [DOI] [PubMed] [Google Scholar]; b) Bordeaux M, Galarneau A, Drone J. Angew. Chem. 2012;124:10870–10881. [Google Scholar]
- 8.Sorokin AB, Kudrik EV, Bouchu D. Chem. Commun. 2008:2562–2564. doi: 10.1039/b804405h. [DOI] [PubMed] [Google Scholar]
- 9.Dydio P, Key HM, Hayashi H, Clark DS, Hartwig JF. J. Am. Chem. Soc. 2017;139:1750–1753. doi: 10.1021/jacs.6b11410. [DOI] [PubMed] [Google Scholar]
- 10.Hyster TK, Knorr L, Ward TR, Rovis T. Science. 2012;338:500–503. doi: 10.1126/science.1226132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Breslow R, Huang Y, Zhang X, Yang J. Proc. Natl. Acad. Sci. USA. 1997;94:11156–11158. doi: 10.1073/pnas.94.21.11156. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen MS, White MC. Science. 2007;318:783–787. doi: 10.1126/science.1148597. [DOI] [PubMed] [Google Scholar]
- 12.Clardy J, Walsh C. Nature. 2004;432:829–837. doi: 10.1038/nature03194. [DOI] [PubMed] [Google Scholar]
- 13.Rapp C, Jung G, Isselhorstscharr C, Zahner H. Liebigs Ann. Chem. 1988:1043–1047. [Google Scholar]
- 14.a) Heath RJ, Rock CO. Curr. Opin. Investig. Drugs. 2004;5:146–153. [PMC free article] [PubMed] [Google Scholar]; b) Jackowski S, Murphy CM, Cronan JE, Jr, Rock CO. J. Biol. Chem. 1989;264:7624–7629. [PubMed] [Google Scholar]; c) Young K, et al. Antimicrob. Agents Chemother. 2006;50:519–526. doi: 10.1128/AAC.50.2.519-526.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kremer L, et al. J. Biol. Chem. 2000;275:16857–16864. doi: 10.1074/jbc.M000569200. [DOI] [PubMed] [Google Scholar]
- 16.a) Jones SM, Urch JE, Brun R, Harwood JL, Berry C, Gilbert IH. Bioorg. Med. Chem. 2004;12:683–692. doi: 10.1016/j.bmc.2003.11.023. [DOI] [PubMed] [Google Scholar]; b) Jones SM, Urch JE, Kaiser M, Brun R, Harwood JL, Berry C, Gilbert IH. J. Med. Chem. 2005;48:5932–5941. doi: 10.1021/jm049067d. [DOI] [PubMed] [Google Scholar]
- 17.Martins-Duarte ES, Jones SM, Gilbert IH, Atella GC, de Souza W, Vommaro RC. Parasitol. Int. 2009;58:411–415. doi: 10.1016/j.parint.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 18.Tang X, Li J, Millan-Aguinaga N, Zhang JJ, O'Neill EC, Ugalde JA, Jensen PR, Mantovani SM, Moore BS. ACS Chem. Biol. 2015;10:2841–2849. doi: 10.1021/acschembio.5b00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tao W, et al. Chem. Sci. 2016;7:376–385. doi: 10.1039/c5sc03059e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a) Havemann J, Yurkovich ME, Jenkins R, Harringer S, Tao W, Wen S, Sun Y, Leadlay PF, Tosin M. Chem. Commun. 2017;53:1912–1915. doi: 10.1039/c6cc09933e. [DOI] [PubMed] [Google Scholar]; b) Tang X, Li J, Moore BS. Chembiochem. 2017;18:1072–1076. doi: 10.1002/cbic.201700090. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Yurkovich ME, Jenkins R, Sun Y, Tosin M, Leadlay PF. Chem. Commun. 2017;53:2182–2185. doi: 10.1039/c6cc09934c. [DOI] [PubMed] [Google Scholar]
- 21.Gust B, Challis GL, Fowler K, Kieser T, Chater KF. Proc. Natl. Acad. Sci. USA. 2003;100:1541–1546. doi: 10.1073/pnas.0337542100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.a) Ro DK, et al. Nature. 2006;440:940–943. doi: 10.1038/nature04640. [DOI] [PubMed] [Google Scholar]; b) Schrewe M, Julsing MK, Lange K, Czarnotta E, Schmid A, Buhler B. Biotechnol. Bioeng. 2014;111:1820–1830. doi: 10.1002/bit.25248. [DOI] [PubMed] [Google Scholar]; c) Du L, et al. Proc. Natl. Acad. Sci. USA. 2017;114:E5129–E5137. doi: 10.1073/pnas.1702317114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Larsen TM, Boehlein SK, Schuster SM, Richards NG, Thoden JB, Holden HM, Rayment I. Biochemistry. 1999;38:16146–16157. doi: 10.1021/bi9915768. [DOI] [PubMed] [Google Scholar]
- 24.Agarwal V, El Gamal AA, Yamanaka K, Poth D, Kersten RD, Schorn M, Allen EE, Moore BS. Nat. Chem. Biol. 2014;10:640–647. doi: 10.1038/nchembio.1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.a) Dormann KL, Bruckner R. Angew. Chem. Int. Ed. 2007;46:1160–1163. doi: 10.1002/anie.200603562. [DOI] [PubMed] [Google Scholar]; b) Dormann KL, Bruckner R. Angew. Chem. 2007;119:1178–1182. doi: 10.1002/anie.200603562. [DOI] [PubMed] [Google Scholar]
- 26.El Gamal A, et al. Proc. Natl. Acad. Sci. USA. 2016;113:3797–3802. doi: 10.1073/pnas.1519695113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.a) Cravatt BF, Prospero-Garcia O, Siuzdak G, Gilula NB, Henriksen SJ, Boger DL, Lerner RA. Science. 1995;268:1506–1509. doi: 10.1126/science.7770779. [DOI] [PubMed] [Google Scholar]; b) Eipper BA, Milgram SL, Husten EJ, Yun HY, Mains RE. Protein Sci. 1993;2:489–497. doi: 10.1002/pro.5560020401. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Eipper BA, Stoffers DA, Mains RE. Annu. Rev. Neurosci. 1992;15:57–85. doi: 10.1146/annurev.ne.15.030192.000421. [DOI] [PubMed] [Google Scholar]; d) Prigge ST, Kolhekar AS, Eipper BA, Mains RE, Amzel LM. Science. 1997;278:1300–1305. doi: 10.1126/science.278.5341.1300. [DOI] [PubMed] [Google Scholar]
- 28.a) Eipper BA, Mains RE. Annu. Rev. Physiol. 1988;50:333–344. doi: 10.1146/annurev.ph.50.030188.002001. [DOI] [PubMed] [Google Scholar]; b) Merkler DJ. Enzyme Microb. Technol. 1994;16:450–456. doi: 10.1016/0141-0229(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 29.U.S. FDA. [Accessed July 21st, 2017]; www.fda.gov/Drugs/DevelopmentApprovalProcess/DrugInnovation/ucm483775.htm.
- 30.a) Chen Y, Wendt-Pienkowski E, Ju J, Lin S, Rajski SR, Shen B. J. Biol. Chem. 2010;285:38853–38860. doi: 10.1074/jbc.M110.147744. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Dunbar KL, Mitchell DA. ACS Chem. Biol. 2013;8:473–487. doi: 10.1021/cb3005325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.a) Silakowski B, et al. J. Biol. Chem. 1999;274:37391–37399. doi: 10.1074/jbc.274.52.37391. [DOI] [PubMed] [Google Scholar]; b) Weinig S, Hecht HJ, Mahmud T, Muller R. Chem. Biol. 2003;10:939–952. doi: 10.1016/j.chembiol.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 32.Yu Y, Guo H, Zhang Q, Duan L, Ding Y, Liao R, Lei C, Shen B, Liu W. J. Am. Chem. Soc. 2010;132:16324–16326. doi: 10.1021/ja106571g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liao R, Liu W. J. Am. Chem. Soc. 2011;133:2852–2855. doi: 10.1021/ja1111173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.