

Abstract
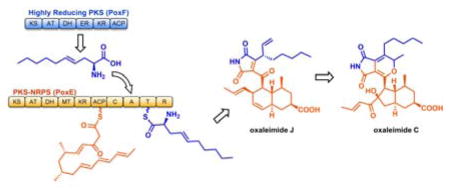
Fungal polyketide synthases (PKSs) can function collaboratively to synthesize natural products of significant structural diversity. Here we present a new mode of collaboration between a highly reducing PKS (HRPKS) and a PKS-nonribosomal peptide synthetase (PKS-NRPS) in the synthesis of oxaleimides from the Penicillium species. The HRPKS is recruited in the synthesis of an olefin-containing free amino acid, which is activated and incorporated by the adenylation domain of the PKS-NRPS. The precisely positioned olefin from the unnatural amino acid is proposed to facilitate a scaffold rearrangement of the PKS-NRPS product to forge the maleimide and succinimide cores of oxaleimides.
Graphical Abstract
Fungal polyketides, synthesized by iterative polyketide synthases (IPKSs), comprise a structurally diverse family of natural products, displaying a wide range of biological activities.1 Standalone IPKSs are capable of assembling complete polyketide scaffolds, such as aromatic compounds by nonreducing PKSs (NRPKS),1c or more reduced products by highly reducing PKSs (HRPKSs).1b Nature can increase structural diversity of polyketides by collaboratively pairing IPKSs in the same biosynthetic pathway.1a For example, pairing of a HRPKS and a NRPKS in series can lead to the synthesis of compounds with both aromatic and reduced structural features, such as radicicol2 and chaetoviridin A (Figure 1A).3 Collaborative pairing of two HRPKSs also enables the parallel construction of two separate parts of the final natural product, as illustrated in the biosynthesis of lovastatin.4 Collaborative IPKSs are frequently colocalized in the same biosynthetic gene cluster, a genetic feature that can be utilized in the genome mining of new natural products.5
Figure 1.
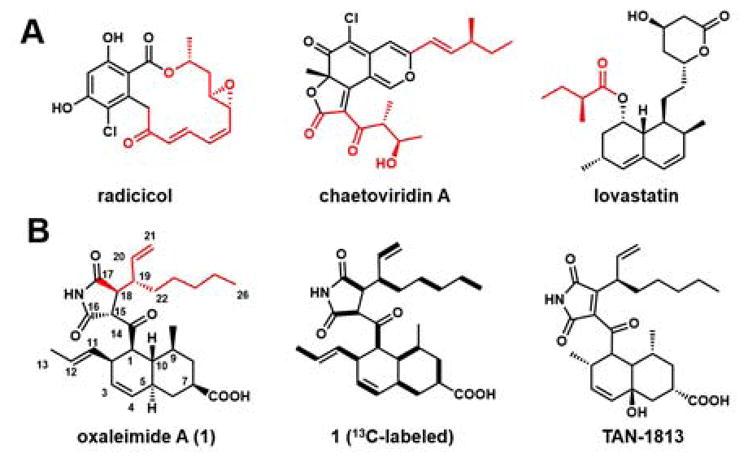
Structures of (A) fungal polyketides derived from collaborative actions of two IPKSs. (B) oxaleimide 1 and TAN-1813. Bold lines show enrichment from [1,2-13C]-acetate.
One strategy to install nitrogen-containing functional groups in fungal polyketides is used by hybrid megasynthases known as IPKS-nonribosomal peptide synthetases (PKS-NRPSs).6 The cis-acting NRPS module can amidate the completed polyketide acyl chain with a specific amino acid selected by the adenylation domain. The aminoacyl polyketide can then be cyclized into compounds that contain a nitrogen heterocycle, such as tetramate. Hence, one would expect pairing a PKS-NRPS with other IPKSs can lead to additional structural diversity. Here we demonstrate a new mode of collaboration between a HRPKS and a PKS-NRPS, in which the HRPKS is responsible for biosynthesis of a free, unnatural amino acid that is subsequently incorporated by the PKS-NRPS to generate new succinimide and maleimide natural products.
During our genome mining efforts, we found two homologous biosynthetic gene clusters in Penicillium oxalicum (pox) and Penicillium aethiopicum (pae) that encode a conserved pair of HRPKS (poxF) and PKS-NRPSs (poxE) (Figure 2A and Table S2). In addition, several other notable tailoring enzymes are also conserved (>80% identity), including a dissociated enoylreductase (ER) as partner of the PKS-NRPS (poxP), a decalin-forming Diels-Alderase (poxQ),7 an α/β-hydrolase (poxO), three P450 monooxygenases (poxC, poxD and poxM), and an aminotransferase (poxL). RT-PCR analysis showed that both clusters were transcriptionally silent when cultured under laboratory conditions (See SI and Figure S6). To activate transcription of the pox cluster, we overexpressed a Gal4-like transcription factor, PoxB, that is encoded in the gene cluster.8 The poxB gene was placed under the PgpdA promoter and was ectopically integrated into the genome of P. oxalicum to give the strain POX3. RT-PCR analysis of POX3 grown on PDA showed that the pox genes were transcribed (Figure S6). Subsequently, metabolites from POX3 were extracted and analyzed by LC/MS. Compared to the wild type strain, which produced secalonic acid D (*)9 and linolenic acid (**) as major products (Figure 2B, ii), POX3 produced an array of compounds (1–10) upon PoxB overexpression (Figure 2B, i). We then selectively inactivated either the HRPKS poxF or the PKS-NRPS poxE in the POX3 strain (Supporting Information). Both strains lost the ability to produce all of the compounds (Figure 2B, iii and iv), establishing the necessity of both megasynthases in the biosynthesis of 1–10.
Figure 2.

Characterization of the natural products from the pox cluster in P. oxalicum. (A) The gene clusters of interest. (B) HPLC traces of the metabolic extracts from different P. oxalicum cultures: (i) POX3 (transcriptionally activated through overexpression of PoxB) (ii) wild-type, (iii) ΔpoxE of POX3, (iv) ΔpoxF of POX3 and (v) ΔpoxO of POX3. *: secalonic acid D; **: linolenic acid that overlaps with 12. (C) X-ray crystal structure of 11a, (R)-PGME-amide of 1. (D) Structures of oxaleimide 2, 3, 9 and 10. (E) LC/MS analysis of the metabolic extracts from the culture of (i) A. nidulans A1145 (An), (ii) An expressing poxF, (iii) An expressing poxF and poxM, (iv) An expressing poxF, poxM and poxL, and (v) standard of 14, containing trace of olefin isomer. Extracted LC traces corresponding to the m/z+ for 14 (m/z+ = 186), m/z− for 15 (m/z− = 185) and m/z− for 16 (m/z− = 183). (F) Chemical complementation of POX3/ΔpoxF using 14 or 15. Extracted LC traces corresponding to the m/z− for 2 (m/z− = 500) and 4–8 (m/z− = 484) of (i) POX3/ΔpoxF, (ii) POX3/ΔpoxF supplemented with 15, (iii) POX3/ΔpoxF supplemented with 14 and (iv) POX3. (G) Proposed biosynthesis of 14.
Each of the ten compounds were characterized to be new natural products. The most abundant compounds oxaleimides A (1), B (2) and C (3) all contain an unusual disubstituted succinimide moiety.10 At the 3 position of the succinimide ring in 1 and 2 (or C18 in Figure 1), both compounds contain a branched aliphatic chain that has a terminal olefin (oct-1-en-3-yl). 1 contains a substituted trans-decalin ring that is carboxylated at C7. To determine the absolute stereochemistry at C7 in 1, the carboxylate group was converted into (R)- and (S)-PGME amides (11a and 11b),11 which allowed assignment of the absolute configuration at C7 to be R (Figure S7). This stereochemistry was further confirmed by X-ray crystal structure of 11a (Figure 2C). The absolute stereochemistry of the remaining compounds was assigned based on that of 1. In contrast to 1, the southern portion of 2 is a 5–6 bicyclic ring system. Compound 3 was isolated as a diastereomeric pair (3 and 3′) and contains a pyran ring fused to the succinimide.
Oxaleimide I (9) is nearly identical to 1, except that the succinimide core is oxidized to a maleimide. A related compound TAN-1813 isolated from Phoma sp. FL-41510 (Figure 1B) is a Ras-farnesyltransferase inhibitor.12 Compared to TAN-1813, 9 differs in substituents around the trans-decalin ring. Oxaleimide J (10) results from migration of the maleimide double bond in 9. Lastly, compounds 4–8 are likely derived from oxidation of the decalin ring in 1 (Figure 3). We tested the cytotoxicity of the compounds toward HeLa cells using a cell viability assay (Supporting Information). Whereas 1 and 2 exhibited no activity, the maleimide 9 displayed cytotoxicity with an IC50 of 0.32 μM. Compounds 3 and 10 showed weaker toxicity with IC50 values ~ 4.0 μM (Figure S8).
Figure 3.

Proposed biosynthetic pathway of compound 1 to 10. PKS-NRPS domain abbreviations: KS: ketosynthase; MAT: malonyl-CoA:ACP transacylase; DH: dehydratase; MT: methyltransferase; KR: ketoreductase; ACP: acyl carrier protein; C: condensation; A: adenylation; T: thiolation; R: reductase; ER: enoylreductase. To determine the biosynthetic origin of the C18 substituent, we looked for a blocked mutant that may accumulate a precursor to 1 and 9 prior to rearrangement. Among the mutant constructs, we isolated a new compound 12 upon knockout of the α/β-hydrolase poxO (Figure 2b, v). 12 is the trans-decalin containing alcohol depicted in Figure 3. The alcohol can be formed from the over-reduction of the aldehyde 13. This could occur when the Knoevenagel condensation of 13 to form 18 does not take place. Formation of similar over-reduced products from PKS-NRPKS has been observed.14 Hence, this suggests that PoxO is responsible for the Knoevenagel condensation. More importantly, the structure of 12, and that inferred for 13, reveal that the aliphatic chain may be derived from incorporation of a nonproteinogenic amino acid, (S, E)-2-aminodec-4-enoic acid (14), by the NRPS domain of PoxE (Figure 3). Condensation between the octaketide product of the PKS and the amino group of 14 yields an aminoacyl product 17, which can be cyclized by the Diels-Alderase PoxQ and reductively released by the reductive (R) domain of PoxE to yield 13.
The trans-decalin compounds isolated in this study, such as 1, 9, and 10 are biosynthetically related to other compounds derived from the combined actions of PKS-NRPSs and Diels-Alderases, including equisetin13 and myceliothermophin.7b The key difference, is the replacement of the tetramate or 3-pyrrolin-2-one, derived from Dieckmann or Knoevenagel condensation of the aminoacyl precursor, respectively, with succinimide or maleimide. Additionally, the substituted allyl group at C18 is an unusual feature not seen among fungal polyketides. To probe the origin of the aliphatic group, we performed a feeding experiment using [1,2-13C2]-acetate and determined the 13C enrichment of 1 (Figure 1B, S17 and Table S4). The labeling studies showed that the substituent at C18 is of polyketide origin. Notably, the terminal olefin is derived from one intact unit of acetate, suggesting that a carbon skeletal rearrangement is required in the biosynthesis of 1.
To test this hypothesis, we chemically synthesized 14 using a two-step metathesis, deprotection reaction sequence from N-Fmoc-(L)-allylglycine, and supplemented it to either the ΔpoxE or ΔpoxF mutant. Whereas no oxaleimide production was restored in the former, addition of 14 led to near complete restoration of oxaleimide production in ΔpoxF (Figure 2F, iii). These results suggested that the HRPKS PoxF is directly involved in the biosynthesis of 14. A notable example of a HRPKS involved in the biosynthesis of an unnatural amino acid is that of 4R-4-[(E)-2-butenyl]-4-methyl-threonine (Bmt) synthase found in the cyclosporine pathway (Figure S9).15 A P450 and an aminotransferase, encoded in tandem in the cyclosporine cluster,16 were proposed to catalyze α-oxidation and transamination of the polyketide to yield Bmt, respectively. In the pox pathway, the same tandem arrangement of genes encoding a P450 (poxM) and an aminotransferase (poxL) is also observed (Figure 2A), providing further support for a dedicated pathway in the synthesis of 14.
The functions of PoxF, M and L were assayed using A. nidulans as a heterologous host (Figure 2E). Expressing the HRPKS alone did not yield any detectable product (Figure 2E, ii), while expressing PoxF and PoxM led to the formation of two new products: 2-hydroxydec-4-enoic acid (15) and 2-oxodec-4-enoic acid (16) (Figure 2E, iii). Further addition of the aminotransferase PoxL led to production of 14 (Figure 2E, iv and v). The role of the PoxL was verified in an enzyme assay during which 16 was reductively aminated to 14 using aspartic acid as the optimal amine donor (Figure S10). The intermediacy of 15 was confirmed when addition of 15 to P. oxalicum ΔpoxF restored the biosynthesis of 1–10 (Figure 2F, ii). We propose a biosynthetic pathway to 14 as shown in Figure 2G. The HRPKS PoxF is recruited by the pathway to synthesize an ACP-bound dec-4-enoate, which was not detected as a free acid in the extract of A. nidulans expressing PoxF only. PoxM-catalyzed oxidation at the α-position creates the enzyme-bound 2-hydroxydec-4-enoyl-ACP thioester, which may be prone to spontaneous hydrolysis to yield 15 due to increased electrophilicity of the carbonyl. 15 can then be further oxidized by PoxM to yield the α-ketoacid 16, which is reductively aminated by PoxL to yield 14.
The HRPKS PoxF hence serves in a new mode of collaborative biosynthesis with the PKS-NRPS PoxE, by providing the olefin containing amino acid 14. The internal olefin is essential, as feeding of the saturated 2-aminodecanoic acid to P. oxalicum ΔpoxE did not restore biosynthesis of 1–10. Therefore, the pathway strategically uses the highly programmed nature of PoxF to introduce a single double bond at a specific position. The importance of the double bond in the subsequent steps is shown in the proposed biosynthetic pathway (Figure 3). Following release of aldehyde 13, the Knoevenagel condensation affords 18 which is then oxidized at the 5-position of the pyrrolidone ring to give 19. This modification is observed in several related natural products.7b, 17 We propose the presence of the olefin from the amino acid building block allows for migration of the substituted allyl group to occur. This allylic transposition reaction takes place in a conjugate addition, semipinacol-like fashion to yield the succinimide 21. This rearrangement is consistent with the labeling pattern established through [1,2-13C2]-acetate feeding experiments. Iterative two-electron oxidations of the C7 methyl in 21 to the carboxylic acid can be catalyzed by one of two remaining P450s (PoxC or PoxD) to yield 1. Subsequent oxidation yields the maleimide scaffold 9. Both 1 and 9 can undergo oxidative modifications in the decalin ring to yield the series of products 2–8. As shown for 9, we propose ring contraction of the trans-decalin ring can be initiated by oxidation at C2 and C3 to give 22, which can undergo a semipinacol rearrangement to afford 23. Isomerization of the terminal olefin in 23 to 24, followed by 6π-electrocyclization to give the pyran 3 (and 3′). 2 is likely the terminal product of the same oxidation/rearrangement cascade starting from 1, although a number of shunt products 4–8 were isolated.
In summary, we have discovered a set of new natural products, named oxaleimides, of which the maleimide members are cytotoxic. Maleimide containing natural products are rare and only a few examples are known.12, 18 Formation of the maleimide requires a new mode of collaboration between two fungal megasynthases. Such collaboration expands the biosynthetic capabilities of fungal PKSs and the chemical space of polyketide natural products.
Supplementary Material
Acknowledgments
This work was supported by the NIH (1DP1GM106413 and 1R35GM118056 to YT, R01GM117016 to NKG and T32GM067555 to JED) and JSPS Program for Advancing Strategic International Networks to Accelerate the Circulation of Talented Researchers (No. G2604 to K.W.). M.S. is supported by Fellowship of Astellas Foundation for Research on Metabolic Disorders. We thank S.I. Khan for assistance in solving the X-ray structures. Chemical characterization studies were supported by shared instrumentation grants from the NSF (CHE-1048804) and the NIH NCRR (S10RR025631).
Footnotes
Notes
The authors declare no competing financial interests.
Experimental details, spectroscopic and computational data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Chooi YH, Tang Y. J Org Chem. 2012;77:9933. doi: 10.1021/jo301592k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cox RJ. Org Biomol Chem. 2007;5:2010. doi: 10.1039/b704420h. [DOI] [PubMed] [Google Scholar]; (c) Crawford JM, Townsend CA. Nat Rev Microbiol. 2010;8:879. doi: 10.1038/nrmicro2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Zhou H, Qiao K, Gao Z, Vederas JC, Tang Y. J Biol Chem. 2010;285:41412. doi: 10.1074/jbc.M110.183574. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Xu Y, Zhou T, Zhou Z, Su S, Roberts SA, Montfort WR, Zeng J, Chen M, Zhang W, Lin M, Zhan J, Molnar I. Proc Natl Acad Sci U S A. 2013;110:5398. doi: 10.1073/pnas.1301201110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Winter JM, Sato M, Sugimoto S, Chiou G, Garg NK, Tang Y, Watanabe K. J Am Chem Soc. 2012;134:17900. doi: 10.1021/ja3090498. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chiang YM, Szewczyk E, Davidson AD, Keller N, Oakley BR, Wang CC. J Am Chem Soc. 2009;131:2965. doi: 10.1021/ja8088185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campbell CD, Vederas JC. Biopolymers. 2010;93:755. doi: 10.1002/bip.21428. [DOI] [PubMed] [Google Scholar]
- 5.Ziemert N, Alanjary M, Weber T. Nat Prod Rep. 2016;33:988. doi: 10.1039/c6np00025h. [DOI] [PubMed] [Google Scholar]
- 6.Boettger D, Hertweck C. Chembiochem. 2013;14:28. doi: 10.1002/cbic.201200624. [DOI] [PubMed] [Google Scholar]
- 7.(a) Sato M, Yagishita F, Mino T, Uchiyama N, Patel A, Chooi YH, Goda Y, Xu W, Noguchi H, Yamamoto T, Hotta K, Houk KN, Tang Y, Watanabe K. Chembiochem. 2015;16:2294. doi: 10.1002/cbic.201500386. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Li L, Yu P, Tang MC, Zou Y, Gao SS, Hung YS, Zhao M, Watanabe K, Houk KN, Tang Y. J Am Chem Soc. 2016;138:15837. doi: 10.1021/jacs.6b10452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bergmann S, Schumann J, Scherlach K, Lange C, Brakhage AA, Hertweck C. Nat Chem Biol. 2007;3:213. doi: 10.1038/nchembio869. [DOI] [PubMed] [Google Scholar]
- 9.Steyn PS. Tetrahedron. 1970;26:51. doi: 10.1016/0040-4020(70)85006-2. [DOI] [PubMed] [Google Scholar]
- 10.Li XW, Ear A, Nay B. Nat Prod Rep. 2013;30:765. doi: 10.1039/c3np70016j. [DOI] [PubMed] [Google Scholar]
- 11.Yabuuchi T, Kusumi T. J Org Chem. 2000;65:397. doi: 10.1021/jo991218a. [DOI] [PubMed] [Google Scholar]
- 12.Ishii T, Hayashi K, Hida T, Yamamoto Y, Nozaki Y. J Antibiot. 2000;53:765. doi: 10.7164/antibiotics.53.765. [DOI] [PubMed] [Google Scholar]
- 13.Kato N, Nogawa T, Hirota H, Jang JH, Takahashi S, Ahn JS, Osada H. Biochem Biophys Res Commun. 2015;460:210. doi: 10.1016/j.bbrc.2015.03.011. [DOI] [PubMed] [Google Scholar]
- 14.(a) Fujii R, Minami A, Gomi K, Oikawa H. Tetrahedron Lett. 2013;54:2999. [Google Scholar]; (b) Song Z, Bakeer W, Marshall JW, Yakasai AA, Khalid RM, Collemare J, Skellam E, Tharreau D, Lebrun MH, Lazarus CM, Bailey AM, Simpson TJ, Cox RJ. Chem Sci. 2015;6:4837. doi: 10.1039/c4sc03707c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Offenzeller M, Santer G, Totschnig K, Su Z, Moser H, Traber R, Schneider-Scherzer E. Biochemistry. 1996;35:8401. doi: 10.1021/bi960224n. [DOI] [PubMed] [Google Scholar]
- 16.Bushley KE, Raja R, Jaiswal P, Cumbie JS, Nonogaki M, Boyd AE, Owensby CA, Knaus BJ, Elser J, Miller D, Di Y, McPhail KL, Spatafora JW. PLoS Genet. 2013;9:e1003496. doi: 10.1371/journal.pgen.1003496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh SB, Goetz MA, Jones ET, Bills GF, Giacobbe RA, Herranz L, Stevensmiles S, Williams DL. J Org Chem. 1995;60:7040. [Google Scholar]
- 18.(a) Bush JA, Long BH, Catino JJ, Bradner WT. J Antibiot. 1987;40:668. doi: 10.7164/antibiotics.40.668. [DOI] [PubMed] [Google Scholar]; (b) Elsebai MF, Nazir M, Kehraus S, Egereva E, Ioset KN, Marcourt L, Jeannerat D, Gütschow M, Wolfender J, König GM. Eur J Org Chem. 2012:6197. [Google Scholar]; (c) Nakagawa Y, Kano H, Tsukuda Y, Koyama H. Tetrahedron Lett. 1967;42:4105. doi: 10.1016/s0040-4039(01)89700-8. [DOI] [PubMed] [Google Scholar]; (d) Steglich W, Steffan B, Kopanski L, Eckhardt G. Angew Chem Int Ed. 1980;19:459. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.