

Abstract
Due to the ability of ovarian cancer (OCa) to acquire drug resistance, it has been difficult to develop efficient and safe chemotherapy for OCa. Here, we examined the therapeutic use of a new self-assembled core–shell nanoscale coordination polymer nanoparticle (NCP-Carbo/GMP) that delivers high loadings of carboplatin (28.0 ± 2.6 wt %) and gemcitabine monophosphate (8.6 ± 1.5 wt %). A strong synergistic effect was observed between carboplatin and gemcitabine against platinum-resistant OCa cells, SKOV-3 and A2780/CDPP, in vitro. The coadministration of carboplatin and gemcitabine in the NCP led to prolonged blood circulation half-life (11.8 ± 4.8 h) and improved tumor uptake of the drugs (10.2 ± 4.4% ID/g at 24 h), resulting in 71% regression and 80% growth inhibition of SKOV-3 and A2780/CDDP tumors, respectively. Our findings demonstrate that NCP particles provide great potential for the codelivery of multiple chemotherapeutics for treating drug-resistant cancer.
Keywords: nanoscale coordination polymers, carboplatin, gemcitabine, synergistic effect, platinum-resistant ovarian cancer
Graphical Abstract
INTRODUCTION
Ovarian cancer (OCa) is the deadliest of all gynecological cancers, accounting for 5% of all cancer deaths among women.1 Despite progress made with the introduction of a platinum–taxane combination as first-line treatment for OCa, the development of platinum (Pt) resistance often leads to relapse and patient’s eventual succumbing to tumors.2–4 The ability of OCa cells to resist multiple chemotherapeutics hinders successful treatment of OCa, with 85% of OCa patients relapsing following their successful first-line treatment.5,6 Pt-resistant disease is often seen when patients develop recurrence within six months of the initial Pt treatment.7,8 Treating the disease with Pt/taxane or other Pt based therapies again following first relapse has been shown to be ineffectual.9 The mechanisms of drug resistance include the decreased metabolism of drugs to the active chemotherapeutics,10 decreased cell uptakes and increased eflux of the drugs,11,12 increased cellular tolerance to the drugs13 or increased DNA repair.14–16 Combination therapy can counter these mechanisms of resistance without increasing dosages to the point of unacceptable toxicity.
The combination of carboplatin (Carbo) and gemcitabine was approved by the FDA in 2006 for the treatment of advanced recurrent ovarian cancer occurring at least 6 months after completion of Pt-based (e.g., Carbo or cisplatin) therapy. Gemcitabine, a nucleotide analogue, induces apoptosis by replacing cytidine during DNA replication.17 Together with Carbo, this combination therapy has been shown to work effectively against drug-resistant tumors without the added toxicity of other Pt cytotoxins (e.g., cisplatin).18–21 However, gemcitabine is often limited by its poor pharmacokinetics and rapid metabolic deactivation.22–24 Furthermore, the combination with Pt often enhances gemcitabine’s hematological toxicity.25,26 Therefore, there is an urgent need to develop a delivery system for combination drugs that can overcome the limitations imposed by free platinum drugs and gemcitabine.
Nanoparticle-based drug delivery systems can more easily control the many relevant properties of drugs than traditional small molecule agents. These include physical properties such as size and surface charge that are dictated by nanoparticle synthesis or assembly, and important pharmacokinetic parameters such as circulation half-life and drug release kinetics.27–33 In particular, nanoparticle drug delivery systems that are able to deliver multiple therapeutics agents have been shown to enhance antitumor efficacy, overcome multidrug resistance, and reduce addictive toxicity.34–42 We have developed a novel delivery vehicle, nanoscale coordination polymers (NCPs), for various cancer therapies,31,43–45 including a combination therapy delivery system carrying oxaliplatin and gemcitabine for the synergistic treatment of pancreatic cancer.46 These nanoparticles combine the organic and inorganic properties of nanoparticles, as well as their intrinsic biodegradability, to prolong blood circulation half-lives and effectively treat tumors with minimal side effects.27,28 Herein, we report that NCPs can codeliver Carbo and gemcitabine for synergistic therapy of Pt-resistant OCa. More importantly, a superior synergistic effect was found in vitro against two Pt-resistant OCa cell lines, SKOV-3 and A2780/CDDP. NCP-Carbo/GMP shows enhanced cellular uptake, prolonged blood circulation, and elevated tumor uptake, resulting in improved antitumor efficacy in Pt-resistant OCa tumor xenograft models.
RESULTS
Synthesis and Characterization of NCP-Carbo/GMP
The Carbo prodrug, Carbo-bis(phosphonic acid), was synthesized and characterized by NMR spectroscopy and mass spectrometry (Figures S1–S6). DOPA-capped NCP-Carbo/GMP nanoparticles (DOPA-NCP-Carbo/GMP) containing the Carbo prodrug and gemcitabine monophosphate (GMP) were initially synthesized by reverse microemulsion (Figure S7). The drug-containing core was capped by DOPA via Zn–phosphate interactions. DOPA further self-assembled into a monolayer via hydrophobic–hydrophobic interactions, making the particles dispersible in organic solvents. The surface was further coated with DSPC, cholesterol, and DSPE-PEG2k at a 1:1:0.75 molar ratio. The Z-average, number-average, PDI, and zeta-potential for NCP-Carbo/GMP were 85.3 ± 0.7 nm, 64.9 ± 1.7 nm, 0.069 ± 0.013, and −6.02 ± 0.55 mV, respectively, by dynamic light scattering (DLS) measurement (Figure 1B and Table 1). The near-neutral surface charge indicated that PEG chains were successfully coated on the particle surface. Transmission electronic microscopy (TEM) showed uniform spherical nanoparticles with a diameter of ~20 nm (Figure 1C), implying that the lipid coating did not disrupt the NCP core. DOPA-NCP-Carbo/GMP gave Carbo loadings of 28.0 ± 2.6 wt % (54.4 ± 5.1 wt % prodrug loading) by ICP-MS. GMP loading was found to be 8.6 ± 1.5 wt % by UV–vis (Figure S12). No change was seen in the particle size upon exposure in 30 nM bovine serum albumin in phosphate buffer saline at 37 °C after 24 h (Figure S13). The in vitro study showed that NCP-Carbo/GMP exhibited excellent stability.
Figure 1.
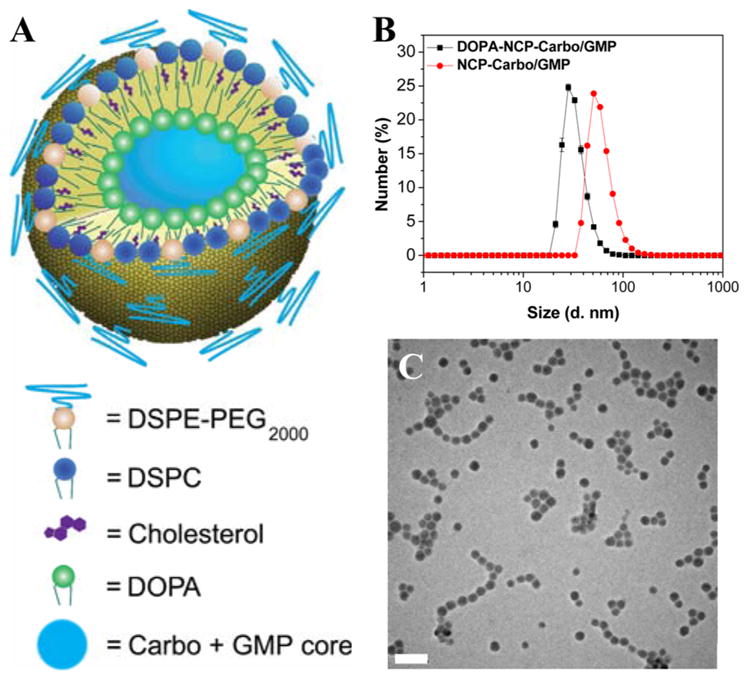
(A) Schematic of NCP-Carbo/GMP. (B) Number-average size distribution of NCP-Carbo/GMP particles. Bare and lipid coated particles were measured in THF and PBS, respectively. (C) TEM micrographs of NCP-Carbo/GMP. Scale bar = 100 nm.
Table 1.
Sizes, Polydispersities, and Zeta Potentials of NCP-Carbo/GMP Particlesa
| NCPs | Z-av diam (nm) | number-av diam (nm) | PDI | zeta potential (mV) |
|---|---|---|---|---|
| DOPA-NCP-Carbo/GMP | 56.9 ± 0.2b | 37.1 ± 0.4b | 0.112 ± 0.003 | na |
| NCP-Carbo/GMP | 85.3 ± 0.7c | 64.9 ± 1.7c | 0.069 ± 0.013c | −6.02 ± 0.55c |
Data are expressed as means ± SD.
Measured in THF.
Measured in PBS.
In Vitro Drug Release
The release profiles of Carbo and GMP from DOPA-NCP-Carbo/GMP and NCP-Carbo/GMP were investigated in PBS with or without 5 mM cysteine at 37 °C at pH 7.4 (Figure S14). In the absence of cysteine, DOPA-NCP-Carbo/GMP revealed rapid burst release due to the dissociation of phosphate groups from Zn2+, with 57% and 33% cumulative release of Pt and GMP before 2 h, respectively. In contrast, only 19% Pt and 21% GMP release were observed for NCP-Carbo/GMP after 24 h. 5 mM cysteine was added to PBS in order to investigate drug release from DOPA-NCP-Carbo/GMP in a simulated intracellular environment. These conditions accelerated the drug release of DOPA-NCP-Carbo/GMP with 82% of Pt and 57% of GMP release after 2 h, indicating that cysteine can penetrate into the NCP core and reduce the Carbo prodrug into active Carbo. Both the reduction of Pt–O bonds and the dissociation of phosphate groups with Zn2+ accelerated the drug release. However, NCP-Carbo/GMP exhibited similarly slow drug release patterns in PBS and PBS supplemented with 5 mM cysteine. The lipid coating on the particle surface prevented the penetration of cysteine into the NCP core, thereby rendering the particle more stable. The external lipid layer may be incorporated into the cell or plasma membrane during and after endocytosis, disrupting the lipid coatings, allowing cysteine to penetrate the particle core, and triggering the reductive degradation of NCPs to release the therapeutic payloads.
In Vitro Cytotoxicity and the Synergistic Effect
To evaluate the potency of NCP-Carbo/GMP, in vitro cytotoxicity was performed on SKOV-3 (Figure 2A,B) or A2780/CDDP (Figure 2D,E) ovarian cancer cells treated with nanoparticles or free drugs at different Carbo or GMP concentrations for 72 h. The cell viability was measured by MTS assay. As seen in Figure 2 and Table 2, the IC50 values of Carbo and GMP against SKOV-3 were 24.21 ± 0.96 μM and 1.89 ± 0.26 μM, respectively. When free Carbo and GMP were combined (Carbo&GMP), the Carbo and GMP IC50 values were dramatically decreased by 12-fold and 2.5-fold, respectively (Carbo IC50 = 2.01 ± 0.62 μM or GMP IC50 = 0.74 ± 0.23 μ M). NCP-Carbo/GMP showed comparable cytotoxicity as their corresponding free drug counterparts, with Carbo IC50 = 1.91 ± 0.55 μM and GMP IC50 = 0.70 ± 0.20 μM. In contrast, monotherapeutic NCP IC50 values of Carbo and GMP were 22.82 ± 2.49 μM and 1.98 ± 0.34 μM, respectively. Similar results were shown with A2780/CDDP cells with NCP-Carbo/GMP, with IC50 values that were about 7.6-fold, 14.4-fold, 2.3-fold, and 2.4-fold lower than those of free Carbo, NCP-Carbo, free GMP, and NCP-GMP, respectively.
Figure 2.

In vitro cytotoxicity plots and combination indices (CI) of Carbo/GMP combinations on SKOV-3 (A–C) and A2780/CDDP (D–F) cells. The cell viabilities on SKOV-3 and A2780/CDDP cells were measured after a 72 h exposure to NCP-Carbo, NCP-GMP, NCP-Carbo/GMP, or free drugs (Carbo, or GMP). Data are mean ± SD (n = 6).
Table 2.
IC50 Values of Carbo, GMP, NCP-Carbo, NCP-GMP, and NCP-Carbo/GMP against SKOV-3 and A2780/CDDP Cells
| Carbo (μM) | GMP (μM) | Carbo&GMP (μM) | NCP-Carbo (μM) | NCP-GMP (μM) | NCP-Carbo/GMP (μM) | |
|---|---|---|---|---|---|---|
| SKOV-3 | 24.2 ± 1.0 | (1.9 ± 0.3) | 2.0 ± 0.6 (0.7 ± 0.2) | 22.8 ± 2.5 | (2.0 ± 0.3) | 1.9 ± 0.6 (0.7 ± 0.2) |
| A2780/CDDP | 9.9 ± 2.2 | (1.1 ± 0.2) | 0.7 ± 0.2 (0.3 ± 0.1) | 18.7 ± 1.0 | (1.2 ± 0.2) | 1.3 ± 0.2 (0.5 ± 0.1) |
Data are expressed as means ± SD. The numbers in parentheses refer to GMP concentrations.
The combination index (CI) provides a quantitative measure of synergism (CI < 1), additivity (CI = 1), or antagonism (CI >1) for the drug combinations. The CI was around 0.5 for NCP-Carbo/GMP against the monotherapeutic NCP and free drugs over a large range of drug effect levels for both SKOV-3 (Figure 2C) and A2780/CDDP (Figure 2F). Synergy was thus seen between Carbo and GMP in both Pt-resistant ovarian cancer cells. These results suggest that codelivery of Carbo and
In Vitro Cell Apoptosis
To investigate the synergistic effect of Carbo and GMP on cell apoptosis, annexin V staining and flow cytometry analysis were performed to investigate cell apoptosis induced by free drugs or nanoparticle formulations. After incubation for 24 h, SKOV-3 and A2780/CDDP cells were stained with Alexa Fluor 488 conjugated annexin V and observed using CLMS. NCP-Carbo/GMP induced highest level of cell apoptosis, as evidenced by the presence of the most and brightest green fluorescence. Carbo&GMP, NCP-GMP, and GMP also induced high levels of cell apoptosis, while Carbo and NCP-Carbo resulted in much less cell apoptosis (Figures S16 and S17).
We further quantified cell apoptosis by flow cytometry (Figure S18). Similar to the cytotoxicity assay and CLSM analysis, NCP-Carbo/GMP showed the highest ability to induce cell apoptosis, resulting in 62.05% and 65.94% apoptosis for SKOV-3 and A2780/CDDP cells, respectively. Car-bo&GMP also showed significantly increased cell apoptosis in both ovarian cancer cell lines with 52.67% and 35.43% of cells undergoing apoptosis in SKOV-3 and A2780/CDDP, respectively. Free GMP and NCP-GMP showed comparable apoptotic cells, ranging from 47 to 49% in SKOV-3 and 26–35% in A2780/CDDP. Little or no apoptotic cells were seen in PBS, Carbo, and NCP-Carbo for both ovarian cancer cell lines.
In Vitro Cellular Uptakes
Rhodamine B-doped NCP-Carbo/GMP (RhB-NCP-Carbo/GMP) nanoparticles were synthesized for confocal microscopy studies. As seen in the DLS (Table S3) and TEM images (Figures S19 and S20), no difference was observed in the size distribution and morphology between dye-doped particles and NCP-Carbo/GMP. To directly observe the NCP-Carbo/GMP uptake, Rhodamine B-dyed nanoparticles were incubated with either SKOV-3 (Figure 3A) or A2780/CDDP (Figure 3B) cells and then observed under CLSM. After a 1 h incubation, the nanoparticles were readily taken up by the cells, as evidenced by the colocalization of green fluorescence (LysoTracker Green-stained endosome) and red fluorescence (Rhodamine B-dyed nanoparticle) seen around the exterior of the nucleus. The red fluorescence signal was greatly enhanced after 4 and 24 h, indicating an increased uptake of this nanoparticle over time.
Figure 3.

CLSM images of colocalization of RhB (red) from NCP-Carbo/GMP particles with a late endosome and lysosome marker, LysoTracker (green), in SKOV-3 (A) and A2780/CDDP (B) cells after incubation for different times. Scale bars: 50 μm. Cellular uptake of Carbo&GMP and NCP-Carbo/GMP in SKOV-3 (C, D) and A2780/CDDP (E, F) cells determined by ICP-MS (Pt uptake) and UV–vis (GMP uptake). Data are expressed as means ± SD (n = 3).
This result was further supported by time-dependent cellular uptake of NCP-Carbo/GMP in SKOV-3 (Figure 3C,D) and A2780/CDDP (Figure 3E,F). Free Carbo showed the highest cellular uptake at 1 h, but it decreased by ~50% at 24 h, indicating that the uptake of free Carbo is rapid. In contrast, the cellular uptake of NCP-Carbo/GMP increased gradually over time and was significantly higher compared to the free combination drugs. At 24 h, the Pt uptake was 4 times and 5 times higher than the free combination treatments in SKOV-3 and A2780/CDDP, respectively. The cellular uptake of free GMP remained constant throughout the 24 h experiment in A2780/CDDP cells, but decreased by ~50% at 24 h in SKOV-3 cells. The uptake of NCP-Carbo/GMP, in terms of GMP, also showed a slight gradual increase over time, but no significant difference was observed.
Pharmacokinetics and Biodistribution
The pharmaco-kinetics and biodistribution of NCP-Carbo/GMP were investigated to determine the extent of MPS clearance and the relative uptake of major MPS organs versus tumor on CT26 tumor bearing mice (Figure 4A). The Pt distribution was quantified by ICP-MS, and the GMP amount in the blood was quantified by HPLC-MS/MS. By ip injection, the blood circulation half-lives were fitted with a one-compartment model using PK solver. NCP-Carbo/GMP resulted in Pt and GMP blood circulation half-lives of 11.8 ± 4.8 h (Figure 4B) and 9.4 ± 1.4 h (Figure 4C), respectively. There were low percentages of injected dose per gram of tissue (% ID/g) in organs associated with MPS activity such as the liver (3.8 ± 2.8% ID/g), spleen (3.7 ± 3.6% ID/g), and kidney (4.8 ± 2.2% ID/g), supporting the ability of NCP-Carbo/GMP to evade MPS clearance. The slow blood clearance and low MPS uptake led to the high tumor accumulation of the drug. The Pt distribution in tumor tissue increased over time, reaching a maximum at 24 h (10.2 ± 4.4% ID/g), which indicated the high accumulation and long retention of NCP-Carbo/GMP in tumor tissue.
Figure 4.

Percentage injected dose per gram (% ID/g) (A) of Pt in tissues after intraperitoneal administration of NCP-Carbo/GMP in CT26 tumor bearing mice at time points 5 min and 1, 3, 5, 8, 24, and 48 h. Data are mean ± SD (n = 3). Average observed and predicted time-dependent Pt (B) and GMP (C) distributions in blood after administration of NCP-Carbo/GMP (n = 3). A one-compartment model was used in determining the Pt and GMP distribution in blood.
Antitumor Activity in Vivo
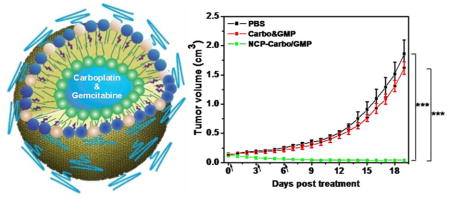
The antitumor activity of NCP-Carbo/GMP was evaluated on SKOV-3 and A2780/CDDP subcutaneous xenograft murine models. Mice bearing SKOV-3 or A2780/CDDP tumors were treated once every 3 days for a total of three ip injections at a dose of 10 mg of Carbo/kg and 2.4 mg of GMP/kg. All of the mice were sacrificed at day 19 for SKOV-3 and day 8 for A2780/CDDP, when the control tumors reached above 2000 mm3. As shown in Figure 5A, NCP-Carbo/GMP showed significant tumor regression in SKOV-3 tumors, with a reduction of 71% compared to the initial tumor volume. For the more aggressive A2780/CDDP tumor, NCP-Carbo/GMP also demonstrated dramatic tumor inhibition, with a tumor inhibitory rate (TIR) of 80% (Figure 5C). The tumor weight of NCP-Carbo/GMP-treated group was 90-fold and 12-fold smaller than that of control in SKOV-3 (Figure 5B) and A2780/CDDP (Figure 5E) tumors, respectively. On the contrary, mice treated with Carbo&GMP showed a similar tumor growth pattern as PBS, suggesting that free combination drugs caused no antitumor efficacy, possibly because free Carbo and GMP do not significantly accumulate in tumor tissue.
Figure 5.

Subcutaneous SKOV-3 xenografts: (A) In vivo tumor growth inhibition. Carbo (dose, 10 mg/kg) and gemcitabine (4.6 mg/kg) and NCP-Carbo/GMP (doses, 10 mg/kg and 4.6 mg/kg) were administered on days 0, 3, and 6. Data are expressed as means ± SD (n = 5). (B) End-point tumor weights. Data are expressed as means ± SD (n = 5). Subcutaneous A2780/CDDP xenografts: (C) In vivo tumor growth inhibition. Carbo (dose, 10 mg/kg) and gemcitabine (4.6 mg/kg) and NCP-Carbo/GMP (doses, 10 mg/kg and 4.6 mg/kg) were administered on days 0, 3, and 6. Data are expressed as means ± SD (n = 5). (D) End-point tumor weights. Data are expressed as means ± SD (n = 5), ***p < 0.001. gemcitabine could overcome drug resistance, leading to much enhanced anticancer efficacy against ovarian tumor models.
TUNEL assay was performed on the resected tumors to further substantiate and quantify the in vivo apoptosis. Apoptotic cells showing high fluorescence intensity indicative of DNA fragmentation were seen at a higher proportion of total cells in tumors treated with NCP-Carbo/GMP (Figures 6B and 6D). NCP-Carbo/GMP induced 92.7 ± 2.4% and 89.3 ± 2.8% tumor cell apoptosis for SKOV-3 and A2780/CDDP, respectively, whereas Carbo&GMP caused 4.0 ± 0.4% and 3.6 ± 0.5% apoptosis for SKOV-3 and A2780/CDDP, respectively. NCP-Carbo/GMP thus exhibits superior anti-cancer efficacy as compared to free drugs. Further analysis of the tumor tissue by H&E staining supported the TUNEL results of NCP-Carbo/GMP inducing the most apoptosis and necrosis compared to low or no apoptosis and necrosis in Carbo&GMP and PBS-treated tumors (Figures 6A and 6C).
Figure 6.

(A) Representative CLSM images of TUNEL assays and H&E staining of SKOV-3 tumor tissues. DNA fragment in apoptotic cells was stained with fluorescein-conjugated deoxynucleotides (green), and the nuclei were stained with DAPI (blue). Top bar = 50 μm. Bottom bar = 100 μm. (B) The percentages of TUNEL-positive cells in SKOV-3 tumor tissues. (C) Representative CLSM images of TUNEL assays and H&E staining of A2780/CDDP tumor tissues. DNA fragment in apoptotic cells was stained with fluorescein-conjugated deoxynucleotides (green), and the nuclei were stained with DAPI (blue). Top bar = 50 μm. Bottom bar = 100 μm. (D) The percentages of TUNEL-positive cells in A2780/CDDP tumor tissues. Data are expressed as means ± SD (n = 3), ***p < 0.001.
No obvious signs of toxicity were observed in vivo after treatment with NCP-Carbo/GMP, as evaluated by body weight evolution, immunogenic response, and histology of major organs. No significant weight loss was observed in the NCP-Carbo/GMP treated group, indicating the absence of severe systemic toxicity (Figures S24 and S25). Negligible differences were observed across treatment groups in the levels of proinflammatory cytokines assessing acute immunogenic response (Figure S26) and histological assessments (Figure S27). No statistically significant differences were observed between the control and NCP-Carbo/GMP groups in terms of the proinflammatory cytokine production. From the H&E-stained sectioned tissues of the heart, liver, lung, spleen, and kidney, no acute pathological change was detected in NCP-Carbo/GMP-treated groups.
DISCUSSION
Combinations of two or more chemotherapeutic agents are often employed as first-line therapy in the clinical treatment of a variety of cancers. The different drugs can either affect distinct molecular targets in order to delay molecular and genetic mutations by cancer cells to resist the chemotherapy, or target similar cellular mechanisms for synergistically enhanced therapy.47,48 However, significant differences in physical and chemical properties of chemotherapeutic drugs cause difficulties in optimizing treatment regimens. The pharmacokinetics and pharmacodynamics of each combination therapy component are unique, which results in inconsistent drug concentrations in tumor sites and suboptimal efficacy.49,50 Codelivering multiple chemotherapeutics within a single nanocarrier provides distinct advantages over administering multiple single-agent nano-particles or free drugs. Physical and chemical differences in solubility and charge of individual drugs are nullified when incorporated together onto a nanoparticle, allowing for a consistent ratio of drug at the tumor site.51–53 Specifically, the modular synthesis of NCP allows for highly reproducible drug loadings, and the core–shell structure endows nanoparticle stability and controlled release kinetics for both drugs.
Here, we utilized the NCP platform that combines the high drug loadings of chemotherapeutics carboplatin and gemcitabine at a precise ratio to elicit a therapeutic effect in a single nanocarrier for Pt-resistant OCa treatment. A major benefit of using nanoparticles of about 10–100 nm size for drug delivery is their intrinsic ability to target tumors by the enhanced permeability and retention (EPR) effect.54,55 NCP-Carbo/GMP has an optimal size that takes advantage of the leaky vasculature to accumulate in the tumor, while sufficiently avoiding both renal filtration which removes particles less than 40 nm and mononuclear phagocytic system (MPS)-mediated clearance of particles over 200 nm. In addition, the near-neutral surface charge of NCP-Carbo/GMP provides the possibility for the particles to resist opsonization. NCP-Carbo/GMP also showed very high stability under physiological conditions, such that drug leakage during systemic circulation was minimized and drug effects were primarily focused at the tumor site to reduce side effects.56–58 All of these characteristics contribute to the particles’ prolonged circulation time and improved tumor accumulation.
Once the particles enter the tumor cells, the lipid bilayer on the surface likely incorporates into the lipid assemblies of cell and plasma membranes, exposing the core of the particle to endogenous reducing agents, such as cysteine, for degradation of the particle to release the therapeutic payloads. The incorporation of lipid bilayer into the cell membranes might be able to prevent the eflux of the released drugs, as shown by gradual increased cellular uptake over time, while the cellular uptake rapidly decreased for free drugs over time due to the eflux effect of cells. As a result, NCP-Carbo/GMP has 5-times higher cellular uptake as compared to the free drugs. The enhanced tumor uptake led to effective regression of the Pt resistant SKOV-3 tumors and dramatic inhibition of the aggressive A2780/CDDP tumors in vivo. The tumor weights were ~90-fold and ~12-fold lower as compared to the control and free combination drugs, respectively.
CONCLUSION
We developed an NCP platform that combines Carbo and gemcitabine for successful treatment of Pt-resistant OCa. We show that Carbo and gemcitabine produce a synergistic effect in SKOV-3 and A2780/CDDP, and this synergy is retained when this combination is codelivered in an NCP. NCP-Carbo/GMP showed a long blood circulation half-life and high drug accumulation in the tumor, resulting in superior tumor regression in vivo when compared to free combination drugs. Equipped with high antitumor therapeutic potential and low systemic toxicity, NCPs possess the ideal characteristics to codeliver multiple chemotherapies to tumors for enhanced cancer therapy.
EXPERIMENTAL PROCEDURES
Materials, Cell Lines, and Animals
All starting materials were purchased from Sigma-Aldrich (Louis, MO) and Fisher Scientific (Pittsburgh, PA) unless otherwise noted and used without further purification. 1,2-Dioleoyl-sn-glycero-3-phosphate (DOPA), 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), cholesterol, and 1,2-distearoyl-sn-glycero-3-phosphoe-thanolamine-N-[amino(polyethylene glycol)2000] (DSPE-PEG2k) were purchased from Avanti Polar Lipids (Alabaster, AL). Gemcitabine monophosphate (GMP) was synthesized from gemcitabine following the literature.59
1H NMR spectra were recorded on a Bruker NMR 400 DRX spectrometer at 400 MHz. The mass spectra of Carbo prodrug, Carbo-bis(phosphonic acid), and its intermediates were determined on an electrospray ionization mass spectrometry (ESI-MS, Agilent 6130) by dissolving the prodrug in water, deprotonated with excess 3 M NaOH, and then filtered through 0.2 μm syringe filter. ESI-MS was taken in positive mode, with a fragmentation voltage of 100 V from 620 to 720 MW/z.
Human ovarian cancer cells SKOV-3 and murine colon cancer cells CT26 were purchased from the American Type Culture Collection (Rockville, MD) and grown in McCoy’s 5A and RPMI 1640, respectively, supplemented with 10% fetal bovine serum (FBS, Gibco, Grand Island, NY). Cisplatin-resistant human ovarian cancer cells A2780/CDDP were obtained from Developmental Therapeutics Core, Northwestern University, and cultured in RPMI 1640 containing 10% FBS. All cells were cultured in a humidified atmosphere containing 5% CO2 at 37 °C.
BALB/c female mice (6 weeks, 18–22 g) and athymic female nude mice (6 weeks, 18–22 g) were provided by Harlan Laboratories, Inc. (USA). The study protocol was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Chicago.
Carbo Prodrug Synthesis
Carbo synthesis was based on previous literature.60–62 Carbo was synthesized by adding equivalent amounts of Ag salt of cyclobutanedicarboxylic acid (Ag-Carbo, 1.51 g) to Pt(NH3)2I2 (2.05 g) in minimal H2O and stirred at room temperature and in darkness for 48 h. The solution was filtered, and the filtrate was evaporated, resulting in a white solid. The product cis-diammine(1,1-cyclobutanedicarboxylato)platinum(II) (Carbo) was recrystallized in 1:1 H2O:EtOH. Yield: 80%. 1H NMR in D2O: δ2.8 (t, 4H); δ 1.8 (quintet, 2H).
Carbo (1.04 g) was reacted with 30% H2O2 (8 mL) in H2O (16 mL) at 70 °C for 5 h in darkness. Addition of EtOH precipitated a white solid, Carbo-(OH)2, which was washed twice with EtOH. Yield: 80%. 1H NMR in D2O: δ 2.67 (t, 4H); δ 2.0 (quintet, 2H). The m/z of [M + H]+ was determined to be 406.1 (expected m/z = 406.1).
Carbo-(OH)2 (0.8 g) was dissolved in minimal DMF, to which was added 4 equiv of diethoxyphosphinyl isocyanate (1.2 mL). The mixture was allowed to react overnight in darkness. The solution was filtered, and the resulting lightly yellow Carbo-bis(phosphoester) product was precipitated by addition of Et2O. The product was then washed twice with Et2O. Yield: 78%. 1H NMR in D2O: δ 4.05 (m, 8H); δ 2.60 (m, 3H); δ 2.39 (m, 1H); δ 1.93 (m, 2H). The m/z of [M + H]+ was determined to be 764.2 (expected m/z = 764.1).
Trimethylsilyl bromide (TMSBr, 800 μL) was added slowly to the Carbo-bis(phosphoester) (0.8 g) dissolved in minimal DMF at 4 °C, and the solution was stirred for 18 h at rt in darkness under N2 protection. A lightly yellow solid was collected after addition of DCM and then washed twice with additional DCM. The solid was dissolved in MeOH and stirred overnight to hydrolyze the acid. The final product, cis, trans-[Pt(1,1-cyclobutanedicarboxylato)(NH3)2(OCONHP(O)-(OH)2)2] [Carbo-bis(phosphonic acid) prodrug] was collected by precipitation by DCM and then washed twice with additional DCM. Yield: 73%. 1H NMR in D2O: δ 2.57 (t, 3H), 2.43 (m, 1H), and 1.91 (m, 2H). 13C NMR in D2O: δ 179.9; 179.5; 55.7; 35.1; 28.0; and 15.4. 195Pt in D2O: δ 174 using K2PtCl4 as reference. The m/z of [M + MeOH]+ was determined to be 684.2 (expected m/z = 684.0).
Particle Synthesis and Characterization
DOPA-NCP-Carbo/GMP was synthesized in reverse microemulsions. To synthesize DOPA-NCP-Carbo/GMP, Carbo prodrug (331.3 μL, 11.5 μmol), GMP sodium salt solution (68.7 μL, 5.0 μmol), and DOPA (44 μmol) were added to a 10 mL aliquot of Triton X-100 (0.3 M in 1.5 M hexanol/cyclohexane) solution to form a W = 7.4 microemulsion. Another microemulsion of 10 mL of Triton X-100 (0.3 M, 1.5 M hexanol/cyclohexane) containing Zn(NO3)2·6H2O (262 μmol) was also prepared. The two microemulsions were stirred vigorously at room temperature for 15 min. The microemulsion containing the Zn complex was added to the other microemulsion dropwise. The combined emulsion was stirred for an additional 30 min at room temperature. After the addition of 20 mL of ethanol, the particles were washed once with ethanol and twice with 50% (v/v) ethanol/tetrahydrofuran (THF) before being redispersed in THF.
Carbo-loaded nanoparticles were dried, weighed, digested in concentrated nitric acid overnight, and diluted with water to determine the Carbo loading by inductively coupled plasma mass spectrometry (ICP-MS, Agilent 7700x ICP-MS). GMP loading was determined by UV–vis spectroscopy and thermogravimetric analyses (TGA). Particles were digested overnight in 6 M hydrochloric acid, and the concentration of GMP in the solution was determined by the absorbance at 275 nm using a Shimadzu UV-2401PC UV–vis spectrophotometer. A baseline spectrum was recorded using 6 M hydrochloric acid. Using the Pt–drug loading from ICP-MS and corresponding to the standards of carboplatin, the absorbance from Pt was subtracted from the total absorbance to determine the GMP–drug loading.
DOPA-capped NCP-Carbo/GMP nanoparticles were further coated with DSPC, cholesterol, and DSPE-PEG2k to increase their stability and allow them to circulate longer in the blood. A THF solution of DSPC, cholesterol, DSPE-PEG2k (molar ratio 1:1:0.75), and DOPA-capped NCP nanoparticles was added dropwise to 500 μL of 30% (v/v) ethanol/H2O at 60 °C under strong stirring. THF was evaporated, and the dispersion was allowed to cool to room temperature before use. NCP-Carbo nanoparticles carrying only Carbo and NCP-GMP particles carrying gemcitabine monotherapy were prepared similarly as NCP-Carbo/GMP.
In Vitro Stability Studies
Particle stability was evaluated in vitro in phosphate buffered saline (PBS) buffer with bovine serum albumin (BSA) binding and time-dependent drug release. BSA binding analysis was done by dispersing 0.45 mg of NCP-Carbo/GMP in 1 mL of PBS containing BSA (30 nM) at 37 °C. DLS measurements were detected every hour for 24 h to determine the size of nanoparticles in suspension over time.
In Vitro Drug Release
In vitro release profiles of Carbo and GMP from NCP-Carbo/GMP were performed in 250 mL of 1× PBS buffer with or without 5 mM cysteine at 37 °C and pH 7.4. DOPA-NCP-Carbo/GMP or NCP-Carbo/GMP (0.75 mg) was suspended in 4 mL of 1× PBS buffer solution with or without 5 mM cysteine in a 10,000 MWCO pleated dialysis bag. The dialysis bag containing the nanoparticle suspension was added into the beaker containing 250 mL of 1× PBS buffer, incubated at 37 °C at pH 7.4, while stirring. Periodically, 1 mL aliquots of solution were taken from the solution, and a fresh 1 mL of buffer solution with or without cysteine was added to the beaker. The removed aliquot was digested in nitric acid or HCl and analyzed by ICP-MS for Pt or UV–vis for GMP, respectively.
In Vitro Cytotoxicity and Synergistic Effect
In vitro cytotoxicity assays were carried out on SKOV-3 and A2780/CDDP ovarian cancer cells. In 96-well plates, SKOV-3 or A2780/CDDP ovarian cancer cells were seeded at a density of 1000 cells/well and 750 cells/well, respectively, in a total of 100 μL of McCoy or RPMI-1640 containing 10% FBS. The cells were incubated at 37 °C for 24 h prior to drug treatment. The culture medium was replaced by fresh medium. Different concentrations of Carbo, GMP, free Carbo plus GMP (Carbo&GMP) mixture (at the same NCP-Carbo and NCP-GMP drug dose), NCP-Carbo, NCP-GMP, and NCP-Carbo/GMP were added and incubated at 37 °C and 5% CO2 for 72 h, and cell viability was measured by MTS assay (Promega, USA) based on the manufacturer’s manual. IC50 values were calculated from curves constructed by plotting cell viability (%) versus drug concentration (μM).
An additional in vitro cytotoxicity study was performed on A2780/CDDP with similar conditions as above in which different concentrations of free drugs or particles were added for 4 h. The culture medium was replaced with fresh medium, and the cells were incubated for an additional 44 h. Cell viability was measured by MTS assay, and the IC50 values were determined.
The CI was calculated using the equation
where D1 and D2 are concentrations of drug 1 and drug 2 in combination at a specific drug effect level (e.g., 50% inhibition concentration), while Dm1 and Dm2 are the concentrations of single agent to reach the same drug effect level. CI values were plotted against drug effect level (ICx values). CI values lower than 1 indicate synergism.
Cell Apoptosis by Confocal Microscopy
For CLSM imaging, cells were seeded on 10 mm2 glass coverslips placed in 6-well plates at a density of 5 × 104 cells per well and incubated with free drugs or particles at a Carbo concentration of 1.9 μM for SKOV-3 and 1.3 μM for A2780/CDDP and/or a GMP concentration of 0.7 μM for SKOV-3 and 0.5 μM for A2780/CDDP for 24 h. After fixing with 4% paraformaldehyde, cells were stained with 10 μg/mL of DAPI and Alexa Fluor 488 conjugated annexin V and observed using CLSM at excitation wavelengths of 405 and 488 nm to visualize nuclei (blue fluorescence) and cell apoptosis (green fluorescence), respectively.
Cell Apoptosis by Flow Cytometry
SKOV-3 or A2780/CDDP were seeded at 500,000 cells/well in 6-well plates containing 2 mL total volume of cell culture medium for 24 h at 37°C and 5% CO2. The culture medium was replaced with fresh medium containing different drug treatments at Carbo concentration of 1.9 μM for SKOV-3 and 1.3 μM for A2780/CDDP and/or a GMP concentration of 0.7 μM for SKOV-3 and 0.5 μM for A2780/CDDP. Following a 24 h incubation, the floating and adherent cells were collected and stained with annexin V/dead cell apoptosis kit with Alexa Fluor 488 annexin V and propidium iodide (PI, Invitrogen, USA) based on the manufacturer’s instructions. The apoptosis was analyzed on a flow cytometer (LSRII 3–8, BD, USA).
In Vitro Cellular Uptakes and Intercellular Distribution
SKOV-3 or A2780/CDDP cells were seeded at a cell density of 5 × 105 cells per well and 2 mL total volume of medium containing 10% FBS. Following 24 h of incubation at 37°C and 5% CO2, the culture medium was replaced by 2 mL of fresh medium containing aliquots of free Carbo and GMP or NCP-Carbo/GMP at 0.96 μM Carbo (or 0.35 μM GMP) for SKOV-3 and 0.65 μM Carbo (or 0.24 μM GMP) for A2780/CDDP. The cells were cultured for different time points (1, 2, 4, and 24 h) at 37 °C and 5% CO2. Media were removed, and the cells were washed with 1× PBS three times. The adherent cells were collected by trypsinization and washed further with 1× PBS three times. The cell numbers were counted prior to the last wash. The cells were digested with concentrated nitric acid or 6 M HCl and analyzed using ICP-MS for Pt content or UV–vis for GMP content. Known cell numbers of SKOV-3 or A2780/CDDP with no drug treatments were used as baseline to subtract any interference from cellular components.
To directly observe the internalization and intracellular distribution of particles under CLSM, Rhodamine B (RhB), a fluorescence marker, was doped into the particle by adding it to the prodrug microemulsion during particle preparation. Cells were seeded on 10 mm2 glass coverslips placed in 6-well plates and incubated with RhB-doped NCP-Carbo/GMP for 1, 2, 4, and 24 h. Cells were washed with PBS three times, fixed with 4% paraformaldehyde, stained with DAPI and LysoTracker Green, and observed under CLSM (FV1000, Olympus, Japan).
In Vivo Pharmacokinetic and Biodistribution Studies
To evaluate the pharmacokinetics and biodistribution of NCP-Carbo/GMP, balb/c mice bearing CT26 tumors were intra-peritoneally injected with NCP-Carbo/GMP at 5 mg/kg Carbo dose (or 1.5 mg/kg GMP dose). Mice were sacrificed at 5 min and 1, 3, 5, 8, 24, and 48 h postinjection. Their liver, lung, spleen, kidney, heart, bladder, tumor, and blood were harvested and digested in concentrated nitric acid for 24 h, and the Pt concentrations were analyzed by ICP-MS.
GMP concentrations in plasma were further analyzed using high-performance liquid chromatography tandem mass spectrometry (HPLC-MS/MS, Agilent 6460 QQQ MS-MS). The extraction method followed a previous literature procedure. 200 μL of ice-cold acetonitrile was added to 50 μL of plasma, and the mixture was vortexed, mixed, and centrifuged. The resulting supernatant was evaporated and reconstituted in 100 μL of water. An injection volume of 20 μL of sample was used. The autosampler and column temperatures were kept at 4 and 30 °C, respectively. The samples were separated via a PGC Hypercarb column (100 mm × 2.1 mm i.d., 5 μm, Thermo Fisher Scientific) fitted with a guard column (Hypercarb 10 mm × 2.1 mm, 5 μm, Thermo Fisher Scientific). A gradient mobile phase of (A) 10 mM ammonium acetate at pH 10 and (B) acetonitrile were used with the initial mobile phase of 95% solvent A and 5% solvent B at a flow rate of 0.3 mL/min. After 2 min, solvent A was gradually decreased to 80% over 0.2 min and held at this condition for 5.6 min. The gradient was returned to 95% solvent A over 0.2 min, and this condition was held for an additional 7 min, for a total run time of 15 min. The mass to charge transition was monitored from 342 to 231.
Antitumor Activity in Vivo
The antitumor activity was conducted on two subcutaneous xenograft mouse models. SKOV-3 cells (5 × 106 cells in 100 μL of medium/matrix gel (v/v 1:1)) or A2780/CDDP cells (5 × 106 cells in 100 μL of medium) were subcutaneously injected in the right flank of mice. When the tumor volume reached around 100 mm3, mice were randomly divided into 3 groups (n = 5) and ip injected with PBS, Carbo&GMP, and NCP-Carbo/GMP at a dose of 10 mg of Carbo/kg and 2.4 mg of Gem/kg every 3 days. Tumor sizes were measured every day by a caliper and calculated as follows: (length × width2)/2. Body weight was also recorded every day as an indicator of systemic toxicity. All mice were sacrificed when the tumor volume of PBS group reached 2000 mm3.
In Vivo Immunogenic Response and General Toxicity Evaluation of NCP
At the end point of the in vivo efficacy, blood was collected, and the serum was separated for immunogenic response analysis. TNF-α, IFN-γ, and IL-6 production was determined by ELISA (R&D Systems, USA). Blood from the control group was also analyzed under the same treatment above for comparison. Organs (heart, liver, spleen, lung, and kidney) were also harvested, sectioned at 5 μm thickness, stained with H&E, and observed for histological examination of toxicity with light microscopy (Panoramic Scan Whole Slide Scanner, PerkinElmer, USA).
In Vivo Tumor Cell Apoptosis
SKOV-3 and A2780/CDDP tumors were excised and embedded in optimal cutting temperature (OCT) medium, sectioned at 5 μm thickness. TdT-mediated dUTP nick end labeling (TUNEL) assay was performed using DNA Fragmentation Detection Kit (Invitrogen, USA) and observed under CLSM to quantify in vivo apoptosis. Nuclei were stained with DAPI (10 μg/mL), and DNA fragments in apoptotic cells were stained with fluorescein-conjugated deoxynucleotides (green). The number of TUNEL-positive cells was divided by the total number of cells to calculate the percentage of apoptotic cells in the sample.
STATISTICAL ANALYSIS
Results were expressed as means ± standard deviation (SD). ANOVA was used to determine statistical significance. A P value <0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We acknowledge NIH (UO1-CA198989) and the University of Chicago Medicine Comprehensive Cancer Center (NIH CCSG: P30 CA014599) for funding support.
ABBREVIATIONS USED
- OCa
ovarian cancer
- Pt
platinum
- Carbo
carboplatin
- NCP
nanoscale coordination polymer
- DOPA
1,2-dioleoyl-sn-glyc-ero-3-phosphate
- GMP
gemcitabine monophosphate
- THF
tetrahydrofuran
- PBS
phosphate buffered saline
- CI
combination index
- RhB
rhodamine B
- TUNEL
TdT-mediated dUTP nick end labeling
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharma-ceut.6b00466.
Methods, ESI-MS, 1H, 13C, and 195Pt NMR, and UV–vis spectra, CLSM images, TEM micrographs, flow cytometry analysis, and additional experimental details (PDF)
References
- 1.Arend RC, Londono-Joshi AI, Straughn JM, Jr, Buchsbaum DJ. The Wnt/β-catenin pathway in ovarian cancer: A review. Gynecol Oncol. 2013;131(3):772–779. doi: 10.1016/j.ygyno.2013.09.034. [DOI] [PubMed] [Google Scholar]
- 2.McGuire WP, Markman M. Primary ovarian cancer chemotherapy: current standards of care. Br J Cancer. 2003;89:S3–S8. doi: 10.1038/sj.bjc.6601494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Markman M, Bookman MA. Second-line treatment of ovarian cancer. Oncologist. 2000;5(1):26–35. doi: 10.1634/theoncologist.5-1-26. [DOI] [PubMed] [Google Scholar]
- 4.Neijt JP, ten Bokkel Huinink WW, van der Burg ME, van Oosterom AT, Willemse PH, Vermorken JB, van Lindert AC, Heintz AP, Aartsen E, van Lent M. Long-term survival in ovarian cancer. Mature data from The Netherlands Joint Study Group for Ovarian Cancer. Eur J Cancer Clin Oncol. 1991;27(11):1367–72. doi: 10.1016/0277-5379(91)90011-2. [DOI] [PubMed] [Google Scholar]
- 5.Foley OW, Rauh-Hain JA, del Carmen MG. Recurrent epithelial ovarian cancer: an update on treatment. Oncology. 2013;27:288–94. (298) [PubMed] [Google Scholar]
- 6.Agarwal R, Kaye SB. Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat Rev Cancer. 2003;3(7):502–516. doi: 10.1038/nrc1123. [DOI] [PubMed] [Google Scholar]
- 7.Pujade-Lauraine E, Hilpert F, Weber B, Reuss A, Poveda A, Kristensen G, Sorio R, Vergote I, Witteveen P, Bamias A, Pereira D, Wimberger P, Oaknin A, Mirza MR, Follana P, Bollag D, Ray-Coquard I. Bevacizumab combined with chemotherapy for platinum-resistant recurrent ovarian cancer: the AURELIA open-label randomized phase III trial. J Clin Oncol. 2014;32(13):1302–1308. doi: 10.1200/JCO.2013.51.4489. [DOI] [PubMed] [Google Scholar]
- 8.Bookman MA. Extending the platinum-free interval in recurrent ovarian cancer: the role of topotecan in second-line chemotherapy. Oncologist. 1999;4(2):87–94. [PubMed] [Google Scholar]
- 9.Blackledge G, Lawton F, Redman C, Kelly K. Response of patients in phase II studies of chemotherapy in ovarian cancer: implications for patient treatment and the design of phase II trials. Br J Cancer. 1989;59(4):650–3. doi: 10.1038/bjc.1989.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–627. doi: 10.1146/annurev.med.53.082901.103929. [DOI] [PubMed] [Google Scholar]
- 11.Liu YY, Han TY, Giuliano AE, Cabot MC. Ceramide glycosylation potentiates cellular multidrug resistance. FASEB J. 2001;15(3):719–730. doi: 10.1096/fj.00-0223com. [DOI] [PubMed] [Google Scholar]
- 12.Borges-Walmsley MI, McKeegan KS, Walmsley AR. Structure and function of efflux pumps that confer resistance to drugs. Biochem J. 2003;376(2):313–338. doi: 10.1042/BJ20020957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karran P. Mechanisms of tolerance to DNA damaging therapeutic drugs. Carcinogenesis. 2001;22(12):1931–1937. doi: 10.1093/carcin/22.12.1931. [DOI] [PubMed] [Google Scholar]
- 14.Masuda H, Ozols RF, Lai GM, Fojo A, Rothenberg M, Hamilton TC. Increased DNA repair as a mechanism of acquired resistance to cis-diamminedichloroplatinum(II) in human ovarian cancer cell lines. Cancer Res. 1988;48(20):5713–16. [PubMed] [Google Scholar]
- 15.Chabner BA, Roberts TG. Chemotherapy and the war on cancer. Nat Rev Cancer. 2005;5(1):65–72. doi: 10.1038/nrc1529. [DOI] [PubMed] [Google Scholar]
- 16.Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8(3):193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 17.Hertel LW, Boder GB, Kroin JS, Rinzel SM, Poore GA, Todd GC, Grindey GB. Evaluation of the antitumor activity of gemcitabine (2′,2′-difluoro-2′-deoxycytidine) Cancer Res. 1990;50(14):4417–22. [PubMed] [Google Scholar]
- 18.Pfisterer J, Plante M, Vergote I, du Bois A, Hirte H, Lacave AJ, Wagner U, Staehle A, Stuart G, Kimmig R, Olbricht S, Le T, Emerich J, Kuhn W, Bentley J, Jackisch C, Lueck HJ, Rochon J, Zimmermann AH, Eisenhauer E. Gemcitabine plus carboplatin compared with carboplatin in patients with platinum-sensitive recurrent ovarian cancer: an intergroup trial of the AGO-OVAR, the NCIC CTG, and the EORTC GCG. J Clin Oncol. 2006;24(29):4699–4707. doi: 10.1200/JCO.2006.06.0913. [DOI] [PubMed] [Google Scholar]
- 19.du Bois A, Luck HJ, Pfisterer J, Schroeder W, Blohmer JU, Kimmig R, Moebus V, Quaas J. Second-line carboplatin and gemcitabine in platinum sensitive ovarian cancer–a dose-finding study by the Arbeitsgemeinschaft Gynakologische Onkologie (AGO) Ovarian Cancer Study Group. Ann Oncol. 2001;12(8):1115–20. doi: 10.1023/a:1011605008922. [DOI] [PubMed] [Google Scholar]
- 20.Pfisterer J, Vergote I, Du Bois A, Eisenhauer E. Combination therapy with gemcitabine and carboplatin in recurrent ovarian cancer. Int J Gynecol Cancer. 2005;15(s1):36–41. doi: 10.1111/j.1525-1438.2005.15355.x. [DOI] [PubMed] [Google Scholar]
- 21.Lorusso D, Di Stefano A, Fanfani F, Scambia G. Role of gemcitabine in ovarian cancer treatment. Ann Oncol. 2006;17(Suppl 5):v188–94. doi: 10.1093/annonc/mdj979. [DOI] [PubMed] [Google Scholar]
- 22.Zhou B-BS, Bartek J. Targeting the checkpoint kinases: chemosensitization versus chemoprotection. Nat Rev Cancer. 2004;4(3):216–225. doi: 10.1038/nrc1296. [DOI] [PubMed] [Google Scholar]
- 23.Bergman AM, Pinedo HM, Peters GJ. Determinants of resistance to 2′,2′-difluorodeoxycytidine (gemcitabine) Drug Resist Updates. 2002;5(1):19–33. doi: 10.1016/s1368-7646(02)00002-x. [DOI] [PubMed] [Google Scholar]
- 24.Hung SW, Marrache S, Cummins S, Bhutia YD, Mody H, Hooks SB, Dhar S, Govindarajan R. Defective hCNT1 transport contributes to gemcitabine chemoresistance in ovarian cancer subtypes: Overcoming transport defects using a nanoparticle approach. Cancer Lett (N Y, NY, U S) 2015;359(2):233–240. doi: 10.1016/j.canlet.2015.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burris HA, 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, Cripps MC, Portenoy RK, Storniolo AM, Tarassoff P, Nelson R, Dorr FA, Stephens CD, Von Hoff DD. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15(6):2403–13. doi: 10.1200/JCO.1997.15.6.2403. [DOI] [PubMed] [Google Scholar]
- 26.Sandler AB, Nemunaitis J, Denham C, Von Pawel J, Cormier Y, Gatzemeier U, Mattson K, Manegold C, Palmer MC, Gregor A, Nguyen B, Niyikiza C, Einhorn LH. Phase III trial of gemcitabine plus cisplatin versus cisplatin alone in patients with locally advanced or metastatic non-small-cell lung cancer. J Clin Oncol. 2000;18(1):122–130. doi: 10.1200/JCO.2000.18.1.122. [DOI] [PubMed] [Google Scholar]
- 27.Yavuz MS, Cheng Y, Chen J, Cobley CM, Zhang Q, Rycenga M, Xie J, Kim C, Song KH, Schwartz AG, Wang LV, Xia Y. Gold nanocages covered by smart polymers for controlled release with near-infrared light. Nat Mater. 2009;8(12):935–939. doi: 10.1038/nmat2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JE, Lee N, Kim H, Kim J, Choi SH, Kim JH, Kim T, Song IC, Park SP, Moon WK, Hyeon T. Uniform Mesoporous Dye-Doped Silica Nanoparticles Decorated with Multiple Magnetite Nanocrystals for Simultaneous Enhanced Magnetic Resonance Imaging, Fluorescence Imaging, and Drug Delivery. J Am Chem Soc. 2010;132(2):552–557. doi: 10.1021/ja905793q. [DOI] [PubMed] [Google Scholar]
- 29.Sajja HK, East MP, Mao H, Wang YA, Nie S, Yang L. Development of multifunctional nanoparticles for targeted drug delivery and noninvasive imaging of therapeutic effect. Curr Drug Discovery Technol. 2009;6(1):43–51. doi: 10.2174/157016309787581066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheon J, Lee J-H. Synergistically Integrated Nanoparticles as Multimodal Probes for Nanobiotechnology. Acc Chem Res. 2008;41(12):1630–1640. doi: 10.1021/ar800045c. [DOI] [PubMed] [Google Scholar]
- 31.Rieter WJ, Pott KM, Taylor KML, Lin W. Nanoscale Coordination Polymers for Platinum-Based Anticancer Drug Delivery. J Am Chem Soc. 2008;130(35):11584–11585. doi: 10.1021/ja803383k. [DOI] [PubMed] [Google Scholar]
- 32.Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers as an emerging platform for cancer therapy. Nat Nanotechnol. 2007;2(12):751–760. doi: 10.1038/nnano.2007.387. [DOI] [PubMed] [Google Scholar]
- 33.Li S-D, Huang L. Pharmacokinetics and Biodistribution of Nanoparticles. Mol Pharmaceutics. 2008;5(4):496–504. doi: 10.1021/mp800049w. [DOI] [PubMed] [Google Scholar]
- 34.Pathak RK, Dhar S. A Nanoparticle Cocktail: Temporal Release of Predefined Drug Combinations. J Am Chem Soc. 2015;137(26):8324–8327. doi: 10.1021/jacs.5b03078. [DOI] [PubMed] [Google Scholar]
- 35.Kolishetti N, Dhar S, Valencia PM, Lin LQ, Karnik R, Lippard SJ, Langer R, Farokhzad OC. Engineering of self-assembled nanoparticle platform for precisely controlled combination drug therapy. Proc Natl Acad Sci U S A. 2010;107(42):17939–17944. S17939/1–S17939/3. doi: 10.1073/pnas.1011368107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salzano G, Navarro G, Trivedi MS, De Rosa G, Torchilin VP. Multifunctional Polymeric Micelles Co-loaded with Anti-Survivin siRNA and Paclitaxel Overcome Drug Resistance in an Animal Model of Ovarian Cancer. Mol Cancer Ther. 2015;14(4):1075–1084. doi: 10.1158/1535-7163.MCT-14-0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sarisozen C, Abouzeid AH, Torchilin VP. The effect of co-delivery of paclitaxel and curcumin by transferrin-targeted PEG-PE-based mixed micelles on resistant ovarian cancer in 3-D spheroids and in vivo tumors. Eur J Pharm Biopharm. 2014;88(2):539–550. doi: 10.1016/j.ejpb.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu X, He C, Wu Y, Chen X, Cheng J. Nanogel-Incorporated Physical and Chemical Hybrid Gels for Highly Effective Chemo-Protein Combination Therapy. Adv Funct Mater. 2015;25(43):6744–6755. [Google Scholar]
- 39.Liu T, Yacoub R, Taliaferro-Smith LD, Sun S-Y, Graham TR, Dolan R, Lobo C, Tighiouart M, Yang L, Adams A, O’Regan RM. Combinatorial effects of lapatinib and rapamycin in triple-negative breast cancer cells. Mol Cancer Ther. 2011;10(8):1460–1469. doi: 10.1158/1535-7163.MCT-10-0925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lammers T, Subr V, Ulbrich K, Peschke P, Huber PE, Hennink WE, Storm G. Simultaneous delivery of doxorubicin and gemcitabine to tumors in vivo using prototypic polymeric drug carriers. Biomaterials. 2009;30(20):3466–3475. doi: 10.1016/j.biomaterials.2009.02.040. [DOI] [PubMed] [Google Scholar]
- 41.Tardi PG, Dos Santos N, Harasym TO, Johnstone SA, Zisman N, Tsang AW, Bermudes DG, Mayer LD. Drug ratio-dependent antitumor activity of irinotecan and cisplatin combinations in vitro and in vivo. Mol Cancer Ther. 2009;8(8):2266–2275. doi: 10.1158/1535-7163.MCT-09-0243. [DOI] [PubMed] [Google Scholar]
- 42.Patel NR, Rathi A, Mongayt D, Torchilin VP. Reversal of multidrug resistance by co-delivery of tariquidar (XR9576) and paclitaxel using long-circulating liposomes. Int J Pharm. 2011;416(1):296–299. doi: 10.1016/j.ijpharm.2011.05.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu D, Poon C, Lu K, He C, Lin W. Self-assembled nanoscale coordination polymers with trigger release properties for effective anticancer therapy. Nat Commun. 2014 doi: 10.1038/ncomms5182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.He C, Liu D, Lin W. Self-assembled nanoscale coordination polymers carrying siRNAs and cisplatin for effective treatment of resistant ovarian cancer. Biomaterials. 2015;36:124–133. doi: 10.1016/j.biomaterials.2014.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu D, He C, Poon C, Lin W. Theranostic nanoscale coordination polymers for magnetic resonance imaging and bi-sphosphonate delivery. J Mater Chem B. 2014;2(46):8249–8255. doi: 10.1039/c4tb00751d. [DOI] [PubMed] [Google Scholar]
- 46.Poon C, He C, Liu D, Lu K, Lin W. Self-assembled nanoscale coordination polymers carrying oxaliplatin and gemcitabine for synergistic combination therapy of pancreatic cancer. J Controlled Release. 2015;201:90–99. doi: 10.1016/j.jconrel.2015.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Woodcock J, Griffin JP, Behrman RE. Development of novel combination therapies. N Engl J Med. 2011;364(11):985–987. doi: 10.1056/NEJMp1101548. [DOI] [PubMed] [Google Scholar]
- 48.Jabr-Milane LS, van Vlerken LE, Yadav S, Amiji MM. Multi-functional nanocarriers to overcome tumor drug resistance. Cancer Treat Rev. 2008;34(7):592–602. doi: 10.1016/j.ctrv.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu C-MJ, Aryal S, Zhang L. Nanoparticle-assisted combination therapies for effective cancer treatment. Ther Delivery. 2010;1(2):323–334. doi: 10.4155/tde.10.13. [DOI] [PubMed] [Google Scholar]
- 50.Lehár J, Krueger AS, Avery W, Heilbut AM, Johansen LM, Price ER, Rickles RJ, Short GF, III, Staunton JE, Jin X. Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat Biotechnol. 2009;27(7):659–666. doi: 10.1038/nbt.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen H, Pazicni S, Krett NL, Ahn RW, Penner-Hahn JE, Rosen ST, O’Halloran TV. Coencapsulation of Arsenic-and Platinum-based Drugs for Targeted Cancer Treatment. Angew Chem, Int Ed. 2009;48(49):9295–9299. doi: 10.1002/anie.200903655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kolishetti N, Dhar S, Valencia PM, Lin LQ, Karnik R, Lippard SJ, Langer R, Farokhzad OC. Engineering of self-assembled nanoparticle platform for precisely controlled combination drug therapy. Proc Natl Acad Sci U S A. 2010;107(42):17939– 17944. doi: 10.1073/pnas.1011368107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Duan X, Xiao J, Yin Q, Zhang Z, Yu H, Mao S, Li Y. Smart pH-sensitive and temporal-controlled polymeric micelles for effective combination therapy of doxorubicin and disulfiram. ACS Nano. 2013;7(7):5858–5869. doi: 10.1021/nn4010796. [DOI] [PubMed] [Google Scholar]
- 54.Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46(12 Part 1):6387–92. [PubMed] [Google Scholar]
- 55.Maeda H, Sawa T, Konno T. Mechanism of tumor-targeted delivery of macromolecular drugs, including the EPR effect in solid tumor and clinical overview of the prototype polymeric drug SMANCS. J Controlled Release. 2001;74(1–3):47–61. doi: 10.1016/s0168-3659(01)00309-1. [DOI] [PubMed] [Google Scholar]
- 56.Duan X, Li Y. Physicochemical characteristics of nanoparticles affect circulation, biodistribution, cellular internalization, and trafficking. Small. 2013;9(9–10):1521–1532. doi: 10.1002/smll.201201390. [DOI] [PubMed] [Google Scholar]
- 57.Taylor-Pashow KML, Della Rocca J, Huxford RC, Lin W. Hybrid nanomaterials for biomedical applications. Chem Commun (Cambridge, U K) 2010;46(32):5832–5849. doi: 10.1039/c002073g. [DOI] [PubMed] [Google Scholar]
- 58.Hirsjärvi S, Dufort S, Gravier J, Texier I, Yan Q, Bibette J, Sancey L, Josserand V, Passirani C, Benoit J-P. Influence of size, surface coating and fine chemical composition on the in vitro reactivity and in vivo biodistribution of lipid nanocapsules versus lipid nanoemulsions in cancer models. Nanomedicine. 2013;9(3):375–387. doi: 10.1016/j.nano.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 59.Risbood P, Kane CT, Jr, Hossain T, Vadapalli S, Chadda S. Synthesis of gemcitabine triphosphate (dFdCTP) as a tris-(triethylammonium) salt. Bioorg Med Chem Lett. 2008;18(9):2957–2958. doi: 10.1016/j.bmcl.2008.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu W, Chen X, Ye Q, Hou S, Lou L, Xie C. 3-Hydroxycarboplatin, a simple carboplatin derivative endowed with an improved toxicological profile. Platinum Met Rev. 2012;56(4):248–256. [Google Scholar]
- 61.Rochon FD, Massarweh G. Synthesis, multinuclear magnetic resonance and crystal structures of Pt(II) complexes containing amines and bidentate carboxylate ligands. Inorg Chim Acta. 2006;359(12):4095–4104. [Google Scholar]
- 62.Singh MM, Szafran Z, Pike RM. Microscale synthesis of cis-diamminedihaloplatinum(II) and a corresponding trans isomer. A rapid and convenient method of preparing cisplatin - an anticancer drug. J Chem Educ. 1990;67(10):A261–A262. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.