

Abstract
UV crosslinking immunoprecipitation (CLIP) is an increasingly popular technique to study protein–RNA interactions in tissues and cells. Whole cells or tissues are ultraviolet irradiated to generate a covalent bond between RNA and proteins that are in close contact. After partial RNase digestion, antibodies specific to an RNA binding protein (RBP) or a protein–epitope tag is then used to immunoprecipitate the protein–RNA complexes. After stringent washing and gel separation the RBP–RNA complex is excised. The RBP is protease digested to allow purification of the bound RNA. Reverse transcription of the RNA followed by high-throughput sequencing of the cDNA library is now often used to identify protein bound RNA on a genome-wide scale. UV irradiation can result in cDNA truncations and/or mutations at the crosslink sites, which complicates the alignment of the sequencing library to the reference genome and the identification of the crosslinking sites. Meanwhile, one or more amino acids of a crosslinked RBP can remain attached to its bound RNA due to incomplete digestion of the protein. As a result, reverse transcriptase may not read through the crosslink sites, and produce cDNA ending at the crosslinked nucleotide. This is harnessed by one variant of CLIP methods to identify crosslinking sites at a nucleotide resolution. This method, individual nucleotide resolution CLIP (iCLIP) circularizes cDNA to capture the truncated cDNA and also increases the efficiency of ligating sequencing adapters to the library. Here, we describe the detailed procedure of iCLIP.
Keywords: UV crosslinking immunoprecipitation, RNA binding proteins, Immunoprecipitation, RBP-RNA complex, iCLIP
1 Introduction
RNA–protein interaction plays an important role in posttranscriptional regulation [1]. Currently, different methods study RNA–RBP interactions with varying resolutions. Electrophoretic mobility shift assay (EMSA) is a simple and sensitive technique to identify RNA–RBP interaction based on the observation that RNA–RBP complexes migrate slower in a native gel than the RNA alone. EMSA has many limitations including (1) possible complex disassembly and mis-assembly during electrophoresis, (2) poor sequence resolution to determine interacting nucleotides, and (3) loss of in vivo cellular context [2]. Traditional RNA binding protein immunoprecipitation (RIP) allows for quantifiable identification of RNA targets bound to an RBP in cellular extracts. Immunoprecipitation of endogenous RNA–protein complexes using antibodies targeting the RBP is followed by purification and profiling of the bound RNA by microarray or high-throughput sequencing. When additional RBPs are co-immunoprecipitated with the assayed RBP (e.g. as part of a larger RNP complex), RIP is unable to distinguish indirectly bound RNA [3]. Promiscuous protein–RNA binding in cell lysates is also a significant source of false positives.
To circumvent the limitations of the previously developed methods, crosslinking immunonoprecipitation (CLIP) was developed. Crosslinking RNA–protein complexes using UV irradiation combined with immunoprecipitation has allowed us to map the RNA sequence bound to a RNA binding protein in vivo with high resolution and specificity [4–9]. RNA–RBP complexes are cross-linked using UV irradiation, partially digested with RNase, and purified by immunoprecipitation; the RNA is then extracted after protease digestion, ligated to an adapter, reverse-transcribed to cDNA, and sequenced. Kirk Jensen and Jenej Ule first applied CLIP to the study of RNA binding protein Nova in brain in 2003 [4]. They were able to identify 340 new RNA tags that were cross-linked to Noval, 18 of which flanked alternative exons suggesting a role of Nova in alternative splicing [4]. Various methods of CLIP have since been developed to increase the resolution of RNA–protein binding to the near nucleotide level allowing for the identification of not only specific binding sites but specific motifs or patterns of RNA–protein binding [6].
CLIP experiments often have limited yield partly due to inefficient crosslinking using UV 254 nm. To improve RNA recovery, Thomas Tuschl’s group developed photoactivatable-ribonucleoside-enhanced crosslinking and immunoprecipitation (PAR-CLIP). PAR-CLIP labels living cells with photoactivatable nucleosides such as 4-thiouridine (4-SU) and 6-thioguanosine (6-SG) that are incorporated into nascent RNA prior to irradiation with UV at 365 nm [8, 10]. When crosslinked, incorporation of 4-SU or 6-SG results in U to C and G to A mutations respectively, allowing researchers to use mutational analysis to identify cross-linked sites and filter out noise. An increase in crosslinking efficiency and resolution are the advantages of PAR-CLIP. However, PAR-CLIP relies on metabolic labeling which has inherent biases as well as difficulties for in vivo application.
A third variant of CLIP or iCLIP, developed by Ule’s group, allows identification of crosslinking sites at the near nucleotide level (Fig. 1) [11]. Unlike other CLIP methods that ligate adapters to RNA before reverse transcription, iCLIP introduces a circularization step after reverse transcription to ligate the cleavable 5′ end adapter to the 3′ end of the cDNA [12]. Because reverse transcriptase often stops at the crosslink sites and does not necessarily extend to the 3′ adapter that is used by other CLIP methods, circularization allows iCLIP to capture these truncated cDNA. The cleavable 5′ end adapter includes random barcodes to eliminate PCR artifacts. iCLIP was successfully used to globally identify RNA that were bound to HNRPNPC, TDP-43, FUS, and TIA1/TIAL1 [12–15].
Fig. 1.
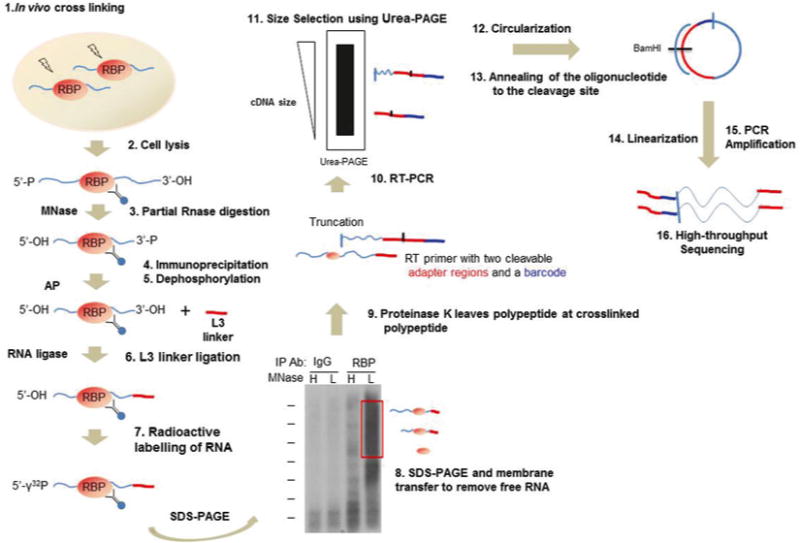
Schematic representation of iCLIP. RNA binding protein (RBP) and RNA are covalently bound in vivo using UV radiation (step 1). Potential RBP–RNA complexes are then purified together (steps 2–5). L3 linker adapter ligation to the 3′ end allows for sequence-specific priming of reverse transcription and the 5′ is radioactively labeled (steps 6 and 7). RBP–RNA complexes are purified from free RNA using SDS-PAGE and wet membrane transfer (step 8). Complexes are recovered from the membrane using Proteinase K (step 9). Reverse transcription truncates at the remaining polypeptide and introduces two cleavable adapter regions and a barcode (step 10). Size selection using Urea-PAGE removes the RT primer prior to circularization of the cDNA (steps 11–13). Linearization generates templates for PCR amplification (steps 14 and 15). High-throughput sequencing generates reads where the barcode sequences are immediately before the last nucleotide of the input cDNA (step 16). This nucleotide is one position upstream of the crosslinked nucleotide, allowing for identification of the RBP–RNA binding site with high resolution
Despite the power of CLIP, cautions should be taken in analyzing any CLIP result in particular due to the sequence-dependent UV crosslinking biases. RBPs are multifunctional. To understand their complete functionality, other genomic methods are needed in conjunction with CLIP to globally identify their functionally relevant RNA targets.
2 Materials
Prepare all reagents in ultrapure, sterile water at room temperature unless stated otherwise. Store stock solutions and buffers at room temperature unless otherwise specified.
2.1 Buffer Components
TE Buffer: 1 M Tris–HCl pH 8.0, 0.5 M ethylenediaminetetraacetic acid (EDTA) pH 8.0, H2O.
Lysis Buffer: 50 mM Tris–HCl pH 7.4, 100 mM NaCl, 1 % Igepal CA-630, 0.1 % SDS, 0.5 % sodium deoxycholate, on the day add 1/100 volume of Protease Inhibitor Cocktail Set III. For tissues use 1/1000 of ANTI-RNase (see Note 1).
High Salt Wash: 50 mM Tris–HCl pH 7.4, 1 M NaCl, 1 mM EDTA, 1 % Igepal CA-630, 0.1 % SDS, 0.5 % sodium deoxycholate.
PNK Buffer: 20 mM Tris–HCl, pH 7.4, 10 mM MgCl2, 0.2 % Tween-20.
5× PNK pH 6.5 Buffer: 350 mM Tris–HCl pH 6.5, 50 mM MgCl2, 5 mM dithiothreitol (DTT). Freeze aliquots of the buffer. Avoid freeze-thaw (see Note 2).
6 4× Ligation Buffer: 200 mM Tris–HCl pH 7.8, 40 mM MgCl2, 4 mM dithiothreitol, Freeze aliquots of the buffer. Avoid freeze-thaw.
PK Buffer: 100 mM Tris–HCl, pH 7.4, 50 mM NaCl, 10 mM EDTA.
PK buffer + 7 M Urea: 10 mM Tris–HCl pH 7.4, 50 mM NaCl, 10 mM EDTA, 7 M Urea.
2.2 UV Crosslinking and Immuno-precipitation
Stratalinker UV Crosslinker (Stratagene, La Jolla, CA).
4-Thiouridine (Sigma, St. Louis, MO) (see Note 3).
Protein G Dynabeads (Life Technologies, Carlsbad, CA).
Protein A Dynabeads (Life Technologies).
Protease Inhibitor Cocktail Set III (Calbiochem/Merck).
ANTI-RNase (Life Technologies).
RNase I (Life Technologies).
Turbo DNase (Life Technologies).
T4 PNK plus 10× PNK Buffer (NEB, Ipswich, MA).
RNasin (Promega, Madison, WI).
Proteus Clarification Mini Spin Columns (Generon, Houston, TX).
T4 RNA Ligase I (NEB).
Pre-adenylated adapter L3-App (IDT, rAppAGATCGGAA-CAGCGGTTCAG/ddC/).
ATP [γ-32P] (Perkin Elmer, Waltham, MA).
2.3 Immunoblotting Components
4–12 % NuPAGE gels (Life Technologies).
Electrophoresis chamber (Life Technologies).
Novex wet transfer apparatus (Life Technologies).
LDS-4× Sample Buffers (Life Technologies).
Pre-stained protein marker (Fermentas, Pittsburg, PA).
Nitrocellulose membrane (VWR, Radnor, PA).
Protran BA85.
Sponge pads for XCell II blotting (Life Technologies).
20× transfer buffer (Life Technologies).
20× MOPS-SDS Running Buffer (Life Technologies).
Whatman filter paper (GE Healthcare, Pittsburg, PA).
Film.
2.4 RNA Isolation Components
Proteinase K (Fisher Scientific, Waltham, MA).
19G syringe needles.
Phenol/Chloroform pH 6.7 (Sigma) (see Note 4).
Phase lock gel heavy tube (VWR).
Glycoblue (Ambion).
3 M sodium acetate pH 5.5 (Life Technologies).
Thermomixer (Eppendorf).
2.5 Reverse Transcription
PCR tubes.
dNTPs (Promega).
Superscript III (Life Technologies).
5× First Strand Buffer (Life Technologies).
DTT (Life Technologies).
1 M HEPES pH 7.3 (Thermo Scientific).
-
TE buffer.
R#Clip Sequences (N denotes any nucleotide).
Rt1clip: /5Phos/NNAACCNNNAGATCGGAAG AGCGTCGTGgatcCTGAACCGC
Rt2clip: /5Phos/NNACAANNNAGATCGGAAG AGCGTCGTGgatcCTGAACCGC
Rt3clip: /5Phos/NNATTGNNNAGATCGGAAG AGCGTCGTGgatcCTGAACCGC
Rt4clip: /5Phos/NNAGGTNNNAGATCGGAAG AGCGTCGTGgatcCTGAACCGC
Rt6clip: /5Phos/NNCCGGNNNAGATCGGAAG AGCGTCGTGgatcCTGAACCGC
Rt7clip: /5Phos/NNCTAANNNAGATCGGAAG AGCGTCGTGgatcCTGAACCGC
Rt8clip: /5Phos/NNCATTNNNAGATCGGAAG AGCGTCGTGgatcCTGAACCGC
Rt9clip: /5Phos/NNGCCANNNAGATCGGAAG AGCGTCGTGgatcCTGAACCGC
Rt11clip: /5Phos/NNGGTTNNNAGATCGGAAG AGCGTCGTGgatcCTGAACCGC
Rt12clip: /5Phos/NNGTGGNNNAGATCGGAAG AGCGTCGTGgatcCTGAACCGC
Rt13clip: /5Phos/NNTCCGNNNAGATCGGAAG AGCGTCGTGgatcCTGAACCGC
Rt14clip: /5Phos/NNTGCCNNNAGATCGGAAG AGCGTCGTGgatcCTGAACCGC
Rt15clip: /5Phos/NNTATTNNNAGATCGGAAG AGCGTCGTGgatcCTGAACCGC
Rt16clip: /5Phos/NNTTAANNNAGATCGGAAG AGCGTCGTGgatc CTGAACCGC
2.6 cDNA Isolation
2× TBE-urea loading buffer (Life Technologies).
6 % TBE-urea (pre-cast gels) (Life Technologies).
Low molecular weight marker (NEB).
TBE Running buffer (Life Technologies).
SYBR Green II (Life Technologies).
Glass Pre-filters (Whatman).
Phase lock gel heavy (VWR).
Costar SpinX Column (Corning Incorporated, Corning, NY).
2.7 5′ End Ligation
10× CircLigase Buffer (Cambio, Cambridge, UK).
CircLigase II (Cambio).
Cut_oligo: GTTCAGGATCCACGACGCTCTTCaaaa [8].
MnCL2 (Cambio).
Bam HI (Fermentas, Pittsburg, PA).
Fast digest Buffer (Fermentas, Pittsburg, PA).
2.8 PCR Amplification
2.9 qPCR Quantification
2.10 Buffers for Stringent-Urea iCLIP Protocol
Urea Cracking buffer: 50 mM Tris–HCl pH 7.4, 6 M Urea, 1 % SDS, 25 % PBS.
T-20 Buffer: 50 mM Tris–HCl pH 7.4, 150 mM NaCl, 0.5 % Tween 20, and 0.1 mM EDTA.
2.11 Stringent-Urea iCLIP
Dynabeads Antibody Coupling Kit (Life Technologies).
SUPERase in RNase inhibitor (Life Technologies).
NuPAGE sample reducing agent (Life Technologies).
NuPAGE antioxidant (Life Technologies).
Linear acrylamide (Life Technologies).
3 Methods
3.1 UV-C Crosslinking
After culturing adherent cells in 10 cm plate, remove medium, add 6 mL ice-cold PBS and place on ice.
Remove lid and irradiate cells with 150 mJ/cm2 (needs to be optimized for different proteins) at 254 nm in a Stratalinker 2400 (see Note 5).
(Optional) Have non-irradiated cells as a negative control of the crosslinking.
Scrape off the cells with cell lifters and transfer the cell suspension into three 2 mL microtubes. Centrifuge at 800 ×g at 4 °C for 1 min to pellet cells, and then remove supernatant.
Snap-freeze the cell pellets on dry ice and store at −80°C until further use.
3.2 4-Thiouridine Labeling and UV-A Crosslinking (Alternative to UV-C)
Add 50 μL of 4SU (stock concentration 100 mM 4SU) to a 10 cm plate of cells culture in 10 mL DMEM supplemented with FBS, PenStrep to get a final concentration of 500 μM 4SU.
Alternatively add 10 μL of 4-thiouridine (stock concentration: 100 mM) to get a final concentration of 100 μM 4SU (see Note 6).
Incubate the cells with 4-thiouridine for 60 min at 500 μM or 8 h at 100 μM: afterward check the viability of your cells. Have cells that are not incubated with 4SU as a negative control.
Aspirate the medium and add 6 mL of ice-cold PBS to cells growing in a 10 cm plate. Remove lid and place on ice.
Irradiate twice at once with 2 × 400 mJ/cm2 in a Stratalinker 2400 with 365 nm bulbs.
Scrape off the cells with cell lifters.
Transfer the cell suspension into three 2 mL microtubes. Centrifuge at 800 ×g at 4 °C for 1 min to pellet cells, and then remove the supernatant.
Snap-freeze the cell pellets on dry ice and store at −80°C until further use.
3.3 Bead Preparation for Immuno-precipitation
Add 100 μL of protein G dynabeads per experiment to a fresh microtube.
Wash beads twice with 900 μL lysis buffer (without protease inhibitor).
Resuspend the beads in 100 μL lysis buffer with 2–10 μg antibody per experiment (see Note 7).
Rotate tubes for 30–60 min at room temperature.
Wash 3 times with 900 μL lysis buffer and leave in the last wash until ready to proceed with immunoprecipitation.
3.4 Cell Lysis and Partial RNA Digestion
Resuspend the cell pellet in 1 mL lysis buffer (with 1:100 Protease Inhibitor Cocktail Set III) and transfer to a 1.5 mL microtube. Prepare enough lysate for two experimental samples and appropriate controls (see Notes 8 and 9).
Sonication of samples (Note: Optional).
Option 1: Sonicate the sample on ice using a probe sonicator. The probe should be approximately 0.5 cm from the bottom of the tube and not touching the tube sides in order to avoid foaming. Sonicate twice with 10 s bursts at 5 dB. Clean the probe by sonicating H2O before and after sample treatment.
Option 2: Use Bioruptor plus for five cycles with alternating 30 s on/30 s off at low intensity.
Prepare two dilutions of RNase I in PBS. 1:50 for High RNase I and 1:250 for low RNAseI. This will need to be carefully optimized (see Note 10).
Add 10 μL of low or high RNAse I dilution and 2 μL Turbo DNase to the cell lysate and immediately place the samples at 37 °C for 3 min shaking at 1100 rpm. Immediately afterward transfer to ice for > 3 min.
Centrifuge for 10 min at 22,000 ×g and at 4 °C to clear the lysate. Carefully collect the supernatant.
Optional: load 500 μL of the lysate onto a Proteus Clarification Mini Spin Column. Centrifuge for 1 min at 22,000 ×g at 4 °C. Transfer flow-through to a new tube and place on ice. Repeat for the rest of the lysate and combine the fractions.
3.5 Immunoprecipitation
Add the lysate to the beads and rotate for 1 h or overnight at 4 °C.
Place on magnet and discard the supernatant and wash three times with high-salt wash (rotate the third wash for at least 1 min at 4 °C) (see Note 11).
Wash twice with PNK Buffer and then leave in 1 mL PNK Buffer and proceed to 3′ end dephosphorylation step.
3.6 RNA 3′ End Dephosphorylation
Discard the supernatant and resuspend the beads in 20 μL of the following mix: 4 μL 5× PNK pH 6.5 Buffer, 0.5 μL PNK, 0.5 μL RNasin, and 15 μL H2O.
Incubate for 20 min at 37 °C in a thermomixer at 1100 rpm.
Wash with PNK Buffer.
Wash with high-salt wash (rotate wash in the cold room for at least 1 min).
Wash twice with PNK Buffer.
3.7 L3 Adapter Ligation
Carefully remove the supernatant and resuspend the beads in 20 μL of the following mix: 8 μL H2O, 5 μL 4× ligation buffer, 1 μL RNA ligase, 0.5 μL RNasin, 1.5 μL pre-adenylated adapter L3-App (20 μM), and 4 μL PEG400.
I ncubate overnight at 16 °C in thermomixer at 1100 rpm.
Add 500 μL PNK Buffer.
Wash twice with high-salt buffer, rotating each wash in the cold room for 5 min, and twice with PNK Buffer.
3.8 RNA 5′ End Labeling
Remove supernatant and resuspend beads in 4 μL Hot PNK mix: 0.2 μL PNK, 0.4 μL γ-[32P]-ATP, 0.4 μL 10× PNK Buffer, and 3 μL H2O.
Incubate for 5 min at 37 °C in a thermomixer at 1100 rpm. Discard the supernatant.
Remove the supernatant as radioactive waste and add 20 μL 1× NuPAGE loading buffer (see Note 12).
Incubate on a thermomixer at 70 °C for 10 min.
Place on the magnet to precipitate the beads, collect the supernatant and load it on a gel.
3.9 SDS-PAGE and Nitrocellulose Transfer
Load the samples on a 4–12 % NuPAGE Bis-Tris gel. Also load 5 μL of a pre-stained protein size marker. Use 500 mL 1× MOPS running buffer.
Run the gel at 180 V for 50 min.
Cut off the dye front and discard it as solid radioactive waste.
Transfer the protein–RNA complexes from the gel to a Nitrocellulose membrane using a Novex wet transfer apparatus.
Transfer at 30 V for 1 h and overnight.
After transfer, rinse the membrane in PBS buffer, then wrap it in saran wrap and expose it to film at −80 °C. Expose for 30 min, 1 h, and overnight.
3.10 RNA Isolation
A smear will be seen in the low RNase condition and not the high RNase condition. Isolate the protein–RNA complexes from the low RNase condition using the autoradiograph as a mask for cutting the respective region (usually 20–60 kDa above the expected molecular weight) of the membrane. Cut into many pieces and place into a 1.5 mL microtube (see Note 13).
Add 10 μL Proteinase K in 200 μL PK buffer to the pieces making sure all pieces are submerged. Incubate pieces for 20 min shaking at 1100 rpm at 37 °C.
Add 200 μL PK Buffer + 7 M Urea and incubate for 20 min at 1100 rpm at 37 °C.
Collect the solution and add it together with 400 μL phenol/chloroform pH 6.7 to a 2 mL Phase Lock Gel Heavy Tube (see Note 14).
Incubate for 5 min at 30 °C shaking at 1100 rpm. Separate the phases by spinning for 5 min at full speed and room temperature.
Transfer the aqueous layer into a new tube. Be careful not to touch the gel matrix with the pipette. Optional: spin again for 1 min and transfer into a new tube.
Precipitate by adding 0.75 μL glycoblue and 40 μL 3 M sodium acetate pH 5.5. Then mix and add 1 mL 100 % ethanol, mix again and place at −20 °C overnight.
Centrifuge at 15,000 rpm at 4 °C for 20 min. Remove the supernatant and wash the pellet with 0.9 mL 80 % ethanol and spin for 5 min (see Note 15).
Resuspend pellet in 5 μL H2O and transfer to a PCR tube.
3.11 Reverse Transcription
Add 1 μL primer Rt#clip (0.5 pmol/μL) and 1 μL dNTP mix (10 mM) to the resuspended pellet. For each experiment or replicate, use a different Rclip primer containing individual barcode sequences (see Note 16).
Incubate at 70 °C for 5 min. Cool to 25 °C. Hold at 25 °C until RT mix is added.
Add RT mix to the resuspended pellet: 7 μL H2O, 4 μL 5× first strand buffer, 1 μL 0.1 M DTT, 0.5 μL RNasin, and 0.5 μL Superscript III.
Run RT Program: 25 °C for 5 min, 43 °C for 20 min, 50 °C for 40 min, 80 °C for 5 min, 4 °C hold.
Add 1.65 μL 1 M NaOH and incubate at 98 °C for 20 min. Then add 20 μL 1 M HEPES-NaOH pH 7.3 to eliminate radioactivity from strongly labeled samples and to prevent RNA from interfering with subsequent reactions.
Add 350 μL TE buffer, 0.75 μL glycoblue, and 40 μL 3 M sodium acetate pH 5.5. Mix and then add 1 mL 100 % ethanol. Mix again and precipitate at −20 °C overnight.
3.12 Gel Purification of cDNA
Centrifuge for 15 min at 15,000 rpm at 4 °C. Remove the supernatant and wash the pellet with 500 μL 80 % EtOH. Centrifuge again, remove supernatant, and resuspend the pellet in 6 μL H2O.
Add 6 μL 2× TBE-urea loading buffer to the cDNA. Recommended: add 6 μL loading buffer to DNA size marker.
Heat samples to 80 °C for 5 min immediately before loading. Optional: leave one lane free between each sample to avoid cross-contamination.
Prepare 800 mL of 1× TBE running buffer and fill the upper chamber with 200 mL and the lower chamber with 600 mL. Use a P1000 pipette to flush remaining urea out of the wells before loading 12 μL of each sample. Load the marker into the last lane.
Run 6 % TBE-urea gel for 40 min at 180 V until the lower dye (dark blue) is close to the bottom.
Cut off the last line containing the size marker and incubate it gently shaking for 10 min in 20 mL TBE buffer with 2 μL SYBR green II. Wash once with TBE and visualize by UV transillumination with 100 % scaling and use as a mask to guide cDNA excision from the rest of the gel.
Together the full L3-App and primer sequence accounts for 52 nt of the cDNA. Cut three bands: at 70–80 nt, 80–100 nt, and 100–150 nt.
Add 400 μL TE to each gel piece and crush gel piece with a 1 mL syringe plunger.
Incubate shaking for 1 h at 1100 rpm at 37 °C, place on dry ice for 2 min, and place back for 1 h at 1100 rpm at 37 °C. Transfer the liquid portion to Costar SpinX column, into which you placed two 1 cm glass pre-filters.
Centrifuge at full speed for 1 min. collect the solution and add it together with 400 μL DNA phenol/chloroform to a 2 mL Phase Lock Gel Heavy Tube.
Incubate for 5 min shaking at 1100 rpm at 30 °C. Separate the phases by spinning 5 min at full speed at RT.
Transfer the aqueous layer into a new tube. Centrifuge again for 1 min and transfer into a new tube.
Add 1 μL glycoblue and 40 μL 3 M sodium acetate, pH 5.5, mix. Then add 1 mL 100 % ethanol. Mix again and precipitate overnight at −20 °C.
3.13 Ligation of Primer to 5′ End of cDNA
Centrifuge the precipitate to pellet the cDNA. Wash pellet with 500 μL 80 % ethanol. Resuspend pellet in 8 μL ligation mix (6.5 μL H2O, 0.8 μL 10× CircLigase buffer II, 0.4 μL 50 mM MnCl2, 0.3 μL CircLigase II) in PCR tubes.
Transfer PCR tube and incubate at 60 °C for 1 h.
Add 30 μL annealing mix: 6 μL H2O, 3 μL Fast digest buffer, 1 μL 10 μM Cut_oligo. Anneal the oligonucleotide using program 95 °C for 2 min, successive 20 s cycles starting at 95 °C and decreasing 1 °C down to 25 °C, hold at 25 °C.
Add 2 μLl BamHI and incubate for 30 min at 37 °C, then incubate for 5 min at 80 °C.
Add 350 μL TE, 0.75 μL glycoblue, and 40 μL 3 M sodium acetate, pH 5.5, and mix. Then add 1 mL 100 % EtOH. Mix again and precipitate at −20 °C overnight.
3.14 PCR Amplification
Centrifuge precipitate and wash with 500 μL 80 % EtOH. Resuspend in 21 μL of H2O.
Prepare PCR mix: 1 μL cDNA, 0.25 μL primer, 5 μL Accuprime Supermix I, and 3.75 μL H2O.
Run PCR reaction: 94 °C for 2 min, [94 °C 15 s, 65 °C 30 s, 68 °C 30 s] for 25–35 cycles, 68 °C for 3 min, 25 °C hold.
Mix 8 μL PCR product with 2 μL 5× TBE Loading Buffer and load on a 6 % TBE gel and stain with SYBR green I (see Note 16).
3.15 qPCR Quantitation
Set up serial dilutions (1:10 to 1:10,000) in a 96-well plate. Barcodes in Rclip primers allow for multiplex.
Prepare master mix: 10 μL Invitrogen Platinum qPCR Supermix w/ROX, 0.5 μL DLP oligo, 0.6 μL Primer 1 (10μM), 0.6 μL Primer 2 (10 μM), and 6.3 μL H2O. Add 18 μL per well in qPCR plate.
Add 2 μL template from serial dilutions and seal the plate with an adhesive film.
Run the following qPCR program (normal setting, detector FAM-TAMRA, passive reference ROX): 54 °C 2 min, 94 °C 10 min, [94 °C 15 s, 62 °C 1 min, 72 °C 30 s] for 40 cycles.
Plot concentrations vs. Ct value for the standard on a log scale and fit a line to the graph. Use standard line to obtain concentrations of the unknown samples using their Ct values.
Dilute the iCLIP library to 10 nM, submit 10 μL for sequencing, and store the rest.
3.16 High-Throughput Sequencing
Libraries can be sequenced using standard Illumina protocols. 50-nt single end runs are recommended.
Acknowledgments
This work was supported by the NIH grant R00 MH 096807.
Footnotes
Lysis Buffer without protease inhibitors can be made in advance. However, Protease Inhibitor Cocktail III or ANTI-RNase should be added on the day of use.
High concentrations of DTT can lower IP efficiency, so lower DTT was used to increase recovery of protein complexes.
Crosslinking with 4-thiouridine allows crosslinking with UV-A light as seen in photoactivatable-ribonucleoside-enhanced CLIP (PAR-CLIP). Addition is optional and used to enhance crosslinking of certain proteins. 4-Thiouridine is light-sensitive and cells should be placed back in the incubator immediately after addition.
Phenol/chloroform is used at a pH of 6.7 to reduce DNA-RNA hybrids in the phenol phase.
Crosslinking depends on the protein that you are working with and method used and should be optimized. However, increasing crosslinking dose could distort library preparation by damaging RNA.
Photoreactive nucleosides are toxic to cells. You should determine optimal duration of incubation prior to use.
The amount of antibody to add depends on quality and purity and will need to be optimized prior to use.
Measure protein concentration using Nanodrop 2000c or a Bradford assay. Normalize samples to lowest concentration. 2 mg/mL protein is recommended.
Recommended controls include absence of RBP in cells, absence of crosslinking, or absence of antibody during IP.
RNase I has been shown to deactivate after prolonged incubation with 0.1 % SDS in lysis buffer.
Save 15 μL from supernatant to monitor depletion efficiency.
SDS-PAGE gels changes it’s pH during its run which can result in alkaline hydrolysis of RNA. The NuPAGE system keeps the pH at around 7 when used with the appropriate buffer.
To determine the specificity of the protein-RNA you will need to check the high RNase I condition. The radioactive band should be ~5 kDa above the MW. The negative controls should have no bands. The band in low RNase I should be diffuse. RNA is usually ~20 kDa. Cutting 15–80 kDa above the expected MW of the protein is recommended.
Try to not disturb the pellet. If you disturb the pellet, repeat spin. Leave on bench for 3 min with cap open to dry.
Use distinct Rtclip primers (Rt1clip–Rt16clip) on the controls and experimental samples. Each Rtclip contains a 4 nt barcode sequences that differs by two nucleotides to ensure that mutations cannot convert one barcode to another. Barcodes enable the user to remove PCR artifacts and control for crosscontamination during multiplexing.
It is recommended that cDNA work be done on a designated bench. Avoid taking cDNA from PCR to an area where iCLIP RNA was used.
References
- 1.Modic M, Ule J, Sibley CR. CLIPing the brain: studies of protein–RNA interactions important for neurodegenerative disorders. Mol Cell Neurosci. 2013;56:429–435. doi: 10.1016/j.mcn.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hellman LM, Fried MG. Electrophoretic Mobility Shift Assay (EMSA) for detecting protein-nucleic acid interactions. Nat Protoc. 2007;2:1849–1861. doi: 10.1038/nprot.2007.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Keene JD, Komisarow JM, Friedersdorf MB. RIP-Chip: the isolation and identification of mRNAs, microRNAs, and protein components of ribonucleoprotein complexes from cell extracts. Nat Protoc. 2006;1:302–307. doi: 10.1038/nprot.2006.47. [DOI] [PubMed] [Google Scholar]
- 4.Ule J, Jensen KB, Ruggiu M, et al. Clip identifies nova-regulated RNA networks in the brain. Science. 2003;302:1212–1215. doi: 10.1126/science.1090095. [DOI] [PubMed] [Google Scholar]
- 5.Darnell RB. HITS-CLIP: panoramic views of protein–RNA regulation in living cells. Wiley Interdiscip Rev RNA. 2010;1:266–286. doi: 10.1002/wrna.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Licatalosi DD, Aldo M, Fak JJ, et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008;456:464–470. doi: 10.1038/nature07488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Milek M, Wyler E, Landthaler M. Transcriptome-wide analysis of protein–RNA interactions using high-throughput sequencing. Semin Cell Dev Biol. 2012;23:206–212. doi: 10.1016/j.semcdb.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 8.Ascano M, Hafner M, Cekan P, et al. Identification of RNA–protein networks using PAR-CLIP. Wiley Interdiscip Rev RNA. 2012;3:159–177. doi: 10.1002/wrna.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huppertz I, Attig J, D’Ambrogio A, et al. iCLIP: protein–RNA interactions at nucleotide resolution. Methods. 2014;65:274–287. doi: 10.1016/j.ymeth.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hafner M, Lanthaler M, Burger L, et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Konig J, Zarnack K, Rot G, et al. iCLIP-transcriptome-wide mapping of protein-RNA interactions with individual nucleotide resolution. J Vis Exp. 2011 doi: 10.3891/2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Konig J, Rot G, Curk T, et al. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat Struct Mol Biol. 2010;17:909–915. doi: 10.1038/nsmb.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tollervey JR, Curk T, Rogelj B, et al. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat Neurosci. 2011;14:452–458. doi: 10.1038/nn.2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rogelj B, Easton LE, Gireesh KB, et al. Widespread binding of FUS along nascent RNA regulates alternative splicing in the brain. Sci Rep. 2012 doi: 10.1038/srep00603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Z, Kayikci M, Briese M, et al. iCLIP predicts the dual splicing effects of TIA-RNA interactions. PLoS Biol. 2010 doi: 10.1371/journal.pbio.1000530. [DOI] [PMC free article] [PubMed] [Google Scholar]