

Abstract
Colorectal cancer (CRC) is one of the leading causes of cancer-deaths worldwide. Methods for the early in situ detection of colorectal adenomatous polyps and their precursors – prior to their malignancy transformation into CRC – are urgently needed. Unfortunately at present, the primary diagnostic method, colonoscopy, can only detect polyps and carcinomas by shape/morphology; with sessile polyps more likely to go unnoticed than polypoid lesions. Here we describe our development of polyp-targeting, fluorescently-labeled mesoporous silica nanoparticles (MSNs) that serve as targeted endoscopic contrast agents for the early detection of colorectal polyps and cancer. In vitro cell studies, ex vivo histopathological analysis, and in vivo colonoscopy and endoscopy of murine colorectal cancer models, demonstrate significant binding specificity of our nanoconstructs to pathological lesions via targeting aberrant α-L-fucose expression. Our findings strongly suggest that lectin-functionalized fluorescent MSNs could serve as a promising endoscopic contrast agent for in situ diagnostic imaging of premalignant colonic lesions.
Keywords: Colorectal Tumors, Endoscopic, Mesoporous Silica Nanoparticle, Ulex Europaeus Agglutinin 1 (UEA1)
Background
Colorectal cancer (CRC) is the third most commonly diagnosed cancer and one of the leading causes of cancer death in both men and women.1 Most CRCs generally follow similar course of development: evolving from normal colon, to well differentiated adenoma with low-grade dysplasia, to highly dysplastic advanced adenoma to invasive carcinoma. The most frequently found, early neoplasms in CRC screenings of individuals over age 50 are adenomatous polyps.1–3 Although the overwhelming majority of these polyps are not malignant, more than 95% of colorectal cancers develop from polyps. Recent research has also shown that some polyps, long considered to be benign, may become cancerous as well.4 To reduce the incidence of colorectal cancer, early in situ methods of detection of colorectal adenomatous polyps and their precursors are urgently needed, prior to polyp malignancy transformation. Unfortunately the current primary diagnostic methods – sigmoidoscopy, colonoscopy, CT colonography, and double-contrast barium enema – can still only detect polyps and cancers by shape/morphology. Smaller/flatter polyps, such as flat/sessile serrated polyps, as well as nascent neoplasms of recurrent disease, have been shown to be easily overlooked during conventional colonoscopy.5–8 Suitable biomarkers are needed to address this lack of detection sensitivity.
Glycosylation drives the specific arrangement of oligosaccharides linked to glycoproteins or glycolipids in mammalian systems and has been well documented to play important roles in modulating critical physiologic functions and their alterations that both impact and drive pathologic processes. Specifically, changes in the composition and quantity of cell surface glycosylation-associated molecules are common features of malignant transformation and progression. Functionally, aberrant glycosylation facilitates tumor invasion, metastasis, and evasion of host immuno-surveillance.9 Alterations in the amount, composition, and linkage configurations of glycosylation-associated molecules are known to correlate with malignant transformation, tumor progression and poor prognosis of cancer patients.10, 11 A specific switch in glycosylation patterns has recently been reported to signal the transformation of breast cancer to metastatic phenotypes, and glycosylation is known to play a direct functional role in cancer cell motility and invasiveness.12, 13 In CRC a growing body of evidence suggests that the aberrant glycosylation of normally occurring mucins (high molecular weight glycoproteins that protect the GI mucosa) and the expression of abnormal glycoproteins on colorectal polyps and neoplasms play significant roles in the malignant transformation of normal colorectal mucosa.10, 13–15
α-L-fucose (C6H12O6), a monosaccharide component of glycosylation, is utilized exclusively in the L-configuration in mammals. The sugar is enzymatically synthesized in mammalian cells and recovered by cells from extracellular sources by a specific transmembrane carrier and intracellular salvage pathway. Significantly elevated expression of α-L-fucose is routinely observed during tumorogenesis and neoplastic progression, and has been shown to be highly expressed on the luminal surfaces of glycoproteins of colorectal dysplastic adenomas and early cancers.16–19 The α-L-fucose targeting lectin Ulex Europaeus Agglutinin-1 (UEA1) has been shown to bind human colorectal adenocarcinomas, adenomas, and polyposis coli, but not to normal epithelium.16, 20–22 Watanabe et al. reported 83% positive rate of UEA1 binding on the apical surfaces of human carcinoma cells, compared to no detectable UEA1 binding on the apical surfaces of non-neoplastic mucosa adjacent to carcinomas.23 Roney et al. developed UEA1-labeled liposomes that carried the fluorescent dye rhodamine in their interiors and bound to adenomatous polyps ex vivo using an adenomatous polyposis coli (Apc)Min/+ mouse model. And high-resolution co-registered optical coherence tomography, fluorescence molecular imaging, and multispectral optical imaging has revealed that UEA1 labeled liposomes possess significantly greater targeting efficiency than control liposomes.24, 25 These ex vivo findings suggest that the fucose-binding lectin UEA1 might be useful in the early detection of colorectal carcinomas by in vivo imaging techniques.
In the current study we developed novel, polyp-targeting, fluorescently-labeled mesoporous silica nanoparticles (MSNs) to serve as targeted endoscopic contrast agents for early detection of polyps and nascent colorectal cancers. MSNs possess high surface areas, low systemic toxicities, and flexible functionalization of their three topologically unique domains.26–28 For our studies, FITC was first co-condensed into the silica framework of MSNs to enable their fluorescence tracking both in and ex vivo. FITC was selected not only for its intrinsic brightness but also for its potential ease of clinical translation. Moreover, unlike liposomal conveyance of fluorescent reporters, the rigid silica framework of MSNs inherently limits proximity-induced self-quenching of reporter fluorescence, thereby yielding a much brighter contrast agent, as well as greater nanoplatform stability in vivo. MSN exteriors were then coated with two different lengths of polyethylene glycol (PEG) polymers, to increase their water solubility and aid their diffusion through mucus. Lastly, fluorescent/PEGylated MSNs were labeled with UEA1 for targeting premalignant lesions, as shown in Figure 1. We posit that these α-L-fucose targeted MSN contrast agents, when combined with fluorescent endoscopic techniques, will permit more sensitive detection of early stage colorectal polyps than can be presently obtained by conventional, white-light colonscopy or other clinically available, contrast-enhanced imaging modalities.
Figure 1.

Schematic illustration of UEA1-functionalized FITC-labeled mesoporous silica nanoparticles (MSNs) for polyp and early colorectal cancer detection.
Methods
Materials
Tetraethyl orthosilicate (TEOS, 99.0%), fluorescein isothiocyanate (FITC), 3-aminopropyl-trimethoxysilane (APTMS, 97%), 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N–lissamine rhodamine B sulfonyl ammonium salt (Rhodamine B PE) and fluorescein isothiocyanate labeled Ulex Europaeus Agglutinin (FITC-UEA1) were obtained from Sigma-Aldrich, USA. Other materials used include Silane-Polyethylene glycol-succinimidyl ester (Silane-PEG-NHS, MW 5000 Nanocs), 2-[Methoxy(polyethyleneoxy)propyl] trimethoxysilane, (PEG-silane, MW 596–725 g/mol, 9–12 EO, Gelest), Hexadecyltrimethylammoniumbromide (CTAB, 99%, Acrôs organics), Diacetylenic phosphatidylcholine 1,2-bis(tricosa-10,12-diynoil)-sn-glycero-3-phosphocholine (DAPC), N-glutaryl-L-α-phosphatidylethanolamine dimyristoyl (DMPC-GLU) (Avanti Polar Lipids, Alabaster, AL), Wheat Germ Agglutinin Alexa Fluor® 594 Conjugate (WGA594, Invitrogen), and 4',6-diamidino-2-phenylindole (DAPI, Invitrogen). Deionized water (18 MΩ) was used in all the experiments.
Preparation of Fluorescently labeled Mesoporous Silica Nanoparticles (FMSNs)
Fluorescent mesoporous silica nanoparticles were prepared as described in our previous studies.26, 27 Briefly, 2 mg of FITC were stirred in an APTMS-ethanol solution (0.1 M in 10 mL of ethanol) in complete darkness for 24 hrs, to synthesize FITC-APTMS. Separately, 0.58 g of CTAB were dissolved in NH4OH (0.17 M, 300 mL) at 40°C, and then added to a dilute solution of TEOS (0.2 M in 5.0 mL of ethanol), followed by vigorous stirring for 5 hrs. The above FITC-APTMS (5.0 mL) and TEOS (1.0 M in 5.0 mL of ethanol) solutions were then mixed together with vigorous stirring for another 1 hr. The resulting solution was then aged at 40°C for 24 hrs. Solid samples were collected via centrifuging at 12,000 rpm for 20 minutes, washing, and re-dispersing the resulting precipitate with deionized water and ethanol several times.
Synthesis of FITC-MSN-PEG-UEA1
To increase the water solubility and mucus mobility of our nanoparticles, Polyethylene glycol (PEG) was employed to modify the surfaces of FMSNs. 75 mg FMSN were re-suspended in ethanol and stirred with 25 mg of silane-(PEG)5000-NHS (Nanocs) and 50 µg of 2-[Methoxy(polyethyleneoxy)-propyl]9–12 triethoxysilane (Gelest) at 60°C for 24 hrs. The nascent PEGylated FMSNs were then centrifuged and re-suspended in ethanol several times, to remove excess/free PEG. 25 mg of PEGylated FMSNs were then reacted with 200 µL of 1 mg mL−1 UEA1 in phosphate buffered saline (PBS) for 2 hrs at 4°C. The final product was then collected by 12,000 rpm centrifuging and re-dispersed in 1x PBS buffer.
Synthesis of fluorescence-labeled liposome
Rhodamine B PE-labeled liposomes were synthesized according to a previously reported procedure.24 Briefly, 4 µmole of DAPC and DMPE were mixed with 0.08 µ mole of Rhodamine B PE in 2 mL chloroform. Lipid film was formed after removing chloroform via rotory evaporation. After PBS hydration, liposomes were extruded using a 1 mL Mini Extruder (Avanti Polar Lipids Inc, Alabaster, USA) and polycarbonate filters with a pore size of 100 nm. Extrusion was performed for 10 cycles in a water bath at 70°C. The extruded liposomes were polymerized on ice using a Strategene Stratalinker 1800 UV Crosslinker (La Jolla, CA). Cross-linking was performed at 3600 mJ per cycle for 20 cycles.
Characterization
MSN morphology was characterized via transmission electron microscopy (TEM) (FEI Tecnai F30), operating at an acceleration voltage of 120 kV. Thermogravimetric analysis (TGA) (Q500, TA Instruments) was used to assess the degree of MSN-PEG/UEA1 conjugation. Approximately 10 mg of sample were dried in an 80°C oven overnight prior to TGA. During TGA, samples were first heated up to 50°C for 30 min for equilibration and then monitored for weight loss over the temperature span 50–550°C. Static/dynamic/electrophoretic light scattering (Nano ZS Zetasizer, Malvern, UK) was used to measure the MSN’s surface charge in solution at pH 7.4. Zeta potential distributions were obtained by averaging ten sequential measurements. Specimens were prepared at concentration of 2 mg per 1 mL of ddH2O. pH values were adjusted by the addition of HCl or NaOH (0.02 M). Before each measurement, each sample was sonicated for 1 min, to preclude aggregation.
Collection and preparation of native mucus samples
Native murine colon mucus was collected via blunt instrument aspiration for assessment of nanoparticle-mucus dispersion and stability studies. Mice that had been fasted overnight were sacrificed and their colons immediately harvested. Isolated colons were then longitudinally opened via surgical scissors and any remnant fecal contents removed by brief cold saline irrigation. Mucus collection then proceeded by gently scraping the exposed luminal surface of each colon with spatula and stored in small eppendorf vials. The mucus was then immediately stored at −80°C until needed, at which time they were thawed for approximately 15 minutes at 37°C.29
Solution transmission electron microscopy
Liquid-state TEM is of great utility in characterizing a nanomaterial’s dispersibility, aggregation, and agglomeration in commonly employed/encountered solutions such as solvents, buffers, and serums. For the studies reported here, liquid-state TEM was performed by use of K-kit (BioMA-Tek, Taiwan) specimen holders. 1 mg/ml nanoparticles were incubated with either PBS or native murine mucus for 0/30/60 minutes, and then loaded into the 2 µm filter of a K-kit before imaging by TEM (FEI Tecnai F20, 120 KeV). Over 100 particles were calculated and analyzed statistically by NOAA (Nano-Objects, and their Aggregates and Agglomerates, ISO/TR13014).
Fluorescence stability in mucus
We spectroscopically monitored (Fluorolog 3) the fluorescence emission stability of our nanoparticles in the presence of PBS and murine mucus. To accomplish this task, 1 mg/mL of FMSN-PEG-UEA1 or Rhodamine-labeled liposomes were incubated for the indicated time in either PBS or native murine mucus, while the fluorescence intensity of the nanoparticle’s incorporated FITC (excitation 470, emission 520; for FMSN-PEG-UEA1) or Rhodamine B PE (excitation 540, emission 625; for Rhodamine labeled liposomes) was monitored. Samples were maintained at 37°C throughout the detection process. Individual experiments were normalized by the fluorescence intensity measured at the initial (t=0) time-point.
Cell culture
Human colon cancer cells, Caco-2 and HCT116, were purchased from American Tissue Culture Collection (ATCC). Cells were cultured in a humidified 37°C atmosphere comprised of 5% CO2 and 95% room air. Caco-2 cells were maintained in DMEM HI glucose medium supplemented with 2 mM L-glutamine, 100 U/ml penicillin/streptomycin, and 20% fetal bovine serum (Gibco BRL-Life Technologies, Grand Island, NY). HCT116 cells were cultured in McCoy’s medium (Gibco) that had been supplemented with 10% fetal bovine serum.
In vitro Binding and UEA1 Competition Assay
Cells were seeded 24 hrs before experiments and fixed with 4% paraformaldehyde on ice for 15 min. Fixed cells were then washed three times with 1x PBS and blocked via 5% bovine serum albumin (BSA) solutions for 60 minutes. FITC-UEA1 staining for α-L-fucose expression was performed for 2 hrs at 37°C. FITC-UEA1 was diluted to a final concentration of 250 ng mL−1. Cells were then washed with 1x PBS twice and fluorescently stained with Hoechst 33342 (excitation 350 nm, emission 460 nm) and Wheat Germ Agglutinin (WGA) (excitation 488 nm, emission 520 nm), for cell nuclei and membrane labeling, respectively. To assess the degree of cellular binding of UEA1 labeled nanoparticles, cells were incubated with 50 µg mL−1 FMSNs for 2 hrs, followed by staining of both nucleus and membrane. For competition assays, 25 µg mL−1 of non-fluorescent UEA1 was used for pre-treatment blocking, 2 hrs prior to UEA1-MSN introduction – an approximately 100-fold higher concentration of UEA1 than was present in the 50 µg mL−1 UEA1-MSNs that were subsequently added. Confocal fluorescence microscopy (Leica TCS SPE) was then performed on the treated cells, to ascertain the affinity and localization of fluorescent moieties while flow cytometry was used to quantify cell binding. Cells were harvested and their fluorescence intensity detected by FACS LSR Fortessa Flow Cytometer (BD Bioscience).
AOM/DSS colorectal cancer mice model
Animals used in these studies were approved under the guidelines of the Institutional Animal Care and Use Committee (IACUC) of the University of Chicago, which complies with the guidelines outlined by the National Institutes of Health. The details of the specific protocols followed in these studies have been described previously.30–32 Six-week-old A/J male mice were administered a single dose of 10 mg/kg body wt. azoxymethane (AOM) i.p. and then provided with 3% dextran sodium sulfate (DSS) in drinking water for 7 days, followed by a 2 week recovery period during which animals received no DSS. This DSS dosing was then repeated once more. As A/J mice are highly sensitive to AOM exposure31, only one dose of AOM was needed, followed by two cycles of DSS, to induce colorectal tumors in the mice. Five weeks after the second cycle of DSS treatment, the presence of colorectal tumors was verified by in vivo whit-elight colonoscopy.
Ex vivo specificity of FMSN-UEA1
Animals were sacrificed 10 weeks after initiating AOM treatment. To assess ex vivo binding specificity, colon segments were soaked in 1x PBS and then cut to multiple sections. Each section of colon tissue was then briefly washed with Mucomyst (N-acetylcysteine), incubated with FMSN-UEA1 for 3 minutes, washed again with 1x PBS, and then surveyed for fluorescence using an IVIS Spectrum (Perkin-Elmer, USA) in vivo optical imaging system. For ex vivo specimen analyses, murine colon segments were fixed in formalin and sectioned into 7 µm thickness of specimens. The specimens were stained with hematoxylin and eosin (H&E) and DAPI to visualize cell morphology and nuclei location.
In vivo colonoscopy
The foregoing AOM/DSS-induced murine model of colorectal cancer was used for all in vivo nanoplatform binding assessment studies. For colonoscopies, mice were fasted 12 hrs and then anesthetized via i.p. injection with 80 mg/kg ketamine and 13 mg/kg xylazine. Mouse colons were then irrigated with 1x Mucomyst (N-acetylcysteine) enemas and topically treated with FMSN-UEA1 for 3 minutes. White-light colonoscopy (Karl Storz-Endoskope) and fluorescence endomicroscopy (Cellvizio, with Coloflex UHD-type probe; Mauna Kea Technologies, France) were used to obtain in vivo images of neoplastic changes. Fluorescence endomicroscopy images were recorded at a scan rate of 12 frames per second, with a scanning field of 30,000 pixels per frame. The field of view was 240 × 200 µm, with a lateral resolution of 1 µm.
Results
As briefly noted earlier, MSNs were synthesized via conventional sol-gel chemistry so as to incorporate FITC within the MSN’s silica framework during co-condensation – to protect the fluorophore from proximity/self and O2-quenching, and to maximize the nanoparticle’s available surface area for conjugating the α-L-fucose targeting ligand UEA1. To penetrate the adhesive, viscoelastic mucus in the colonic lumen and reach colonic endothelium cells, MSNs were coated with a mixture of polyethylene glycol (PEG) molecules, comprised of low and high molecular weight PEG chains. PEGylated nanoparticles are known to greatly increase the diffusion of nanoparticles in mucus.33 The combined use of low and high molecular weight PEG chains for nanoparticle coating also provides excellent colloidal stability, as well as numerous sites for biomacromolecule conjugation.34, 35 To achieve dual PEGylation of MSNs, we employed high molecular weight silane-PEG-NHS (molecular weight: 5K) and low molecular weight PEG-silane (molecular weight: 596–725). The ratio of low to high molecular weight PEG was empirically optimized and fixed at 15 (data not shown). NHS ester groups of silane-PEG-NHS were used to link the lysine residues of UEA1 peptides and form stable amide bond crosslinks.
TEM of our nanoplatforms revealed the characteristic morphology of mesoporous silica, with nanoparticles averaging approximately 70 nm in diameter, as shown in Figure 2a. Conjugation of PEG/UEA1 to these fluorescent MSNs, confirmed by TGA, demonstrated weight losses of FMSN, FMSN-PEG and FMSN-PEG-UEA1 of 8%, 16%, and 24% respectively, as shown in Figure 2b. Mean nanoparticle diameters and zeta-potentials were determined by use of a Zetasizer (Figure 2c). Hydrodynamic diameters of FMSNs were found to step-wise increase with the conjugation of PEG and UEA1 to the nanoplatform: from 61.6±7 nm for bare FMSN to 70.3±3 nm for FMSN-PEG to 75.4±5 nm for FMSN-PEG-UEA1. Surface charge measurements of nanoparticles revealed an average decrease of 22.5 mV during exterior functionalization: from 9.68 ±0.6 mV for FMSN to 9.03±0.2 mV for FMSN-PEG to −13.5±0.4 mV for FMSN-PEG-UEA1.
Figure 2.
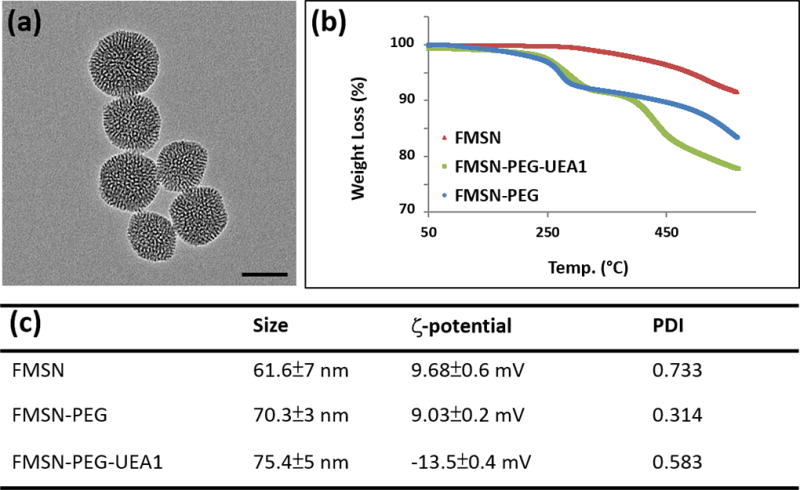
(a) TEM of FMSNs. Scale bar: 50 nm. (b) Thermogravimetric analysis of FMSN, FMSN-PEG and FMSN-PEG-UEA1 (c) Dynamic light scattering and zeta-potential measurements of pre/post UEA1-labeled FMSNs.
To mimic the colorectal environment, we also characterized the dispersion stability of our nanoplatforms in murine mucus, since colonic mucus presents a highly significant barrier to nanoparticles reaching the targeted epithelial cells. Native murine colon mucus was collected and incubated with our nanoplatforms (please refer Experimental Section for details). Nanoparticle aggregation, agglomeration, and dispersibility studies were conducted in both PBS and murine mucus using K-kit, a new and novel approach for nanoparticle TEM characterization in which nanoparticles can be imaged in liquid suspension, due to their highly constrained Brownian motion (and thus free from motion-induced imaging artifacts) imposed by the narrowness of their confines. Unlike conventional drying/freezing of samples prior to electron microscopy, which can induce structural damage and artificially alter nanoparticle aggregation and agglomeration, the K-kit approach enables TEM of the native morphology of colloidal nanoparticles and bio-nanomaterials in liquid phases.36, 37 For these studies 1 mg/ml FMSN-PEG-UEA1 was mixed and incubated with PBS/mucus at 37°C for 0/30/60 minutes and loaded into K-kit chambers before TEM observation. Particle morphology and dynamics during TEM were then analyzed statistically by NOAA (Nano-Objects, and their Aggregates and Agglomerates, ISO/TR13014). Figure 3a shows the images of nanoparticles as individual nano-objects (upper row) and aggregates/agglomerates (lower row). More than 100 particles were imaged and analyzed statistically by NOAA (Figure 3b). No obvious aggregation/agglomeration of our nanoplatforms were observed within the first 60 minutes of study.
Figure 3.
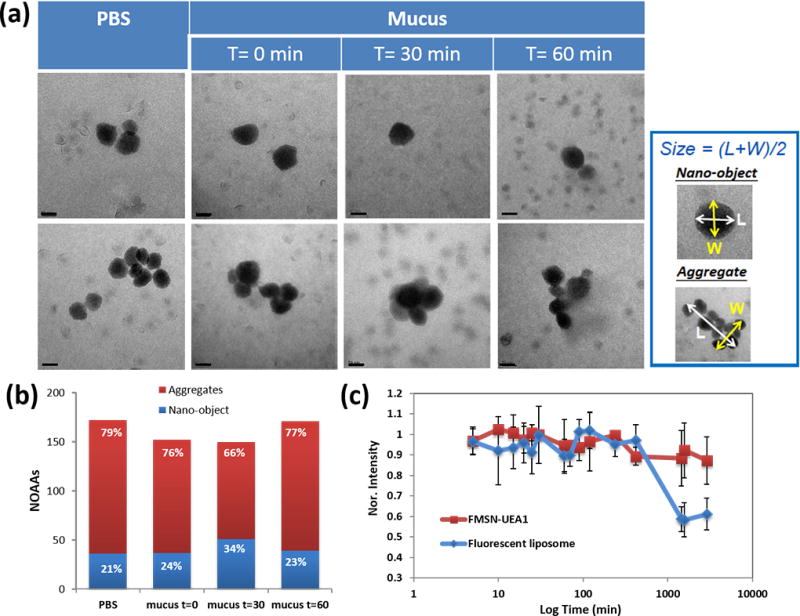
Stability of nanoparticle dispersion in murine mucus and 1x PBS. (a) Liquid-state TEM images of FMSN-PEG-UEA1 in 1x PBS and mucus for 0, 30 and 60 min. Scale bar: 50 nm. (b) Nano-Objects and their Aggregates/Agglomerates (NOAA) analysis of individual FMSN-PEG-UEA1 morphology in PBS and murine mucus. X-axis denotes the NOAA value of each condition while column height gives the percentage of nano-objects (blue)/aggregates and agglomerates (red) at the corresponding condition. (c) Nanoparticle fluorescence stability of FMSN-PEG-UEA1 (red) and fluorescent liposome (blue) in mucus, as a function of time.
Similarly, we assessed the optical reporter stability of our nanoparticles by continuously monitoring their fluorescence intensity in the PBS or murine mucus environments. Rhodamine-labeled liposomes, similar to those used by others in the ex vivo study of UEA1 targeting, were also synthesized and placed in PBS/mucus, for comparison.24 As shown in Figure 3c, the fluorescence intensities of both FMSN-PEG-UEA1 and Rhodamine-labeled liposomes remained stable for the first 4 hrs of their respective exposures. However, after a little over 24 hrs, the fluorescence intensity of Rhodamine liposomes in mucus dropped to nearly one-half its initial value, reflecting significant compromises of the liposome membrane’s integrity, as shown in Figure 3c. By comparison, the fluorescence intensity of our FMSN-PEG-UEA1 in murine mucus diminished by only 10% a little more than 24 hrs post incubation. Both FMSN-PEG-UEA1 and Rhodoamin liposome fluorescence intensity in PBS remained comparably stable for well over 24 hrs (data not shown).
For in vitro assessment of our nanoplatform’s α-L-fucose targeting efficacy, comparisons were made between human colorectal adenocarcinoma cell lines that are deficient α-L-fucose (e.g., HCT116 cells) and those that overexpress α-L-fucose (e.g., Caco-2 cells).38 Nanoparticles were incubated with either HCT116 or Caco-2 cells for 2 hrs at 37°C and then twice washed with 1x PBS, to remove any unbound nanoparticles. The newly washed cells were then stained with DAPI (blue) and WGA594 (red), to enable the localization of nuclei and plasma membranes, respectively, via fluorescence confocal microscopy (Figure 4). To better quantitate our confocal microscopy findings, >10,000 cells were harvested from culture and subjected to flow cytometey and statistical analysis using FlowJo (FlowJo, LLC) software, with the intensity of FITC being used to approximate nanoplatform binding efficacy. UEA1 peptides were conjugated onto FMSNs at a ratio of 1:200, with 250 ng mL−1 of UEA1 binding to the surface of 50 ug mL−1 of FMSN. The resulting FITC-labeled UEA1 (FITC-UEA1) was then added to 80% confluent HCT116 and Caco-2 cells in culture. FITC-UEA1 revealed affinity only for α-L-fucose-expressing Caco-2 cells, as shown in Figure 4e, with 84.6% of FITC-UEA1 binding to Caco-2 (Figure 4m) compared to 1.23% of FITC-UEA1 binding to HCT116 (Figure 4b and 4j). A similar scenario also observed when cell were treated non-fluorescent UEA1 peptides conjugated FITC-labeled MSNs (FMSN-UEA1) (Figure 4c and 4g), with 80.6% of FMSN-UEA1 binding to Caco-2 (Figure 4o) compared to 13.6% of FMSN-UEA1 binding to HCT116 (Figure 4k). Untargeted FITC-labeled MSNs (FMSNs) exhibited little affinity, with 17.2% binding to Caco-2. (Figure 4f and n). Furthermore, incubation of Caco-2 cells with non-fluorescent UEA1 for 1 hr completely eliminated/blocked our fluorescent nanoplatform’s binding (Figure 4h), with 21.3% binding left (Figure 4p). Controls for HCT116 and Caco-2 are shown in Figures 4a, j and 4d, l, respectively.
Figure 4.
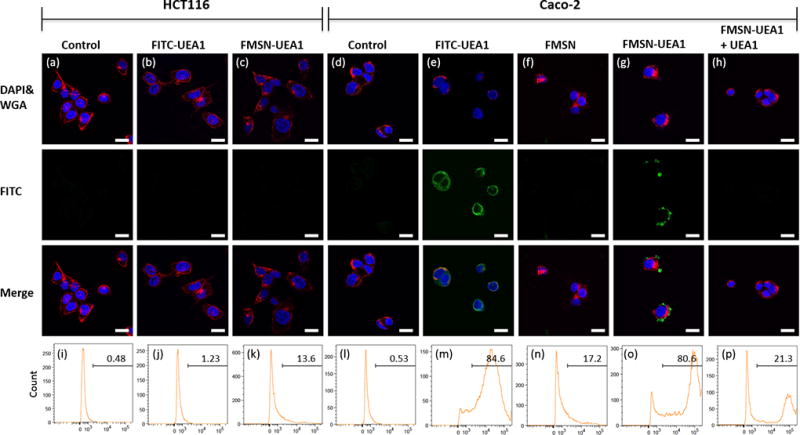
In vitro UEA1 / α-L-fucose binding affinity in human colorectal adenocarcinoma. α-L-fucose negative HCT116 (a-c) and α-L-fucose positive Caco-2 (d-h) human colorectal cancer cells were used to demonstrate binding affinity. Scale bar: 10 µm. Qualitative confocal fluorescence microscopy (a–h) and quantitative flow cytometry analyses (i–p) of the UEA1 binding were performed. HCT116 cells were incubated with PBS (a, i), FITC-UEA1 (b, j) and FMSN-UEA1 (c, k) while Caco-2 were treated with PBS (d, l), FITC-UEA1 (e, m), FMSN (f, n) and FMSN-UEA1 (g, o). Caco-2 were pre-treated non-fluorescence UEA1 for 1 hr and then treated FMSN-UEA1 (h, p). FITC (green) represented the location of FITC labeled probes. DAPI (blue) and WGA594 (red) were used to stain nuclei and cell membranes, respectively.
To more closely mimic human colon cancer and assess the targeting specificity of FMSN-UEA1 for polyps and early colorectal cancers, azoxymethane (AOM) / dextran sulfate sodium (DSS) treated mice – a standard model for colitis-associated colorectal tumorogenesis – were used in a series of in vivo and ex vivo imaging studies and histological analyses.30, 31 Animals were sacrificed 10 weeks after initiating tumorigenesis with AOM and age-matched with untreated mice that served as controls. The colons of euthanized mice were then harvested, circumferentially cut into short segments, and then sliced open along their longitudinal axis. Colon segments were next soaked once in PBS and cut to smaller sub-sections. Each sub-section of colon tissue was then washed with Mucomyst (N-acetylcysteine), to remove any retained mucus, and incubated with 10 mg mL−1 of FMSN-UEA1 for 3 minutes. Tissues were then washed twice in 1x PBS and surveyed for fluorescence using an IVIS Spectrum optical imaging system.
Figures 5a and 5b illustrate the binding affinity of FMSN-UEA1 to the surface of AOM/DSS-treated and healthy/control colon tissues, respectively, with the color barscale quantifying the fluorescent photon flux. As the figure shows, AOM/DSS mice demonstrated ~5x greater fluorescence intensity (and thus FMSN-UEA1 binding) than control animal tissues. The same tissues were then fixed, wax-embedded, block-sectioned vertically to a 7 µm depth, and stained by hematoxylin and eosin (H&E) and DAPI. Figures 5c and 5d show the H&E staining of AOM/DSS mice and control mice tissues, respectively. AOM/DSS specimens showed abnormal lumen architectures with large irregular nuclei and increased nuclear-to-cytoplasmic ratios. By contrast, control animal specimens showed well-organized crypt structures on their luminal sides (upper side: lumen, lower side: muscle). Tissue specimens stained with DAPI and analyzed by fluorescence microscopy (Figures 5e and 5f) revealed FMSN-UEA1 staining (green) the luminal side of colons from AOM/DSS treated mice but not the corresponding controls.
Figure 5.
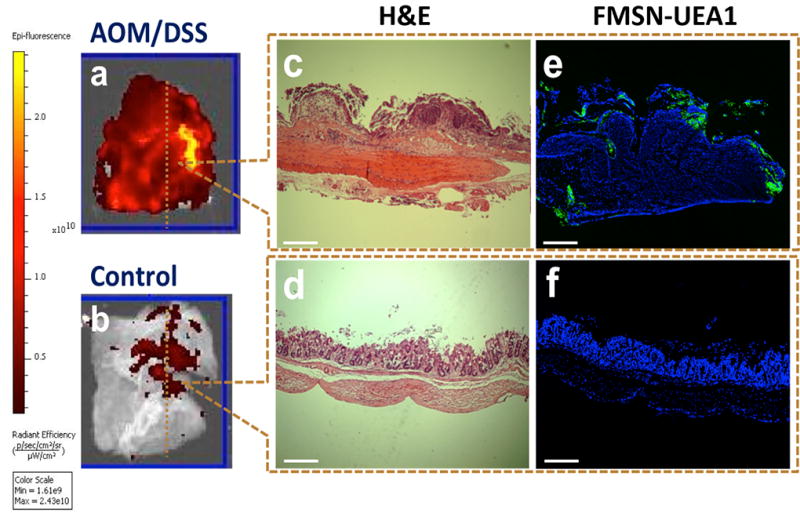
Ex vivo UEA1 and α-L-fucose binding specificity in murine colorectal adenocarcinoma. Colon segments from AOM/DSS treated A/J mice and control mice, incubated with FMSN-UEA1. (a, b) Vertically-sectioned specimens (1 cm × 1 cm) of AOM/DSS treated and control mice stained with H&E (c, d) for delineating tissue morphology and DAPI (e, f) for nuclei localization and green fluorescence for FMSNUEA1 (e,f). Scale bar: 200 µm.
To more readily assess the targeting specificity of our nanoplatforms via a well-controlled ex vivo setting, colon sections of AOM/DSS-treated mice were incubated with either 0.5 mg mL−1 of FMSN-PEG or 0.5 mg mL−1 of FMSN-UEA1 for 2 hrs, counterstained with DAPI, and microscopically examined. As shown in Figure 6a, no signal was observed in colon tissues that had been incubated with FMSN-PEG. FMSN-UEA1, however, was found to be aggregated solely on the luminal surfaces of aberrant crypts – regions known to overexpress α-L-fucose – albeit with some parasitic accumulation of contrast agent within adjacent normal appearing mucus-filled crypts (Figure 6b). 50 µg mL−1 of F-UEA1 (approximately 20-fold higher concentration of UEA1 than that conjugated onto the surfaces of MSNs) was used as a positive control, to confirm the presence of α-L-fucose, and demonstrated significant binding (Figure 6c) to colonic tissues only from AOM/DSS-treated mice.
Figure 6.
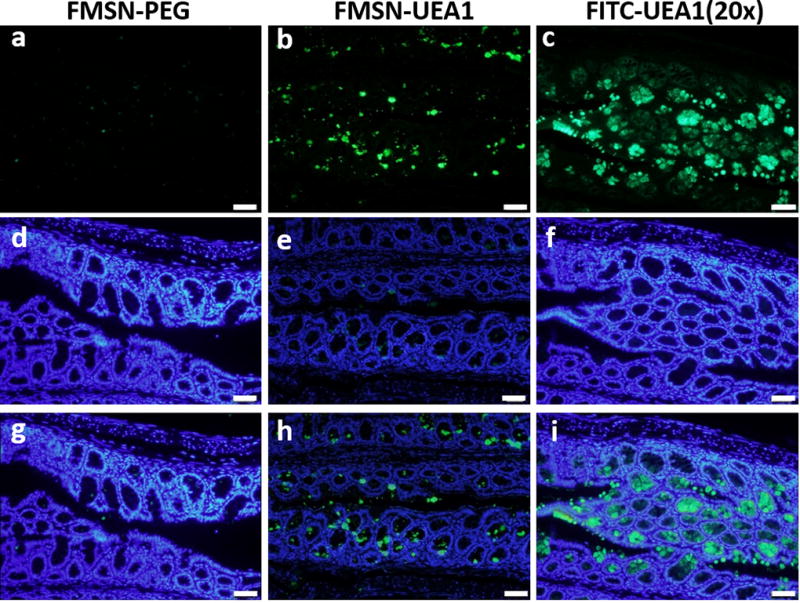
Ex vivo binding specificity of FMSN-PEG-UEA1. Harvested AOM/DSS mouse polyp/tumor specimens incubated with FMSN-PEG (a, d and g) and FMSN-UEA1 (b, e and h). 50 µg mL−1 FITC-UEA1 was used as a positive control for staining (c, f and i). Fluorescence channels: FITC (a–c, green), DAPI (d–f, blue) and merge (g–i). Scale bar 50 µm.
In vivo endoscopic exams were performed using a commercially available, clinical colonoscope and probe-based laser confocal fluorescence endomicroscope. Following 3 days maintenance of mice on a low autofluorescence diet (Teklad 2916, Harlan Laboratories) and 8 hrs fasting immediately prior to imaging – to minimize the background fecal fluorescence – mice were anesthetized with 80 mg kg−1 ketamine and 13 mg kg−1 xylazine via i.p. injection. White-light colonoscopies were first conducted on AOM/DSS-treated and healthy control mice, to determine the presence/absence and anatomical location of colorectal polyps/tumors (Figures 7a and b). Multiple intraluminal masses were readily observed in AOM/DSS–treated mice (Figure 7a) whereas colons from control animals showed no pathological lesions. Colonoscopies were immediately followed by repeated 10% Mucomyst (N-acetylcysteine) colonic irrigations, to remove residual mucus and stool. The re-prepped colons were then irrigated with 10 mg mL−1 of FMSN-UEA1 for 3 minutes and repeatedly washed with 1x PBS, to minimize the presence of unbound probe; a protocol routinely used in the endoscopically approached, luminal targeting of murine colon pathologies via contrast agents.39 Mice were then examined by probe-based laser confocal fluorescence endomicroscopy, to visualize nanoplatform co-registration with noted pathologies. As Figures 7a and 7c show, pathological regions of AOM/DSS-treated mouse colons demonstrated marked nanoparticle binding relative to healthy adjacent colon regions of the same animal and negative controls (Figure 7d).
Figure 7.
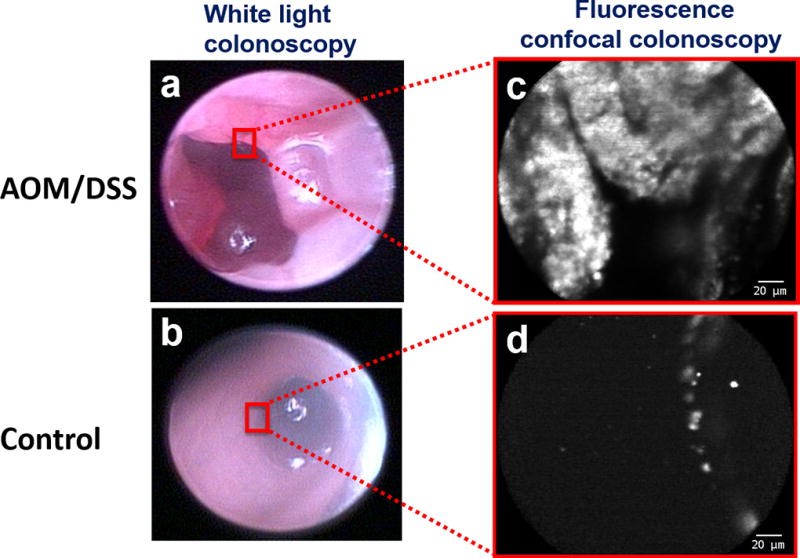
In vivo FMSN-PEG-UEA1 -guided confocal laser endomicroscopy. AOM/DSS treated (a, c) and control (b, d) mice were insufflated and irrigated via enema with FMSN-PEG-UEA1 for 3 minutes prior to evacuation and saline irrigation, to reduce unbound nanoparticle background fluorescence. Images were obtained by white light colonoscopy (a, b) and probe-based laser confocal fluorescence endomicroscopy (c, d). Scale bar: 20 µm.
Discussion
At present there is considerable interest in developing in situ methods for the early detection of colorectal adenomatous polyps and their precursors – prior to polyp malignant transformation. Such interest arises, in part, from the limitations of current clinical practice in which polyps and cancers are primarily detected by shape/morphology, with smaller/flatter polyps being frequently overlooked even during conventional colonoscopy – the diagnostic gold standard. To address this urgent need, we have designed, synthesized, and evaluated both in vitro and in/ex vivo, fluorescent MSNs that target the α-L-fucose that is known to be expressed on the luminal surfaces of membrane-bound glycoproteins of dysplastic colorectal adenomas. The imaging studies described in this paper are the first to employ α-L-fucose targeting fluorescent nanoparticles in vivo as endoscopic contrast agents.
The α-L-fucose-binding lectin UEA1, as noted above, has recently been postulated to be a promising marker in the early detection of colorectal carcinomas23, 24, 40. In a recent ex vivo study UEA1-labeled fluorescent liposomes, similar to those used in our work for comparison with our FMSNs, demonstrated significant affinity for adenomatous polyps in colon tissues that had been harvested from APCmin/+ mice.24 In those studies, however, the liposome’s cavity was reserved for the conveyance of other moieties, relegating the incorporation of limited numbers of fluorescent reporters to the liposome’s membrane and thereby constraining the nanoplatform’s brightness and thus detectability. Such membrane localization of fluorophores can also potentially afford environmental mitigation of fluorescence intensity (e.g., O2 quenching), as well as result in decreased structural integrity of the liposome’s membrane. Utilization of liposome interiors for the transport of greater numbers of fluorophores can lead to substantial proximity-induced self-quenching that also diminishes nanoplatform detectability. To enable more optimal packing of fluorescent reporters – at number densities approaching, but not surpassing, those at which reporter self-quenching occurs – requires more precise localization and spatial stabilization of fluorophores in the nanomaterial.
FMSNs circumvent this constraint while, at the same time, affording the option of having much larger volumes for the conveyance of additional moieties for therapy (e.g., controlled drug release41, photodynamic therapy27, 42–44, radiotherapy45) and/or multimodality imaging (e.g., iron/manganese oxides and Gd for MRI46, radionuclides for PET/SPECT47–49, Au/Pt for CT50). By incorporating fluorophores directly within the FMSN’s optically transparent silica matrix, reporters can be precisely, spatially fixed and separated within the nanoparticle at densities just below those that would cause self-quenching, resulting in ultra-bright fluorescence emission. And via their easily adjusted nanopore diameter, wall thickness, and particle size, FMSNs intrinsically support great customizability in their structure and functionality. FMSNs also are not inherently susceptible to degradation by enzymes (e.g., lipases) and bile salts that exist in the colonic luminal environment51, 52.
While the FMSNs employed in this study were delivered just prior to endoscopy, oral delivery of these contrast agents during the patient’s colon prep hours earlier could, potentially, offer greater time/opportunity for the contrast agent to diffuse throughout the colon than either enema or endoscopic administration. Oral delivery of contrast agents, unlike i.v. administration, obviates their adsorption of serum proteins like albumin that can promote uptake by the reticuloendothelial system and subsequent shuttling from circulation to the liver, spleen and lung. In vivo imaging studies of orally administered MSNs by our group53 – labeled with 99mTc for real-time quantitation and tracking – revealed MSNs to be remarkably stable throughout GI tract despite wide variations in pH and enzymatic activities including those capable of carbohydrate synthesis and catabolism, with no absorption or leakage into systemic circulation.
Whether delivered rectally by enema/endoscope or orally, all luminal approaches to the colon’s epithelium must pass through an intervening, dual-layer, dynamic mucosal barrier – even when the colon has been properly prepped for colonoscopy. In the healthy colon the dual-layer mucus gel is 1–2 mm in thickness on the luminal surface of the epithelium The innermost layer, closest to the apical colonic epithelial cell surface is compact and generally impermeable to bacteria; approximately 50–100 microns thick in mice and rats, but thicker in humans. Some viruses like polio, however, can get through it to infect the host suggesting that there might be small (<80nm) pores/channels in the striated/compact inner mucus layer. Overlying the inner layer is the outermost layer of mucus which is much thicker (several hundred microns in mice and rats) and much more porous. Mucus is continuously secreted, released from the cell surface, and digested/recycled, with lifetimes measured in minutes to hours. In addition to mucins, mucus gels contain cells, bacteria, lipids, salts, proteins, macromolecules, and cellular debris; by weight these gels are 90–95% water yet 103 to 104 times more viscous than water, at low/diffusion velocities.
Despite possessing negative surface charges that might be expected to repel negatively charged mucins, virus-sized carboxyl-modified polystyrene nanoparticles have been shown to form multiple hydrophobic adhesive interactions with mucin fibers that are strong enough to bundle mucin strands into thick ropes.33, 54, 55 Potentially harmful bacteria like E. coli are immobilized by mucus via hydrophobic interactions. Polyvalent adhesive interactions with mucus can be achieved via electrostatic interactions too. Since the glycosylated regions of mucin fibers are densely coated with negative charges, mucus can also bind positively charged particles with high avidity. The dual capacity to form polyvalent adhesive interactions via both hydrophobic and anionic forces represents a challenging problem for nanoparticle diffusion to the colonic epithelial cell.
Ultra-dense, short-chain PEGylation of nanoparticles has shown considerable promise in promoting the diffusion of nanoplatforms through mucus; with particles 100–500 nm in diameter demonstrating up to 3 orders-of-magnitude greater velocities in human mucus than non-PEGylated versions, and effective diffusion coefficients reduced only 4–6 fold from those of identical particles in water.33 As described earlier, we observed some parasitic accumulation of FMSNs in mucus-filled crypts both in vivo and ex vivo despite our use of 2 different forms of PEG. We posit that, even with such dense PEGylation, our FMSNs were still not adequately shielded as to preclude non-trivial mucosal impediment and entrapment. To improve mucosal transport of our nanoparticles, ongoing studies are aimed at fine-tuning the surface charge distribution of our FMSNs so as to enable still denser post-synthesis PEGylation – and employing yet lower M.W. PEG coatings, especially for smaller diameter FMSNs where smaller radii of curvature adversely affect PEGylation shielding.
Fluorescent in/ex vivo endoscopic and microscopic imaging studies, confirmed by subsequent histological analyses of harvested tissues, demonstrated significant targeting specificity of PEGylated UEA1-labeled FMSNs for polyps and nascent colorectal adenomas. As such, these lectin-functionalized fluorescent MSNs hold great promise in serving as bright endoscopic contrast agents for the in situ diagnostic imaging of polyps and early colorectal cancers. Further development of these nanoplatforms, exploiting the three topologically distinct domains to enhance particle transport in mucus and differentially target stage-dependent pathogenesis in malignant transformation, could provide a functionality of potentially significant clinical value.
Acknowledgments
We would like to thank Dr. Chung-Shi Yang from Bio Materials Analysis Technology Inc. for his advice on the synthesis and characterization of fluorescence-labeled liposomes.
Funding: The study was funded in part by the following grants: BN-103-PP-04 and NM-103-PP-01 (National Health Research Institutes of Taiwan), NSC-100-2911-I-400-502, NSC-100-2917-I-002-030, NSC102-2113-M-400-001-MY3, MOST105-2113-M-400-007-MY3, MOST106-2113-M-039-002-MY2 (National Science Council/Ministry of Science and Technology of Taiwan), CMU105-N-11 (China Medical University, Taiwan), NIH/NCI grant R01CA171785, CA036745, NIH/NCCAM grants AT004418 and AT005362, Samuel Freedman Research Laboratories for Gastrointestinal Cancer Research and the University of Chicago Comprehensive Cancer Center.
Footnotes
Statement of Competing Interests: The authors declare no conflict of interest.
References
- 1.American Cancer Society. Cancer Facts & Figures 2011. American Cancer Society; Altanta: 2011. [Google Scholar]
- 2.American Cancer Society. Colorectal Cancer Facts & Figures 2011–2014. American Cancer Society; Atlanta: 2014. [Google Scholar]
- 3.Levin B, Lieberman DA, Mcfarland B, Andrews KS, Brooks D, Bond J, Dash C, Giardiello FM, Glick S, Johnson D, Johnson CD, Levin TR, Pickhardt PJ, Rex DK, Smith RA, Thorson A, Winawer SJ, American Cancer Society Colorectal Cancer Advisory G, Force USM-ST, American College of Radiology Colon Cancer C Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: a joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. Gastroenterology. 2008;134(5):1570–1595. doi: 10.1053/j.gastro.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 4.American Cancer Society. Colorectal Cancer Prevention and Early Detection. American Cancer Society; Atlanta: Feb 5, 2015. [Google Scholar]
- 5.Hetzel JT, Huang CS, Coukos JA, Omstead K, Cerda SR, Yang S, O'brien MJ, Farraye FA. Variation in the detection of serrated polyps in an average risk colorectal cancer screening cohort. Am J Gastroenterol. 2010;105(12):2656–2664. doi: 10.1038/ajg.2010.315. [DOI] [PubMed] [Google Scholar]
- 6.Liang J, Kalady MF, Appau K, Church J. Serrated polyp detection rate during screening colonoscopy. Colorectal Dis : the Official Journal of the Association of Coloproctology of Great Britain and Ireland. 2012;14(11):1323–1327. doi: 10.1111/j.1463-1318.2012.03017.x. [DOI] [PubMed] [Google Scholar]
- 7.Kudo S, Lambert R, Allen JI, Fujii H, Fujii T, Kashida H, Matsuda T, Mori M, Saito H, Shimoda T, Tanaka S, Watanabe H, Sung JJ, Feld AD, Inadomi JM, O'brien MJ, Lieberman DA, Ransohoff DF, Soetikno RM, Triadafilopoulos G, Zauber A, Teixeira CR, Rey JF, Jaramillo E, Rubio CA, Van Gossum A, Jung M, Vieth M, Jass JR, Hurlstone PD. Nonpolypoid neoplastic lesions of the colorectal mucosa. Gastrointest Endosc. 2008;68(4 Suppl):S3–47. doi: 10.1016/j.gie.2008.07.052. [DOI] [PubMed] [Google Scholar]
- 8.Soetikno RM, Kaltenbach T, Rouse RV, Park W, Maheshwari A, Sato T, Matsui S, Friedland S. Prevalence of nonpolypoid (flat and depressed) colorectal neoplasms in asymptomatic and symptomatic adults. JAMA : the Journal of the American Medical Association. 2008;299(9):1027–1035. doi: 10.1001/jama.299.9.1027. [DOI] [PubMed] [Google Scholar]
- 9.Dennis JW, Granovsky M, Warren CE. Glycoprotein glycosylation and cancer progression. Biochim Biophys Acta. 1999;1473(1):21–34. doi: 10.1016/s0304-4165(99)00167-1. [DOI] [PubMed] [Google Scholar]
- 10.Orntoft TF, Vestergaard EM. Clinical aspects of altered glycosylation of glycoproteins in cancer. Electrophoresis. 1999;20(2):362–371. doi: 10.1002/(SICI)1522-2683(19990201)20:2<362::AID-ELPS362>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 11.Couldrey C, Green JE. Metastases: the glycan connection. Breast Cancer Res : BCR. 2000;2(5):321–323. doi: 10.1186/bcr75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ono M, Hakomori S. Glycosylation defining cancer cell motility and invasiveness. Glycoconj Journal. 2004;20(1):71–78. doi: 10.1023/B:GLYC.0000018019.22070.7d. [DOI] [PubMed] [Google Scholar]
- 13.Niv Y. Mucin and colorectal cancer. Isr Med Assoc J : IMAJ. 2000;2(10):775–777. [PubMed] [Google Scholar]
- 14.Even-Desrumeaux K, Baty D, Chames P. State of the art in tumor antigen and biomarker discovery. Cancers. 2011;3(2):2554–2596. doi: 10.3390/cancers3022554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drake PM, Cho W, Li B, Prakobphol A, Johansen E, Anderson NL, Regnier FE, Gibson BW, Fisher SJ. Sweetening the pot: adding glycosylation to the biomarker discovery equation. Clin Chem. 2010;56(2):223–236. doi: 10.1373/clinchem.2009.136333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rhodes JM, Black RR, Savage A. Glycoprotein abnormalities in colonic carcinomata, adenomata, and hyperplastic polyps shown by lectin peroxidase histochemistry. J Clin Pathol. 1986;39(12):1331–1334. doi: 10.1136/jcp.39.12.1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haltiwanger RS. Fucose is on the TRAIL of colon cancer. Gastroenterology. 2009;137(1):36–39. doi: 10.1053/j.gastro.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moriwaki K, Miyoshi E. Fucosylation and gastrointestinal cancer. World J Hepatol. 2010;2(4):151–161. doi: 10.4254/wjh.v2.i4.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miyoshi E, Moriwaki K, Terao N, Tan CC, Terao M, Nakagawa T, Matsumoto H, Shinzaki S, Kamada Y. Fucosylation is a promising target for cancer diagnosis and therapy. Biomolecules. 2012;2(1):34–45. doi: 10.3390/biom2010034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yonezawa S, Nakamura T, Tanaka S, Maruta K, Nishi M, Sato E. Binding of Ulex europaeus agglutinin-I in polyposis coli: comparative study with solitary adenoma in the sigmoid colon and rectum. J Natl Cancer Inst. 1983;71(1):19–24. [PubMed] [Google Scholar]
- 21.Yonezawa S, Nakamura T, Tanaka S, Sato E. Glycoconjugate with Ulex europaeus agglutinin-I-binding sites in normal mucosa, adenoma, and carcinoma of the human large bowel. J Natl Cancer Inst. 1982;69(4):777–785. [PubMed] [Google Scholar]
- 22.Yonezawa S, Nakamura T, Irisa S, Otsuji Y, Sato E. Ulex europaeus agglutinin I staining of human glomerular lesions using a highly sensitive immunoperoxidase method in paraffin sections. Nephron. 1983;35(3):187–189. doi: 10.1159/000183072. [DOI] [PubMed] [Google Scholar]
- 23.Watanabe M, Ohtani H, Tanaka M, Ikehara H. Appearance of Ulex europaeus agglutinin-1 and Griffonia simplicifolia agglutinin-1 binding sites on cancer cells in sigmo-rectal polyps. Acta Pathol Jpn. 1992;42(11):800–806. [PubMed] [Google Scholar]
- 24.Roney CA, Xu B, Xie J, Yuan S, Wierwille J, Chen CW, Chen Y, Griffiths GL, Summers RM. Rh-I-UEA-1 polymerized liposomes target and image adenomatous polyps in the APC(Min/+) mouse using optical colonography. Mol Imaging. 2011;10(4):305–316. doi: 10.2310/7290.2010.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roney CA, Xie J, Xu B, Jabour P, Griffiths G, R.M S. Glycoprotein expression by adenomatous polyps of the colon. 2008;6916(69161O):1–10. [Google Scholar]
- 26.Cheng S-H, Hsieh C-C, Chen N-T, Chu C-H, Huang C-M, Chou P-T, Tseng F-G, Yang C-S, Mou C-Y, Lo L-W. Well-defined mesoporous nanostructure modulates three-dimensional interface energy transfer for two-photon activated photodynamic therapy. Nano Today. 2011;6(6):552–563. [Google Scholar]
- 27.Cheng S-H, Lee C-H, Chen M-C, Souris JS, Tseng F-G, Yang C-S, Mou C-Y, Chend C-T, Lo L-W. Tri-functionalization of mesoporous silica nanoparticles for comprehensive cancer theranostics—the trio of imaging, targeting and therapy. J Mater Chem. 2010;20:6149–6157. [Google Scholar]
- 28.Chen NT, Cheng SH, Liu CP, Souris JS, Chen CT, Mou CY, Lo LW. Recent advances in nanoparticle-based forster resonance energy transfer for biosensing, molecular imaging and drug release profiling. Int J Mol Sci. 2012;13(12):16598–16623. doi: 10.3390/ijms131216598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li X, Chen D, Le C, Zhu C, Gan Y, Hovgaard L, Yang M. Novel mucus-penetrating liposomes as a potential oral drug delivery system: preparation, in vitro characterization, and enhanced cellular uptake. Int J Nanomed. 2011;6:3151–3162. doi: 10.2147/IJN.S25741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neufert C, Becker C, Neurath MF. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat Protoc. 2007;2(8):1998–2004. doi: 10.1038/nprot.2007.279. [DOI] [PubMed] [Google Scholar]
- 31.Dougherty U, Mustafi R, Wang Y, Musch MW, Wang CZ, Konda VJ, Kulkarni A, Hart J, Dawson G, Kim KE, Yuan CS, Chang EB, Bissonnette M. American ginseng suppresses Western diet-promoted tumorigenesis in model of inflammation-associated colon cancer: role of EGFR. BMC Complement Altern Med. 2011;11:111. doi: 10.1186/1472-6882-11-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nambiar PR, Girnun G, Lillo NA, Guda K, Whiteley HE, Rosenberg DW. Preliminary analysis of azoxymethane induced colon tumors in inbred mice commonly used as transgenic/knockout progenitors. Inter J Oncol. 2003;22(1):145–150. [PubMed] [Google Scholar]
- 33.Lai SK, Wang YY, Hanes J. Mucus-penetrating nanoparticles for drug and gene delivery to mucosal tissues. Advanced drug delivery reviews. 2009;61(2):158–171. doi: 10.1016/j.addr.2008.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu T, Thierry B. A solution to the PEG dilemma: efficient bioconjugation of large gold nanoparticles for biodiagnostic applications using mixed layers. Langmuir. 2012;28(44):15634–15642. doi: 10.1021/la301390u. [DOI] [PubMed] [Google Scholar]
- 35.Backmann N, Kappeler N, Braun T, Huber F, Lang HP, Gerber C, Lim RY. Sensing surface PEGylation with microcantilevers. Beilstein J Nanotech. 2010;1:3–13. doi: 10.3762/bjnano.1.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu KL, Wu CC, Huang YJ, Peng HL, Chang HY, Chang P, Hsu L, Yew TR. Novel microchip for in situ TEM imaging of living organisms and bio-reactions in aqueous conditions. Lab Chip. 2008;8(11):1915–1921. doi: 10.1039/b804986f. [DOI] [PubMed] [Google Scholar]
- 37.Tai LA, Kang YT, Chen YC, Wang YC, Wang YJ, Wu YT, Liu KL, Wang CY, Ko YF, Chen CY, Huang NC, Chen JK, Hsieh YF, Yew TR, Yang CS. Quantitative characterization of nanoparticles in blood by transmission electron microscopy with a window-type microchip nanopipet. Anal Chem. 2012;84(15):6312–6316. doi: 10.1021/ac301523n. [DOI] [PubMed] [Google Scholar]
- 38.Kalischuk LD, Leggett F, Inglis GD. Campylobacter jejuni induces transcytosis of commensal bacteria across the intestinal epithelium through M-like cells. Gut pathog. 2010;2:14. doi: 10.1186/1757-4749-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Miller SJ, Joshi BP, Feng Y, Gaustad A, Fearon ER, Wang TD. In vivo fluorescence-based endoscopic detection of colon dysplasia in the mouse using a novel peptide probe. PLoS One. 2011;6(3):e17384. doi: 10.1371/journal.pone.0017384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsushita Y, Yonezawa S, Nakamura T, Shimizu S, Ozawa M, Muramatsu T, Sato E. Carcinoma-specific Ulex europaeus agglutinin-I binding glycoproteins of human colorectal carcinoma and its relation to carcinoembryonic antigen. J Natl Cancer Inst. 1985;75(2):219–226. [PubMed] [Google Scholar]
- 41.Lee CH, Cheng SH, Huang IP, Souris JS, Yang CS, Mou CY, Lo LW. Intracellular pH-responsive mesoporous silica nanoparticles for the controlled release of anticancer chemotherapeutics. Angew Chem Int Edit. 2010;49(44):8214–8219. doi: 10.1002/anie.201002639. [DOI] [PubMed] [Google Scholar]
- 42.Cheng S, Hsieh C, Chen N, Chu C, Huang C, Chou P, Tseng F, Yang C, Mou C, Lo L. Well-defined mesoporous nanostructure modulates three-dimensional interface energy transfer for two-photon activated photodynamic therapy. Nano Today. 2011;6(6):552–563. [Google Scholar]
- 43.Cheng S, Lee C, Chen M, Souris JS, Tseng F, Yang C, Mou C, Chend C, Lo L. Tri-functionalization of mesoporous silica nanoparticles for comprehensive cancer theranostics—the trio of imaging, targeting and therapy. J Mater Chem. 2010;20:6149–6157. [Google Scholar]
- 44.Chen NT, Tang KC, Chung MF, Cheng SH, Huang CM, Chu CH, Chou PT, Souris JS, Chen CT, Mou CY, Lo LW. Enhanced plasmonic resonance energy transfer in mesoporous silica-encased gold nanorod for two-photon-activated photodynamic therapy. Theranostics. 2014;4(8):798–807. doi: 10.7150/thno.8934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen F, Hong H, Shi S, Goel S, Valdovinos HF, Hernandez R, Theuer CP, Barnhart TE, Cai W. Engineering of hollow mesoporous silica nanoparticles for remarkably enhanced tumor active targeting efficacy. Sci Rep. 2014;4:5080. doi: 10.1038/srep05080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu SH, Lin YS, Hung Y, Chou YH, Hsu YH, Chang C, Mou CY. Multifunctional mesoporous silica nanoparticles for intracellular labeling and animal magnetic resonance imaging studies. Chembiochem. 2008;9(1):53–57. doi: 10.1002/cbic.200700509. [DOI] [PubMed] [Google Scholar]
- 47.Chakravarty R, Goel S, Hong H, Chen F, Valdovinos HF, Hernandez R, Barnhart TE, Cai W. Hollow mesoporous silica nanoparticles for tumor vasculature targeting and PET image-guided drug delivery. Nanomedicine (Lond) 2015;10(8):1233–1246. doi: 10.2217/nnm.14.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen F, Nayak TR, Goel S, Valdovinos HF, Hong H, Theuer CP, Barnhart TE, Cai W. In vivo tumor vasculature targeted PET/NIRF imaging with TRC105(Fab)-conjugated, dual-labeled mesoporous silica nanoparticles. Mol Pharm. 2014;11(11):4007–4014. doi: 10.1021/mp500306k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee SB, Kim HL, Jeong HJ, Lim ST, Sohn MH, Kim DW. Mesoporous silica nanoparticle pretargeting for PET imaging based on a rapid bioorthogonal reaction in a living body. Angew Chem Int Edit. 2013;52(40):10549–10552. doi: 10.1002/anie.201304026. [DOI] [PubMed] [Google Scholar]
- 50.Song JT, Yang XQ, Zhang XS, Yan DM, Wang ZY, Zhao YD. Facile Synthesis of Gold Nanospheres Modified by Positively Charged Mesoporous Silica, Loaded with Near-Infrared Fluorescent Dye, for in Vivo X-ray Computed Tomography and Fluorescence Dual Mode Imaging. ACS Appl Mater Interfaces. 2015;7(31):17287–17297. doi: 10.1021/acsami.5b04359. [DOI] [PubMed] [Google Scholar]
- 51.Chen Y, Chen H, Shi J. In vivo bio-safety evaluations and diagnostic/therapeutic applications of chemically designed mesoporous silica nanoparticles. Adv Mater. 2013;25(23):3144–3176. doi: 10.1002/adma.201205292. [DOI] [PubMed] [Google Scholar]
- 52.Souris JS, Lee CH, Cheng SH, Chen CT, Yang CS, Ho JA, Mou CY, Lo LW. Surface charge-mediated rapid hepatobiliary excretion of mesoporous silica nanoparticles. Biomaterials. 2010;31(21):5564–5574. doi: 10.1016/j.biomaterials.2010.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shih M-H, Liao W-N, Chen M-N, Yang C-S. Acid resistant Tc-99m labeling of mesoporous silica nanoparticles for imaging in GI tract. J Nucl Med. 2010;51 [Google Scholar]
- 54.Lai SK, O'hanlon DE, Harrold S, Man ST, Wang YY, Cone R, Hanes J. Rapid transport of large polymeric nanoparticles in fresh undiluted human mucus. Proc Natl Acad Sci USA. 2007;104(5):1482–1487. doi: 10.1073/pnas.0608611104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Johansson ME, Larsson JM, Hansson GC. The two mucus layers of colon are organized by the MUC2 mucin, whereas the outer layer is a legislator of host-microbial interactions. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4659–4665. doi: 10.1073/pnas.1006451107. [DOI] [PMC free article] [PubMed] [Google Scholar]