

Abstract
Alternative splicing is a fundamental regulatory process of gene expression. Defects in alternative splicing can lead to various diseases, and modification of disease-causing splicing events presents great therapeutic promise. Splicing outcome is commonly affected by extracellular stimuli and signaling cascades that converge on RNA-binding splicing regulators. These trans-acting factors recognize cis-elements in pre-mRNA transcripts to affect spliceosome assembly and splice site choices. Identification of these splicing regulators and/or upstream modulators has been difficult and traditionally done by piecemeal. High-throughput screening strategies to find multiple regulators of exon splicing have great potential to accelerate the discovery process, but typically confront low sensitivity and low specificity of screening assays. Here we describe a unique screening strategy, IRAS (identifying regulators of alternative splicing), using a pair of dual-output minigene reporters to allow for sensitive detection of exon splicing changes. Each dual-output reporter produces green fluorescent protein (GFP) and red fluorescent protein (RFP) fluorescent signals to assay the two spliced isoforms exclusively. The two complementary minigene reporters alter GFP/RFP output ratios in the opposite direction in response to splicing change. Applying IRAS in cell-based high-throughput screens allows sensitive and specific identification of splicing regulators and modulators for any alternative exons of interest. In comparison to previous high-throughput screening methods, IRAS substantially enhances the specificity of the screening assay. This strategy significantly eliminates false positives without sacrificing sensitive identification of true regulators of splicing.
1. INTRODUCTION
Most mammalian multiexon genes produce multiple mRNA isoforms because of pre-mRNA alternative splicing, which dramatically increases transcriptome complexity and proteome diversity. Many pre-mRNA splicing patterns are highly regulated to produce functionally distinct gene products during development or in response to extracellular stimuli (Chen & Manley, 2009; Zheng & Black, 2013). The selection of alternative splice sites is typically determined by the interactions between cis-regulatory elements in the pre-mRNA and trans-acting protein factors that can affect spliceosome assembly (Black, 2003; Fu & Ares, 2014). A single alternative exon can contain multiple cis-elements in itself or in its surrounding introns, and thus can be regulated by multiple trans-factors. Meanwhile each trans-factor can control a multitude of target exons. Only tens of RNA-binding proteins have been described to modulate alternative splicing in eukaryotic cells. Given the myriad genes subject to alternative splicing, many splicing factors probably have not been identified and await discovery. Factors controlling the activity of known splicing regulators are also mostly unclear.
Genome-wide sequencing-based methods allow for identifying many targets of individual splicing factors (Chen & Zheng, 2009; Hartmann & Valcárcel, 2009; Witten & Ule, 2011); however, identifying individual positive and negative regulators of an alternative exon has been challenging and often involved laborious intensive workflow and needed to be done individually. Previous strategies using various cell-based screening techniques were able to identify few new factors that target exons, however, were overall insensitive for identifying a larger sets of regulators of splicing (Kar, Havlioglu, Tarn, & Wu, 2006; Oberdoerffer et al., 2008; Topp, Jackson, Melton, & Lynch, 2008; Wu, Kar, Kuo, Yu, & Havlioglu, 2006). Most of these strategies designed a single output of splicing, such that splicing changes were measured by changes in expression of one of the two isoforms which were then converted into either a fluorescence-based or a luminescence-based readout. These single-output reporters, however, would also directly measure the overall expression, in addition to splicing, of the reporter transcripts. Additionally, the alternative isoform generating the readout must be produced at a very low basal level for any changes in splicing to be measurable, which has occluded the application of reporters on exons spliced at intermediate levels. Because the single-output reporter has to be spliced at either very low or very high level for the screening strategy to work, individual screens can only identify activators or repressors of splicing, but not both. The use of a pair of single-output reporters that encoded two different fluorescent proteins could overcome some of these limitations (Kuroyanagi, Ohno, Sakane, Maruoka, & Hagiwara, 2010). However, the requirement for integrating two minigene reporters into genomes as well as the possibilities of multiple insertion loci and different copy numbers can impose difficulty in analyzing results.
A dual-output reporter, in which both splicing isoforms are assayed, allows for screening of changes in isoform ratio and can detect both increases and decreases in exon inclusion with higher sensitivity. This screening strategy allows targeted exons spliced at intermediate levels and can also reduce the amount of false positives that alter overall reporter expression independently of splicing. Because the expression of the two alternative isoforms is negatively correlated, using the output ratio of the two isoforms to measure splicing level squares the dynamic range given by individual output. One example of dual-output reporters contained open-reading frame (ORF) of green fluorescent protein (GFP) and red fluorescent protein (RFP) in each of the mutually exclusive exons (Kuroyanagi, Kobayashi, Mitani, & Hagiwara, 2006). Because the large GFP and RFP ORFs were in place of the alternative exons, this reporter was tailored to insert an intronic cis-element of interest and identify its cognate trans-factors and was not designed to capture most regulatory elements within and flanking the exon of interest.
Another dual-fluorescence splicing reporter was successfully used to identify chemicals modulating alternative splicing of MAPT exon 10 (Stoilov, Lin, Damoiseaux, Nikolic, & Black, 2008). This reporter (pflareA) contains tandem ORFs of GFP and RFP. The GFP ORF is split between two constitutive exons by the alternative cassette exon of interest with its flanking introns. The GFP ORF is initiated for translation when the alternative exon is skipped, and the RFP translation will not be initiated. When the alternative exon is included, the GFP ORF loses its start codon, and the downstream RFP is then translated. Some alternative exons may have in-frame start codon for the GFP reading frame. In that case, ATG start codons within the alternative exon are mutated. The RFP/GFP ratio can then be used as a proxy of splicing level and a change in RFP/GFP ratio indicates alteration of splicing. Because the GFP ORF is closer to the 5′ end of the mRNA than the RFP ORF, modulators of translation would likely have an impact on the RFP/GFP ratio. As seen for many other screening strategies mentioned earlier, variables affecting the readout (ie, RFP/GFP ratio for this reporter) without an impact on pre-mRNA splicing constitute a majority of (often about 90%) hits.
In summary, genome-wide screens for splicing modulators of any single alternative exon confront a multitude of challenges including low sensitivity, narrow dynamic range, substantial false positives, low throughput, and limited applicability. Previous screening strategies using different types of splicing reporters have each addressed a few of these challenges to some degree. To provide a universal screening method broadly applicable to any cassette exon with high sensitivity and specificity, we have designed a novel strategy named IRAS, identifying regulators of alternative splicing, which significantly improves global identification of true positives (Fig. 1).
Fig. 1.
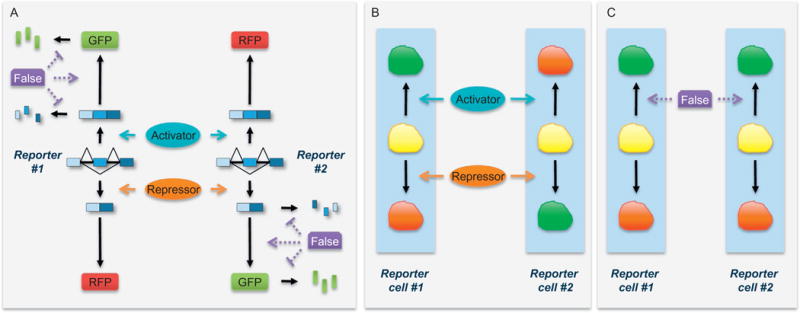
IRAS strategy illustrated by dual-fluorescence reporters. (A) Two complementary reporters differ in several nucleotides. Reporter #1 translates GFP when the alternative exon is included and RFP when the alternative exon is skipped. Reporter #2 translates RFP from the included isoform and GFP from the skipped isoform. Therefore, the ratios of GFP/RFP or RFP/GFP represent the inclusion ratio of the alternative exon in reporters #1 and #2, respectively. Factors that alter these ratios without affecting the inclusion ratio are false positives in screens. Possible actions of false positives that increase GFP/RFP ratio are labeled in purple (dark gray in the print version). False positives that increase RFP/GFP ratio are not drawn. (B and C) Expected phenotypes induced by true splicing activators and repressors and false positives in screens using cells stably express either reporters #1 or #2. A true splicing regulator changes GFP/RFP ratios in the two reporter cells in the opposite direction, whereas a false positive more likely affects the GFP/RFP ratios in the same direction. False positives that increase RFP/GFP ratio are not drawn.
2. METHOD DESIGN
IRAS uses two complementary dual-output minigene reporters exhibiting opposite changes of the readouts in response to splicing alternation (Fig. 1A). These outputs can be fluorescence based or luminescence based. For the simplicity of description, we herein use fluorescence-based reporters to explain the concept of IRAS, as we have successfully screened with these reporters. For IRAS to work, one reporter produces RFP when an exon is included and GFP from the mRNA lacking the exon, whereas a second reporter produces GFP and RFP to represent exon inclusion and exclusion respectively. The first reporter is based on the pflareA minigene for which the RFP/GFP ratio represents the exon inclusion ratio (Fig. 2A). Any ATG start codons present within the alternative exon of interest and in frame with the GFP ORF need to be mutated for pflareA minigene to translate RFP when the exon is included. The second minigene (pflareG) reporter is constructed by having the start codon of GFP in the alternative exon such that when the alternative exon is included GFP is translated (Fig. 2B) (Zheng, Damoiseaux, Chen, & Black, 2013). This can be easily done if the alternative exon has an in-frame start codon. Otherwise, an ATG start codon for GFP within the alternative exon can be engineered by site-directed mutagenesis or insertion. When the alternative exon is skipped, GFP does not have an initiation codon and the downstream RFP ORF is used. Therefore, GFP/RFP ratio reflects the exon inclusion ratio in the pflareG reporter.
Fig. 2.
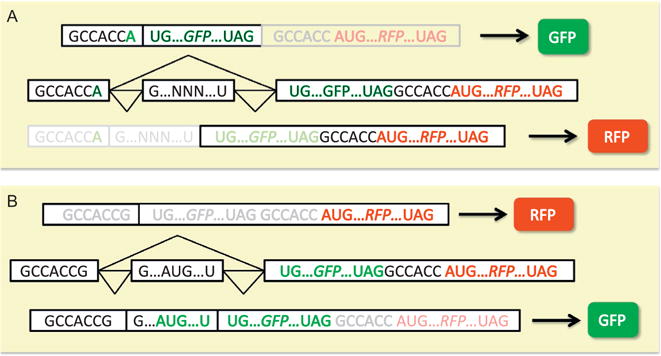
Examples of two dual-fluorescence reporters that produce opposite splicing-dependent readouts. (A) Reporter translates GFP when the alternative exon is included and RFP when the alternative exon is skipped. (B) Reporter translates RFP from the included isoform and GFP from the skipped isoform.
The idea behind IRAS is the following. Screening with individual reporters can identify hundreds and thousands of hits, many of which are false positives affecting the GFP/RFP ratio without altering the splicing of the exon. Some false positives may have preferentially stabilized one of the fluorescent proteins. Others may differentially affect ORF translation efficiency between GFP and RFP (Fig. 1A). A true splicing modulator should change the GFP/RFP ratios of the two reporters in the opposite direction (Fig. 1B). Because pflareA and pflareG minigenes differ in only a few nucleotides, a false-positive hit that alters GFP/RFP ratio of the first minigene without affecting splicing is very unlikely to change GFP/RFP ratio of the second minigene in the opposite direction. Instead, this false positive, if affecting only GFP ORF or RFP ORF, could change GFP/RFP ratios of the two reporters in the same direction (Fig. 1C). Using these two dual-fluorescence minigene reporters, we minimize systematic variations associated with fluorescent screening to enable more sensitive and accurate detection of moderate splicing changes. IRAS can simultaneously identify multiple activators and repressors of an alternative exon. Combining the screening results from these two reporters significantly eliminates false positives and enriches for identification of true splicing regulators.
Multiple technologies allow fluorescence-based selection of positive hits. Fluorescence-activated cell sorting (FACS) enriches cell population that deviates from control cells in fluorescence gating. The FACS procedure is relatively fast but requires subsequent profiling of the sorted cells, thus is most suitable for genetic screens and not for chemical screens. Genetic alterations (eg, cDNA overexpression or shRNA knockdown) within the sorted cells can then be amplified by PCR using library-specific primers and identified by next-generation sequencing. Another technique to select positive hits is fluorescence imaging. The versatility of fluorescence imaging has been aided by the development of cataloged libraries and high-throughput robotic liquid-handling systems which has enabled screening at greater depth and the identification of multiple regulators from a single screen. These libraries (eg, cDNA, shRNA, siRNA) are typically prearrayed in multiwell plates in a format of one well one known candidate; therefore, the identities of positive hits are known right after screening.
Here in detail we describe the application of IRAS using a gain-of-function genetic screen as an example (Fig. 3). In addition to cDNAs, IRAS can be used to screen libraries of siRNAs or shRNAs, and of small molecules, and we will discuss issues for different libraries. Overall the method described here greatly reduces false positives while maintaining high sensitivity in detecting regulators. This system allows for genome-wide screening for factors regulating splicing of any exon of interest.
Fig. 3.
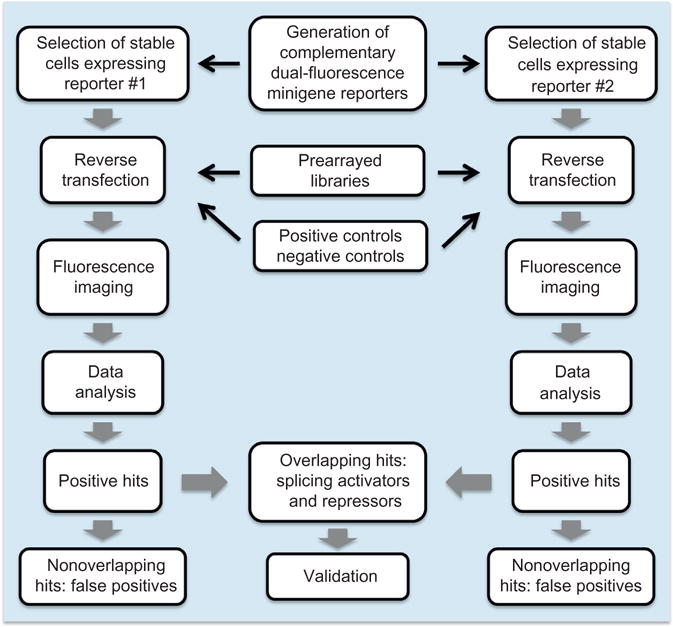
Workflow of a fluorescent cell-based high-throughput IRAS genetic screen.
3. CONSTRUCTION OF DUAL-FLUORESCENCE MINIGENE REPORTERS
To construct the minigene reporters, the alternative exon and its flanking intronic sequences will need to be inserted into the EcoRI and BamHI sites of either the pflareA or pflareG vectors. The selection of intronic sequence can be optimized with prior knowledge. Conserved intronic sequence is usually indicative of regulation and is recommended to be included (Chen & Zheng, 2008). In most cases, different length of intronic sequences is chosen to generate multiple versions of minigenes. The one with a mid-range inclusion ratio is recommended because this allows screening for both positive and negative regulators simultaneously.
3.1 Materials and Equipment
Pflare A reporter and plfareG reporter
Phusion High-Fidelity DNA polymerase (NEB)
Purified genomic DNA containing the gene and exon of interest
QuikChange II XL Site-Directed Mutagenesis Kit (Catalog# 200521, Agilent)
QIAquick Gel Extraction Kit (Qiagen)
NanoDrop (ThermoFisher Scientific)
Alkaline phosphatase (NEB)
T4 DNA Ligase (NEB)
TOP10 Competent Cells (ThermoFisher Scientific)
EcoRI (NEB)
BamHI (NEB)
LB-kanamycin agar plate
Plasmid Miniprep Kit (Qiagen)
Agarose gel electrophoresis setup
3.2 Protocol
Design forward and reverse primers for cloning the alternative exon of interest and its flanking introns.
Synthesize the forward primer with an EcoRI recognition site at the 5′ end. Synthesize the reverse primer with a BamHI recognition site at the 5′ end.
PCR amplification using the above forward and reverse primers, Phusion High-Fidelity DNA polymerase, and genomic DNA as the template.
Confirm the PCR amplicon by agarose gel electrophoresis.
Excise the band of the PCR amplicon and purify the PCR product with QIAquick Gel Extraction Kit.
Measure the concentrations of the recovered PCR products with NanoDrop.
Restriction enzyme double digestion of the PCR product with EcoRI and BamHI for 1 h at 37°C. Meanwhile linearize the pflareA and pflareG vectors with EcoRI and BamHI for 1 h at 37°C. Dephosphorylate the linearized vectors with alkaline phosphatase for 0.5 h at 37°C.
Confirm the linearized backbone vectors by agarose gel electrophoresis.
Gel-purify the digested PCR products and vectors using QIAquick Gel Extraction Kit.
Ligate the PCR product to the linearized vector with T4 DNA ligase at 16°C overnight.
Transform the ligated products into TOP10 competent cells.
Plate the transformed competent cells on LB-kanamycin agar plates and incubate at 37°C overnight.
Select several colonies for amplification and purification of plfareA-exon and plfareG-exon reporters using Plasmid Miniprep Kit.
For the plfareA-exon vector, mutate any ATG codons present within the alternative exon of interest to ATA using the QuikChange II XL Site-Directed Mutagenesis Kit.
For the pflareG-exon reporter, an ATG start codon within the alternative exon in frame with the GFP ORF is required for expression of GFP when the exon is included. If missing, a start codon needs to be created using the QuikChange II XL Site-Directed Mutagenesis Kit. The Kozak consensus sequence gccRccAUGG is also recommended to promote translation initiation for the GFP ORF.
3.3 Considerations
After construction the reporters should be transfected in the cells of choice to confirm their applicability for the screen. The choice of cells will be discussed later. The two reporters should exhibit similar splicing levels of the inserted alternative exons. They should also express both GFP and RFP but with opposite preference. For example, if one expresses a high level of GFP and a low level of RFP, the other one should express a high level of RFP and a low level of GFP. Any known positive or negative modulator of the inserted alternative exon should also be tested on the reporters for its ability to change the GFP/RFP ratio, and more importantly in the opposite direction for the two reporters. Such tests not only examine the performance of the reporters but also confirm a known modulator suitable as a spike-in control in the final screen.
4. GENERATION OF STABLE CELL CLONES
Two criteria need to be met for the parental cell line. First, the cell line ideally needs to contain characteristics of or be derived from the primary cells of interest, so that the identified hits are more likely physiologically relevant. This is particularly important for loss-of-function screens (eg, with shRNA or siRNA libraries) where the most interesting cell-type-specific splicing factors to be targeted are sometimes expressed only in a highly relevant cell line. Second, for genetic screens, the cell line needs to be highly transfectable to increase the signal-to-noise ratio of the fluorescence readouts, because both transfected and untransfected cells within a well contribute to the final fluorescence signals.
4.1 Materials and Equipment
A parental cell line of choice
QIAquick Gel Extraction Kit (Qiagen)
DraIII (NEB)
Lipofectamine 2000 (ThermoFisher Scientific)
G418 or Geneticin (ThermoFisher Scientific)
Phenol red-free media (ThermoFisher Scientific)
FACS machine
4.2 Protocol
Linearize the pflareA-exon vector by DraIII digestion and gel purify the vector with QIAquick Gel Extraction Kit.
Transfect the linearized pflareA-exon vector into the target cell line of choice using Lipofectamine 2000.
Maintain the transfected cells in G418-containing media for 2 weeks to enrich stable cell clones.
Sort double fluorescent (ie, GFP+RFP+) single cells to individual wells of multiwell plates with a FACS machine and allow cell clones to grow and expand. Some cell clones may lose GFP and RFP expression during the selection process and can be discarded. From now on, the cells need to be maintained in phenol red-free media for enhanced fluorescence visualization and imaging.
Repeat steps 1–4 to generate stable cell clones expressing pflareG-exon reporter.
4.3 Considerations
The stable cell clones need to be tested for their responsiveness to splicing changes before high-throughput screens. An appropriate clone should not change GFP/RFP ratio when transfected with an empty control vector and should do so when transfected with a known regulator of the exon. An RT-PCR assay to measure the splicing changes at the RNA level is highly recommended. This step will narrow down the number of appropriate cell clones.
We tested the “transfectability” of each selected stable cell clone using a GFP expression plasmid. A highly transfectable cell clone would show very high GFP signals without affecting the RFP signals. We also compared different transfection reagents and optimized transfection reagent to GFP plasmid ratios as well as cell densities of selected clones to finalize the transfection conditions for the screen. Once an optimal stable cell clone was identified, we expanded them in a large scale.
5. LIBRARY AND ARRAY CONSTRUCTION
Although IRAS is broadly applicable to screen any library, considerations should be taken in light of the library attributes. For example, auto-fluorescent chemicals interfere with the readouts and are technically not “screenable,” even although these false-positive ones can be filtered by IRAS. Loss-of-function screens depend on the specificity of shRNA and siRNA. Multiple shRNA or siRNA hits targeting the same gene are often required to declare a true target. Many splicing regulators have paralogs that cross-regulate each other, such that RNAi depletion of one paralog induces expression of the other paralogs. Paralogous splicing regulators often have similar effects on a target exon, and thus need to be all depleted for a significant splicing change of the target exon. As a result, these regulatory paralogs may not be easily identified in loss-of-function screens. Loss-of-function screens may also miss true regulators that are not expressed in the screened cells. Gain-of-function screen has less concern on off-target effects, paralogous compensation, or missing expression, but overexpression may induce artificial splicing changes. Therefore, the physiological relevance of the hits will require loss-of-function validation. In this report, we use gain-of-function screens to demonstrate the preparation of the arrayed library.
5.1 Materials and Equipment
The mammalian gene collection (MGC) library: developed by NIH and representing the most extensive collection of mammalian cDNA clones available. All cDNA clones are in the pCMVsport6.0 or pCMVsport6.1 backbone vectors prearrayed in 96-well plates (Life Technology).
Genetix Qbot (Molecular Devices)
Biomek FX robot (Beckman Coulter)
5.2 Protocol
Prepare an “assay ready” cDNA library by duplicating the MGC collection bacteria plates into 384-well plates using a Genetix Qbot (Molecular Devices). A library of about 16 thousand clones would occupy 45 384-well plates.
Prepare the plasmid DNA of the library using plasmid prep kit (Macherey-Nagel) on a Biomek FX robot (Beckman Coulter).
Normalize plasmid concentration and spot them into assay plates. We typically spot 40 ng cDNA per well (except the wells of A-H23 and A-H24) in the 384-well plate format. We also spot 40 ng negative control (pCMV sport6.0) plasmids to wells of C23, C24, D23, and D24; 40 ng plasmids of a positive regulator (if available) to wells of E23, E24, F23, and F24; as well as a negative regulator (if available) to wells of G23, G24, H23, and H24 (16).
Two identical sets of “assay ready” library plates are prepared for pflareA and pflareG screens, respectively. The assay plates can take days to prepare and should be kept in −80°C until use.
6. CELL-BASED HIGH-THROUGHPUT SCREENS
The number of cells and the transfection conditions need to be optimized. We have been successful in reverse-transfecting 8000 N2a cells per 384-well with 40 ng plasmid DNA and 0.12 μL Lipofectamine 2000. Typically, about 150 million cells and 2 mL Lipofectamine 2000 are needed to screen about 16 thousand cDNA candidates once.
6.1 Materials and Equipment
Trypsin (ThermoFisher Scientific)
Report cell lines
Arrayed library plates
Lipofectamine 2000 (ThermoFisher Scientific)
Opti-MEM (ThermoFisher Scientific)
Multidrop 384 (Thermo LabSystems)
6.2 Protocol
Scale up the reporter cell cultures to 10 15-cm culture dishes by the day of performing the screens.
Thaw the cDNA plates while the reporter cells and transfection reagents are being prepared.
Dissociate the cells by trypsin.
Resuspend and dilute the cells in phenol red-free feeding media to be 400 thousand cells per mL.
Mix 2 mL Lipofectamine 2000 with 166 mL Opti-MEM.
Dispense 10 μL Opti-MEM containing 0.12 μL Lipofectamine 2000 into each well with Multidrop 384 to mix with the plasmid DNA in the plates.
Dispense 8000 reporter cells in 20 μL feeding media to every well except A23, A24, B23, and B24 wells using Multidrop 384.
Dispense cell-free feeding media into A23, A24, B23, and B24 of all plates. These four wells will be used to derive a scale factor for interplate normalization (see later).
Dispense cell-free media to one (or multiple) new 384-well plate as a background plate to measure background GFP and RFP fluorescence intensity of the cell media.
7. DATA ACQUISITION
Multiple fluorescence imagers can be used to obtain the fluorescence signals. We used a Typhoon imager rather than a conventional fluorescence microplate reader. The confocal optics of the Typhoon imager detects light from a limited depth at the bottom of the culture well. This yields an improved signal-to-noise ratio by reducing background signals from media above the cells. Each data point gained from Typhoon imaging represents overall fluorescence intensity of the whole population of cells within individual wells. By contrast, high-content fluorescence imager produces richer data including fluorescence intensity, cell shape, size, and cell count at a single-cell resolution. Despite the versatility of a high-content imager, its imaging time and data processing speed dramatically decrease the throughout and much of its data are probably irrelevant in evaluating splicing changes.
7.1 Imaging Resolution and Speed
Typhoon imagers use laser scanning technology. The imaging resolution and speed are intrinsically inversely correlated. Imaging at a higher resolution generates better signal-to-noise ratio, but slow down scanning and increase the lagging time of image acquisition between the first and the last plate. We have optimized the scanning speed and resolution. The Typhoon 9410 can scan at pixel sizes of 10, 25, 50, 100, 200, 500, and 1000 μm. The corresponding pixel numbers per well and total scanning times for forty-five 384-well plates containing the MGC cDNA library are listed in Table 1. To examine the effect of pixel size, we measured the correlation of signal intensities acquired at different pixel sizes. We have found that the dynamic range of fluorescence changes for our reporter cells can be fully captured at 200 μm, probably because the signal-to-noise ratio starts to plateau at the resolution of 200 μm pixels (or 256 pixels per well). Scanning at larger pixel sizes, eg, 500 μm (or 49 pixels per well) significantly increased data variance at low fluorescence intensities. At a pixel size of 200 μm, the whole MGC library can be scanned within 100 min, which is a reasonable lagging time between the first and last plate. Therefore, 200 μm is optimal for 384-well plates. To meet the similar data quality, 100 μm pixel size should be needed to screen 1536-well plates.
Table 1.
Scanning Pixel Size of Typhoon Imager Determines Signal Intensity and Scanning Speed
| Pixel Size (μm) | Number of Pixels Per Individual 384-Well | Total Scanning Time for the MGC Plates (min) |
|---|---|---|
| 10 | 102,400 | 2000 |
| 25 | 16,384 | 1600 |
| 50 | 4096 | 400 |
| 100 | 1024 | 210 |
| 200 | 256 | 100 |
| 500 | 49 | 50 |
| 1000 | 16 | 30 |
7.2 The Uniformity of Fluorescence Signals Across Wells and Plates
Because all candidates are compared to control wells, systematic noises across wells and plates should be kept at minimum relative to the signals, ie, changes in fluorescence intensity induced by splicing alternation. The sources of systematic noises include plates, wells, the scanner, media, and cell variability. To measure the systematic variance of the fluorescence signals, we added the same amount of media to multiple 384-well plates and measured fluorescence intensities of each well at different position of the scanner, which allowed us to quantify noises derived from plates, wells and the scanner. Satisfyingly, each individual wells varied in fluorescence signals by only 4–7% at different positions on the scanner. Therefore, scanning positions and the scanner only contribute minor variance to the system.
Surprisingly the largest contributor to systemic noises is the well-to-well variance, as the fluorescence intensities fluctuate significantly between wells. In particular, wells on the edge of a plate had (up to 30%) higher signals than inner wells for both GFP and RFP. Such an edge effect was likely due to faster media evaporation along the edges, because the fluorescence difference amplified over time. However, the edge effect was very reproducible across plates. Indeed, wells of the same coordinates varied in fluorescence intensity by only about 5% between plates. In summary, when we compared uniform plates containing the same cell-free media, the fluorescence signal from a specific well coordinate was much similar to the same coordinate in a different plate than to a different coordinate in the same plate.
We also compared well intensities between cell-free media and media-containing parental nonfluorescent N2a cells. The cellular autofluorescence of the parental N2a cells was about on average 1.9% and 6.6% the level of the media fluorescence in the GFP channel and the RFP channel, respectively. Therefore, the native cellular autofluorescence is negligible.
7.3 Materials and Equipment
Typhoon laser scanner equipped with lasers and filters for GFP and RFP (GE Healthcare Life Sciences, PA, USA)
ImageQuant TL Software (GE Healthcare Life Sciences, PA, USA)
7.4 Protocol
Fourty-eight hours after transfection, arrange and scan the plates on the Typhoon scanner. GFP signals are acquired using the blue laser (488 nm) and 520 nm band pass emission filter. RFP signals are obtained using the green laser (532 nm) and 610 nm band pass emission filter. Keep the scanning parameters (including the PMT voltages) consistent across all plates.
Use ImageQuant TL software to analyze the scanned images (.gel files) and obtain raw GFP and RFP fluorescence intensity of every well. Export the GFP/RFP signal values into Data/Text/Excel files.
8. BACKGROUND FLUORESCENCE CORRECTION AND NORMALIZATION
Because the largest source of background comes from the coordinates of the wells rather than the order of the plates or the scanning position, we perform interplate well-by-well background correction using a background plate that contains uniform cell-free media. Nevertheless, the variance originated from plates and scanning positions averaged around 7% and should be taken into consideration. Therefore, we normalized all plates before background correction.
To normalize the plates, we purposefully dispense cell-free media into wells A23, A24, B23, and B24 of each plate to derive a scaling factor for that plate. The GFP and RFP signal intensity of each of the four media-alone wells (A23, A24, B23, and B24) are acquired similarly as any other wells in all cell plates and the background plate. For example, XFPijk is the raw GFP or RFP intensities of a well at row “i” column “j” in plate k. We calculate the scale factor Sk for plate k based on the A23, A24, B23, and B24 wells by the following equation:
where is the average values across all the plates. The normalized and background-corrected GFP and RFP expression values (XFPx) of the stable cells after transfection with cDNA x at row “i” column “j” in plate c are calculated with the following equation:
where Sc and Sb are the scale factors for the cell plates and the background plates, respectively, and nb is the total number of background plates used.
9. DATA ANALYSIS
To estimate the splicing ratio of the reporter upon expression of cDNA x, we use for the pflareG minigene reporter and for the pflareA minigene reporter. To calculate the basal splicing level of the reporter, we used the four wells (ie, C23, C24, D23, and D24) that were transfected with empty control vector in every plate and derive the mean in the pflareG-exon cells and the mean in the pflareA-exon cells. To examine the effect of cDNA x on the splicing of the pflareG reporter, we used the following formula to approximate the change in the splicing ratio (Mx):
where was derived from the same plate as cDNAx. Similarly, to examine the action of cDNA x on the splicing of the pflareA reporter, we calculated the change in the splicing ratio (Mx) as followed:
where was derived from the same plate as cDNAx. A value of Mx>0 indicates a possible increase in splicing by cDNAx. A value of Mx>0 indicates a possible decrease in splicing. To assess the overall fluorescence expression of cells after transfection, or the “A” value, we use the following equation:
Note that multiple variables, including live cell numbers, transcriptional activity, and translational activity of the reporter, can have an impact on A values. The M values, indicative of splicing changes, can be scatter plotted against the A values to visualize the performance of the whole library.
With the R package locfdr, we calculated z score and local false discovery rate (FDR) for individual cDNA by comparing their M values to the population distribution of M values. The cutoff of the M values in calling a positive hit, as usual, could be arbitrary. We have found that a typical FDR threshold of 0.05 has yielded relatively high sensitivity (80–88%) for our reporter screens.
The two parallel screens using pflareA and pflareG reporters produce two different sets of possible splicing regulators. A simple overlap of the two target sets will eliminate a large proportion of false positives. A gene identified by both screens is much more likely to be the real regulators. In our past experience, overlapping candidates constitute about 5–20% of total hits and the sensitivity of the combined screens is roughly equal to the product of the sensitivity of the individual screens.
10. VALIDATION
The overlapping hits can be subjected to various kinds of secondary validation and gene enrichment analysis. RNA splicing factors and RNA-binding proteins are expected to be enriched in comparison to the library input. More importantly, the candidates need to be examined for their actions on the splicing of the endogenous target exon, because minigene reporters do not necessarily recapitulate the regulation of endogenous exons. For example, the minigene may not contain all of the relevant cis-regulatory elements, or the heterologous genomic context may alter its splicing pattern. Cells used for screening may have different transcriptomes from the cells in the organism. Thus, molecules that affect the splicing of the minigene reporter will need to be confirmed on endogenous transcripts in the correct cell type in subsequent analyses. This can be achieved by either over-expressing the cDNA candidate or RNAi depletion of the candidate gene followed by RT-PCR assay of the endogenous spliced variants. Note that gain-of-function and loss-of-function validations have various limitations as mentioned previously.
11. LIMITATIONS AND PERSPECTIVES
Fluorescence-based screening assays have both advantages and disadvantages over luminescence-based screening assays. Chemiluminescence assays typically have a much lower background and allow for a larger dynamic range of screening but require additional steps of cell lysis and enzymatic reactions before data acquisition. Fluorescence-based screening by contrast is noninvasive and allows temporal profiling of the readouts. Although we have used a fluorescence-based genetic screen as an illustrating example, luminescence-based minigenes can be designed in certain circumstances.
Several features of this screen require careful attention. First, reporter minigene expression can vary substantially among stable cell clones. Cell clones of higher expression are not necessarily better, because they may not be as efficiently transfectable or responsive to known regulators, as we have seen. Furthermore, some clones can change GFP/RFP ratios even when transfected with an empty control vector. Therefore, multiple clonal lines should be tested to find the optimal one. Second, the screening parameters and the kinetics of reporter expression need to be examined for screening different libraries or in different cell types. For example, we have seen that cDNA overexpression plateaus at 2 days posttransfection but RNAi depletion typically requires 3 days or longer. To avoid overconfluency within a well, the number of cells initially plated needs to be optimized; this can affect the transfection condition.
The IRAS system can accommodate nearly any cassette exon and its flanking introns. However, mutagenesis in one or both reporters is often required and must be assessed carefully, as it may generate or alter cis-acting splicing regulatory elements. Because a mutation is introduced into only one of the two vectors, a gain of false positives and/or a loss of true positives will manifest in only one of the two screens. Since the candidate regulators are selected from the overlap from both screens, the mutagenesis, if causing any effect, will most likely lead to a loss of true positives rather than the gain of false positives. Finally, altering the amount of flanking intron sequence included with the test exon can affect the basal splicing level of the reporter. A minigene exhibiting an intermediate level of exon inclusion (eg, 15–85%) will allow more effective simultaneous screening for both activators and repressors.
References
- Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annual Review of Biochemistry. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. http://dx.doi.org/10.1146/annurev.biochem.72.121801.161720. [DOI] [PubMed] [Google Scholar]
- Chen M, Manley JL. Mechanisms of alternative splicing regulation: Insights from molecular and genomics approaches. Nature Reviews Molecular Cell Biology. 2009;10(11):741–754. doi: 10.1038/nrm2777. http://dx.doi.org/10.1038/nrm2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Zheng S. Identify alternative splicing events based on position-specific evolutionary conservation. PLoS One. 2008;3(7):e2806. doi: 10.1371/journal.pone.0002806. http://dx.doi.org/10.1371/journal.pone.0002806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Zheng S. Studying alternative splicing regulatory networks through partial correlation analysis. Genome Biology. 2009;10(1):R3. doi: 10.1186/gb-2009-10-1-r3. http://dx.doi.org/10.1186/gb-2009-10-1-r3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Ares M., Jr Context-dependent control of alternative splicing by RNA-binding proteins. Nature Reviews Genetics. 2014;15(10):689–701. doi: 10.1038/nrg3778. http://dx.doi.org/10.1038/nrg3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann B, Valcárcel J. Decrypting the genome’s alternative messages. Current Opinion in Cell Biology. 2009;21(3):377–386. doi: 10.1016/j.ceb.2009.02.006. http://dx.doi.org/10.1016/j.ceb.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Kar A, Havlioglu N, Tarn W, Wu JY. RBM4 interacts with an intronic element and stimulates tau exon 10 inclusion. The Journal of Biological Chemistry. 2006;281(34):24479–24488. doi: 10.1074/jbc.M603971200. http://dx.doi.org/10.1074/jbc.M603971200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroyanagi H, Kobayashi T, Mitani S, Hagiwara M. Transgenic alternative-splicing reporters reveal tissue-specific expression profiles and regulation mechanisms in vivo. Nature Methods. 2006;3(11):909–915. doi: 10.1038/nmeth944. http://dx.doi.org/10.1038/nmeth944. [DOI] [PubMed] [Google Scholar]
- Kuroyanagi H, Ohno G, Sakane H, Maruoka H, Hagiwara M. Visualization and genetic analysis of alternative splicing regulation in vivo using fluorescence reporters in transgenic. Caenorhabditis elegans Nature Protocols. 2010;5(9):1495–1517. doi: 10.1038/nprot.2010.107. http://dx.doi.org/10.1038/nprot.2010.107. [DOI] [PubMed] [Google Scholar]
- Oberdoerffer S, Moita LF, Neems D, Freitas RP, Hacohen N, Rao A. Regulation of CD45 alternative splicing by heterogeneous ribonucleoprotein, hnRNPLL. Science (New York, NY) 2008;321(5889):686–691. doi: 10.1126/science.1157610. http://dx.doi.org/10.1126/science.1157610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoilov P, Lin C, Damoiseaux R, Nikolic J, Black DL. A high-throughput screening strategy identifies cardiotonic steroids as alternative splicing modulators. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(32):11218–11223. doi: 10.1073/pnas.0801661105. http://dx.doi.org/10.1073/pnas.0801661105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topp JD, Jackson J, Melton AA, Lynch KW. A cell-based screen for splicing regulators identifies hnRNP LL as a distinct signal-induced repressor of CD45 variable exon 4. RNA (New York, NY) 2008;14(10):2038–2049. doi: 10.1261/rna.1212008. http://dx.doi.org/10.1261/rna.1212008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witten JT, Ule J. Understanding splicing regulation through RNA splicing maps. Trends in Genetics: TIG. 2011;27(3):89–97. doi: 10.1016/j.tig.2010.12.001. http://dx.doi.org/10.1016/j.tig.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JY, Kar A, Kuo D, Yu B, Havlioglu N. SRp54 (SFRS11), a regulator for tau exon 10 alternative splicing identified by an expression cloning strategy. Molecular and Cellular Biology. 2006;26(18):6739–6747. doi: 10.1128/MCB.00739-06. http://dx.doi.org/10.1128/MCB.00739-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Black DL. Alternative pre-mRNA splicing in neurons: Growing up and extending its reach. Trends in Genetics: TIG. 2013;29(8):442–448. doi: 10.1016/j.tig.2013.04.003. http://dx.doi.org/10.1016/j.tig.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Damoiseaux R, Chen L, Black DL. A broadly applicable high-throughput screening strategy identifies new regulators of Dlg4 (Psd-95) alternative splicing. Genome Research. 2013;23(6):998–1007. doi: 10.1101/gr.147546.112. http://dx.doi.org/10.1101/gr.147546.112. [DOI] [PMC free article] [PubMed] [Google Scholar]