

Abstract
Objective(s):
Epidemiological and biochemical studies conducted over the past two decades have established a strong link between type 2 diabetes mellitus (T2DM) and Alzheimer’s disease (AD). However, the exact mechanisms through which aberrations in insulin signaling associated with T2DM contribute to cognitive decline are not yet known.
Materials and Methods:
In an effort to explore possible molecular links between T2DM and AD, the present study investigated the status of neurodegeneration, adult hippocampal neurogenesis, and nitrosative stress induced protein S-nitrosylation in streptozotocin (STZ) induced mice models of T2DM. Morris water maze task and subsequent histological and immunohistochemical assessment were conducted. Expression of neurogenesis markers (Ki67, DCX, and NeuN) and APP 770 was determined by qRT-PCR.
Results:
A significant decline in spatial learning and reference memory was observed with consequent neurodegeneration in brain cortex and hippocampus in the diabetic group as compared to the control group. A subsequent increase in expression of APP 770 was also observed in T2DM brain regions. Moreover, a significant decrease in transcriptional expression of Ki67, DCX, and NeuN was also evident in T2DM brain regions, which indicated possible aberrations in adult hippocampal neurogenesis in T2DM. Furthermore, an increased immunohistochemical signal for S-nitrosylation was also observed in T2DM, which also suggested its potential contribution in T2DM associated neuronal deterioration.
Conclusion:
It is suggested that these identified aberrations in the diabetic brain may communally increase the susceptibility of developing AD in patients with T2DM. Further studies of the underlying molecular mechanisms may help to strategize a combination therapy for these debilitating disorders.
Keywords: Alzheimer’s disease, Neurodegeneration Neurogenesis, S-nitrosylation, Type 2 diabetes mellitus
Introduction
Diabetes mellitus type 2 (T2DM) is a chronic metabolic disorder that affects about 422 million people around the globe annually (1). Initiated by cellular resistance to insulin, this disease leads to a state of hyperglycemia in the body that ultimately causes cytotoxicity in the brain, kidneys, blood vessels, nerves, eyes, and the gastrointestinal tract (2). Various molecular players, including peptide hormones and proinflammatory cytokines, come into action upon its onset, leading to reduced cellular insulin sensitivity and beta-cell dysfunction–a combination that disrupts cross-talk between endocrine pancreas, liver, skeletal muscle, adipose tissue, gastrointestinal tract, and the central nervous system aggravating hyperglycemia and activating multiple pathways that lead to cell death (3). Of all its complications, its effects on the central nervous system are the most debilitating and have grim effects on quality of life.
Metabolic disturbances associated with diabetes, especially insulin resistance, have been observed to contribute towards the development of pathological hallmarks of Alzheimer’s disease (AD), making T2DM a major risk factor for AD (4). These common pathogenic mechanisms have led to AD being referred to as ‘type 3 diabetes’ and indicate that it is basically the type of diabetes that selectively involves brain (5). Although AD is the most common form of dementia (50–60% cases) and affects 20% people above the age of 80, there is still no clue about the cause of this devastating illness and hence, no cure (1). The economic burden of T2DM and AD care is increasing every year, triggering the need to understand their biochemical basis and to find potential drug targets.
Extensive studies have been conducted to understand the effect of T2DM on the functioning of the brain and have surprisingly found that the effect of T2DM is similar to that of AD. Geroldi et al. (6) reported that cognitive impairment correlated With biochemical and clinical features of insulin resistance syndrome. Studies have revealed inflammation, insulin resistance and mitochondrial dysfunction as common pathogenic mechanisms shared by AD and diabetes Mellitus (DM). Impaired glucose metabolism, even in the absence of T2DM, causes a decrease in hippocampal volume and microstructure along with changes in microvasculature which affect learning ability, neurotransmission, and memory consolidation (7-9). Neuronal cell death has been reported to increase in patients with both AD and DM (in comparison to those with AD or T2DM only) due to generation of oxidative stress by advanced glycation end products (AGEs) (10, 11). In addition to altering the physiology of the brain and causing neuronal apoptosis, T2DM has also been reported to alter neuronal migration, one of the key processes involved in adult neurogenesis (12). Neurogenesis is necessary for learning, memory, and healing from injuries and continues throughout the life. It has already been established that irregular neurogenesis accompanies neurodegeneration in AD brains (13).
Nitrosative stress, a redox mediated post trans-lational modification (14), is another key player in the pathophysiology of both AD and DM and imparts its effects through aberrant S-nitrosylation of several metabolic, apoptotic, and structural proteins (15). The alterations in protein S-nitrosylation may underlie the adverse effect of hyperglycemia and may be the causative factor for cognitive impairment associated with AD. Although aberrant S-nitrosylation has been reported in various complications of T2DM, its role in cognitive decline and dementia associated with diabetes has not been explored.
In the present study, memory impairment was studied in streptozotocin (STZ)-induced mice models of T2DM through Morris water maze test. We also quantified the extent of neurodegeneration and neurogenesis through the expression of amyloid precursor protein 770 (APP770), Ki67 (a proliferating cell marker), doublecortin (DCX; an immature progenitor cell marker), and neuronal marker (NeuN), respectively. Additionally, the S-nitrosylation status of proteins in the diabetic brain was also targeted using immunohistochemistry.
Materials and Methods
Induction of type 2 diabetes in mice
Male BALB/c mice (n=20) bred and housed in the animal house of Atta-ur-Rahman School of Applied Biosciences (ASAB), National University of Sciences and Technology (NUST), were used in this study. Four mice were housed per cage and kept at constant temperature (25±2 °C) under natural light-dark cycle (12-12 hr). Diabetes was induced in the diabetic group (n=10) by switching the mice to high-fat diet (HFD) (basic mice feed 59%, sugar 20%, animal fat 18%, and egg yolk 3%) after weaning and rendering two low doses of STZ (100 mg/kg) at 6 and 9 weeks of age in 0.1 M citrate buffer (pH 4.5) after overnight fasting through intraperitoneal injections. The control group (n=10) were fed basic mice feed (crude protein 30%, crude fat 9%, crude fiber 4%, and moisture 10.4%) and injected with citrate buffer only. The fasting blood glucose (FBG) levels of animals were assessed after eight days of STZ injection using On-Call® EZ II blood glucose monitoring system (Blood ACON International, USA). Mice with FBG levels higher than 12 mmol/l were considered diabetic (16) and used for further analysis. The details of FBG levels are provided as supplementary data. The detailed experimental protocol is illustrated in Figure 1.
Figure 1.
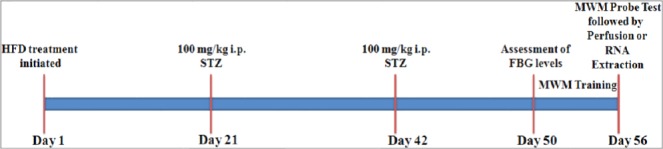
Schematic diagram of the experimental protocol
Morris water maze test
The Morris water maze (MWM) test was conduct-ed with slight modifications in the protocol described (7). Briefly, a 120×60 cm circular pool was divided into four quadrants and filled with water (20 °C±2 °C) to a depth of 33 cm. The water was made opaque by addition of blue dye and a transparent platform (13×32 cm) was placed in the North-Western qua-drant. Each mouse was given five acquisition trials per day for five consecutive days with a minimum inter-trial interval of 10 min. Mice were released from one of the four defined starting positions and allowed to navigate around the pool for 90 sec to locate the hidden platform. The mice that located the platform within 90 sec were left there for 5 sec while those that failed to locate the platform within this time were manually guided towards the platform and left there for 20 sec. The average escape latency for five trials of each day was calculated. Spatial probe trial test was conducted on the sixth day; the platform was removed and mice were allowed to swim for 90 sec. The time spent in each quadrant was recorded along with the number of times each mouse crossed the position where the platform was located.
Histological and immunohistochemical examination
The protocol established by Gage et al. (17) was used for heart perfusion. The brain tissue (n=4, each group) was excised and incubated in 4% paraformaldehyde (PFA) for 24 hr at 4 °C and later dehydrated using 70% isopropanol (1 hr), 95% isopropanol (1 hr), and 100% isopropanol (1 hr) followed by incubation in xylene for 4 hr. Paraffin embedding was performed by placing the tissue in molten paraffin for 4 hr at 60 °C and then allowing it to solidify at 4 °C for block formation. Tissue sections (5 µ) were mounted on slides and subjected to hematoxylin and eosin (H&E) staining. The slides were visualized using an inverted microscope (Labomed, USA) at 4X, 10X, and 40X resolutions. The images were captured by Pixel Pro™ image analysis software (Labomed, USA).
For immunohistochemical analysis, sagittal sec-tions (5 µ) were mounted on poly lysine coated adhesive slides. Graded concentrations of ethanol were used to rehydrate the sections followed by heat mediated antigen retrieval that was performed by incubating sections for 35 min in sodium citrate (pH: 6) in a pressure cooker. The sections were washed subsequently and incubated in 35% H2O2 to quench endogenous H2O2. The slides were incubated in 5% bovine serum albumin in phosphate buffered saline (PBS) for 10 min to avoid non-specific binding of antibody followed by overnight incubation in 4 °C in 0.1% bovine serum albumin in PBS containing rabbit polyclonal antibody for S-nitrosocysteine (1:100; ab50185). The sections were washed and incubated in horseradish peroxidase (HRP) conjugated anti-rabbit IgG (1:100; ab97051) for 1 hr at room temperature. The sections were placed in a solution containing 0.025% of 3,3′ diaminobenzidine (DAB, ab50185) for 10 min to visualize the peroxidase reaction product. The sections were counter stained with hematoxylin and visualized.
Quantitative real –time PCR
RNA extraction from total hippocampal and cortical regions (n=6, each group) was carried out according to the manufacturer’s protocol using Tri-reagent. The extracted RNA was quantified using Nanodrop 2000 (Thermoscientific, USA) and an equal amount of RNA (2 µg) was transcribed into cDNA.
A reaction mixture containing 4 μl of HOT FIREPol® EvaGreen® qPCR Mix Plus, 1 μl of forward and reverse primers and 1 µl of cDNA template was prepared. The expression of β-actin, APP770, DCX, NeuN, and Ki67 was quantified using ABI Prism 7300 sequence detection system (Applied Biosystems, 7300) using specific forward and reverse primers (supplementary Table 1). Dissociation curves and agarose gel electrophoresis were used to access the quality of PCR product. All values were normalized to those obtained for β-actin and the values obtained from these experiments were analyzed relative to gene expression data using the 2-∆∆CT method (18). The data was statistically analyzed by Student’s t-test and a P<0.05 was considered to be statis-tically significant. The results were presented as expression graphs using GarphPad Prism (v5).
Ethics statement
All the experiments performed were in compli-ance with the rulings of the Institute for Laboratory Animal Research, Division on Earth and Life Sciences, National Institute of Health, USA (Guide for the Care and Use of Laboratory Animals: Eighth Edition, 2011). The study has the ethical approval of the Internal Review Board (IRB) of ASAB, NUST.
Results
Hyperglycemia impairs cognitive functions
MWM maze test was employed to assess the cognitive decline in response to hyperglycemia in STZ induced mice models of T2DM. Average escape latency, recorded over a period of five days, reflected a decrease in spatial learning in diabetic mice in comparison to the control group (P<0.05 as analyzed by two-way ANOVA). The control depicted a stable learning curve whereas escape latency of diabetic mice kept fluctuating but remained higher than the control group throughout the experimental period (Figure 2a). Statistical analysis of the data obtained for day 5 of MWM provided further evidence for a statistically significant decline (P<0.001) in spatial learning and memory (Figure 2b). Assessment of reference memory using probe trial test showed a significant decrease in reference memory in the diabetic group (Figures 3a & b). Owing to the lack of memory, the diabetic mice spent less time in the target quadrant (P<0.05) and crossed the location of the previously hidden platform for fewer times (P<0.01) as compared to control group.
Figure 2.
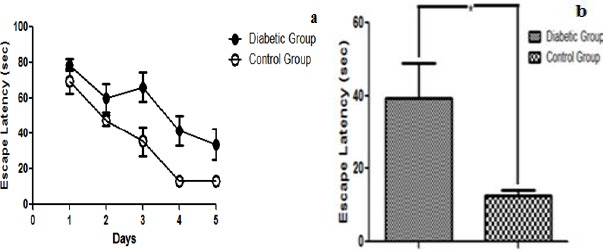
(a) Effect of Hyperglycemia on learning and memory using MWM test: Graph depicts escape latency (sec) to assess formation of spatial memory between control and diabetic group statistically analyzed by two-way ANOVA. Diabetic group depicted lesser retention of spatial memory as compared to control thereby finding platform much later than normal mice. Error bars represent mean+ SEM. (b) Comparison between cognitive performances of different groups on day 5: In comparison to control, hyperglycemic animals showed a significant decrease in escape latency indicating a decline in learning and memory. Data was analyzed using Student’s t-test (shown as mean±SEM, *P<0.05)
Figure 3.
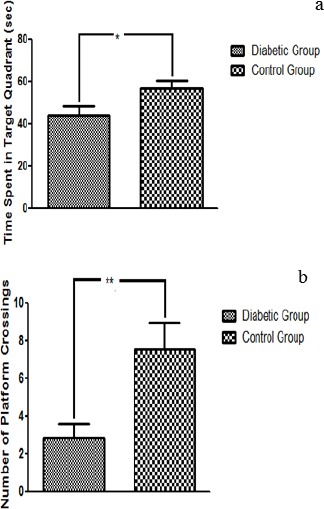
Effect of Hyperglycemia on Reference Memory: (a) The Graph indicates that the control mice centered their search around the position where platform was located previously and crossed its location several times while the diabetic mice spent more time along the perimeter. The data is shown as mean±SEM, *P<0.05. (b) The Graph indicates that the control mice recalled the position of the platform and crossed its location several times while the diabetic mice spent more time along the perimeter indicating a deficit in memory of diabetic mice. The data is shown as mean±SEM, **P<0.001
Histological examination of brain
Neurodegeneration was observed in both cortical and hippocampal regions of the diabetic mice. The H&E stained brain sections showed the classical appearance of shrunk neurons and a decrease in neuronal density in diabetic brains as compared to control sections. A significant reduction in cell density in the cortical region of the diabetic group (25.00±2.51) as compared to the control group (39.67±3.38, P<0.02) was observed. Reduction in the cell density was also observed in the hippocampus of the diabetic group (4.00±1.00) as compared to the control group (8.66 ± 0.33, P<0.01) (Figures 4, 5).
Figure 4.
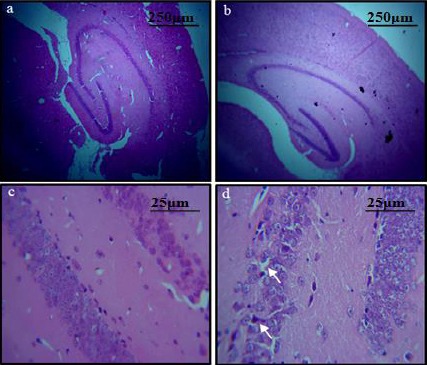
H&E stained sections of C- shaped hippocampus: (a) Pattern of dark neurons within the pyramidal layer of the hippocampus of control group. (b) Diabetic group (original magnification 4X) (c) Pattern of dark neurons within the CA3 of the hippocampus of control group (d) diabetic group. Shrunken cell bodies and neurodegeneration can be observed in the latter slide (original magnification 40X)
Figure 5.
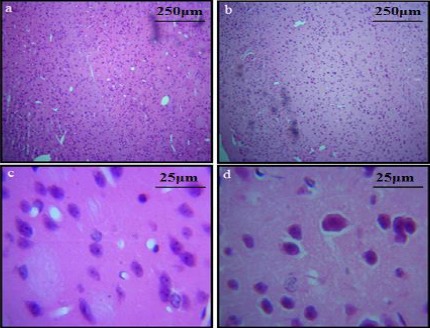
H&E stained sections of cortex: (a) control group (b) diabetic group (Layers I to VI; original magnification 10X) (c) unaffected neurons of layer 2 of cerebral cortex in control group and (d) diabetic group (original magnification 40X). A decrease in neuronal density can also be observed in cortex of diabetic group
Immunohistochemical analysis of S-nitrosylated proteins
DAB staining using Anti S-nitrosocysteine antibody indicated a clear increase in S-nitrosylated proteins in CA1 region of the hippocampus in diabetic mice (Figure 5). Similar results were obtained for the cortical region as well, suggesting an increased nitrosylation of cellular and synaptic proteins.
Transcriptional analysis of APP770 and neurogenesis markers
Quantitative PCR indicated an overall increase in the expression of APP 770 in response to hyperglycemia in both cortical and hippocampal tissue from diabetic mice indicating neurodegeneration in both of these regions. However, statistical analysis revealed this difference to be statistically significant in the hippocampal region only (P<0.05). Moreover, the transcriptional expression of Ki67, a marker for neuronal proliferation, was significantly decreased in diabetic mice in comparison to control suggesting an overall decrease in neuronal proliferation in response to hyperglycemia. The downregulation of Ki67 was statistically significant in both hippocampal and cortical regions (P<0.001) however it was slightly higher in the hippocampus region suggesting a severe damage to this part of the brain.
Quantitative PCR results for the expression of DCX also showed a significant decreased in both hippocampal and cortical (P<0.001) regions of the diabetic mice. On comparison among the two regions, its expression in the hippocampus was significantly lower (P<0.05) suggesting an increased damage to neuronal migration in the hippocampus.
Gene expression analysis of Neu N, a marker for mature neurons, also demonstrated a significant downregulation in both cortex and hippocampus (P<0.001) adding further evidence to the findings that the neurogenesis is affected in response to hyperglycemia (Figure 7).
Figure 6.

DAB stained sections representing the status of S-nitrosylated proteins: A comparison of (a) control group and (b) diabetic Group indicates an increase in the amount of S-nitrosylated proteins in response to hyperglycemia in hippocampal region of brain. Original magnification 10X. Similar results were obtained for cortical regions of (c) control group and (d) diabetic group. Original Magnification 40X
Figure 7.
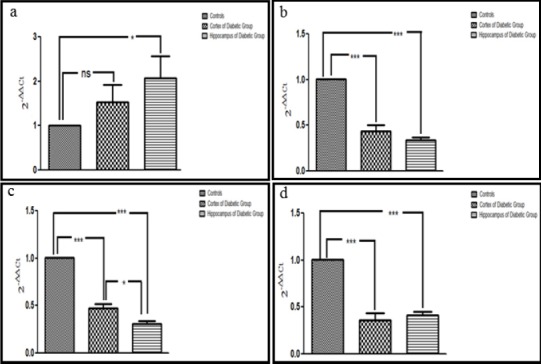
Transcriptional expression of APP770, Ki67, DCX and NeuN in diabetic mice compared with control: a: Histogram representing an increased transcriptional expression of APP 770 in cortex and hippocampus of diabetic mice in comparison to control indicates its contribution towards neurodegeneration. b-d: Decrease in expression of Ki67, DCX and NeuN is evident in cortex and hippocampus of diabetic mice, suggesting aberrations in adult neurogenesis. The data is shown as mean ± SEM, *P<0.05, ns= non-significant
Discussion
The current study was conducted with the objective of investigating various contributing factors of cognitive impairment in a mouse model of T2DM that may contribute towards the development of AD.
The findings of the assessment of spatial memory, conducted through MWM paradigm, suggested a significant cognitive deficit in hyperglycemic mice. The present findings are in agreement with the previously reported studies that demonstrated a significant cognitive decline in diabetic mice (7, 19) while assessing the role of insulin treatment; it failed to reverse cognitive decline and memory impairment in diabetic rats (20). Based on these findings it can be postulated that cognitive decline is one of the major complications of T2DM where hyperglycemia causes a permanent deficit in learning and information processing ability, which makes patients of T2DM vulnerable to developing AD and associated dementia (21-23).
The observed neuronal shrinkage and degradation in the CA3 region that marked a severe structural disruption of the hippocampus and an overall decrease in cortical neuronal density of diabetic mice, provides substantial evidence of neurodegeneration associated with T2DM. As the alterations in glucose and insulin signaling cause changes in tau phosphor-rylation and amyloid beta (Aβ) metabolism where insulin accelerates Alzheimer-related pathology specifically neurodegeneration (24, 25), these observations further substantiate our understanding about the linking pathological features among T2DM and AD. In this context, expression of APP770 in diabetic and control groups was investigated that showed a substantial rise in its expression levels in the diabetic group. Accumulation of Aβ peptide, a derivative of APP, is one of the pathological hallmarks of AD (26). An increased expression in the diabetic group may suggest that manipulation of the expression of APP770 could be one of the several ways in which insulin resistance is contributing towards neurodegeneration. However, the exact mechanism of T2DM associated neuro-degeneration further needs to be explored.
Furthermore, an accumulation of S-nitrosylated proteins in the hippocampus and cortical regions of the diabetic brain also indicated the effect of nitro-sative stress on the brain, which may also evidence that diabetes brings an overall increase in S-nitrosylation. A similar increase was also documented in the brains of diabetic animals within few weeks of diabetes induction (27), which also supports our findings. Furthermore, the alterations in protein S-nitrosylation due to hyperglycemia also contribute to endothelial dysfunction and mediate the development of diabetic associated consequences (28). In T2DM and AD, the synaptic disruption is documented as a combined effect of hyperglycemia and oligomeric Aβ, linked through aberrant protein S-nitrosylation (29). It is suggested that the nitrosative stress during early stages of this disease leads to an overall increase in S-nitrosylation of various protein targets. A proteome wide study to find affected proteins would provide a further insight into the role of these proteins in the pathophysiology of both of these disorders.
In order to justify the extent of neuronal loss, as observed in histochemical studies, the state of neurogenesis in T2DM was also studied. Previous studies suggested that although hyperglycemic environment promotes the growth of progenitor cells, it reduces their survival thereby impairing neurogenesis and causing cognitive impairment (30). However, the effect of hyperglycemia on individual stages of neurogenesis has not been reported previously. We found a significant decrease in all three stages of neurogenesis in the diabetic brain. Neuronal proliferation, migration, and maturation (studied through transcriptional expression of Ki67, DCX, and NeuN) were significantly reduced in the diabetic brain as compared to the control group. Other researchers also reported a similar reduction in neurogenesis markers in STZ model of type 1 diabetes (31). Moreover, reduced cell proliferation and neuroblast differentiation were also observed in diabetic models exposed to aluminum (Al) which further aggravated the diabetes associated altered neurogenesis (32). Also, there is a significant decrease in transcriptional expression of neurogenesis mar-kers after Al exposure, however, the damage was reduced by treatment with metformin (a biguanide drug commonly used to treat T2DM). This showed the possible association of metformin in mediating the adult hippocampal neurogenesis (33). In addition, liraglutide and lixisenatide, available for treating diabetes, potentially enhance neurogenesis. However, the neurogenic and neuroprotective effects of these anti-diabetics need further in-depth understanding.
In addition, the possible influence of APP on neurogenesis cannot be ruled out. APP conferred its effects differently by two of its domains i.e. soluble secreted APPs (sAPPs) which are neuroprotective and significantly associated with neurogenesis while APP intracellular domain (AICD) negatively modulates neurogenesis (34, 35). It is postulated that the altered expression of APP770 (a sAPP) during a pathological condition, as observed in the present study, disrupts its neuro protective potential and may influence neurogenesis in T2DM.
Conclusion
Our findings suggest memory impairment is one of the major consequences of T2DM that can be attributed to neurodegeneration via upregulation of APP770, aberrant protein S-nitrosylation, and downregulation of neurogenesis. As a combinatorial effect, all these observed alterations increase the susceptibility of developing AD in patients with T2DM. Further studies are required to identify and understand aberrations of proteins involved in affected pathways along with the role of other post-translation modifications that may be helpful in strategizing a combination therapy for both of these debilitating disorders.
Acknowledgment
This research was supported through an MS student’s research grant by National University of Sciences and Technology (NUST), Islamabad, Pakistan. We also thank Ms. Sanila Amber, PhD scholar, ASAB, NUST for helping in the quantitative assessment of histological data.
Conflict of interest
The authors declare that they have no conflicts of interest.
References
- 1.Smith L. Global report on Diabetes. United States: World health organization; 2016. [Google Scholar]
- 2.Rossetti L, Giaccari A, DeFronzo RA. Glucose toxicity. Diabetes Care. 1990;13:610–630. doi: 10.2337/diacare.13.6.610. [DOI] [PubMed] [Google Scholar]
- 3.Scheen AJ. Pathophysiology of type 2 diabetes. Acta Clin Belg. 2014;58:335–341. doi: 10.1179/acb.2003.58.6.001. [DOI] [PubMed] [Google Scholar]
- 4.Verdile G, Fuller SJ, Martins RN. The role of type 2 diabetes in neurodegeneration. Neurobiol Dis. 2015;84:22–38. doi: 10.1016/j.nbd.2015.04.008. [DOI] [PubMed] [Google Scholar]
- 5.De la Monte SM, Wands JR. Alzheimer's disease is type 3 diabetes-evidence reviewed. J Diabetes Sci Technol. 2008;2:1101–1113. doi: 10.1177/193229680800200619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geroldi C, Frisoni GB, Paolisso G, Bandinelli S, Lamponi M, Abbatecola AM, et al. Insulin resistance in cognitive impairment: the InCHIANTI study. Arch Neurol. 2005;62:1067–1072. doi: 10.1001/archneur.62.7.1067. [DOI] [PubMed] [Google Scholar]
- 7.Biessels GJ, Kamal A, Urban IJ, Spruijt BM, Erkelens DW, Gispen WH. Water maze learning and hippocampal synaptic plasticity in streptozotocin-diabetic rats: effects of insulin treatment. Brain Res. 1998;800:125–135. doi: 10.1016/s0006-8993(98)00510-1. [DOI] [PubMed] [Google Scholar]
- 8.Kerti L, Witte AV, Winkler A, Grittner U, Rujescu D, Flöel A. Higher glucose levels associated with lower memory and reduced hippocampal microstructure. Neurology. 2013;81:1746–1752. doi: 10.1212/01.wnl.0000435561.00234.ee. [DOI] [PubMed] [Google Scholar]
- 9.van Bussel FC, Backes WH, Hofman PA, van Oostenbrugge RJ, Kessels AG, van Boxtel MP, et al. On the interplay of microvasculature, parenchyma, and memory in type 2 diabetes. Diabetes Care. 2015;38:876–882. doi: 10.2337/dc14-2043. [DOI] [PubMed] [Google Scholar]
- 10.Valente T, Gella A, Fernàndez-Busquets X, Unzeta M, Durany N. Immunohistochemical analysis of human brain suggests pathological synergism of Alzheimer's disease and diabetes mellitus. Neurobiol Dis. 2010;37:67–76. doi: 10.1016/j.nbd.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 11.Takeda S, Sato N, Uchio-Yamada K, Sawada K, Kunieda T, Takeuchi D, et al. Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Aβdeposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci U S A. 2010;107:7036–7041. doi: 10.1073/pnas.1000645107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramos-Rodriguez JJ, Molina-Gil S, Ortiz-Barajas O, Jimenez-Palomares M, Perdomo G, Cozar-Castellano I, et al. Central proliferation and neurogenesis is impaired in type 2 diabetes and prediabetes animal models. PloS One. 2014;9:89229. doi: 10.1371/journal.pone.0089229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haughey NJ, Nath A, Chan SL, Borchard AC, Rao MS, Mattson MP. Disruption of neurogenesis by amyloid beta-peptide, and perturbed neural progenitor cell homeostasis, in models of Alzheimer's disease. J Neurochem. 2002;83:1509–1524. doi: 10.1046/j.1471-4159.2002.01267.x. [DOI] [PubMed] [Google Scholar]
- 14.Foster MW, Liu L, Zeng M, Hess DT, Stamler JS. A genetic analysis of nitrosative stress. Biochemistry. 2009;48:792–799. doi: 10.1021/bi801813n. [DOI] [PubMed] [Google Scholar]
- 15.Nakamura T, Lipton SA. Redox regulation of mitochondrial fission, protein misfolding, synaptic damage, and neuronal cell death: potential implications for Alzheimer's and Parkinson's diseases. Apoptosis. 2010;15:1354–1363. doi: 10.1007/s10495-010-0476-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dong J, Xu H, Wang PF, Cai GJ, Song HF, Wang CC, et al. Nesfatin-1 stimulates fatty-acid oxidation by activating AMP-activated protein kinase in STZ-induced type 2 diabetic mice. PloS One. 2013;8:83397. doi: 10.1371/journal.pone.0083397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gage GJ, Kipke DR, Shain W. Whole animal perfusion fixation for rodents. J Vis Exp. 2012;30:3564. doi: 10.3791/3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2? ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 19.Manschot SM, Biessels GJ, Cameron NE, Cotter MA, Kamal A, Kappelle LJ, et al. Angiotensin converting enzyme inhibition partially prevents deficits in water maze performance, hippocampal synaptic plasticity and cerebral blood flow in streptozotocin-diabetic rats. Brain Res. 2003;966:274–282. doi: 10.1016/s0006-8993(02)04211-7. [DOI] [PubMed] [Google Scholar]
- 20.Zheng H, Zheng Y, Zhao L, Chen M, Bai G, Hu Y, et al. Cognitive decline in type 2 diabetic db/db mice may be associated with brain region-specific etabolic disorders. Biochim Biophys Acta. 2017;1863:266–273. doi: 10.1016/j.bbadis.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 21.Ohara T, Doi Y, Ninomiya T, Hirakawa Y, Hata J, Iwaki T, et al. Glucose tolerance status and risk of dementia in the community. The Hisayama Study. Neurology. 2011;77:1126–1134. doi: 10.1212/WNL.0b013e31822f0435. [DOI] [PubMed] [Google Scholar]
- 22.Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol. 2006;5:64–74. doi: 10.1016/S1474-4422(05)70284-2. [DOI] [PubMed] [Google Scholar]
- 23.Kawamura T, Umemura T, Hotta N. Cognitive impairment in diabetic patients: Can diabetic control prevent cognitive decline?J Diabetes Investig. 2012;3:413–423. doi: 10.1111/j.2040-1124.2012.00234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Umegaki H. Neurodegeneration in diabetes mellitus. Adv Exp Med Biol. 2012;724:258–265. doi: 10.1007/978-1-4614-0653-2_19. [DOI] [PubMed] [Google Scholar]
- 25.Moran C, Beare R, Phan TG, Bruce DG, Callisaya ML, Srikanth V. Alzheimer's disease neuroimaging initiative (ADNI). Type 2 diabetes mellitus and biomarkers of neurodegeneration. Neurology. 2015;85:1123–1130. doi: 10.1212/WNL.0000000000001982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pham E, Crews L, Ubhi K, Hansen L, Adame A, Cartier A, et al. Progressive accumulation of amyloid-beta oligomers in Alzheimer's disease and in amyloid precursor protein transgenic mice is accompanied by selective alterations in synaptic scaffold proteins. FEBS J. 2010;277:3051–3067. doi: 10.1111/j.1742-4658.2010.07719.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Noriega-Cisneros R, Cortés-Rojo C, Manzo-Avalos S, Clemente-Guerrero M, Calderón-Cortés E, Salgado-Garciglia R, et al. Mitochondrial response to oxidative and nitrosative stress in early stages of diabetes. Mitochondrion. 2013;13:835–840. doi: 10.1016/j.mito.2013.05.012. [DOI] [PubMed] [Google Scholar]
- 28.Wadham C, Parker A, Wang L, Xia P. High glucose attenuates protein S-nitrosylation in endothelial cells: role of oxidative stress. Diabetes. 2007;56:2715–2721. doi: 10.2337/db06-1294. [DOI] [PubMed] [Google Scholar]
- 29.Akhtar MW, Sanz-Blasco S, Dolatabadi N, Parker J, Chon K, Lee MS, et al. Elevated glucose and oligomeric β-amyloid disrupt synapses via a common pathway of aberrant protein S-nitrosylation. Nat Commun. 2016;7:102–142. doi: 10.1038/ncomms10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lang BT, Yan Y, Dempsey RJ, Vemuganti R. Impaired neurogenesis in adult type-2 diabetic rats. Brain Res. 2009;1258:25–33. doi: 10.1016/j.brainres.2008.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang WJ, Tan YF, Yue JT, Vranic M, Wojtowicz JM. Impairment of hippocampal neurogenesis in streptozotocin-treated diabetic rats. Acta Neurol Scand. 2008;117:205–210. doi: 10.1111/j.1600-0404.2007.00928.x. [DOI] [PubMed] [Google Scholar]
- 32.Nam SM, Kim JW, Yoo DY, Jung HY, Choi JH, Hwang IK, et al. Reduction of adult hippocampal neurogenesis is amplified by aluminum exposure in a model of type 2 diabetes. J Vet Sci. 2016;17:13–20. doi: 10.4142/jvs.2016.17.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahmed S, Mahmood Z, Javed A, Hashmi SN, Zerr I, Zafar S, et al. Effect of metformin on adult hippocampal neurogenesis: Comparison with donepezil and links to cognition. J Mol Neurosci. 2017;62:88–98. doi: 10.1007/s12031-017-0915-z. [DOI] [PubMed] [Google Scholar]
- 34.Plummer S, Van den Heuvel C, Thornton E, Corrigan F, Cappai R. The neuroprotective properties of the amyloid precursor protein following traumatic brain injury. Aging Dis. 2016;7:163–179. doi: 10.14336/AD.2015.0907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou ZD, Chan CH, Ma QH, Xu XH, Xiao ZC, Tan EK. The roles of amyloid precursor protein (APP) in neurogenesis: Implications for pathogenesis and therapy of Alzheimer disease. Cell Adh Migr. 2011;5:280–292. doi: 10.4161/cam.5.4.16986. [DOI] [PMC free article] [PubMed] [Google Scholar]