

Abstract
A single dose of the apolipoprotein (apo)A‐I mimetic peptide D‐4F rendered high‐density lipoprotein (HDL) less inflammatory, motivating the first multiple‐dose study. We aimed to assess safety/tolerability, pharmacokinetics, and pharmacodynamics of daily, orally administered D‐4F. High‐risk coronary heart disease (CHD) subjects added double‐blinded placebo or D‐4F to statin for 13 days, randomly assigned 1:3 to ascending cohorts of 100, 300, then 500 mg (n = 62; 46 men/16 women). D‐4F was safe and well‐tolerated. Mean ± SD plasma D‐4F area under the curve (AUC, 0–8h) was 6.9 ± 5.7 ng/mL*h (100 mg), 22.7 ± 19.6 ng/mL*h (300 mg), and 104.0 ± 60.9 ng/mL*h (500 mg) among men, higher among women. Whereas placebo dropped HDL inflammatory index (HII) 28% 8 h postdose (range, 1.25–0.86), 300–500 mg D‐4F effectively halved HII: 1.35–0.57 and 1.22–0.63, respectively (P < 0.03 vs. placebo). Oral D‐4F peptide dose predicted HII suppression, whereas plasma D‐4F exposure was dissociated, suggesting plasma penetration is unnecessary. In conclusion, oral D‐4F dosing rendered HDL less inflammatory, affirming oral D‐4F as a potential therapy to improve HDL function.
Study Highlights
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
✓ Single‐dose apoA‐I mimetic peptide D‐4F rendered HDL less inflammatory. Orally available peptides are preferable, avoiding injections.
WHAT QUESTION DID THIS STUDY ADDRESS?
✓ Whether chronic oral D‐4F would safely and durably improve HDL HII following typical dose‐responsive plasma pharmacokinetics.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
✓ Oral D‐4F stably improved HII additively to statin therapy; counter‐intuitively, improvement was strongly related to oral dosing as opposed to plasma exposure, now providing human data congruent with emerging preclinical studies showing intestinal apoA‐I mimetic exposure alters HII absent systemic absorption.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE
✓ Decoupling apoA‐I peptides from the vagaries of systemic absorption unexpectedly opens up a wide opportunity for oral peptide dosing, reviving hope that oral peptides will improve HDL function, obviating injections, and thereby renewing the prospects for development of apoA‐I mimetics.
High‐density lipoprotein cholesterol (HDL‐C) is inversely related to coronary heart disease (CHD).1 Its major protein, apolipoprotein A‐I (apoA‐I), predicts CHD better,2 having proven atheroprotective by transgenic overexpression,3 somatic gene transfer,4 or intravenous infusion5 in animal models. As part of a broader effort to enhance HDL function,7 infusing the full apoA‐I molecule yielded mixed results on atherosclerosis (review by Chyu & Shah).6 Although these efforts have not validated apoA‐I as a therapy, they have not ruled it out. The size of the apoA‐I molecule itself poses a logistical/financial barrier to development. The difficulty synthesizing such a large molecule (243 aa) inspired smaller peptides mimicking apoA‐I function.
Encouragingly, apoA‐I mimetics reduced8 or regressed9 atherosclerosis in animals, altering HDL function without changing HDL‐C mass. Proposed mechanisms include accelerating HDL‐mediated cholesterol efflux/reverse cholesterol transport and enhancing HDL's anti‐oxidant/anti‐inflammatory properties.10 Indeed, HDL's anti‐inflammatory properties outperform HDL‐C mass in identifying high‐risk patients.11 Moreover, injecting human apoA‐I into mice rendered HDL less inflammatory, as with human volunteers.12 Statins present a clinically relevant benchmark for improving the HDL inflammatory index (HII), lowering HII 28% in patients with CHD.11 However, this drop failed to render high‐risk patients’ HDL anti‐inflammatory, motivating other approaches to augment the partial benefit of statins.13, 14 A key property for an apoA‐I mimetic is the ability to lower HII, especially beyond statin.
D‐4F is a peptide comprising 18 D‐amino acids whose tertiary structure resembles apoA‐I, without sequence homology.8 We conducted the first human studies wherein a single oral dose of D‐4F was minimally absorbed in healthy volunteers (BP‐01) and patients with CHD (BP‐02). Despite minimal plasma exposure, D‐4F improved HII,15 posing a conundrum: If so little oral D‐4F reaches the circulation, how is it suppressing HDL inflammation so profoundly? Intriguingly, this seeming disconnect is congruent with subsequent animal studies revealing the intestine as D‐4F's site of action.
Here, we report the first multiple‐dose study of oral D‐4F in man (BP‐03). The primary objective was to assess D‐4F's safety, tolerability, and pharmacokinetics among statin‐treated subjects with stable CHD or equivalent risk. Secondarily, we assessed pharmacodynamics by HII, using a cell‐based assay, the most sensitive indicator for D‐4F's biological activity.15
METHODS
Trial design
This was a proof‐of‐concept, single‐center, double‐blinded/placebo‐controlled parallel clinical trial of 13 daily doses of placebo/active D‐4F randomized 1:3 in three sequential, ascending‐dose cohorts: 100 mg, 300 mg, or 500 mg D‐4F/placebo. Additional methodologic details appear in Supplementary Text S1.
Participants
We recruited patients aged 21–80 years with stable CHD or equivalent risk per adult treatment panel‐III criteria, on stable‐dose statin (>4 weeks; Supplementary Table S1). Subjects gave written informed consent. Visits took place at the University of Pennsylvania (UPenn) General Clinical Research Center. The UPenn Institutional Review Board approved the study, conducted per Good Clinical Practice.
Interventions
D‐4F peptide was diluted in 60 mL 25% sucrose‐water as described.11 Cohorts of 20 were randomized 15 D‐4F/5 placebo in 3 ascending‐dose groups: 100 mg, 300 mg, and 500 mg, dosed after the overnight fast but ≥2 h before breakfast and concomitant medications. For example, the first cohort (n = 20) was assigned placebo/D‐4F 1:3, yielding 5 placebo/15 D‐4F. When the 100 mg group nearly completed 13 days, the principal investigator (R.L.D.) reviewed safety/tolerability before approving the next group, 300 mg. The same randomization/review process was conducted with the 300 mg and 500 mg groups. Seven visits included screening and two safety visits at 21 days and 42 days. Subjects reported on days 1, 3, 7, and 13 for supervised dosing, and otherwise took D‐4F at home. Blood samples were drawn for pharmacokinetics/pharmacodynamics before (time 0) and 15 min, 30 min, 1 h and 2 h after the first and last dose (days 1 and 13). Samples were obtained 2 h postdose on days 3 and 7, extending to 8 h on day 13. The latter provided the most extensive serial observations: 0, 2, 4, and 8 h; accordingly, pharmacokinetics/pharmacodynamics focus on day 13.
Outcomes
The primary outcomes were safety/tolerability and D‐4F pharmacokinetics, and secondarily pharmacodynamics: HII by monocyte chemotactic activity (MCA).
Assays
Plasma D‐4F levels were assayed as described,11 detecting ≥1.0 ng/mL. For HII, samples were sucrose cryopreserved and analyzed blinded to treatments as described,11 as were ancillary lipid or anti‐inflammatory biomarkers based on prior animal or human studies.10, 15, 16, 17, 18, 19 MCA values in the absence of HDL were normalized to one (i.e., HII is the ratio [MCALDL+HDL]/[MCALDL alone], reduced to a unit ratio (UR). Thus, 1.0 UR is neutral/normal, indicating added HDL was neither pro‐inflammatory nor anti‐inflammatory, because MCALDL+HDL = MCALDL. If added HDL were pro‐inflammatory, MCALDL+HDL >MCALDL, so HII >1.0 UR. Conversely, if added HDL were anti‐inflammatory, MCALDL+HDL <MCALDL, so HII <1.0 UR. Based on prior studies, we did not expect ancillary tests of lipid or anti‐inflammatory biomarkers to change, but assayed several in case multiple‐daily dosing caused unanticipated emergent benefits.
STATISTICAL METHODS
Because the primary objective of this proof‐of‐concept experiment was to assess safety, tolerability, and pharmacokinetics, no formal power calculations were performed. By clinical judgment, the enrollment goal was 60 completers. Comparative pharmacokinetic/pharmacodynamic changes were assessed between dose groups, and within dose groups predose/postdose, and between dose groups vs. placebo. Gender was considered a potential interacting factor. A linear mixed model of repeated measurements tested differences between D‐4F dose groups and placebo at each time point. Pairwise comparisons of least square means between D‐4F groups and placebo at each time point were examined. Statistical analyses were two‐tailed (α = 0.05), and results unadjusted unless indicated.
Pharmacokinetic and pharmacodynamic calculations
Pharmacokinetic parameters, including the area under the curve (AUC) from 0 h to the last measurable concentration (AUC(0–t)), or extrapolated to infinity (AUC(0–inf)), apparent elimination half‐life, maximum concentration (Cmax), and time of maximum (Tmax) concentration postdose, were computed by noncompartmental analysis using Stata version 14. Integrating time points from 0–8 h, the resulting area is less dependent on individual time points than constraining analysis to a particular time point. Since most subjects’ D‐4F was close to/approaching zero at 8 h, extrapolating to infinity scarcely changed the area. Therefore, we used the actual data from 0–8 h rather than extrapolate. A woman on 500 mg had extreme plasma D‐4F concentrations confirmed by retesting; hence, we consider her a bona fide biological outlier, and censored the data set by simply dropping her data. Therefore, pharmacokinetics conservatively, if grossly, underestimate D‐4F levels, especially among women. On an exploratory basis, we used regression to evaluate the pharmacodynamic curve between D‐4F dose and censored plasma D‐4F AUC over 8 h on day 13, and to evaluate the curve between D‐4F dose and primary pharmacodynamic outcome, postdose change in HII by MCA.
RESULTS
Participants
We recruited 47 eligible men and 16 sterile women from July to December 2005, enrolling 62 receiving ≥1 dose of D‐4F/placebo (46 men/16 women). Altogether, 59 subjects completed ≥12 visits, mean age 60.5 years (range, 28–78 years), weight 93.5 kg, and body mass index 31.6 kg/m2 (Table 1). Enrollees taking ≥1 dose comprised the safety analysis (n = 62). Pharmacokinetic/pharmacodynamic analysis was per protocol (n = 59) by the three completer cohorts taking ≥12/13 daily doses of D‐4F/placebo: (1) 20 completers/21 enrollees (100 mg group); (2) 20/21 (300 mg group); and (3) 19/20 (500 mg group), totaling 59 of 62. Additional details are outlined in Supplementary Text S2.
Table 1.
Baseline characteristics
| Characteristic | Placebo | 100 mg | 300 mg | 500 mg | Pooled | |
|---|---|---|---|---|---|---|
| Disposition and analytical population | ||||||
| Total enrolled: safety population | 16 | 16 | 15 | 15 | 62 | |
| Withdrew before 12/13 visits | 1 (6%) | 1 (6%) | 0 (0%) | 1 (7%) | 3 (5%) | |
| Completed 12/13 visits: unabbridged efficacy population | 15 (94%) | 15 (94%) | 15 (100%) | 14 (93%) | 59 (95%) | |
| Severe outlier on plasma D‐4F PKs | 0 (0%) | 0 (0%) | 0 (0%) | 1 (7%) | 1 (2%) | |
| PK outlier Dropped: Censored PKs population | 15 (94%) | 15 (94%) | 15 (100%) | 13 (87%) | 58 (94%) | |
| Demographics | ||||||
| Men | 14 (88%) | 12 (75%) | 9 (60%) | 11 (73%) | 46 (74%) | |
| Women, uncensored | 2 (13%) | 4 (25%) | 6 (40%) | 4 (27%) | 16 (26%) | |
| Age, years | 61.0 (11.8) | 59.2 (9.5) | 59.9 (8.4) | 62.0 (8.0) | 60.5 (9.4) | |
| Race/ethnicity (nonexclusive categories) | ||||||
| Native American | 1 (6%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (2%) | |
| African American | 2 (13%) | 3 (19%) | 5 (33%) | 1 (7%) | 11 (18%) | |
| White | 12 (75%) | 13 (81%) | 10 (67%) | 14 (93%) | 49 (79%) | |
| Multiracial | 1 (6%) | 0 (0%) | 0 (0%) | 0 (0%) | 1 (2%) | |
| Hispanic | 1 (6%) | 0 (0%) | 1 (7%) | 0 (0%) | 2 (3%) | |
| Anthropometrics | ||||||
| Height, cma | Men | 174.3 (4.9) | 176.8 (7.3) | 176.1 (7.1) | 178.1 (5.9) | 176.2 (6.2) |
| Women | 159.5 (7.8) | 157.1 (6.0) | 164.6 (4.1) | 156.0 (7.0) | 159.9 (6.5) | |
| Total body mass, kg | 94.7 (15.5) | 96.7 (16.1) | 92.2 (19.7) | 92.4 (20.2) | 94.0 (17.6) | |
| IBM, kga | Men | 69.8 (4.4) | 72.1 (6.7) | 71.5 (6.4) | 73.2 (5.3) | 71.5 (5.6) |
| Women | 51.9 (7.0) | 49.8 (5.5) | 56.5 (3.7) | 48.7 (6.4) | 52.3 (5.9) | |
| Lean body mass, kga | Men | 66.8 (6.8) | 69.1 (7.6) | 65.4 (9.0) | 67.4 (8.9) | 67.3 (7.8) |
| Women | 47.0 (3.4) | 47.1 (5.2) | 52.7 (7.4) | 46.2 (7.0) | 49.0 (6.6) | |
| Body surface area, m2 b | Men | 1.98 (0.16) | 2.05 (0.20) | 1.97 (0.22) | 2.02 (0.20) | 2.00 (0.19) |
| Women | 1.74 (0.08) | 1.76 (0.17) | 1.92 (0.27) | 1.73 (0.24) | 1.81 (0.22) | |
| Body mass index, kg/m2 | 31.8 (4.2) | 32.6 (3.8) | 31.2 (5.9) | 31.2 (6.2) | 31.7 (5.0) | |
| Major sources of risk (nonexclusive categories) | ||||||
| CHD | 11 (69%) | 6 (38%) | 8 (53%) | 10 (67%) | 35 (56%) | |
| DM | 8 (50%) | 11 (69%) | 8 (53%) | 7 (47%) | 34 (55%) | |
| Other ASCVD (e.g., PAD, AAA, CVD) | 0 (0%) | 1 (6%) | 0 (0%) | 0 (0%) | 1 (2%) | |
| FRS >20% absent CHD, ASCVD, or DM | 1 (6%) | 0 (0%) | 0 (0%) | 1 (7%) | 2 (3%) | |
| CHD and DM | 3 (19%) | 1 (6%) | 1 (7%) | 2 (13%) | 7 (11%) | |
| CHD absent other ASCVD and DM | 7 (44%) | 4 (25%) | 8 (53%) | 7 (47%) | 26 (42%) | |
| Hypertension | 12 (75%) | 13 (81%) | 9 (60%) | 10 (67%) | 44 (71%) | |
| HDLs | ||||||
| HDL‐C, mg/dLc | Men | 41.3 (7.4) | 45.3 (12.0) | 42.4 (11.8) | 42.2 (7.8) | 42.8 (9.6) |
| Women | 76.0 (17.0) | 48.3 (12.8) | 54.0 (12.8) | 58.8 (14.4) | 56.5 (14.9) | |
| ApoA‐I, mg/dLa | Men | 109.6 (14.2) | 113.5 (16.5) | 104.9 (15.4) | 115.7 (11.1) | 111.2 (14.5) |
| Women | 148.5 (16.3) | 124.0 (16.3) | 120.3 (10.4) | 140.3 (14.1) | 131.9 (16.7) | |
| Predose HDL inflammatory index, UR | 1.25 (0.36) | 1.21 (0.36) | 1.35 (0.46) | 1.22 (0.22) | 1.26 (0.36) | |
| Atherogenic lipoproteins | ||||||
| Non‐HDL‐C, mg/dL | 102.1 (28.8) | 107.6 (32.9) | 112.7 (34.1) | 99.5 (23.5) | 105.5 (29.8) | |
| Apo B100, mg/dL | 72.2 (18.3) | 74.1 (18.7) | 77.4 (22.7) | 69.7 (12.3) | 73.2 (17.9) | |
| TG, mg/dL, median, PSD | 143 (86.3) | 127 (54.8) | 99 (66.7) | 92 (71.1) | 110 (71.9) | |
| VLDL‐C, mg/dL | 25.0 (16.4) | 23.3 (12.8) | 20.6 (12.7) | 25.7 (19.7) | 23.6 (15.4) | |
| LDL‐C UC, mg/dL | 77.1 (24.9) | 84.4 (27.8) | 92.1 (35.8) | 73.9 (18.6) | 81.8 (27.7) | |
AAA, abdominal aortic aneurysm; Apo, apolipoprotein; ASCVD, atherosclerotic cardiovascular disease; CHD, coronary heart disease; CVD, cardiovascular disease; DM, diabetes mellitus; FRS, Framingham risk score; HDL, high‐density lipoprotein; HDL‐C, high‐density lipoprotein‐cholesterol; IBM, ideal body mass; PAD, peripheral artery disease; PK, pharmacokinetic; PSD, pseudo‐SD; TG, triglyceride; UC, by ultracentrifugation; UR, unit rate; VLDL‐C, very low‐density lipoprotein‐cholesterol.
Unless otherwise stated, continuous data are presented as mean (SD). Skewed data are presented as median (pseudo‐SD [PSD]). Significance test for interaction by gender: ± <0.1, a P < 0.05, b P < 0.01, c P < 0.001, a P < 0.0001.
Safety and tolerability
Vitals, safety laboratory tests, and examinations were unremarkable at all visits, as were electrocardiograms save a placebo‐exposed subject with atrial fibrillation. Altogether, 74 adverse events (AEs) affected 53%, chiefly nasopharyngitis (11% of total events), urinary tract infections (8%), extremity pain (7%), and myalgia (5%; Table 2). Affected subjects were similar by group: 7 (100 mg), 9 (300 mg), and 10 (500 mg; 44–67% of subjects receiving any D‐4F), and 7 on placebo (44%). However, AE counts were dose‐related: 12 (100 mg), 20 (300 mg), and 26 (500 mg), most mild to moderate (64 events). Six subjects experienced 10 AEs of severe intensity. Only one was possibly drug‐related, a subject on 500 mg in the D‐4F group with vasovagal syncope. However, none of the severe AEs warranted medication withdrawal. There was one serious AE requiring hospitalization: 9 days after completing D‐4F, a patient with CHD had angina following a heated workplace dispute; investigations ruled out myocardial infarction. No other serious AEs occurred.
Table 2.
Adverse events by dose group and intensity
| Dose | Mild | Moderate | Severe | Total | |
|---|---|---|---|---|---|
| No. of subjects with AEs | 28 (100%) | 9 (100%) | 6 (100%) | 33 (100%) | |
| Cardiac disorders | |||||
| Angina pectoris | 300 mg | 1 (3.6%) | 0 (0) | 1 (16.7%) | 2 (6.1%) |
| Atrial fibrillation | Placebo | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) |
| Eye disorders | |||||
| Arcus lipoids | Placebo | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) |
| Vision blurred | 300 mg | 0 (0) | 0 (0) | 1 (16.7%) | 1 (3.0%) |
| Gastrointestinal disorders | |||||
| Abdominal distension | 300 mg | 2 (7.1%) | 0 (0) | 0 (0) | 2 (6.1%) |
| Abdominal pain upper | 100 mg | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) |
| 300 mg | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) | |
| 500 mg | 0 (0) | 1 (11.1%) | 0 (0) | 1 (3.0%) | |
| Diarrhea | 100 mg | 2 (7.1%) | 0 (0) | 0 (0) | 2 (6.1%) |
| Dry mouth | 300 mg | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) |
| Dyspepsia | Placebo | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) |
| 100 mg | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) | |
| Flatulence | 300 mg | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) |
| Gastric disorder | 500 mg | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) |
| Gastro‐esophageal reflux disease | 500 mg | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) |
| Hematochezia | Placebo | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) |
| Nausea | 300 mg | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) |
| 500 mg | 0 (0) | 1 (11.1%) | 0 (0) | 1 (3.0%) | |
| Toothache | 100 mg | 0 (0) | 1 (11.1%) | 0 (0) | 1 (3.0%) |
| General disorders and administration site conditions | |||||
| Chest pain | Placebo | 0 (0) | 2 (22.2%) | 0 (0) | 2 (6.1%) |
| 300 mg | 0 (0) | 0 (0) | 1 (16.7%) | 1 (3.0%) | |
| Fatigue | 500 mg | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) |
| Edema | Placebo | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) |
| 300 mg | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) | |
| Pain | Placebo | 0 (0) | 1 (11.1%) | 0 (0) | 1 (3.0%) |
| 500 mg | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) | |
| Immune system disorders | |||||
| Hypersensitivity | 500 mg | 0 (0) | 1 (11.1%) | 0 (0) | 1 (3.0%) |
| Infections and infestations | |||||
| Ear infection | Placebo | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) |
| Influenza | 100 mg | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) |
| Nasopharyngitis | 100 mg | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) |
| 300 mg | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) | |
| 500 mg | 5 (17.9%) | 0 (0) | 0 (0) | 5 (15.2%) | |
| Urinary tract infection | Placebo | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) |
| 100 mg | 2 (7.1%) | 0 (0) | 0 (0) | 2 (6.1%) | |
| 300 mg | 2 (7.1%) | 0 (0) | 0 (0) | 2 (6.1%) | |
| 500 mg | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) | |
| Injury, poisoning, and procedural complications | |||||
| Animal bite | 500 mg | 0 (0) | 1 (11.1%) | 0 (0) | 1 (3.0%) |
| Contusion | 500 mg | 0 (0) | 0 (0) | 1 (16.7%) | 1 (3.0%) |
| Limb injury | 500 mg | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) |
| Investigations | |||||
| Cardiac murmur | 100 mg | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) |
| Musculoskeletal and connective tissue disorders | |||||
| Arthralgia | Placebo | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) |
| Back pain | 300 mg | 0 (0) | 1 (11.1%) | 0 (0) | 1 (3.0%) |
| 500 mg | 0 (0) | 0 (0) | 1 (16.7%) | 1 (3.0%) | |
| Chest wall pain | Placebo | 0 (0) | 1 (11.1%) | 0 (0) | 1 (3.0%) |
| Muscle spasms | Placebo | 0 (0) | 1 (11.1%) | 0 (0) | 1 (3.0%) |
| 300 mg | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) | |
| Musculoskeletal pain | 500 mg | 0 (0) | 0 (0) | 1 (16.7%) | 1 (3.0%) |
| Myalgia | Placebo | 0 (0) | 1 (11.1%) | 0 (0) | 1 (3.0%) |
| 500 mg | 1 (3.6%) | 1 (11.1%) | 0 (0) | 2 (6.1%) | |
| Pain in extremity | 300 mg | 1 (3.6%) | 0 (0) | 2 (33.3%) | 3 (9.1%) |
| 500 mg | 2 (7.1%) | 0 (0) | 0 (0) | 2 (6.1%) | |
| Shoulder pain | Placebo | 0 (0) | 1 (11.1%) | 0 (0) | 1 (3.0%) |
| Nervous system disorders | |||||
| Headache | 100 mg | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) |
| Syncope vasovagal | 500 mg | 0 (0) | 0 (0) | 1 (16.7%) | 1 (3.0%) |
| Respiratory, thoracic, and mediastinal disorders | |||||
| Rhinorrhea | 100 mg | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) |
| Throat irritation | 300 mg | 1 (3.6%) | 0 (0) | 0 (0) | 1 (3.0%) |
| Skin and subcutaneous tissue disorders | |||||
| Pruritus | Placebo | 0 (0) | 1 (11.1%) | 0 (0) | 1 (3.0%) |
AEs, adverse events.
Pharmacokinetic results
In the 500 mg group, we censored pharmacokinetic analyses, eliminating the woman with extremely high D‐4F: Cmax 640 ng/mL (vs. 49 for the other women on 500 mg), and AUC(0–8h) 1,437 ng/mL*h (vs. 246; Supplementary Table S2 vs. Table 3). For the remaining, censored population, postdose plasma D‐4F levels rose rapidly, peaking 1.3–2.4 h across groups (Figure 1). On day 13, predose/trough D‐4F was undetectable in all the study groups (<1 ng/mL), except two subjects on 500 mg achieving low concentrations: 1.06 and 4.41 ng/mL. Pooling genders, after 13 days, the mean confidence interval (CI) Cmax (ng/mL) from 100 mg was 2.8 (CI = 2–3.6), from 300 mg 8.1 (CI = 5.2–11), and from 500 mg 37.1 (CI = 20.3–53.9) when D‐4F was detectable postdose. The Cmax remained relatively stable from days 1–13, as was the AUC(0–t) from 0–2 h postdose (Table 3). The D‐4F levels 2 h postdose peaked at 3–7 days, and did not increase thereafter. Plasma D‐4F AUC increased from 100 mg to 500 mg (P < 0.0001 by nonparametric test for trend). Typically, women achieved higher plasma levels than men (Figure 1 a,b). Unsurprisingly, women had higher effective doses on adjusting nominal dose for body size. Ideal body mass (IBM)20 was particularly informative, women having higher D‐4F/IBM than men (Supplementary Figure S2).
Table 3.
Pharmacokinetics of D‐4F in plasma
| Characteristic | 100 mg | 300 mg | 500 mg uncensored | 500 mg after censoring women | |
|---|---|---|---|---|---|
| I. Pharmacokinetics including subjects with undetectable plasma D‐4F treated as zero values | |||||
| A. Plasma D‐4F Cmax, where Cmax = 0 when D‐4F undetectable, ng/mL, mean (95% CI) | |||||
| Day 1 (0–2 h) | Men | 2.37 (1.31–3.43) | 4.43 (1.17–7.69) | 23.39 (6.32–40.45) | 23.39 (6.32–40.45) |
| Women | 4.03 (‐2.55 to 10.61) | 6.86 (3.70–10.02) | 177.98 (‐43.36 to 399.32) | 67.30 (5.05–129.55) | |
| Pooled across interactiona | [2.70 (1.27–4.14)] | [5.40 (3.07–7.74)] | [64.61 (‐0.46 to 129.68)] | [32.80 (12.80–52.79)] | |
| Day 13 (0–8 h) | Men | 2.27 (1.31–3.23) | 5.38 (2.54–8.21) | 33.56 (14.81–52.30) | 33.56 (14.81–52.30) |
| Women | 2.25 (‐0.15 to 4.65) | 10.80 (5.83–15.77) | 196.80 (‐94.21 to 487.80) | 49.06 (7.98–90.15) | |
| Pooled across interactionb | 2.27 (1.40–3.13) | 7.55 (4.68–10.41) | 80.20 (‐5.62 to 166.01) | 37.14 (20.36–53.91) | |
| B. Plasma D‐4F AUC, where AUC = 0 when D‐4F undetectable, ng/mL × hour, mean (95% CI) | |||||
| Day 1 (0–2 h) | Men | 2.82 (1.05–4.60) | 4.56 (1.57–7.54) | 32.94 (7.82–58.07) | 32.94 (7.82–58.07) |
| Women | 6.42 (‐4.15 to 16.98) | 7.00 (3.79–10.21) | 246.59 (‐37.00 to 530.18) | 107.66 (‐4.40 to 219.73) | |
| Pooled across interactiona | [3.54 (1.14–5.93)] | [5.53 (3.31–7.75)] | [89.92 (4.03–175.80)] | [48.95 (16.19–81.72)] | |
| Day 13 (0–8 h) | Men | 6.94 (3.70–10.18) | 22.70 (9.85–35.54) | 104.06 (66.41–141.71) | 104.06 (66.41–141.71) |
| Women | 11.60 (‐2.82 to 26.02) | 47.37 (27.06–67.68) | 522.93 (‐39.03 to 1084.89) | 246.76 (33.16–460.36) | |
| Pooled across interactiona | [7.87 (4.20–11.55)] | [32.57 (20.09–45.04)] | [223.74 (44.56–402.91)] | [136.99 (75.92–198.06)] | |
| II. Pharmacokinetics excluding subjects with undetectable plasma D‐4F | |||||
| A. Excluded population: D‐4F‐exposed subjects with undetectable plasma D‐4F (day 1 5/45 = 11%, day 13 4/44 = 9%) | |||||
| Day 1 (0–2 h) | 3/15 (20%) | 2/15 (13%) | 0/15 (0%) | ||
| Day 13 (0–8 h) | 3/15 (20%) | 1/15 (7%) | 0/14 (0%) | ||
| B. Plasma D‐4F time of events when D‐4F detected on day 13, pooling genders, hours, mean (95% CI) | |||||
| Time of peak | Mean (CI) | 2.25 (1.61–2.89) | 2.39 (1.47–3.31) | 1.65 (0.89–2.42) | |
| Median (range) | 2.00 (1.00–4.00) | 2.00 (0.50–7.92) | 1.27 (0.25–4.02) | ||
| T‐Half | Mean (CI) | 0.81 (0.29–1.32) | 1.18 (0.60–1.77) | 1.99 (1.38–2.60) | |
| Median (range) | 0.46 (0.37–3.04) | 0.45 (0.33–2.95) | 1.88 (0.35–4.83) | ||
| C. Plasma D‐4F Cmax when D‐4F detected, ng/mL, mean (95% CI) | |||||
| Day 1 (0–2 h) | Men | 2.84 (1.81–3.88) | 5.70 (2.01–9.38) | 23.39 (6.32–40.45) | 23.39 (6.32–40.45) |
| Women | 6.05 (‐3.06 to 15.16) | 6.86 (3.70–10.02) | 177.98 (‐43.36 to 399.32) | 67.30 (5.05–129.55) | |
| Pooled across interactiona | [3.38 (1.80–4.95)] | [6.23 (3.85–8.62)] | [64.61 (‐0.46 to 129.68)] | [32.80 (12.80–52.79)] | |
| Day 13 (0–8 h) | Men | 2.72 (1.82–3.63) | 6.05 (3.20–8.89) | 33.56 (14.81–52.30) | 33.56 (14.81–52.30) |
| Women | 3.37 (1.72–5.03) | 10.80 (5.83–15.77) | 196.80 (‐94.21 to 487.80) | 49.06 (7.98–90.15) | |
| Pooled across interactionb | 2.83 (2.04–3.62) | 8.09 (5.22–10.95) | 80.20 (‐5.62 to 166.01) | 37.14 (20.36–53.91) | |
| D. Plasma D‐4F observed AUC when D‐4F detected, ng/mL × hour, mean (95% CI) | |||||
| Day 1 (0–2 h) | Men | 3.38 (1.43–5.34) | 5.86 (2.63–9.09) | 32.94 (7.82–58.07) | 32.94 (7.82–58.07) |
| Women | 9.62 (‐5.08 to 24.33) | 7.00 (3.79–10.21) | 246.59 (‐37.00 to 530.18) | 107.66 (‐4.40 to 219.73) | |
| Pooled across interactiona | [4.42 (1.64–7.21)] | [6.38 (4.17–8.59)] | [89.92 (4.03–175.80)] | [48.95 (16.19–81.72)] | |
| Day 13 (0–8 h) | Men | 8.33 (5.09–11.56) | 25.54 (12.41–38.67) | 104.06 (66.41–141.71) | 104.06 (66.41–141.71) |
| Women | 17.40 (2.03–32.76) | 47.37 (27.06–67.68) | 522.93 (‐39.03 to 1084.89) | 246.76 (33.16–460.36) | |
| Pooled across interactiona | [9.84 (6.00–13.68)] | [34.89 (22.42–47.36)] | [223.74 (44.56–402.91)] | [136.99 (75.92–198.06)] | |
| E. Plasma D‐4F extrapolated AUC when D‐4F detected on day 13, ng/mL × hour, mean (95% CI) | |||||
| Day 13 (0–∞) | Men | 8.47 (4.87–12.08) | 27.24 (12.83–41.66) | 111.07 (71.91–150.22) | 111.07 (71.91–150.22) |
| Women | 19.94 (‐0.40 to 40.28) | 50.21 (28.84–71.59) | 574.85 (12.12–1137.59) | 304.27 (38.08–570.47) | |
| Pooled across interactiona | [10.56 (5.71–15.41)] | [38.73 (24.69–52.77)] | [243.58 (57.54–429.61)] | [155.65 (79.95–231.36)] | |
AUC, area under the curve; CI, confidence interval; Cmax, peak plasma concentration.
Italics indicate a mean that was arrived at by dropping observations from a woman with an extraordinarily robust plasma response to D‐4F, who was considered a biological outlier. Censored means thereby conservatively err on the side of underestimating the effect of D‐4F, especially among women. Significance test for interaction by gender (a.k.a. effect modification): a P < 0.05; b P < 0.1. When interaction is present, the pooled results should be treated with special caution, as results may not be readily generalized to mixed‐gender populations, unless they match the ratio of men to women within the particular group being generalized. Some authorities suggest accepting P < 0.1 as evidence for significant interaction. Pooled results affected by significant effect modification at P < 0.05 are enclosed by brackets. Parameters incorporating time were based on actual times rather than target times.
Figure 1.
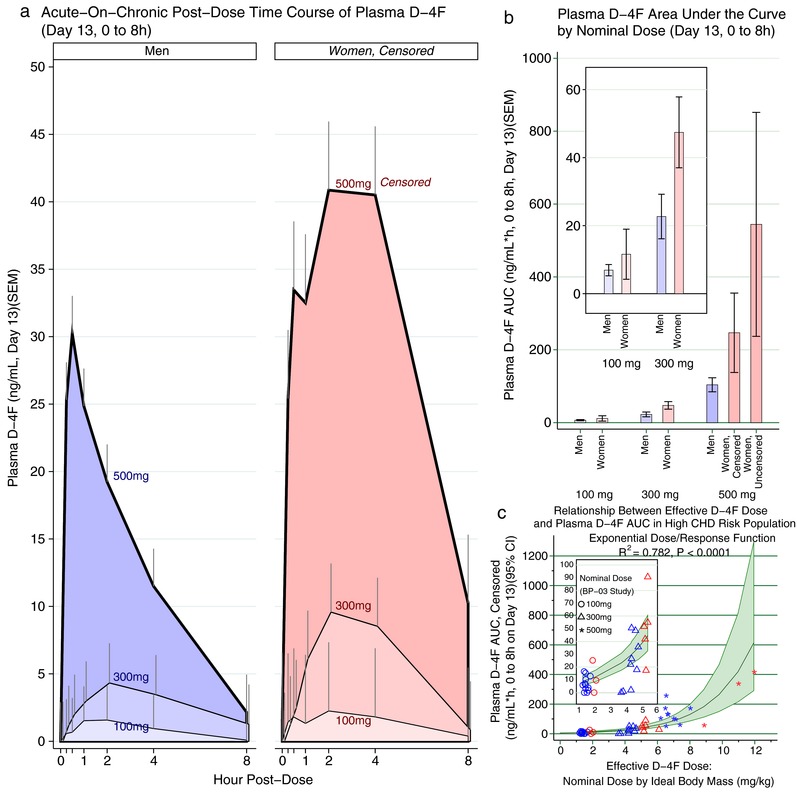
(a) The time course of plasma D‐4F is presented for subjects with detectable levels over 8 h on day 13 by nominal D‐4F dose and gender. D‐4F was given at hour 0. As expected, exposure to higher nominal doses of D‐4F resulted in greater plasma D‐4F levels. Concentrations also differed between men and women, the latter achieving higher D‐4F levels, especially at the higher doses. Among women, the 500 mg group is censored, having excluded a woman with extraordinarily high D‐4F levels, thus underestimating the full effect of 500 mg on women. (b) The area under the curve (AUC) was determined over 8 h on day 13, and is again presented by nominal D‐4F dose and gender. Here, it was practical to present the women exposed to 500 mg as censored and uncensored bars for comparison. Consistent with a, women had higher D‐4F AUC. Because the uncensored analysis compresses the other bars, the inset presents the 100 mg and 300 mg groups separately for clarity. (c) We modeled the dose/response curve between effective D‐4F oral dose and plasma exposure as D‐4F AUC. The optimal fit was obtained by effective D‐4F dose in mg D‐4F per kg ideal body mass, rather than the nominal D‐4F dose (P < 0.0001; R2 = 0.78). See Supplementary Table S3 for details. The dose/response relationship was greater than linear with no evidence of a plateau at the upper end of the dosing range; as such, an exponential function provided a better fit than a straight‐line function. The blue markers indicate men and the red indicate women. CHD, coronary heart disease; CI, confidence interval.
Affirming the nonparametric test, we used regression to model the dose/response curve, allowing us to gauge whether other factors might influence the dose/response relationship, particularly gender. Often, plasma D‐4F was undetectable, resulting in a large number of zero values in the dependent variable, motiving two‐part regression,21 consisting of (1) logistic regression modeling presence/absence of detectable D‐4F, and (2) generalized linear modeling conditional on D‐4F detection. The dose/response curve was greater than linear (i.e., higher doses prompted greater plasma D‐4F exposure than rectilinear projections based on the increase in dose). Accordingly, the generalized linear modeling residuals had a log‐normal right skew, so an exponential function provided the best fit (Supplementary Table S3).
Although the nominal D‐4F dose predicted plasma D‐4F exposure (overall R2 = 0.64/P < 0.0001), a much better fit was achieved by adjusting D‐4F dose by body weight, henceforth, the effective dose. Specifically, IBM achieved the best fit (R2 = 0.78/P < 0.0001; Figure 1 c and Supplementary Table S3); moreover, relative likelihood suggested the model based on nominal D‐4F dose was only 50% as informative as D‐4F/IBM. Regardless of how the D‐4F dose was modeled (nominal vs. effective dose), there was a strong dose/response relationship between D‐4F dose and (1) absence/presence of detectable plasma D‐4F, and (2) plasma D‐4F AUC (all P < 0.0001). Hence, with greater D‐4F doses, D‐4F was more likely detected, and among those achieving detectable levels, the plasma D‐4F AUCs increased more than dose‐proportional with increasing doses.
Neither the discrete nor continuous aspects of the dose/response relationship were influenced by gender. This suggests greater plasma D‐4F exposure among women follows their higher effective D‐4F doses.
Pharmacodynamic results
Acute‐on‐chronic dosing lowers HDL inflammatory index
Following 13 days of D‐4F, predose baseline HII ranged from 1.2–1.4 UR across groups, confirming our high‐risk subjects started out with pro‐inflammatory HDL, this despite statin therapy. In all groups, HII fell 2 h postdose. For example, in those assigned placebo, HII was 1.25 UR predose, then reached to 0.82 UR by 2 h (–31%; P = 0.0001 vs. predose; Table 4). The drop ranged from ‒15.8% (100 mg) to ‒40.5% (500 mg). We can only speculate to what study procedure caused a universal drop in HII 2 h postdose. This could reflect diurnal changes, prolonged fasting, or enforced bed rest. Greater drops accompanied 300 mg and 500 mg 2, 4, and 8 h postdose vs. 100 mg (P < 0.05; Table 4 and Figure 2 a). More importantly, higher doses differed from placebo. Notably, HII fell more on 300 mg vs. placebo 4–8 h postdose and 8 h postdose on 500 mg (all P < 0.05). At best, by 8 h 300 mg dropped HII from 1.35 to 0.57 UR (–57.8%) and 500 mg 1.22–0.63 UR (–48.4%); thus, despite different baselines, at higher D‐4F doses, HII settled to a very similar level: 0.60 UR vs. 0.86 UR for placebo. At best, 300 and 500 mg D‐4F approximately halved HII vs. ∼30% less on placebo; thus, placebo‐corrected suppression was –27% beyond placebo for 300 mg (P < 0.005 vs. placebo) and –21% for 500 mg (P < 0.03). Placebo correction simply subtracts the %change in the placebo group from the gross %change in a given active D‐4F group. This net change reflects the added contribution of D‐4F. For 4 h and 8 h, the “placebo‐corrected” change is somewhat a misnomer, because patients took their usual medications at 2 h with breakfast. Importantly, this included their morning regimen to prevent atherosclerotic cardiovascular disease (ASCVD). Thus, the net contribution of D‐4F is the added contribution of D‐4F beyond any benefits of subjects’ ASCVD medications. This is an important distinction, as the goal of development is to improve HII beyond any benefits of usual clinical care to prevent ASCVD. On the lowest D‐4F dose, 100 mg, the drop in HII was indistinguishable from that of placebo, except at 4 h, when HII fell less than placebo (P = 0.04). This suggests ≥300 mg D‐4F is required to suppress HII to around 0.60 UR.
Table 4.
HDL inflammatory index pharmacodynamics: postdose time course over 8 h on day 13
| Parameter | Sucrose Placebo | 100 mg D‐4F + Sucrose | 300 mg D‐4F + Sucrose | 500 mg D‐4F + Sucrose |
|---|---|---|---|---|
| 0 h postdose | ||||
| HII | 1.25 UR (1.09–1.41) | 1.21 UR (1.05–1.37) | 1.35 UR (1.19–1.51) | 1.22 UR (1.05–1.39) |
| 2 h postdose | ||||
| HII | 0.82 UR (0.66–0.98) | 0.99 UR (0.83–1.15) | 0.79 UR (0.63–0.95) | 0.73 UR (0.56–0.90) |
| Delta‐HII | ‒0.43 UR (‒0.25,‒0.61) | ‒0.22 UR (‒0.04,‒0.39) | ‒0.56 UR (‒0.39,‒0.74) | ‒0.50 UR (‒0.31,‒0.68) |
| Percent change | ‒31.1% (‒43.7,‒18.6%) | ‒15.8% (‒28.4,‒3.2%) | ‒37.8% (‒50.3,‒25.2%) | ‒40.5% (‒53.5,‒27.5%) |
| P = 0.0001 | P = 0.0153 | P = 0.0001 | P = 0.0001 | |
| Placebo‐adjusted percent change | +15.3% (‒2.6,+33.3%) | ‒6.7% (‒24.6,+11.3%) | ‒9.4% (‒27.6,+8.9%) | |
| P = 0.0936 | P = 0.4648 | P = 0.3118 | ||
| Fast ended and subjects took daily medications, including morning ASCVD‐prevention regimen | ||||
| 4 h postdose / 2 h post‐ASCVD regimen and breakfast | ||||
| HII | 0.91 UR (0.75–1.07) | 1.10 UR (0.94–1.26) | 0.71 UR (0.55–0.87) | 0.74 UR (0.57–0.91) |
| Delta‐HII | ‒0.34 UR (‒0.17,‒0.52) | ‒0.12 UR (+0.06,‒0.29) | ‒0.64 UR (‒0.47,‒0.82) | ‒0.48 UR (‒0.30,‒0.66) |
| Percent change | ‒26.1% (‒38.6,‒13.5%) | ‒7.1% (‒19.6,+5.5%) | ‒44.5% (‒57.1,‒32.0%) | ‒38.3% (‒51.3,‒25.3%) |
| P = 0.0001 | P = 0.2714 | P = 0.0001 | P = 0.0001 | |
| Placebo‐adjusted percent change | +19.0% (+1.0,+36.9%) | ‒18.5% (‒36.4,‒0.5%) | ‒12.2% (‒30.5,+6.0%) | |
| P = 0.0386 | P = 0.0441 | P = 0.1875 | ||
| 8 h postdose / 4 h post‐ASCVD regimen and breakfast | ||||
| HII | 0.86 UR (0.70–1.02) | 1.00 UR (0.84–1.16) | 0.57 UR (0.41–0.73) | 0.63 UR (0.46–0.80) |
| Delta‐HII | ‒0.39 UR (‒0.21,‒0.56) | ‒0.22 UR (‒0.04,‒0.39) | ‒0.79 UR (‒0.61,‒0.96) | ‒0.59 UR (‒0.41,‒0.77) |
| Percent change | ‒27.9% (‒40.5,‒15.4%) | ‒16.6% (‒29.2,‒4.0%) | ‒54.7% (‒67.2,‒42.1%) | ‒48.5% (‒61.5,‒35.5%) |
| P = 0.0001 | P = 0.0109 | P = 0.0001 | P = 0.0001 | |
| Placebo‐adjusted percent change | +11.3% (‒6.6,+29.3%) | ‒26.8% (‒44.7,‒8.8%) | ‒20.6% (‒38.9,‒2.3%) | |
| P = 0.2140 | P = 0.0039 | P = 0.0276 | ||
ASCVD, atherosclerotic cardiovascular disease; HDL, high‐density lipoprotein; HII, high‐density lipoprotein inflammatory index; UR, unit ratio, the native units for HII.
ASCVD Delta‐HII is simply the postdose HII minus predose HII on the UR scale. Percent change is Delta‐HII/predose HII × 100% on the percent scale. The placebo‐adjusted percent change subtracts the percent change from the placebo group for each group on active D‐4F, on the percent scale. Data are presented as mean (95% confidence interval).
Figure 2.
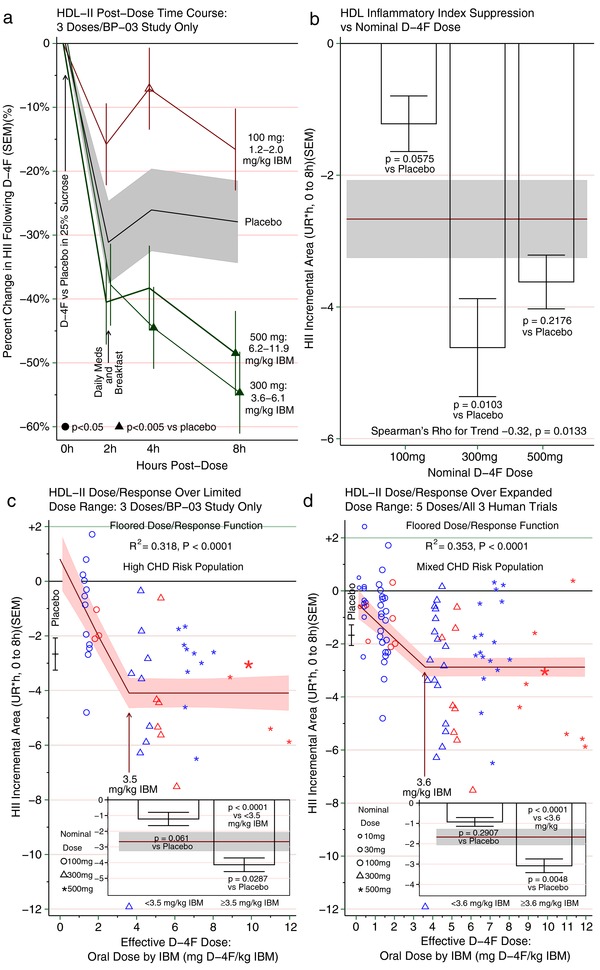
(a) The time course for the change in high‐density lipoprotein (HDL) inflammatory index (HII) is presented over 8 h on day 13 as mean and SEM. Subjects drank D‐4F in a sucrose solution at hour 0, and withheld their usual daily medications and fasted for at least 2 h postdose. By 2 h postdose, HII was uniformly lower than baseline in all groups (i.e., no D‐4F group varied from the placebo group). Then subjects ate breakfast and took their usual medications, including their usual regimen to prevent atherosclerotic vascular disease (ASCVD). Thereafter, higher D‐4F groups ultimately diverged from the placebo group. Among those receiving 300 mg or more of D‐4F, the HII dropped below that of the placebo group at either 4 h or 8 h postdose. In contrast, those receiving 100 mg of D‐4F were no better than placebo, and were even worse than placebo at one point (hour 4). The range for the effective dose of D‐4F is provided as mg D‐4F/ideal body mass (mg/kg). The data from this panel are also shown in Table 4 using various scales, including the absolute scale in unit ratio (UR), change from baseline in UR, and the raw and placebo‐adjusted percent change. (b) The time course curves for each subject on day 13 are presented as HII incremental area under the curve (incAUC 0–8 h), as described in the Methods and illustrated in Supplementary Figure S1. Briefly, the incAUC is the difference between the postdose HII absolute AUC (absAUC) and the predose baseline projected over 8 h (T0 × 8 h). Because the HII typically drops below baseline, the difference (HII absAUC — T0 × 8 h) results in a negative number. A negative HII incAUC is interpreted as HII suppression over 8 h compared to predose HII, that is, a less inflammatory HII. Conversely, a positive HII incAUC would have been interpreted as HII potentiation, that is, a more inflammatory HII. Each subject's incAUC was calculated from HII on its native/absolute scale, the UR; thus, the units for the incAUC are UR*hour over 8 h. We then calculated the mean and SEM of all the individual incAUCs for each group, and present the aggregated HII suppression as incAUC by group. The mean and SEM for the group receiving placebo + ASCVD medications is presented as a horizontal band, and groups receiving D‐4F + ASCVD medications are presented as bars with mean and SEM. The test for trend by nominal dose supported an inverse relationship (rho ‒0.32; P = 0.01), suggesting higher D‐4F doses are generally more apt to render HII less inflammatory than lower doses. That said, the 300 mg and 500 mg doses were not significantly different from each other, suggesting a threshold dose/response relationship rather than a linear relationship. Were a linear relationship operative at the highest dose, one would instead expect the 500 mg dose to vary from the 300 mg, and in the direction of greater HII suppression. Instead, they seem to provide similar HII suppression, more consistent with a flat dose/response relationship. For comparison, the insets within c and d pool the 300 mg and 500 mg dose groups. (c) Dose/response curve between effective D‐4F dose and HII incAUC on day 13 as described in b. The effective D‐4F dose is the nominal D‐4F dose in mg per kg ideal body mass (IBM; mg/kg). As effective dose increased, the HII incAUC declined, consistent with more anti‐inflammatory HDL. This continued until an effective dose of 3.5 mg/kg. At higher effective doses, the dose‐response curve hit a floor, beyond which higher doses did not lower HII incAUC further, as suggested by the similar HII suppression from the 300 mg and 500 mg dose groups in b. A flat/floored dose/response relationship suggests there is a limit to which D‐4F can render HII more anti‐inflammatory rather than continued linear dose responsiveness. For more details on the curve analysis, see Supplementary Table S4. The inset compares HII suppression as incAUC below and above the threshold. Below 3.5 mg/kg, the HII incAUC was numerically worse than placebo, but not significantly so, whereas above 3.5 mg/kg, the HII suppression was significantly improved beyond placebo (P = 0.0287). (d) This panel takes the same approach as c, but expands the dose range by adding two doses in the linear descent portion (10 mg and 30 mg) by combining the day 13 data from c with single‐dose human studies of D‐4F. Doing so enhances the sample size for the 100 mg, 300 mg, and 500 mg doses, augments the number of women, and broadens the population by including subjects whose risk does not exceed the threshold for high ASCVD risk. Again, with increasing effective exposures to D‐4F, HII suppression by incAUC improves to a point, but then flattens out to a floor effect. The data in the inset show that those with effective doses above the threshold have HII that is significantly less inflammatory than those on placebo (P = 0.0048). CHD, coronary heart disease.
Indeed, a dose/response relationship between D‐4F and HII was apparent, as D‐4F dose inversely related to HII (P = 0.0018 by nonparametric trend test). There was a mild but significant inverse correlation between D‐4F AUC and HII %change 8 h postdose (r = –0.38/P = 0.0360), and D4‐F incremental area under the curve (incAUC) and HII incAUC negatively correlated (Spearman's rho = –0.37/P = 0.01). Similarly, there was a modest trend for nominal D‐4F dose to predict lower HII incAUC, as higher D‐4F doses predicted enhanced HII suppression (rho = –0.32/P = 0.01; Figure 2 b). These nonparametric tests suggest greater D‐4F doses, and, to some extent, plasma D‐4F exposure portends lower/improved HII.
We then modeled the dose/response curve between D‐4F and HII. As effective dose rose (D‐4F mg/kg IBM), HII fell linearly. However, upon reaching an effective dose of 3.5 mg/IBM, the curve flattened to a floor (Figure 2 c and Supplementary Table S4, model 4, R2 = 0.32/P = 0.0004). Importantly, individuals dosed ≥3.5 mg/IBM had a lower HII compared to placebo (P = 0.03), whereas dosing <3.5 mg/IBM was statistically no worse than the diurnal drop from placebo (P = 0.06). As a practical matter, all subjects on 100 mg received <3.5 mg/IBM. Accounting for variability, the 90%/95% tolerance interval (TI) provides a range that 90% of the underlying population should fall within, at 95% CI. The TI provides a “reality check” on whether a particular dose might bring the overwhelming majority (90%) of subjects to a goal. For D‐4F, a dose achieving a lower TI boundary >3.5 mg/IBM would assure the vast majority of subjects were “on the floor” of the dose/response curve. On 300 mg, effective dose ranged 3.6–6.1 mg/IBM, averaging 4.7. Although all were dosed to ≥3.5 mg/IBM, the TI ranged 2.9–6.5 mg/IBM. Thus, in a larger study, 300 mg will probably fall short. Likewise, all on 500 mg received ≥3.5 mg/IBM (range, 6.2–11.9 mg/IBM), but TI ranged 2.8–12.9 mg/IBM. Accordingly, if 300 or 500 mg were set as fixed doses, their corresponding TIs suggest not all would receive ≥3.5 mg/IBM. Thus, higher fixed doses may be needed to maximally suppress HII.
This analysis also suggested that gender does not influence the dose/response, either by (1) shifting the entire curve up/down the y‐axis (P = 0.5 for intercept) or (2) altering the shape of the curve (P = 0.6 for effect modification). Hence, the effective dose in mg D‐4F/IBM adequately incorporates the apparent gender interaction, probably because IBM itself is gender‐dependent. If affirmed, more consistent results might be obtained by dosing D‐4F by IBM in mechanistic studies, and larger fixed doses in studies reducing D‐4F to practice.
We also evaluated models including plasma D‐4F exposure (Supplementary Table S4, models 5/6). Unlike oral dose, modeling HII by D‐4F levels revealed plasma exposure was profoundly weaker and, indeed, unrelated (model 5; P = 0.3; Supplementary Figure S3). Tellingly, when oral dose was added to the model with D‐4F exposure, oral dose remained highly predictive of HII (model 6; P < 0.0005) with even less support for any influence by plasma D‐4F (P = 0.6). This implies oral peptide dose is an overwhelmingly dominant HII predictor, excluding any apparent role for plasma D‐4F exposure.
As a sensitivity analysis, we included data from the two other human studies, BP‐01 and BP‐02. Baseline characteristics for BP‐01 are provided in Supplementary Table S5, and BP‐02 is described elsewhere.15 This extended the dose range on the rapid‐descent phase (i.e., before the pre‐floor breakpoint), by including 10 mg and 30 mg doses. These studies also provided more data on 100–500 mg D‐4F and extended the risk profile into the primary prevention realm. Perhaps most appealing, broadening the analysis added more women. The expanded analysis affirmed the floored shape of the dose/response curve (R2 = 0.35/P < 0.0001; Figure 2 d), and, again, those receiving a dose above the breakpoint had a significantly better drop in HII vs. placebo (P = 0.005). The depth to the floor was shallower upon including lower‐risk subjects. This is intuitive, as healthier subjects are less apt to have pro‐inflammatory HDL at baseline. Conversely, the deeper floor for high‐risk subjects suggests greater therapeutic opportunity for the sicker population. Unsurprisingly, HII was the only pharmacodynamic parameter altered by D‐4F, and the ancillary lipid or anti‐inflammatory biomarkers remained stable as in the prior study; therefore, repeated dosing did not lead to emergent changes, as expected.
DISCUSSION
This is the first multiple‐dose study of oral D‐4F in humans, and, encouragingly, chronic daily dosing was generally well‐tolerated. Overall, the frequency and types of SEs in this study population were not unexpected, and not even AEs of severe intensity necessitated D‐4F withdrawal. Echoing our prior single‐dose study,15 all subjects in the present study had elevated baseline HII. Previously, HII significantly improved 4 h after a single dose of 300 mg, and 2 h after a single dose of 500 mg vs. placebo.15 Here, we report after 13 consecutive days, doses of ≥300 mg D‐4F significantly lowered HII 4‒8 h postdose. HII returns to baseline by 24 h, suggesting prolonged suppression might require multiple daily dosing or much higher doses. We gave unformulated D‐4F, making no attempt to prolong delivery; doing so might reduce the dosing interval.
It is important to appreciate the drop in HII from D‐4F occurred in patients who were already on statins and other ASCVD‐preventing medications, indicating an additive effect rather than an isolated effect. For comparison, Ansell et al.11 also studied a population with CHD/equivalent risk with pro‐inflammatory HII (1.38 UR), except their subjects were statin‐naïve. After 6 weeks, simvastatin 40 mg prompted a 28% drop in HII. In a separate cohort, HII better distinguished CHD cases from controls than HDL‐C. The statin results provide clinical context for our acute‐on‐chronic study of D‐4F, in which 300 mg after 8 h lowered HII 55% and 500 mg 49%, compared to a 28% drop in the control group (Figure 3). Intriguingly, the control group also had a drop in HII over 8 h that was nearly identical to the magnitude of the statin benchmark. Indeed, we designed our experiment so our patients with CHD took their morning ASCVD‐preventive medications during the extended observation period (day 13, 2 h postdose). This likely contributed to the prolonged (8 h) drop in HII following D‐4F or placebo. The improvement may also involve a diurnal phenomenon or an effect of prolonged fasting or bed rest, as HII also improved 2 h postdose. Although these factors may have initiated the drop in HII across all groups, we speculate the drop was perpetuated over 8 h by the ASCVD medications taken by all groups. However, as we cannot parse out the causes for the universal drop, we can deduce it involves a 28% drop by the end of 8 h based on the placebo group. Subtracting this effect from the total drop in HII from D‐4F gives an idea of the apparent drop attributable to D‐4F (i.e., the effect D‐4F superimposed upon any cumulative effects of diurnal, fasting, or unknown effects and ASCVD medications). Thus, 300 mg D‐4F prompted a placebo‐adjusted drop in HII of ‒27% and 500 mg dropped it ‒21%. Thus, these doses lower HII by a similar magnitude as simvastatin 40 mg, but more importantly, are additive to chronic statin therapy, because statin use was mandatory in our experiment. Interestingly, adding the statin in the Ansell et al.11 study lowered HII, but failed to get it “across the finish line” that is, statins failed to lower HII from a pro‐inflammatory baseline to the anti‐inflammatory range of <1.0 UR. In contrast, adding 300‒500 mg D‐4F to statin lowered HII to a decisively anti‐inflammatory level, around 0.60 UR. A novel implication is that using both lipid‐altering medications together can achieve such a profound drop in HII that HDL is actually rendered anti‐inflammatory compared, which typically does not happen with statin monotherapy. Beyond lipid‐altering medications, others have shown that diet and exercise can lower HII 21%,13 and anti‐inflammatory medications can lower HII among patients with rheumatoid arthritis 17‒18%.14 It would be interesting to determine whether diet and aerobic exercise are additive to lowered HII from D‐4F, as exercise alters apoA‐I and HDL function, opening up the possibility of overlapping mechanisms. If so, perhaps D‐4F could be used as a gymnomimetic to mimic or augment a benefit of exercise when activity is limited.22
Figure 3.
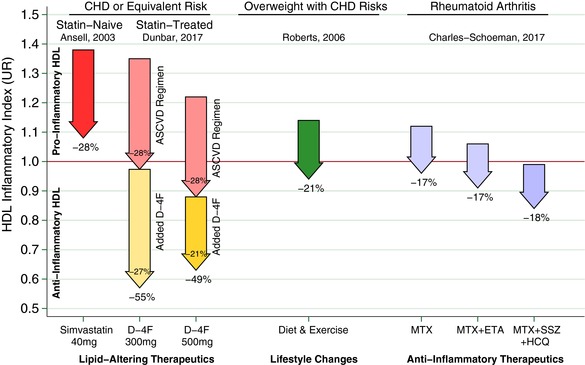
Arrows represent the change in high‐density lipoprotein (HDL) inflammatory index (HII) from various interventions, where the arrow base is that population's aggregated mean HII pre‐intervention, expressed as a unit ratio (UR). The arrow tip is that population's mean HII following the intervention. An individual HII above 1.0 UR is pro‐inflammatory, indicating monocyte chemotactic activity (MCA) increased when HDL was added to low‐density lipoprotein in that individual's culture. Conversely, an HII below 1.0 R is anti‐inflammatory, that is, MCA decreased upon adding HDL. The statins represent a benchmark as a lipid‐altering drug that can lower HII. Ansell et al.11 showed that adding simvastatin 40 mg daily to a population of statin‐naïve patients with coronary heart disease (CHD) or equivalent risk can lower HII as much as 28%. Thus, on average, the statin rendered patients with CHD HIIs less inflammatory than they were before starting that statin. Unfortunately, this was not sufficient to render most patients’ HDL anti‐inflammatory, as the mean HII remained >1.0 UR, albeit, much closer to 1.0 UR than before. The inability of the statin to convert most patients with CHD HII from pro‐inflammatory to anti‐inflammatory motivates other approaches that could be added to statins, including D‐4F. Hence, as a complementary lipid‐altering approach, we present the results of superimposing 300‒500 mg D‐4F upon chronic statin therapy in our population with CHD or equivalent risk, at 8 h post D‐4F on day 13. We use a pair of arrows to depict the D‐4F results to distinguish the “placebo‐corrected” drop associated with D‐4F as the lower/yellow arrows, and the drop in the control group in the upper/pink arrows. This is an important distinction, as, on average, both groups experienced a drop from baseline. Importantly, patients took their usual preventive medications for atherosclerotic cardiovascular disease (ASCVD) at 2 h. Those who added the ASCVD medications to placebo had a 28% drop in HII. Those on 300 mg D‐4F + ASCVD medications had a 55% drop in HII, and after subtracting the 28% from the control group, a net drop of 27% attributable to D‐4F beyond that of the control group. Likewise, those on 500 mg D‐4F + ASCVD medications had a 49% drop in HII, and after subtracting the 28% drop from the control group, a net drop of 21% drop attributable to D‐4F. For both D‐4F doses shown, superimposing D‐4F upon the usual ASCVD regimen was sufficiently additive to robustly render the vast majority of our high‐risk patients’ HII anti‐inflammatory. Indeed, the average HII was suppressed to 0.57 UR on 300 mg and 0.63 UR on 500 mg. For comparison, others have shown interventions outside of lipid‐altering medications can lower HII. Roberts et al.13 demonstrated HII suppression among overweight/obese men with CHD risk factors from a high‐fiber/low‐fat diet and aerobic exercise, which lowered HII 21%. Charles‐Schoeman et al.14 demonstrated several intensive anti‐inflammatory regimens lowered HII 17‒18% among patients with rheumatoid arthritis: methotrexate monotherapy (MTX), MTX with etanercept (ETN), and triple therapy with MTX, sulfasalazine (SSZ), and hydroxychloroquine (HCQ).
The dose/response relationship between nominal D‐4F dose and HII did not seem linear, motiving exploratory analyses to better define the relationship. As in the single‐dose study,15 the HII response seemed dose‐responsive to a point. Specifically, ≥3.5 mg/IBM orally administered D‐4F significantly lowered HII vs. placebo. When data from healthier subjects were included, the floor was shallower/closer to baseline, suggesting D‐4F suppresses HII better when starting at a more pro‐inflammatory baseline (e.g., in sicker patients). In retrospect, constraining dosing to once‐daily probably limited D‐4F's efficacy. Conversely, multiple daily dosing or formulating D‐4F to extend delivery throughout the day might influence trough assays. Perhaps this would deepen the HII floor response further; alternatively, the floor may indicate there is simply a limit to how much apoA‐I or its mimetics can suppress HDL's inflammatory properties. In either case, future studies should broaden daily exposure (e.g., thrice daily).
Surprisingly, the dose/response relationship for HII was a function of oral peptide ingestion but not plasma exposure. This would not be expected of a peptide whose efficacy depends on delivery to plasma or downstream; rather, this evokes a presystemic mechanism, which nicely accords with mounting preclinical evidence discussed below. Thus, a novel aspect of the current report is the dose/response analysis supporting the concept that D‐4F peptide need not reach the plasma to influence HDL, provided enteric delivery is sufficient. This is noteworthy as most orally administered peptides, including D‐4F, achieve very low plasma delivery, tempering enthusiasm for development. Conversely, if it altered HDL before even reaching plasma, limited plasma exposure might not present an obstacle for D‐4F to improve this aspect of HDL function, thus encouraging the development of oral apoA‐I mimetic peptides. Patients with CHD or equivalent risk often have pro‐inflammatory HDL by HII,11 which also typifies conditions predisposing to CHD events: type 2 diabetes,18 chronic kidney disease,19 infection,23, 24 and postsurgery.25 Pro‐inflammatory HDL also accompanies advanced heart failure26 and severe pulmonary hypertension.27 The anatomy of the small intestine villus requires lipoproteins (including HDL) synthesized in the enterocytes to pass through the lamina propria of the villus to enter lymphatic or capillary circulation, both accessed in the lamina propria. A plethora of immune cells surrounds the lymphatics and capillary tuft in the lamina propria. Chattopadhyay et al.28 fed mice a Western diet, and found oxidized phospholipids were formed in the lamina propria and provoked an intense inflammatory response in this area of the villus. It is known that HDL assembled in areas of inflammation loses its anti‐inflammatory properties.29 Chattopadhyay et al.28 also demonstrated adding to the Western diet an apoA‐I mimetic peptide resembling 4F, Tg6F, dramatically suppressed oxidized phospholipid production and the related intense inflammatory response in the villus. At least 30% of circulating HDL originates in the small intestine.30 It is entirely possible that much of the dysfunctional HDL in our patients originated in their small intestine, and oral D‐4F acted similarly to Tg6F, improving HII by reducing the inflammatory response in the villi of the small intestine. If so, the proportion of HDL originating from the small intestine might dictate the maximal benefit achieved by delivering D‐4F peptide to the villus. Unless D‐4F stimulates enteric HDL synthesis, even if it rendered enteric HDL completely anti‐inflammatory, because ∼70% of total HDL originates outside the gut, there would be a hard limit to the maximal benefit of D‐4F. If so, adding D‐4F would improve HII up to a point, after which higher D‐4F doses would not improve HII appreciably. We speculate this could cause the floored dose response we saw, and simultaneously explain why plasma exposure seems irrelevant to efficacy. This hypothesis needs to be directly tested in future research.
Regardless of how, when, or where HDL becomes pro‐inflammatory, D‐4F has emerged as a potent tool to suppress HII. Indeed, D‐4F reversed pro‐inflammatory HDL from patients with CHD when added to patients’ plasma in vitro,31 dramatically decreased atherosclerosis in mice8 and rabbits,32 and with statin9 regressed atherosclerosis in mice. D‐4F was also anti‐inflammatory in a murine influenza model,16 reduced murine cerebral33 and renal34 arteriolar inflammation, and improved vascular function, reduced angiostatin, and decreased oxidative stress to improve angiogenic potential in the heart of tight‐skin mice.35 Our protocol required patients to take statins, so the drop in HII may not translate to statin‐averse/statin‐intolerant populations. At this point, preclinical evidence suggests apoA‐I mimetics improve atherosclerosis. However, we made no attempt to measure atherosclerosis in this study, so our data do not inform the larger question of whether D‐4F might prove atheroprotective in humans. That said, given acceptable safety/tolerability and the facility of chronic daily dosing, the theoretical potential for benefit favors further development, especially because oral peptide dosing has not proven a barrier to improving HII.
As expected, the ancillary pharmacodynamic parameters and clinical lipids did not change with D‐4F. This may reflect wide variations in concomitant ASCVD therapies (e.g., beta blockers, nitrates, angiotensin converting enzyme inhibitors, and aspirin). The unknown effect of medications on the other pharmacodynamic parameters may have further confounded the results. Thus, the decision to not standardize concomitant CHD or even diabetes mellitus medications may have limited the ability to detect changes in CHD biomarkers beyond HII.
D‐4F pharmacology
After 13 consecutive daily doses, oral D‐4F yielded low plasma levels (i.e., nanograms/mL of peptide). D‐4F was rapidly absorbed (Tmax 1–2 h across all doses), but minimally so. One woman on 500 mg had plasma concentrations ∼10‐fold above her cohorts at several time points. The remaining women achieved much higher plasma D‐4F levels than men, especially on 500 mg. This may reflect higher effective doses we gave women. Indeed, this study identified major differences in apparent D‐4F absorption between men and women. As this was not anticipated, we made no attempt to enroll similar numbers of women as men. Because the finding is based on a fairly small number of women, it may not generalize to all women, especially heavier women. Furthermore, an obvious shortcoming is that censoring overly influential but valid data post hoc biases our pharmacokinetic results toward underestimating plasma D‐4F exposure, and this is especially true of our data for women. The dose/response analyses were exploratory, and should be considered hypothesis‐generating. To test gender effects, future studies should enroll more women and dose D‐4F by IBM. Pharmacokinetics support future studies dosing D‐4F ≥3.5 mg/IBM to suppress HII, which would probably require doses over 500 mg, based on TIs.
Do plasma pharmacokinetics mediate HDL anti‐inflammatory benefits?
When this study was designed, it was presumed D‐4F altered HDL function upon systemic exposure, which also motivated subsequent studies involving i.v. 4F dosing. Remarkably, plasma D‐4F exposure did not influence the dose/response between D‐4F dose and HII, raising a serious challenge to the presumption that i.v. exposure is even helpful. If plasma levels had mediated the dose/response, this should dominate the dose/response model at the expense of the dose administered. On the contrary, plasma D‐4F seemed completely disconnected from HII suppression, not just in the present study, but also across all three human studies undertaken, justifying skepticism that systemic exposure is necessary or even helpful to improve HII. It is conceivable that the disconnect between plasma levels and altered HDL function could result from a major loss of D‐4F before reaching plasma. Even with first‐pass loss, one might expect even a weak relationship between plasma exposure and improved HDL function, but there was no hint of such. On the other hand, emerging preclinical evidence strongly supports a primarily intestinal site of action, as discussed below, which may explain why systemic exposure is uninformative. Thus, whereas a first‐pass effect is conceivable, we think a presystemic site of action is better supported by current evidence.
L‐4F differs from D‐4F in being composed of L‐amino acids. Injecting patients with CVD with s.c. or i.v. L‐4F at 0.03‒1.1 mg/kg yielded Cmax of 324‒10,062 ng/mL.36 Despite such prodigious plasma concentrations, L‐4F failed to improve their HII.36 In contrast, orally administered D‐4F peptide yielded profoundly lower Cmax, only 2.25‒67.3 ng/mL, yet significantly suppressed HII at doses ≥3.5 mg/kg. One possible explanation is that L‐4F and D‐4F are not comparable. We doubt this, as their efficacy was indistinguishable in vivo 32 when administered s.c. More likely, the initial premise that efficacy would require achieving a critical mass in plasma was misguided.
Where is the site of action for D‐4F to render HDL less inflammatory?
If orally dosed D‐4F peptide does not require plasma exposure to suppress HII, then where does it act? Navab et al.37 gave apoE‐null mice ≤45 mg/kg D‐4F orally or s.c. Plasma levels were ∼1,000‐fold higher after s.c. vs. oral administration. Regardless of route, 4.5–45 mg/kg suppressed HII and serum amyloid A, whereas lower doses did not. Moreover, 45 mg/kg by either route reduced aortic atherosclerosis ∼50% in apoE‐null mice on a Western diet. Tellingly, for each dose, fecal D‐4F recovery was similar whether the peptide was administered orally or s.c.,37 further affirming D‐4F dose rather than plasma exposure drives efficacy.
This study implicated the intestine as a major site of action for the peptide; moreover, this was regardless of administration route.37 Navab et al.38 affirmed this by feeding a Western diet to LDLR‐null mice, and dosing D‐4F orally or s.c. at 45 mg/kg/day. After s.c. injection, plasma D‐4F was 298‐fold higher than after the same oral dose, and hepatic levels were 96‐fold higher after injection compared to oral administration. However, small intestine levels only varied 1.66 ± 0.33‐fold.38 Arachidonic and linoleic acid metabolites were suppressed in the intestine, liver, and hepatic bile, irrespective of administration route, as was plasma serum amyloid A.38 Thus, even though systemic and oral dosing led to major differences in hepatic and plasma exposure, similar intestinal concentrations resulted in similar efficacy.
Zhao et al.39 tested an apoA‐I mimetic peptide with no sequence homology to D‐4F, in LDLR‐null mice on a Western diet. Dosing 40 mg/kg/day by i.p. injection, plasma levels were 22 ± 3 μM and whole‐aorta atherosclerosis was reduced 55%, and aortic‐sinus atherosclerosis was reduced by 61%. After administering 75 mg/kg/day of the peptide orally, none was detectable in plasma, and yet, whole‐aorta atherosclerosis decreased 58% and aortic‐sinus atherosclerosis decreased 71%,39 elegantly demonstrating plasma exposure of this apoA‐I mimetic peptide was dissociated from its ability to remediate atherosclerosis. Thus, animal studies imply it does not matter whether an apoA‐I mimetic is given by injection or infusion or orally, so long as the enterocytes receive a certain threshold.
Meriwether et al.40 recently showed i.v. administration of both D‐4F and L‐4F in mice selectively targeted the small intestine and the peptides were transported into the intestinal lumen. Intriguingly, the 4F peptide altered transintestinal cholesterol efflux, one of the major processes for clearing excess cholesterol from the body. Why did s.c. or i.v. L‐4F not prove anti‐inflammatory?36 The simplest answer would be that the doses were too low to achieve the requisite enteric exposure. Supporting this hypothesis, we found HII was most consistently improved when ≥3.5 mg/kg IBM oral D‐4F peptide was given, whereas the highest L‐4F dose in the studies by Watson et al.36 was 1.1 mg/kg and only a few subjects received this dose; most received 0.43 mg/kg.
Based on the preclinical evidence, our mechanistic hypothesis is that apoA‐I mimetics require sufficient enteric delivery to prevent villus inflammation and perhaps drive transintestinal cholesterol efflux. This could be achieved by an oral dose (e.g., ≥3.5 mg/IBM) or driving plasma systemic exposure so high it manages to reach the enterocyte from the other side. Logistically, oral dosing would be preferable to patients and its lower doses easier to supply. Regarding HII specifically, as before, D‐4F may dampen inflammation in the villi, thereby reducing the formation of dysfunctional HDL as the lipoprotein transits from the site of synthesis (the enterocyte) through the site of inflammation (the lamina propria of the villus) on its way into the circulation without D‐4F directly associating with HDL or even being absorbed. Alternatively, any D‐4F that does get absorbed may integrate with nascent HDL as it is assembled in the peri‐enterocytic space.
Whither ApoA‐I mimetics?
In an editorial accompanying the report by Zhao et al.,39 Wool, Reardon, and Getz41 offered that this body of work has “raised the exciting possibility of a future orally available agent targeted at improving HDL function, likely through a gut‐mediated mechanism of action.” We believe the data from the current paper add to the hope that such an agent can be developed, with the ultimate goal of developing a new approach to limit atherosclerosis.
Supporting information
Supplemental Text S1: Methodologic Details
Supplementary Text S2: Additional Results Details
Supplementary Figure S1: Calculation of HDL Inflammatory Index Incremental Area Under the Curve
Supplementary Figure S2: Effective D‐4F Exposure by Ideal Body Mass
Supplementary Figure S3: HDL Inflammatory Index in Relation to Plasma D‐4F Exposure
Supplementary Table S1: Exhaustive Enrollment Criteria
Supplementary Table S2: Selected Characteristics for Inliers vs Outlier
Supplementary Table S3: Regression Models of D‐4F Pharmacokinetics: D‐4F Area Under the Curve vs D‐4F Dose
Supplementary Table S4: Regression Models of D‐4F Pharmacodynamics
Supplementary Table S5: Baseline Characteristics for the BP‐01 Study
Acknowledgments
Project support for the clinical experiment was funded by Bruin Pharma, Inc. A.M.F. and M.N. are supported by 2P02 HL‐30568 and a network grant from the Leducq Foundation. R.L.D. received support from K23HL091130 from the NHLBI, and the UPenn clinical research facility received support from UL1RR024134 from the NCRR. The authors thank Jacob Levy and Maria Escobar for assistance with editing the manuscript. We also thank the nurses of the General Clinical Research Center and the study participants. The content is solely the responsibility of the authors and not sponsoring institutions.
Conflict of Interest
A.M.F. and M.N. are principals in Bruin Pharma and A.M.F. is an officer in Bruin Pharma.
Author Contributions
R.L.D., M.V., L.T.B., D.D., R.B.N., M.N., A.M.F., and D.J.R. wrote the manuscript. R.L.D., L.T.B., A.M.F., and D.J.R. designed the research. R.L.D., M.V., L.T.B., D.D., R.B.N., M.N., and D.J.R. performed the research. R.L.D. and A.M.F. analyzed the data.
References
- 1. Gordon, D.J. , Knoke, J. , Probstfield, J.L., Superko, R. & Tyroler, H.A. High‐density lipoprotein cholesterol and coronary heart disease in hypercholesterolemic men: the Lipid Research Clinics Coronary Primary Prevention Trial. Circulation 74, 1217–1225 (1986). [DOI] [PubMed] [Google Scholar]
- 2. Francis, M.C. & Frohlich, J.J. Coronary artery disease in patients at low risk–apolipoprotein AI as an independent risk factor. Atherosclerosis 155, 165–170 (2001). [DOI] [PubMed] [Google Scholar]
- 3. Plump, A.S. , Scott, C.J. & Breslow, J.L. Human apolipoprotein A‐I gene expression increases high density lipoprotein and suppresses atherosclerosis in the apolipoprotein E‐deficient mouse. Proc. Natl. Acad. Sci. USA 91, 9607–9611 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Benoit, P. et al Somatic gene transfer of human ApoA‐I inhibits atherosclerosis progression in mouse models. Circulation 99, 105–110 (1999). [DOI] [PubMed] [Google Scholar]
- 5. Miyazaki, A. et al Intravenous injection of rabbit apolipoprotein A‐I inhibits the progression of atherosclerosis in cholesterol‐fed rabbits. Arterioscler. Thromb. Vasc. Biol. 15, 1882–1888 (1995). [DOI] [PubMed] [Google Scholar]
- 6. Chyu, K.Y. & Shah, P.K. HDL/ApoA‐1 infusion and ApoA‐1 gene therapy in atherosclerosis. Front. Pharmacol. 6, 187 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Barter, P.J. & Rye, K.A. Targeting high‐density lipoproteins to reduce cardiovascular risk: what is the evidence? Clin. Ther. 37, 2716–2731 (2015). [DOI] [PubMed] [Google Scholar]
- 8. Navab, M. et al Oral administration of an Apo AI mimetic peptide synthesized from D‐amino acids dramatically reduces atherosclerosis in mice independent of plasma cholesterol. Circulation 105, 290–292 (2002). [DOI] [PubMed] [Google Scholar]
- 9. Navab, M. et al D‐4F and statins synergize to render HDL antiinflammatory in mice and monkeys and cause lesion regression in old apolipoprotein E‐null mice. Arterioscler. Thromb. Vasc. Biol. 25, 1426–1432 (2005). [DOI] [PubMed] [Google Scholar]
- 10. Navab, M. et al Oral D‐4F causes formation of pre‐beta high‐density lipoprotein and improves high‐density lipoprotein‐mediated cholesterol efflux and reverse cholesterol transport from macrophages in apolipoprotein E‐null mice. Circulation 109, 3215–3220 (2004). [DOI] [PubMed] [Google Scholar]
- 11. Ansell, B.J. et al Inflammatory/antiinflammatory properties of high‐density lipoprotein distinguish patients from control subjects better than high‐density lipoprotein cholesterol levels and are favorably affected by simvastatin treatment. Circulation 108, 2751–2756 (2003). [DOI] [PubMed] [Google Scholar]
- 12. Navab, M. et al Normal high density lipoprotein inhibits three steps in the formation of mildly oxidized low density lipoprotein: step 1. J. Lipid Res. 41, 1481–1494 (2000). [PubMed] [Google Scholar]
- 13. Roberts, C.K. , Ng, C. , Hama, S. , Eliseo, A.J. & Barnard, R.J. Effect of a short‐term diet and exercise intervention on inflammatory/anti‐inflammatory properties of HDL in overweight/obese men with cardiovascular risk factors. J. Appl. Physiol (1985) 101, 1727–1732 (2006). [DOI] [PubMed] [Google Scholar]
- 14. Charles‐Schoeman, C. et al Improvement of high‐density lipoprotein function in patients with early rheumatoid arthritis treated with methotrexate monotherapy or combination therapies in a randomized controlled trial. Arthritis Rheumatol. 69, 46–57 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bloedon, L.T. et al Safety, pharmacokinetics, and pharmacodynamics of oral apoA‐I mimetic peptide D‐4F in high‐risk cardiovascular patients. J. Lipid Res. 49, 1344–1352 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Van Lenten, B.J. et al Influenza infection promotes macrophage traffic into arteries of mice that is prevented by D‐4F, an apolipoprotein A‐I mimetic peptide. Circulation 106, 1127–1132 (2002). [DOI] [PubMed] [Google Scholar]
- 17. Navab, M. , Hama, S.Y. , Hough, G.P. , Subbanagounder, G. , Reddy, S.T. & Fogelman, A.M. A cell‐free assay for detecting HDL that is dysfunctional in preventing the formation of or inactivating oxidized phospholipids. J. Lipid Res. 42, 1308–1317 (2001). [PubMed] [Google Scholar]
- 18. Schillinger, M. et al Inflammation and carotid artery–risk for atherosclerosis study (ICARAS). Circulation 111, 2203–2209(2005). [DOI] [PubMed] [Google Scholar]
- 19. Lee, K.W. , Lip, G.Y. , Tayebjee, M. , Foster, W. & Blann, A.D. Circulating endothelial cells, von Willebrand factor, interleukin‐6, and prognosis in patients with acute coronary syndromes. Blood 105, 526–532 (2005). [DOI] [PubMed] [Google Scholar]
- 20. Devine, B.J. Clinical Pharmacy Case Studies, Case Number 25: Gentamicin Therapy. (ed. McCarron, M.M.) In: Drug Intelligence & Clinical Pharmacy. Annals of Pharmacotherapy http://journals.sagepub.com/doi/abs/10.1177/106002807400801104?journalCode=aopb (1974).
- 21. Belotti, F. , Deb, P. , Manning, W.G. & Norton, E.C. twopm: Two‐part models. Stata J [Internet]. http://www.stata-journal.com/article.html?article=st0368 (2015). Accessed 15 April 2017. [Google Scholar]
- 22. Burke, M.F. , Dunbar, R.L. & Rader, D.J. Could exercise metabolomics pave the way for gymnomimetics? Sci. Transl. Med. 2, 41ps35 (2010). [DOI] [PubMed] [Google Scholar]
- 23. Cruz, D. , Wastson, A. & Miller, C. Host‐derived oxidized phospholipids and HDL regulate innate immunity in human leprosy. J. Clin. Invest. 118, 2917–2928 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kelesidis, T. , Yang, O.O. , Currier, J.S. , Navab, K., Fogelman, A.M. & Navab, M. HIV‐1 infected patients with suppressed plasma viremia on treatment have pro‐inflammatory HDL. Lipids Health Dis. 10, 35 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Van Lenten, B.J. et al Anti‐inflammatory HDL becomes pro‐inflammatory during the acute phase response. Loss of protective effect of HDL against LDL oxidation in aortic wall cell cocultures. J. Clin. Invest. 96, 2758–2767 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim, J.B. et al Heart failure is associated with impaired anti‐inflammatory and antioxidant properties of high‐density lipoproteins. Am. J. Cardiol. 112, 1770–1777 (2013). [DOI] [PubMed] [Google Scholar]
- 27. Ross, D.J. et al Proinflammatory high‐density lipoprotein results from oxidized lipid mediators in the pathogenesis of both idiopathic and associated types of pulmonary arterial hypertension. Pulm. Circ. 5, 640–648 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chattopadhyay, A. et al Tg6F ameliorates the increase in oxidized phospholipids in the jejunum of mice fed unsaturated LysoPC or WD. J. Lipid Res. 57, 832–847 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hewing, B. et al Effects of native and myeloperoxidase‐modified apolipoprotein A‐I on reverse cholesterol transport and atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 34, 779–789 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Brunham, L.R. et al Intestinal ABCA1 directly contributes to HDL biogenesis in vivo. J. Clin. Invest. 116, 1052–1062 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Navab, M. et al Apolipoprotein A‐I mimetic peptides. Arterioscler. Thromb. Vasc. Biol. 25, 1325–1331 (2005). [DOI] [PubMed] [Google Scholar]
- 32. Van Lenten, B.J. et al Lipoprotein inflammatory properties and serum amyloid A levels but not cholesterol levels predict lesion area in cholesterol‐fed rabbits. J. Lipid Res. 48, 2344–2353 (2007). [DOI] [PubMed] [Google Scholar]
- 33. Buga, G.M. et al D‐4F decreases brain arteriole inflammation and improves cognitive performance in LDL receptor‐null mice on a Western diet. J. Lipid Res. 47, 2148–2160 (2006). [DOI] [PubMed] [Google Scholar]
- 34. Buga, G.M. et al D‐4F reduces EO6 immunoreactivity, SREBP‐1c mRNA levels, and renal inflammation in LDL receptor‐null mice fed a Western diet. J. Lipid Res. 49, 192–205 (2008). [DOI] [PubMed] [Google Scholar]
- 35. Weihrauch, D. et al Effects of D‐4F on vasodilation, oxidative stress, angiostatin, myocardial inflammation, and angiogenic potential in tight‐skin mice. Am. J. Physiol. Heart Circ.Physiol. 293, H1432–H1441 (2007). [DOI] [PubMed] [Google Scholar]
- 36. Watson, C.E. et al Treatment of patients with cardiovascular disease with L‐4F, an apo‐A1 mimetic, did not improve select biomarkers of HDL function. J. Lipid Res. 52, 361–373 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Navab, M. et al Intestine may be a major site of action for the apoA‐I mimetic peptide 4F whether administered subcutaneously or orally. J. Lipid Res. 52, 1200–1210 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Navab, M. et al D‐4F‐mediated reduction in metabolites of arachidonic and linoleic acids in the small intestine is associated with decreased inflammation in low‐density lipoprotein receptor‐null mice. J. Lipid Res. 53, 437–445 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhao, Y. et al In vivo efficacy of HDL‐like nanolipid particles containing multivalent peptide mimetics of apolipoprotein A‐I. J. Lipid Res. 55, 2053–2063 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Meriwether, D. et al Transintestinal transport of the anti‐inflammatory drug 4F and the modulation of transintestinal cholesterol efflux. J. Lipid Res. 57, 1175–1193 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wool, G.D. , Reardon, C.A. & Getz, G.S. Mimetic peptides of human apoA‐I helix 10 get together to lower lipids and ameliorate atherosclerosis: is the action in the gut? J. Lipid Res. 55, 1983–1985 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Text S1: Methodologic Details
Supplementary Text S2: Additional Results Details
Supplementary Figure S1: Calculation of HDL Inflammatory Index Incremental Area Under the Curve
Supplementary Figure S2: Effective D‐4F Exposure by Ideal Body Mass
Supplementary Figure S3: HDL Inflammatory Index in Relation to Plasma D‐4F Exposure
Supplementary Table S1: Exhaustive Enrollment Criteria
Supplementary Table S2: Selected Characteristics for Inliers vs Outlier
Supplementary Table S3: Regression Models of D‐4F Pharmacokinetics: D‐4F Area Under the Curve vs D‐4F Dose
Supplementary Table S4: Regression Models of D‐4F Pharmacodynamics
Supplementary Table S5: Baseline Characteristics for the BP‐01 Study