

Abstract
Anesthetic agents provide patient comfort and optimize conditions for surgical and procedural interventions. These agents have been shown to modulate autophagy, which is a cellular mechanism that maintains tissue homeostasis by degrading and recycling excess, aged, or dysfunctional proteins. However, it is not always clear if upregulated autophagy is beneficial or harmful. This review assesses the anesthetic effects on autophagy. In the vast majority of studies, anesthetic modulation of autophagy is beneficial for cell survival.
Keywords: apoptosis, autophagy, intravenous anesthetics, ischemic preconditioning, myocardium, neuroprotection, signaling molecules, volatile anesthetics
INTRODUCTION
Autophagy is a cellular mechanism that helps to maintain normal tissue homeostasis by degrading and recycling aged, excess, or dysfunctional proteins.1,2 However, excessive autophagy may lead to cellular death via induction of apoptosis (type I programmed cell death) or necrosis, or via a separate mechanism known as autophagic cell death (Type II programmed cell death).2,3,4,5 This process plays a key role in neurodegenerative disorders, cancers, cardiomyopathies, diabetes, liver diseases, autoimmune disorders, and infections.6,7 Additionally, there is evidence that autophagy decreases with aging, which may contribute to the development of age-related diseases.8
Anesthetic agents are widely used to provide patient comfort and optimize conditions for procedural or surgical interventions, and are an integral part of modern medicine. Agents used for general anesthesia are typically divided into volatile anesthetics, such as sevoflurane, desflurane, isoflurane, and halothane, and intravenous agents that include benzodiazepines, barbituates, local anesthetics, and opioids. Recently, there has been a growing body of literature on the effects of anesthetic agents on autophagy. This review will briefly describe autophagy and contrast it to apoptosis, provide a discussion on various commonly used anesthetics, and summarize current literature regarding anesthetic effects on autophagy.
AUTOPHAGY
Autophagy, derived from Greek for “self-eating”, is a cellular mechanism in which long-lived, dysfunctional, or excess proteins are degraded to maintain normal tissue homeostasis.1,2,6 Essentially, the cell “eats” itself to preserve energy and provide cellular housekeeping.6 Autophagy functions to 1) provide energy and 2) remove unwanted cellular material.8
In autophagy, a double-membraned structure known as an autophagosome nonspecifically sequesters the cytoplasmic content of a cell and then fuses with a lysosome.2,9 The lysosome is an organelle which allows for recycling of macromolecules by breaking down cellular structures with proteases and hydrolases into individual biochemical components, such as amino acids, fatty acids, sugars, and cholesterol.8 These may then be used to create new cellular structures.8
There are three main types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy.8 In macroautophagy, portions of the cytosol are sequestered inside a autophagosome, which then fuses with a lysosome.8 In microautophagy, the lysosomal membrane itself protrudes to isolate cytoplasmic contents as a single membrane vesicle.8 Macroautophagy and microautophagy may be selective or non-selective.3 Chaperone-mediated autophagy, on the other hand, is a selective pathway in which soluble cytosolic proteins are delivered to the lysosome after protein unfolding and translocation across the lysosomal membrane.3,8
Autophagy is regulated by various signaling pathways. Under normal conditions, autophagy occurs constitutively at basal levels in most cells, which is thought to promote cellular upkeep.2,9 In times of stress or starvation (such as hypoxia), this pathway is upregulated to promote cell survival.4,6,10 One mechanism of this is through 5′AMP-activated protein kinase (AMPK) activation.11 However, excessive and unregulated autophagy may lead to cellular death possibly due to induction of apoptosis or because of too much of the cytosolic contents and organelles of the cell being degraded.2,3,4,12 As such, autophagy must be tightly regulated, present at basal levels most of the time and be induced only when necessary.3 Target of rapamycin (TOR) kinase is a signaling control point that is active and inhibits autophagy during conditions that promote growth (such as nutrient availability).6 In turn, nutrient deprivation, such as during hypoxia, causes repression of TOR kinase.6
Autophagic flux refers to the rate of autophagic degradation, which may be regulated either up or down under various conditions.13 Common approaches to assess it include western blot analysis to measure the presence of specific autophagy related proteins, transmission electron microscope to evaluate presence of autophagosomes and autolysosomes, and fluorescence microscopy.13 Autophagy inhibition may occur in either early stage, during autophagosome formation or at later stages, during fusion between the autophagosome and lysosome.14 Inhibition during early stage autophagy may be done with 3-methyladenine (blocks the type III PI3 kinase complex) or beclin-1 or autophagy-related protein (Atg) 5 knockout with siRNAs, whereas later stage inhibition can be accomplished with chloroquine, bafilomycin A1, or Rab7 knockout.14
Apoptosis, first described by Kerr et al.15 in 1972, is widely known as programmed cell death.4,15 Morphologically, this occurs in two distinct stages: formation of apoptotic bodies followed by phagocytosis and cellular degradation.15 Hallmarks of apoptosis include chromatin condensation, DNA fragmentation, cellular swelling, and activation of specific proteases known as caspases.2,4,15 There are two apoptotic signaling cascade pathways, the intrinsic and extrinsic pathways.16 The intrinsic pathway is triggered from cellular stress, such as DNA damage, starvation, and hypoxia, whereas the extrinsic pathway is initiated through death receptor activation.16,17
Although autophagy and apoptosis are morphologically different processes, the two share several similarities. Both occur under conditions of cellular stress, although autophagy typically happens first to attempt to maintain cellular homeostasis.17,18 Several proteins play a role in both autophagy and apoptosis.16 These include Atgs, Bcl-2/beclin-1, caspases, p53, and FADD-like interleukin-1β-converting enzyme-inhibitory protein (FLIP).16 The Bcl-2/beclin-1 complex inhibits both autophagy and apoptosis.17 While Atgs are necessary for autophagy, Atg5 and Atg12 may induce apoptosis during stress, and in turn, apoptosis inhibits Atg5.17 Caspases and FLIP are involved in the death receptor pathway of apoptosis, and their activation blocks autophagy.17 p53 may promote proteins that trigger either the intrinsic or extrinsic pathway of apoptosis, but also downregulates autophagy via inactivating AMPK and activating mechanistic TOR (mTOR).16
ANESTHETIC AGENTS
Agents used for general anesthesia are divided into two categories: inhalational anesthetics and intravenous anesthetics. There are four components necessary to provide general anesthesia in humans: hypnosis, amnesia, immobility, and analgesia. while inhalational anesthetics may provide all of these components (albeit to different degrees), there is no single intravenous agent that may do the same. Currently, the four most commonly used inhalational anesthetics in the united states are nitrous oxide, isoflurane, sevoflurane, and desflurane. Isoflurane, sevoflurane, and desflurane are all halogenated hydrocarbons, and are derivatives of ether.19 The exact mechanism of action of volatile anesthetics is unknown, although they are thought to modify neuronal function by decreasing excitatory neurotransmission and/or increasing inhibitory neurotransmission through interactions with protein targets, such as γ-aminobutyric acid (GABA) receptor and voltage-gated sodium channels.20,21
Drugs that may be used for intravenous anesthesia include propofol, etomidate, ketamine, sodium thiopental, midazolam, and opioids. These are commonly used for induction of anesthesia but they may be used for maintenance of anesthesia as well. Etomidate, propofol, midazolam, and sodium thiopental produce anesthesia by enhancing the inhibitory neurotransmitter GABA in the central nervous system (CNS). Ketamine primarily antagonizes N-methyl-D-asparate (NMDA) receptors, the site of action of the excitatory neurotransmitter NMDA, but also acts on nicotinic receptors, muscarinic receptors, monoaminergic receptors, opioid receptors, voltage-gated sodium channels, and L-type calcium channels.22 Opioids work on opioid receptors to produce analgesia. These drugs are used in a variety of combinations to produce the desired effects.
A pubmed search was performed using keywords “anesthesia” and “autophagy” as well as “anesthesiology” and “autophagy.” Non-English texts were excluded. Additionally, relevant citations from texts were reviewed and included.
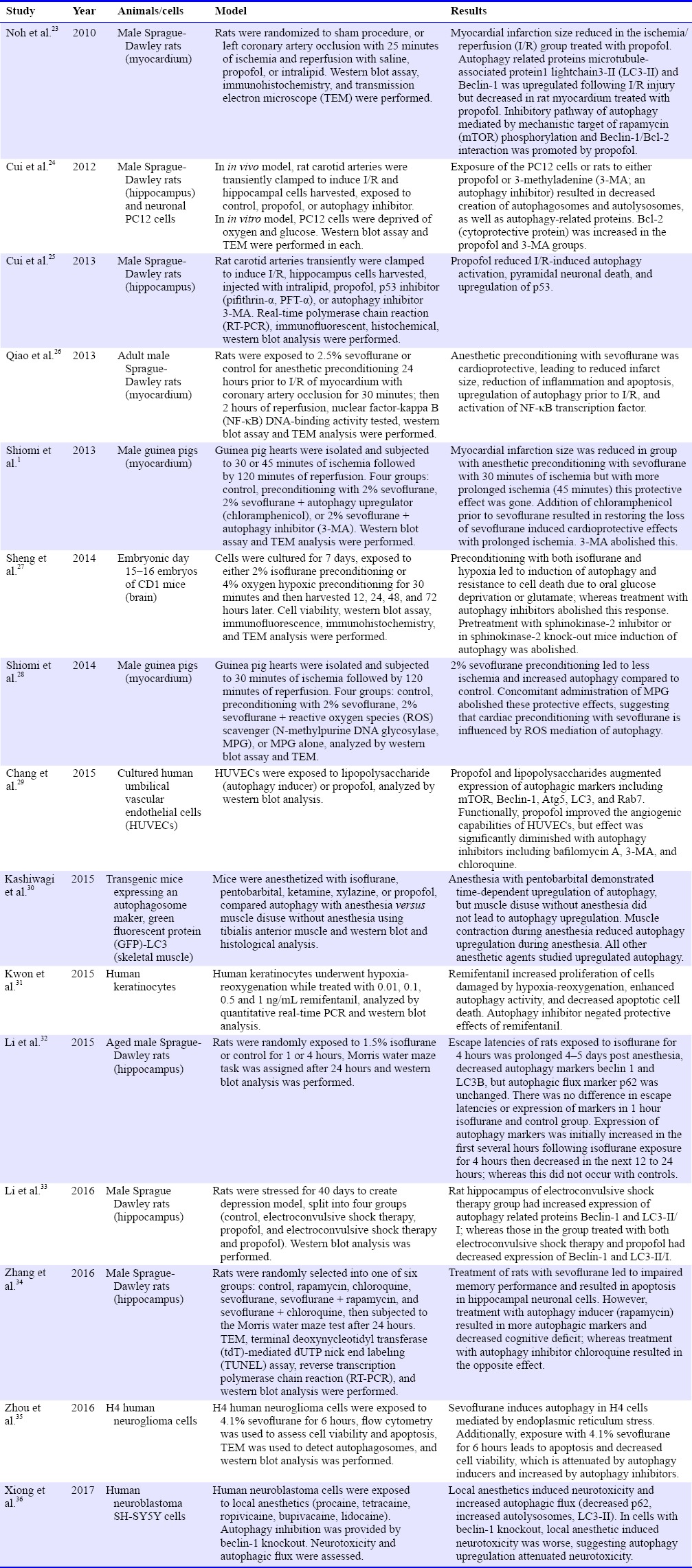
Since 2010, there are 15 studies linking anesthesia with autophagy. Among them, 13 studies were animal studies; whereas the other two used cultured human cells. Six studies utilized intravenous anesthetic agents, seven studies used volatile anesthetic agents, one investigation studied both volatile and intravenous anesthetic agents, and one study determined the effects of local anesthetics (Table 1).
Table 1.
Studies involving anesthesia and autophagy
ANESTHETIC EFFECTS ON CARDIAC AUTOPHAGY
There is a well-established literature that volatile anesthetics result in cardiac preconditioning,37,38,39,40,41 which is a phenomenon in which a short period of ischemia protects against a subsequent more drastic ischemic event.41,42 Similar to ischemic insult, volatile anesthetics induce cardiac preconditioning by activation of protein kinase C (PKC) and modulation of nitric oxide synthase, ultimately stimulating the mitochondrial adenosine triphosphate-sensitive potassium (mitoKATP) channel and triggering reactive oxygen species (ROS) release.41 Sevoflurane preconditioning also enhanced AMPK activation in an ROS-dependent manner, which provides cardioprotection.43 Anesthetic preconditioning, like ischemic reconditioning, provides both early and delayed protection as well.41 However, cardioprotection from anesthetic preconditioning is lost during prolonged ischemic periods.1
Several studies suggest that autophagy plays a role in anesthetic preconditioning of myocardium (Table 1). Qiao et al.26 in 2013 found that sevoflurane preconditioning resulted in not only nuclear factor-kappa B (NF-κB) activation, but also the formation of autophagosomes in rat myocardium, suggesting upregulated autophagy. There was also a concomitant decrease in inflammatory markers tumor necrosis factor-B (TNF-κB) and interleukin-1α (IL-1α), as well as apoptosis markers (active caspase-3). Administration of a NF-κB inhibitor parthenolide prior to anesthetic preconditioning decreased autophagy during delayed anesthetic preconditioning. However, Qiao's group did not use an autophagy inhibitor as part of their study. Shiomi et al.1 published two studies relating to autophagy and anesthetic preconditioning. In one study, they examined the involvement of autophagy in the loss of sevoflurane preconditioning effects during prolonged ischemia. They found that sevoflurane preconditioning in guinea pig hearts was lost during longer ischemic insult (45 minutes of ischemia compared to 30 minutes), but was restored with administration of chloramphenicol, an autophagy inducer. Administration of an autophagy inhibitor, on the other hand, abolished protective effects. In another study, Shiomi et al.28 found that upregulation of autophagy was mediated by ROS during sevoflurane preconditioning.
Only one study in our literature search used an intravenous agent to study ischemia/reperfusion injury in myocardium and autophagic cell death.23 In Noh et al's23 2010 study, they induced ischemia/reperfusion injury in rat myocardium and then randomized the rats to one of four groups, sham operation (no ischemia), and the following three groups had left coronary artery occlusion associated myocardial ischemia for 25 minutes followed by reperfusion and either infusion of saline, propofol, or intralipid. They found that myocardial infarction size was reduced in the ischemia/reperfusion group treated with propofol. The autophagy related proteins LC3-II and beclin-1 were upregulated following ischemia/reperfusion injury but decreased in the myocardium of rats treated with propofol. The inhibitory pathway of autophagy mediated by mTOR phosphorylation and beclin-1/Bcl-2 interaction was also promoted by propofol, suggesting that propofol attenuated autophagic cell death following ischemia/reperfusion.
These studies suggest that autophagy plays a role following ischemia/reperfusion injury in the myocardium. Interestingly, while all studies suggest protection with volatile anesthetics, the role of autophagy differs. With volatile anesthetic preconditioning, autophagy induction was found to be protective whereas Noh's study23 suggests that intravenous anesthetic agents attenuates autophagic cell death. This may be explained by autophagy playing two roles during ischemia/reperfusion; during ischemia, autophagy is protective as a result of AMPK activation and mTOR inhibition; whereas during reperfusion following ischemia autophagy can lead to autophagic cell death.23
Anesthetic effects on neuronal autophagy
Propofol has widely been thought of as a neuroprotective agent.24 Proposed mechanisms for this effect include anti-inflammatory/antioxidant effects against free radicals and reactive oxygen species.23,44 reduction in cerebral metabolic rate of oxygen,24,45 activation of GABAA receptors,46 calcium channel inhibition,25 and inhibition of NMDA subtype of glutamate receptors.47
In the adult hippocampus, studies have suggested that hypoxia-ischemia injury or electroconvulsive shock therapy (ECT) may result in autophagy.25,33 In Cui et al's24 2012 study, treatment of neuronal PC12 cells or rats with an autophagy inhibitor 3-methyladenine (3-MA) resulted in increased cell survival, decreased autophagy related proteins (LC3-II, beclin-1, and class III PI3K), and decreased amounts of autophagosomes and autolysosomes, suggesting that excessive autophagy is deleterious to hippocampal neurons. Additionally, Cui et al.24,25 demonstrated that administration of propofol also resulted in autophagy inhibition and increased cell survival in the hippocampus following hypoxia-ischemia injury. One potential explanation for this effect is inhibition of the p53 pathway by propofol, as a study showed that following ischemia/reperfusion in hippocampal cells, propofol, p53 inhibitor (pifithrin-γ, PFT-α) and autophagy inhibitor (3-methyladenine, 3-MA) all resulted in p53 and autophagy related protein inhibition following ischemia/reperfusion injury.25 ECT is a treatment modality used for depression. Its side effects include memory and learning impairment.48 Propofol has been shown to reduce these side effects.49 Li et al.33 in 2016 showed that propofol resulted in reduction of autophagy nd autophagy related proteins (beclin-1 and LC-II/I) in depressed rats that underwent ECT, suggesting that autophagy may be responsible for attenuation of memory and learning deficits after ECT.
Volatile anesthetics during adulthood are typically considered safe and may even confer neuroprotection in ischemia/reperfusion injury50; however, recently there has been a growing concern that volatile anesthetics used in animals and humans at the extremes of age may be detrimental. Some animal studies have suggested that inhalational anesthetics may increase the risk of postoperative cognitive dysfunction, a temporary or mid-term decline in cognitive function after surgery and anesthesia that primarily affects older individuals.32,34,51,52 The dual effects of volatiles may however depend upon the concentration of volatile anesthetics used53 or the duration of exposure,54 in which deleterious effects occur with high concentration and long duration of use, whereas protective effects are present at low concentration and short term use. Autophagy may play a role in these effects as discussed in the following paragraphs. In this connect, impaired autophagy has been implicated in the pathogenesis of several neurodegenerative diseases due to accumulation of autophagosomes in neurons, such as Alzheimer's disease, Huntington's disease, and Parkinson's disease.55
There are four recent studies27,32,34,35 that more closely examine the relationship between volatile anesthetics, autophagy, and their ties to neurocognitive disorders. Li et al.32 in 2015 conducted a study which investigated hippocampal autophagy of aged rats exposed to 1.5% isoflurane of various durations compared to oxygen. They found that exposure of aged rats to isoflurane for 4 hours resulted in impairment of spatial learning and memory, whereas this effect did not occur with only 1 hour of isoflurane exposure. Additionally, after 4 hours of exposure to isoflurane, there was a transient activation of autophagy evidenced by increased amounts of beclin-1 and LC3B-II, followed by a decrease in levels afterwards indicating autophagy suppression.
Zhang et al.34 in 2016 similarly studied the effects of sevoflurane on aged rats. Their study showed that sevoflurane in aged rats led to impaired memory and induced hippocampal neuronal apoptosis. These processes might involve autophagy. When rats were given an autophagy inhibitor (chloroquine), sevoflurane-induced neuronal apoptosis and memory impairment worsened, while conversely, treatment with an autophagy inducer, rapamycin, reduced the cognitive deficit.
Another study examining sevoflurane and possible neuronal cell damage was performed by Zhou et al.35 in 2016, in which they exposed H4 human neuroglioma cells to various concentrations of sevoflurane (0, 4.1%, or 8%) for 6 hours. They noted that sevoflurane resulted in increased apoptosis, increased expression of C/EBP homologous protein and glucose-regulated protein 78 (markers of endoplasmic reticulum stress), decreased cell viability, and autophagy activation. Autophagy activation with rapamycin led to decreased apoptosis, decreased markers of endoplasmic reticulum stress, and increased cell viability; whereas the opposite effects occurred with autophagy inhibition with 3-MA. Using 4-phenylbutyrate, an inhibitor of endoplasmic reticulum stress, also increased cell viability and decreased sevoflurane-induced autophagy and apoptosis.
Sheng et al.27 discovered in 2014 that autophagy from anesthetic or hypoxic preconditioning in mouse cortical neurons may be mediated by sphinosine kinase 2 (SPK2). Sheng's group demonstrated similar results from the aforementioned studies, and found that preconditioning with either isoflurane or hypoxia led to increased autophagy (evidenced by increased LC3-II/LC3-I ratio) and increased cell viability (resistance to oral glucose deprivation and glutamate). Autophagy plays a key role in the protective effects of preconditioning as autophagy inhibition with 3-MA abrogated the benefits of preconditioning. Additionally, preconditioning upregulated SPK2, and SPK2 inhibitors or SPK2 knockout prevented autophagy from preconditioning, suggesting that SPK2 is essential to autophagy and preconditioning.
These studies suggested that autophagy occurs in conjunction with administration of anesthesia, which indeed is important in modulating cellular survival. However, there is some conflicting data between these studies. In certain studies, autophagy induction resulted in increased cell survival,27,34 whereas in others, cell survival decreased.24,25,33,35 This is consistent with previous research that shows that autophagy is both pro-survival and pro-death, depending on cell type and cellular conditions.56
ANESTHETIC EFFECTS ON AUTOPHAGY IN OTHER CELL TYPES
Chang et al.29 in 2015 investigated the effects of propofol on vascular endothelium. Cultured human umbilical vascular endothelial cells (HUVECs) were exposed to either lipopolysaccharide, which is known to be an autophagy inducer in HUVECs, or propofol. Using western blot analysis, Chang et al.29 found that both propofol and lipopolysaccharides augmented the expression of autophagic markers including mTOR, Beclin-1, Atg5, LC3, and Rab7. The group also showed that propofol improved the angiogenic capabilities of HUVECs. However, this effect was significantly diminished with autophagy inhibitors including bafilomycin A, 3-MA, and chloroquine.
Either prolonged general anesthesia or sedation leads to skeletal muscle atrophy.30 Kashiwagi et al.30 in 2015 studied whether anesthesia or muscle disuse was responsible for the atrophy. They anesthetized mice with a variety of anesthetic agents (pentobarbital, ketamine, ketamine and xylazine combined, isoflurane, and propofol) and detected upregulation of autophagy. Electrical stimulation of muscle during anesthesia attenuated autophagy. However, autophagy was not present when they immobilized muscle by denervation without anesthesia. Their study suggests that both anesthesia and muscle inactivity may be responsible for skeletal muscle autophagy upregulation.
Kwon et al.31 studied effects of remifentanil on human keratinocytes and hypoxia-reoxygenation injury. Human keratinocytes underwent hypoxic injury by being cultured under 1% oxygen tension for 24 hours, exposed to several different concentrations of remifentanil, then reoxygenated over 12 hours. Their group found that remifentanil treatment resulted in autophagy activation, increased keratinocyte proliferation, and decreased apoptosis. 3-MA inhibited autophagy and prevented the protective actions of remifentanil. Their results suggest that remifentanil is protective against hypoxia-reoxygenation and that these effects were mediated through increased autophagy.
Xiong et al.36 in 2017 examined local anesthetic agents-induced neurotoxicity. They used both ester (procaine and tetracaine) and amide-type (bupivacaine, lidocaine and ropivicaine) on human neuroblastoma cells and assessed neurotoxicity and presence of autophagy. Additionally, they inhibited autophagy by transfecting human neuroblastoma cells with siRNA beclin-1, causing knockout of beclin-1. They found that local anesthetics induced neurotoxicity and increased autophagic flux (evidenced by decreased p62, increased autolysosomes, LC3-II). When autophagy was inhibited, local anesthetic-induced neurotoxicity was greater, suggesting that autophagy upregulation attenuated local anesthetic induced neurotoxicity.
CONCLUSION
Autophagy is a cellular mechanism that maintains cellular homeostasis. This is a process that must be closely regulated as too much or too little autophagy may result in cellular death. Dysregulated autophagy may play a role in many different disease processes.6,7 Anesthetic agents, both volatiles and intravenous, are shown to modulate autophagic flux. However, unlike apoptosis, it is not always clear whether upregulated autophagy is good or bad. In the vast majority of studies, which included in vitro and in vivo studies, anesthetic modulation of autophagy is beneficial. The only study where this was not the case was Kashiwagi's,54 in which anesthesia resulted in upregulated autophagy and skeletal muscle atrophy. In some cases, upregulated autophagy was beneficial, whereas in others the opposite is true. Possible explanations include the degree to which autophagy is upregulated, or during when in the autophagy cycle regulation occurs. The mechanism by which anesthetic agents are able to affect autophagic flux is not entirely clear. This may be due to direct cause-effect or indirect effects mediated via other signaling molecules.
There are ample areas for future research. There is a growing area of research connecting autophagy with many disease processes, including neurodegenerative disorders, cancers, cardiomyopathies, diabetes, liver conditions, autoimmune disorders, and infections.6,7 Given that anesthetics may modulate autophagy, anesthetics may alter these disease processes via autophagy as well. Anesthetic modulation may depend on the amount or duration of anesthesia delivered or type of anesthetic agent. In instances where autophagy modulation is beneficial, in the future, autophagy inhibitors or inducers may be given clinically. Additionally, it is not known what the long-term effects of anesthetics are on autophagic flux, as all the studies in this review were over periods of time lasting days at the most. However, we must be cautious in applying data from animal studies and in vitro studies to clinical practice. Nonetheless, autophagy and anesthetic modulation of autophagy are an exciting area for future research.
Footnotes
Conflicts of interest
None declared.
Plagiarism check
Checked twice by iThenticate.
Peer review
Externally peer reviewed.
Open peer reviewer
Lei Huang, Loma Linda University, USA.
Funding: This study was supported by the Robert M. Epstein Professorship endowment, University of Virginia, Charlottesville, VA, USA.
REFERENCES
- 1.Shiomi M, Miyamae M, Takemura G, et al. Induction of autophagy restores the loss of sevoflurane cardiac preconditioning seen with prolonged ischemic insult. Eur J Pharmacol. 2014;724:58–66. doi: 10.1016/j.ejphar.2013.12.027. [DOI] [PubMed] [Google Scholar]
- 2.Martinet W, De Meyer GR. Autophagy in atherosclerosis. Curr Atheroscler Rep. 2008;10:216–223. doi: 10.1007/s11883-008-0034-y. [DOI] [PubMed] [Google Scholar]
- 3.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yousefi S, Simon HU. Apoptosis regulation by autophagy gene 5. Crit Rev Oncol Hematol. 2007;63:241–244. doi: 10.1016/j.critrevonc.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 5.Ginet V, Spiehlmann A, Rummel C, et al. Involvement of autophagy in hypoxic-excitotoxic neuronal death. Autophagy. 2014;10:846–860. doi: 10.4161/auto.28264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12. doi: 10.1002/path.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jung KT, Lim KJ. Autophagy: can it be a new experimental research method of neuropathic pain? Korean J Pain. 2015;28:229–230. doi: 10.3344/kjp.2015.28.4.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cuervo AM. Autophagy and aging: keeping that old broom working. Trends Genet. 2008;24:604–612. doi: 10.1016/j.tig.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mizushima N, Klionsky DJ. Protein turnover via autophagy: implications for metabolism. Annu Rev Nutr. 2007;27:19–40. doi: 10.1146/annurev.nutr.27.061406.093749. [DOI] [PubMed] [Google Scholar]
- 10.Kuma A, Hatano M, Matsui M, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432:1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 11.Meley D, Bauvy C, Houben-Weerts JH, et al. AMP-activated protein kinase and the regulation of autophagic proteolysis. J Biol Chem. 2006;281:34870–34879. doi: 10.1074/jbc.M605488200. [DOI] [PubMed] [Google Scholar]
- 12.Scott RC, Juhász G, Neufeld TP. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr Biol. 2007;17:1–11. doi: 10.1016/j.cub.2006.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Loos B, du Toit A, Hofmeyr JH. Defining and measuring autophagosome flux-concept and reality. Autophagy. 2014;10:2087–2096. doi: 10.4161/15548627.2014.973338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thorburn A. Apoptosis and autophagy: regulatory connections between two supposedly different processes. Apoptosis. 2008;13:1–9. doi: 10.1007/s10495-007-0154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li M, Gao P, Zhang J. Crosstalk between autophagy and apoptosis: potential and emerging therapeutic targets for cardiac diseases. Int J Mol Sci. 2016;17:332. doi: 10.3390/ijms17030332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu G, Pei F, Yang F, et al. Role of autophagy and apoptosis in non-small-cell lung cancer. Int J Mol Sci. 2017;18:367. doi: 10.3390/ijms18020367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mariño G, Niso-Santano M, Baehrecke EH, Kroemer G. Self-consumption: the interplay of autophagy and apoptosis. Nat Rev Mol Cell Biol. 2014;15:81–94. doi: 10.1038/nrm3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chiao S, Zuo Z. A double-edged sword: volatile anesthetic effects on the neonatal brain. Brain Sci. 2014;4:273–294. doi: 10.3390/brainsci4020273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herold KF, Sanford RL, Lee W, et al. Volatile anesthetics inhibit sodium channels without altering bulk lipid bilayer properties. J Gen Physiol. 2014;144:545–560. doi: 10.1085/jgp.201411172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Campagna JA, Miller KW, Forman SA. Mechanisms of actions of inhaled anesthetics. N Engl J Med. 2003;348:2110–2124. doi: 10.1056/NEJMra021261. [DOI] [PubMed] [Google Scholar]
- 22.Kohrs R, Durieux ME. Ketamine: teaching an old drug new tricks. Anesth Analg. 1998;87:1186–1193. doi: 10.1097/00000539-199811000-00039. [DOI] [PubMed] [Google Scholar]
- 23.Noh HS, Shin IW, Ha JH, Hah YS, Baek SM, Kim DR. Propofol protects the autophagic cell death induced by the ischemia/reperfusion injury in rats. Mol Cells. 2010;30:455–460. doi: 10.1007/s10059-010-0130-z. [DOI] [PubMed] [Google Scholar]
- 24.Cui D, Wang L, Qi A, Zhou Q, Zhang X, Jiang W. Propofol prevents autophagic cell death following oxygen and glucose deprivation in PC12 cells and cerebral ischemia-reperfusion injury in rats. PLoS One. 2012;7:e35324. doi: 10.1371/journal.pone.0035324. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Cui DR, Wang L, Jiang W, Qi AH, Zhou QH, Zhang XL. Propofol prevents cerebral ischemia-triggered autophagy activation and cell death in the rat hippocampus through the NF-κB/p53 signaling pathway. Neuroscience. 2013;246:117–132. doi: 10.1016/j.neuroscience.2013.04.054. [DOI] [PubMed] [Google Scholar]
- 26.Qiao S, Xie H, Wang C, Wu X, Liu H, Liu C. Delayed anesthetic preconditioning protects against myocardial infarction via activation of nuclear factor-kappaB and upregulation of autophagy. J Anesth. 2013;27:251–260. doi: 10.1007/s00540-012-1494-3. [DOI] [PubMed] [Google Scholar]
- 27.Sheng R, Zhang TT, Felice VD, et al. Preconditioning stimuli induce autophagy via sphingosine kinase 2 in mouse cortical neurons. J Biol Chem. 2014;289:20845–20857. doi: 10.1074/jbc.M114.578120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shiomi M, Miyamae M, Takemura G, et al. Sevoflurane induces cardioprotection through reactive oxygen species-mediated upregulation of autophagy in isolated guinea pig hearts. J Anesth. 2014;28:593–600. doi: 10.1007/s00540-013-1755-9. [DOI] [PubMed] [Google Scholar]
- 29.Chang CY, Chen PH, Lu SC, et al. Propofol-enhanced autophagy increases motility and angiogenic capacity of cultured human umbilical vascular endothelial cells. Life Sci. 2015;142:49–59. doi: 10.1016/j.lfs.2015.10.014. [DOI] [PubMed] [Google Scholar]
- 30.Kashiwagi A, Hosokawa S, Maeyama Y, et al. Anesthesia with Disuse Leads to Autophagy Up-regulation in the Skeletal Muscle. Anesthesiology. 2015;122:1075–1083. doi: 10.1097/ALN.0000000000000561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kwon JY, Park BS, Kim YH, et al. Remifentanil protects human keratinocytes against hypoxia-reoxygenation injury through activation of autophagy. PLoS One. 2015;10:e0116982. doi: 10.1371/journal.pone.0116982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li ZQ, Li LX, Mo N, et al. Duration-dependent regulation of autophagy by isoflurane exposure in aged rats. Neurosci Bull. 2015;31:505–513. doi: 10.1007/s12264-015-1549-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li P, Hao XC, Luo J, Lv F, Wei K, Min S. Propofol mitigates learning and memory impairment after electroconvulsive shock in depressed rats by inhibiting autophagy in the hippocampus. Med Sci Monit. 2016;22:1702–1708. doi: 10.12659/MSM.897765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang X, Zhou Y, Xu M, Chen G. Autophagy is involved in the sevoflurane anesthesia-induced cognitive dysfunction of aged rats. PLoS One. 2016;11:e0153505. doi: 10.1371/journal.pone.0153505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou YF, Wang QX, Zhou HY, Chen G. Autophagy activation prevents sevoflurane-induced neurotoxicity in H4 human neuroglioma cells. Acta Pharmacol Sin. 2016;37:580–588. doi: 10.1038/aps.2016.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiong J, Kong Q, Dai L, et al. Autophagy activated by tuberin/mTOR/p70S6K suppression is a protective mechanism against local anaesthetics neurotoxicity. J Cell Mol Med. 2017;21:579–587. doi: 10.1111/jcmm.13003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cason BA, Gamperl AK, Slocum RE, Hickey RF. Anesthetic-induced preconditioning: previous administration of isoflurane decreases myocardial infarct size in rabbits. Anesthesiology. 1997;87:1182–1190. doi: 10.1097/00000542-199711000-00023. [DOI] [PubMed] [Google Scholar]
- 38.Kersten JR, Schmeling TJ, Pagel PS, Gross GJ, Warltier DC. Isoflurane mimics ischemic preconditioning via activation of K(ATP) channels: reduction of myocardial infarct size with an acute memory phase. Anesthesiology. 1997;87:361–370. doi: 10.1097/00000542-199708000-00024. [DOI] [PubMed] [Google Scholar]
- 39.Yvon A, Hanouz JL, Haelewyn B, et al. Mechanisms of sevoflurane-induced myocardial preconditioning in isolated human right atria in vitro. Anesthesiology. 2003;99:27–33. doi: 10.1097/00000542-200307000-00008. [DOI] [PubMed] [Google Scholar]
- 40.Zaugg M, Lucchinetti E, Uecker M, Pasch T, Schaub MC. Anaesthetics and cardiac preconditioning. Part I. Signalling and cytoprotective mechanisms. Br J Anaesth. 2003;91:551–565. doi: 10.1093/bja/aeg205. [DOI] [PubMed] [Google Scholar]
- 41.Swyers T, Redford D, Larson DF. Volatile anesthetic-induced preconditioning. Perfusion. 2014;29:10–15. doi: 10.1177/0267659113503975. [DOI] [PubMed] [Google Scholar]
- 42.Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124. [DOI] [PubMed] [Google Scholar]
- 43.Lamberts RR, Onderwater G, Hamdani N, et al. Reactive oxygen species-induced stimulation of 5'AMP-activated protein kinase mediates sevoflurane-induced cardioprotection. Circulation. 2009;120:S10–15. doi: 10.1161/CIRCULATIONAHA.108.828426. [DOI] [PubMed] [Google Scholar]
- 44.Braz MG, Braz LG, Freire CM, et al. Isoflurane and propofol contribute to increasing the antioxidant status of patients during minor elective surgery: a randomized clinical study. Medicine (Baltimore) 2015;94:e1266. doi: 10.1097/MD.0000000000001266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vasileiou I, Xanthos T, Koudouna E, et al. Propofol: a review of its non-anaesthetic effects. Eur J Pharmacol. 2009;605:1–8. doi: 10.1016/j.ejphar.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 46.Hollrigel GS, Toth K, Soltesz I. Neuroprotection by propofol in acute mechanical injury: role of GABAergic inhibition. J Neurophysiol. 1996;76:2412–2422. doi: 10.1152/jn.1996.76.4.2412. [DOI] [PubMed] [Google Scholar]
- 47.Orser BA, Bertlik M, Wang LY, MacDonald JF. Inhibition by propofol (2,6 di-isopropylphenol) of the N-methyl-D-aspartate subtype of glutamate receptor in cultured hippocampal neurones. Br J Pharmacol. 1995;116:1761–1768. doi: 10.1111/j.1476-5381.1995.tb16660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pigot M, Andrade C, Loo C. Pharmacological attenuation of electroconvulsive therapy--induced cognitive deficits: theoretical background and clinical findings. J ECT. 2008;24:57–67. doi: 10.1097/YCT.0b013e3181616c14. [DOI] [PubMed] [Google Scholar]
- 49.Li X, Li W, Luo J, et al. Effects of propofol on the activation of hippocampal CaMKIIalpha in depressed rats receiving electroconvulsive therapy. J ECT. 2012;28:242–247. doi: 10.1097/YCT.0b013e31826140c7. [DOI] [PubMed] [Google Scholar]
- 50.Lai Z, Zhang L, Su J, Cai D, Xu Q. Sevoflurane postconditioning improves long-term learning and memory of neonatal hypoxia-ischemia brain damage rats via the PI3K/Akt-mPTP pathway. Brain Res. 2016;1630:25–37. doi: 10.1016/j.brainres.2015.10.050. [DOI] [PubMed] [Google Scholar]
- 51.Culley DJ, Baxter MG, Crosby CA, Yukhananov R, Crosby G. Impaired acquisition of spatial memory 2 weeks after isoflurane and isoflurane-nitrous oxide anesthesia in aged rats. Anesth Analg. 2004;99:1393–1397. doi: 10.1213/01.ANE.0000135408.14319.CC. table of contents. [DOI] [PubMed] [Google Scholar]
- 52.Culley DJ, Baxter MG, Yukhananov R, Crosby G. Long-term impairment of acquisition of a spatial memory task following isoflurane-nitrous oxide anesthesia in rats. Anesthesiology. 2004;100:309–314. doi: 10.1097/00000542-200402000-00020. [DOI] [PubMed] [Google Scholar]
- 53.Zhou X, Li W, Chen X, et al. Dose-dependent effects of sevoflurane exposure during early lifetime on apoptosis in hippocampus and neurocognitive outcomes in Sprague-Dawley rats. Int J Physiol Pathophysiol Pharmacol. 2016;8:111–119. [PMC free article] [PubMed] [Google Scholar]
- 54.Xiao H, Liu B, Chen Y, Zhang J. Learning, memory and synaptic plasticity in hippocampus in rats exposed to sevoflurane. Int J Dev Neurosci. 2016;48:38–49. doi: 10.1016/j.ijdevneu.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 55.Kiriyama Y, Nochi H. The function of autophagy in neurodegenerative diseases. Int J Mol Sci. 2015;16:26797–26812. doi: 10.3390/ijms161125990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu M, Zhang HL. Death and survival of neuronal and astrocytic cells in ischemic brain injury: a role of autophagy. Acta Pharmacol Sin. 2011;32:1089–1099. doi: 10.1038/aps.2011.50. [DOI] [PMC free article] [PubMed] [Google Scholar]