

Abstract
Vibrio cholerae is responsible for the cause of severe life-threatening infection known as cholera. The study aimed to analyze the genetic make-up of V. cholerae O139 isolates from India and compare its phylogeny with the global strains. The genome data revealed that all isolates were of same sequence type (ST69) which belongs to seventh pandemic clone, with same virulence gene profile and, antimicrobial resistance gene profile except for two isolates. No known CRISPR repeats were identified in any of these isolates. Three different phages were identified among the isolates. All the isolates were found to harbour int SXT and seventh pandemic-specific gene (VC2346). Results from this study enhance our understanding on the persistence of ST69 V. cholerae O139 over 20 years.
Keywords: Vibrio cholerae O139, ST69, MLST, Antimicrobial resistance
Introduction
Historically, only the O1 serogroup of V. cholerae has been the causative agent of epidemic cholera. In late 1992, in India and Bangladesh, a novel serogroup designated V. cholerae O139 emerged, replacing V. cholerae O1, and gave rise to major cholera outbreaks [1]. Based on the whole genome phylogenies of V. cholerae, the relation of isolates responsible for global cholera could be understood [2]. Nearly 141 draft genomes of V. cholerae O139 are accessible in the NCBI genome projects. Evolution and adaptation mechanism of this pathogen can be demonstrated based on genomic diversity of clinical isolates from various geographical regions and timescale [3]. The current study aims to perform and report the whole genome shotgun sequences of V. cholerae O139 isolates for the first time in India. The genomes reported here will help to better understand the evolution and endemicity of cholera in India.
Methods
Isolates selection
Clinical strains of V. cholerae O139 obtained from the past 20 years (FC1105-2005, FC1225-2001, FC1341-2002, FC1384-2000, FC1817-1994, FC1877-1995, FC2271-1997, FC2273-1998, FC3611a-1999 and FC3611b-1997) were revived from the repository at the Department of Clinical Microbiology, Christian Medical College, Vellore, India. Isolates were identified with standard biochemical methods and were confirmed by agglutination with O139 antisera raised in-house. The genomic DNA was extracted using Qiagen automated DNA extraction method with QIAsymphony DSP Virus/Pathogen Mini Kit (QIAsymphony, Qiagen, Germany). All 10 V. cholerae O139 was confirmed based on the 16S rRNA gene, by amplification, sequencing, and BLAST against the NCBI database.
Next generation sequencing
The whole genome shotgun sequencing for the V. cholerae O139 isolates was performed using Ion Torrent (PGM, Life Technologies) with 400-bp read chemistry (Life Technologies, Carlsbad, CA). Genomic DNA libraries were prepared using Ion Plus Fragment Library Kit; (Life Technologies) according to manufacturer’s instructions. AMPure beads were used to purify the genomic libraries and their concentrations were determined using the Qubit 3.0 fluorimeter (Invitrogen, Merelbeke, Belgium). Emulsion PCR was performed on pooled libraries (Ion One Touch Hi-Q 400 Template Kit v2 DL Kit; Life Technologies), and template-positive Ion Sphere particles were enriched using Dynabeads Myone streptavidin C1 beads. Finally, pooled samples were loaded on Ion 318V2 chip for sequencing.
Genome assembly and annotation
The generated whole genome data were assembled de novo using Assembler SPAdes v.5.0.0.0 embedded in Torrent suite server v.5.0.5. The genome sequence was annotated using PATRIC (Pathosystems Resource Integration Center), the bacterial bioinformatics database and analysis resource (http://www.patricbrc.org) [4], and the NCBI Prokaryotic Genome Automatic Annotation Pipeline (PGAAP) (http://www.ncbi.nlm.nih.gov/genomes/static/Pipeline.html).
Downstream genome analysis
The whole genome data were analyzed using open access tools at Center for Genomic Epidemiology (CGE) web-based server. Sequence types for the study isolates were determined using MLST 1.8 (https://cge.cbs.dtu.dk//services/MLST/), and antimicrobial resistance were identified using ResFinder 2.1 (https://cge.cbs.dtu.dk//services/ResFinder/) with the 90% threshold for identity and with 60% of minimum length coverage. Genome data was screened for virulence genes through PATRIC database. The presence of plasmids was analyzed using PlasmidFinder 1.3 (https://cge.cbs.dtu.dk//services/PlasmidFinder/) with 95% threshold for identity. The study isolates were further screened for the presence of prophage sequences within the genome using PHAST (PHAge Search Tool) (http://phast.wishartlab.com) [5].
The web-based MyDbFinder 1.0 (https://cge.cbs.dtu.dk/services/MyDbFinder/) was used, in silico to determine the SXT elements (int SXT), specific integrase genes of class 1 integron (intI gene) and specific gene (VC2346) of the seventh pandemic strains with a selected threshold equal to 98% identity as previously described [6].
Phylogenetic analyses
The phylogenetic analysis was performed based on MLST housekeeping genes (adk, gyrB, mdh, metE, pntA, purM, pyrC) using the MEGA7 software [7]. The evolutionary history was inferred using the Neighbor-Joining method. The evolutionary distances were computed using the Maximum Composite Likelihood method. goEBURST analysis for the V. cholerae O139 strains was performed using PHYLOViZ 1.1 software [8] to identify the relation between study isolates and the global strains with sequence types of V. cholerae (ST168-KC895170.1, ST169-KC895206.1, ST170-KJ657169.1, ST182-KC895159.1, ST429-KC895162.1, N16961-AE003852.1, M2140_ST70-CP013315.1, M802_ST71-LT907989.1, M1616_ST73-KC895170.1, M796_ST75-DQ316973.1).
Results and discussion
Genome features, virulence and antimicrobial resistance determinants
Assembly of the raw reads of the isolates ranged from 69 to 196X coverage (Table 1). NGS revealed that all isolates belong to same sequence type ST69. Similarly in a study by Siriphap et al. [6], most of the seventh pandemic isolates belonged to ST69 and all the clinical strains of O1 and O139 were highly conserved to ST69. Literature study reveals that the Bay of Bengal as a major hub connecting the spread of cholera around the globe and has continually being evolved in South Asia, later spread around the globe in at least three independent but overlapping waves. Besides V. cholerae serogroup O1 is widespread throughout the globe, whereas serogroup O139 is almost solely confined to Asia [9].The probable transmission of V. cholerae O139 sequence types (ST69, ST187, ST161, ST162 and ST163) that reported elsewhere from South Asia was depicted in Fig. 1.
Table 1.
Genome characteristics of Vibrio cholerae O139 Indian isolates
| S. no | Isolate | Year of isolation | GC content (%) | Coverage (X) | CDS | rRNA | tRNA | ARDB | CARD | Sequence type | Confirmed CRISPR | IS elements | Antimicrobial resistance genes | Plasmids | Accession nos. |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | FC1105 | 2005 | 47.5 | 84.7 | 3785 | 12 | 71 | 6 | 17 | ST69 | − | IS3-ISVch4 IS200-IS1004 ISAs1-IS1358 IS481 Tn3-ISShfr9 IS66-ISPsy43 IS5-ISVch8 IS5-ISVch5 IS3-ISVch4 Tn3-ISPsy42 |
strA, strB, floR, catB9, sul2 | − | NGQZ00000000 |
| 2 | FC1225 | 2001 | 47.5 | 81.7 | 3796 | 13 | 71 | 2 | 14 | ST69 | − | IS3-ISVch4 IS481-ISVch1 Tn3-ISShfr9 IS200-IS1004 ISAs1-IS1358 IS5-ISVch5 |
catB9 | − | NGQY00000000 |
| 3 | FC1341 | 2002 | 47.5 | 74.7 | 3763 | 12 | 72 | 2 | 14 | ST69 | − | IS3-ISVch4 IS481-ISVch1 Tn3-ISShfr9 IS200-IS1004 ISAs1-IS1358 IS5-ISVch5 |
catB9 | − | NGQX00000000 |
| 4 | FC1384 | 2000 | 47.5 | 96 | 3797 | 12 | 77 | 7 | 18 | ST69 | − | IS3-ISVch4 IS481-ISVch1 Tn3-ISShfr9 IS200-IS1004 ISAs1-IS1358 IS5-ISVch5 |
strA, strB, floR, catB9, sul2 | − | NGQW00000000 |
| 5 | FC1817 | 1994 | 47.5 | 69.1 | 3845 | 11 | 75 | 6 | 17 | ST69 | − | IS3-ISVch4 IS481-ISVch1 Tn3-ISShfr9 IS200-IS1004 ISAs1-IS1358 IS5-ISVch5 |
strA, strB, floR, catB9, sul2 | − | NGQV00000000 |
| 6 | FC1877 | 1995 | 47.5 | 82.4 | 3750 | 12 | 73 | 6 | 17 | ST69 | − | IS3-ISVch4 IS481-ISVch1 Tn3-ISShfr9 IS200-IS1004 ISAs1-IS1358 IS5-ISVch5 |
strA, strB, floR, catB9, sul2 | − | NGQU00000000 |
| 7 | FC2271 | 1997 | 47.5 | 80.7 | 3785 | 14 | 75 | 6 | 17 | ST69 | − | IS3-ISVch4 IS481-ISVch1 Tn3-ISShfr9 IS200-IS1004 ISAs1-IS1358 IS5-ISVch5 |
strA, strB, floR, catB9, sul2 | − | NGQT00000000 |
| 8 | FC2273 | 1998 | 47.5 | 77.4 | 3797 | 10 | 71 | 6 | 17 | ST69 | − | IS3-ISVch4 IS481-ISVch1 Tn3-ISShfr9 IS200-IS1004 ISAs1-IS1358 IS5-ISVch5 |
strA, strB, floR, catB9, sul2 | − | NGQS00000000 |
| 9 | FC3611a | 1999 | 47.5 | 84.8 | 3801 | 11 | 78 | 6 | 17 | ST69 | − | IS3-ISVch4 IS481-ISVch1 Tn3-ISShfr9 IS200-IS1004 ISAs1-IS1358 IS5-ISVch5 |
strA, strB, floR, catB9, sul2 | − | NGQR00000000 |
| 10 | FC3611b | 1997 | 47.5 | 92.9 | 3819 | 10 | 72 | 6 | 17 | ST69 | − | IS3-ISVch4 IS481-ISVch1 Tn3-ISShfr9 IS200-IS1004 ISAs1-IS1358 IS5-ISVch5 |
strA, strB, floR, catB9, sul2 | − | NGQQ00000000 |
−, negative
Fig. 1.
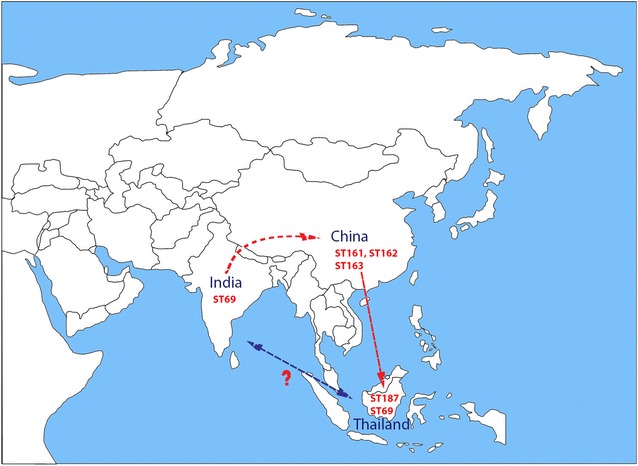
Transmission events inferred for V. cholerae O139 sequence types among South Asia
ResFinder 2.1 returned same antimicrobial resistance genes for all except two isolates (Table 1). catB9 gene responsible for chloramphenicol resistance was found common in all isolates. strA and strB (streptomycin resistance), floR (florfenicol/chloramphenicol resistance), and sul2 (sulphonamide resistance) genes were identified in all except FC1225 and FC1341 strains. The occurrence of these resistance genes were previously been reported in V. cholerae O139 serogroup. Number of antimicrobial resistance genes identified in these genomes as per comprehensive antibiotic resistance database, CARD and antibiotic resistance genes database, ARDB database matching were ~ 17 and ~ 6 genes. PlasmidFinder 1.3 resulted negative for all 10 isolates. Additionally, all 10 isolates were found to possess virulence genes including ctxA, ctxB, rtx, hlyA, zot, ace, tcpT, higB, doc, parE.
Repeat regions and insertional elements
The clusters of regularly interspaced short palindromic repeats (CRISPR) and spacer sequences in the genome were screened using CRISPR finder (http://crispr.u-psud.fr/Server/) [10] and PATRIC database which resulted in no CRISPR regions in these genomes (Table 1).
However, ISfinder (https://www-is.biotoul.fr/blast.php) [11] resulted various IS elements that were listed in Table 1. Most of the IS elements such as IS3-ISVch4, IS200-IS1004, ISAs1-IS1358 (O139), IS481, Tn3-ISShfr9, IS66-ISPsy43, IS5-ISVch8, IS5-ISVch5, IS3-ISVch4, Tn3-ISPsy42 were previously reported from V. cholerae genomes. Of these, ISAs1-IS1358 was reported to be from V. cholerae O139. Prophage screening results showed that the isolates FC1105, FC1225, FC1341, FC1877, FC3611a and FC3611b were found to have V. cholerae phage CTX (NC_015209). The phage K139 (NC_003313) was detected in FC1817, whereas VFJ (NC_021562) was seen in FC2271 and FC2273 respectively.
All study isolates lacked the intI gene but were possessed the SXT element (int SXT) with 99.9% identity to the reference sequence, which was mostly known to be, associated with the sulfamethoxazole and trimethoprim resistant strains [6]. All isolates harboured the seventh pandemic-specific gene (VC2346), suggesting that they belong to the same clonal linage and might originate from a common ancestor of the seventh pandemic strain.
Phylogenetic analyses
The phylogenetic analysis based on MLST housekeeping genes revealed the relation between ST69 Indian strains and global strains (Fig. 2). All the study isolates are closely related to ST429 originated from M802 (ST71) global isolates. Also, M2140 (ST70), M796 (ST75), ST168, M1616 (ST73), ST169, ST170 and ST182 belong to the same clonal complex with ST71 as the founder type. goEBURST analysis of V. cholerae confirmed that ST69 belong to the clonal complex CC0 with the above-mentioned sequence types (Fig. 3).
Fig. 2.
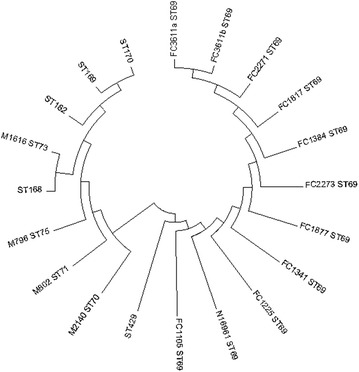
The evolutionary history was inferred using the Neighbor-Joining method. The optimal tree with the sum of branch length = 0.03301416 is shown. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. The evolutionary distances were computed using the Maximum Composite Likelihood method and are in the units of the number of base substitutions per site. The analysis involved 20 nucleotide sequences. Codon positions included were 1st + 2nd + 3rd + Noncoding. All positions containing gaps and missing data were eliminated. There were a total of 3215 positions in the final dataset. Evolutionary analyses were conducted in MEGA7
Fig. 3.
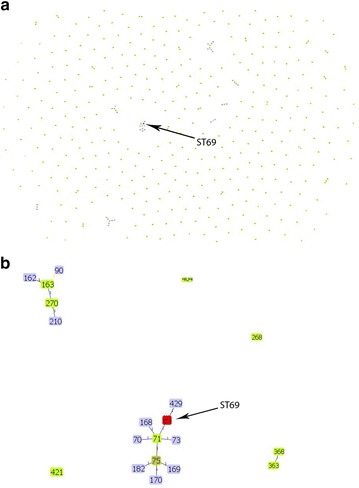
Figure depicting the comparison of V. cholerae O139 strains with a relation of ST69 with the clones at global level b ST69 belongs to one of the few clonal complexes available for V. cholerae rather than a singleton
Accession numbers
The genome sequences of V. cholerae O139 strains have been deposited at DDBJ/ENA/Genbank under the accession numbers NGQQ00000000–NGQZ00000000. The version described in this manuscript is version one.
Conclusion
To the best of our knowledge, this is the first report of ST69 V. cholerae O139 in India. Though there are few reports of clinical V. cholerae isolates from India, our study adds new dimension to the earlier finding to show that the sequence type of V. cholerae O139 continues to persist in India for over 20 years is of ST69. This implies that clonal expansion of V. cholerae O1 seventh pandemic has resulted in ST69 strains. The isolates exhibited same virulence profile but different antimicrobial resistance gene profile. This study helps in understanding the epidemiology of V. cholerae and genomic relationship between these isolates with the strains from China and Thailand. Also a continuous surveillance of V. cholerae O139 serogroup can provide more insights into the evolution of V. cholerae.
Authors’ contributions
DRNK, MSDP, SA and BV designed the study. MSDP and TS performed the laboratory work. DRNK and MSDP analysed the data and wrote the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors gratefully acknowledge the Institutional Review Board of the Christian Medical College, Vellore (83-i/11/13) for approving the study and providing lab space and facilities.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Not applicable.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
The study was funded by Indian Council of Medical Research (ICMR), New Delhi (Ref. No: AMR/TF/55/13ECDII dated 23/10/2013).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Shalini Anandan, Email: shalinianadan@cmcvellore.ac.in.
Naveen Kumar Devanga Ragupathi, Email: speed.naveen1@gmail.com.
Dhiviya Prabaa Muthuirulandi Sethuvel, Email: dhiviyap@gmail.com.
Suji Thangamani, Email: sujit@gmail.com.
Balaji Veeraraghavan, Phone: +91 9442210555, Email: vbalaji@cmcvellore.ac.in.
References
- 1.Jesudason MV, John TJ. The Vellore vibrio watch. Lancet. 1996;347:1493–1494. doi: 10.1016/S0140-6736(96)91733-X. [DOI] [PubMed] [Google Scholar]
- 2.Mutreja A, Kim DW, Thomson NR, Connor TR, Lee JH, Kariuki S, et al. Evidence for several waves of global transmission in the seventh cholera pandemic. Nature. 2011;477:462–465. doi: 10.1038/nature10392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yi Y, Lu N, Liu F, Li J, Zhang R, Jia L. Genome sequence and comparative analysis of a Vibrio cholerae O139 strain E306 isolated from a cholera case in China. Gut Pathog. 2014;6:3. doi: 10.1186/1757-4749-6-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wattam AR, Abraham D, Dalay O, Disz TL, Driscoll T, Gabbard JL, et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2014;42:D581–D591. doi: 10.1093/nar/gkt1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou Y, Liang Y, Lynch K, Dennis JJ, Wishart DS. PHAST: a fast phage search too. Nucleic Acids Res. 2011;39:W347–W352. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Siriphap A, Leekitcharoenphon P, Kaas RS, Theethakaew C, Aarestrup FM, Sutheinkul O, Hendriksen RS. Characterization and genetic variation of Vibrio cholerae isolated from clinical and environmental sources in Thailand. PLoS ONE. 2017;12:e0169324. doi: 10.1371/journal.pone.0169324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 8.Francisco AP, Vaz C, Monteiro PT, Melo-Cristino J, Ramirez M, Carriço JA. PHYLOViZ: phylogenetic inference and data visualization for sequence based typing methods. BMC Bioinform. 2012;13:87. doi: 10.1186/1471-2105-13-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moore S, Thomson N, Mutreja A, Piarroux R. Widespread epidemic cholera caused by a restricted subset of Vibrio cholerae clones. Clin Microbiol Infect. 2014;20:373–379. doi: 10.1111/1469-0691.12610. [DOI] [PubMed] [Google Scholar]
- 10.Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35:W52–W57. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006;34:D32–D36. doi: 10.1093/nar/gkj014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.