

Abstract
Cytosolic sensing of nucleic acids initiates tightly regulated programs to limit infection. Oocyte fertilization represents a scenario when inappropriate responses to exogenous yet non-pathogen-derived nucleic acids would have negative consequences. We hypothesized that germ cells express negative regulators of nucleic acid sensing (NAS) in steady state and applied an integrated data-mining and functional genomics approach to identify a rheostat of DNA and RNA sensing – the inflammasome component NLRP14. We demonstrated that NLRP14 interacted physically with the nucleic acid sensing pathway and targeted TBK1 (TANK Binding Kinase 1) for ubiquitination and degradation. We further mapped domains in NLRP14 and TBK1 that mediated the inhibitory function. Finally, we identified a human nonsense germline variant associated with male sterility that results in loss of NLRP14 function and hyper-responsiveness to nucleic acids. This discovery points to a mechanism of nucleic acid sensing regulation that may be of particular importance in fertilization.
eTOC paragraph
Cytosolic sensing of nucleic acids initiates tightly regulated programs to limit infection. Fertilization represents a scenario when inappropriate responses to exogenous yet non-pathogen-derived DNA/RNA would have negative consequences. We identify an evolutionarily conserved immunological rheostat safeguarding against such responses – a function that may have been a prerequisite for sexual reproduction.
Introduction
Virtually all organisms have some form of system, whether cellular or molecular, that functions to protect their metabolic requirements for life – or in simpler terms, for replicating their genomes. From bacteria and protozoans, to insects and humans, organisms have a means of distinguishing self from non-self and mounting appropriate responses to foreign agents. The innate immune system, which is conserved across virtually all multi-cellular organisms on the planet (from sponges to insects, plants, and vertebrates), includes a diverse set of receptors capable of detecting molecular patterns like sugars, lipids, polymers, and nucleic acids, that are principal in prompting these responses (Kumar et al., 2011a).
In vertebrates, cytosolic detection of nucleic acids is critical for initiating innate (characterized by production of Type I interferons; IFNα/β) and adaptive immune responses to invading pathogens (Wu and Chen, 2014). However, these inflammatory programs must be tightly regulated to prevent aberrant responses to self nucleic acids. Indeed, RIG-I (Retinoic Acid-inducible Gene 1), the cytosolic receptor for RNA, distinguishes self from non-self RNA through interaction with a 5′-triphosphate that is unique to viral RNA (Wu and Chen, 2014). As DNA is not physiologically found in the cytoplasm, cytosolic DNA sensing, mediated by cGAS (Cyclic GMP-AMP Synthase) in collaboration with STING (Stimulator of Interferon Genes), does not distinguish between self and non-self DNA (Nagata et al., 2010). However, during fertilization, sperm cell derived DNA can be found in oocyte cytoplasm, and could a trigger nucleic acid sensing response. We therefore reasoned that germ cells must possess a robust mechanism to negatively regulate cytosolic nucleic acid sensing.
To identify negative regulators of cytosolic nuclei acid sensing, we mined RNA sequencing (RNAseq) data from various human tissues for transcripts found specifically in germ cells that were downregulated following fertilization. We subsequently performed a functional genomics screen on these candidate genes and identified NLRP14 (NACHT, LRR And PYD Domains-Containing 14; a component of the inflammasome family of proteins) as a rheostat for nucleic acid sensing. We demonstrated that ectopic expression of NLRP14 resulted in loss of signaling through the cytosolic nucleic acid sensing pathways. Conversely, CRISPR-mediated (Clustered regulatory interspaced short palindromic repeats) targeting of this gene resulted in enhanced in DNA and RNA sensing and augmented production of pro-inflammatory cytokines. We showed that NLRP14 interacted physically with components of the nucleic acid sensing pathway, including STING, RIG-I, MAVS (Mitochondrial Antiviral Signaling Protein), and TBK1. In addition, we determined that NLRP14 targeted TBK1 for ubiquitination and degradation, and delineated domains in NLRP14 and TBK1 that mediate the interactions as well as the inhibitory function. Finally, we identified a human nonsense germline variant associated with male sterility that results in loss of NLRP14 function and exaggerated responses to nucleic acids.
Results
NLRP14 is a candidate modulator of cytosolic nucleic acid sensing
We reasoned that expression of a potent negative regulator of NAS may be of particular importance for germ cells during fertilization. Indeed, key members of the NAS pathway are expressed in oocytes and likely constitute the same functions they have in detecting nucleic acids in non-germ cells (Supplemental Table 1). We mined the Genotype-Tissue Expression (GTEx) (http://commonfund.nih.gov/GTEx) database to identify genes preferentially expressed in testes and ovary (the tissues most relevant to fertilization). Our analysis yielded 1180 genes with expression in testes and/or ovaries and low or absent expression in all other tissues (Figure 1A; see Materials and Methods for a full description of the analysis). We further limited the candidate gene list by focusing on genes whose expression is down-regulated following fertilization (Yan et al., 2013) (see Materials and Methods), as NAS inhibition would be required during fertilization but dispensable and perhaps even deleterious thereafter. Thus, we identified 20 candidate genes that were preferentially expressed in germ cells and down regulated after fertilization.
Figure 1. Data-driven candidate discovery and targeted functional screen identifies putative regulators of cytosolic nucleic acid sensing in germ cells.
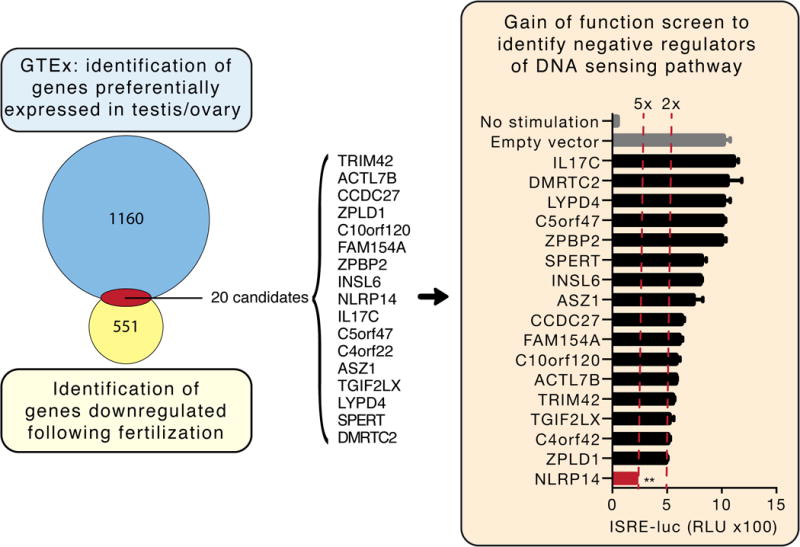
Left panel: Venn diagram of genes preferentially expressed in testis and/or ovary and down-regulated following oocyte fertilization (see Materials for further description of the discovery method). Of the 20 candidates identified, 17 (listed) were successfully cloned and expressed in 293T cells. Right panel: 293T cells stably expressing STING-HA (293T-STING-HA) were cotransfected with cGAS-Flag and each candidate along with the luciferase gene under control of the ISRE promoter and monitored for luciferase expression 24h post transfection. Shown are relative luciferase units (RLU) normalized to Renilla luciferase expression. Dotted red lines indicate 2- and 5-fold suppression. **P<0.05, Student’s test. Error bars indicate the SD. Data is representative of at least two independent experiments.
We next performed an overexpression screen of these candidate genes to determine whether they regulated cytosolic DNA sensing pathways in 293T cells, which have been extensively explored as a model system for nucleic acid sensing. Specifically, 293T cells do not normally express the cytosolic DNA sensors cGAS and its signal adaptor STING. This allowed us to generate cells where this pathway was reconstituted through stable expression of HA-tagged STING (293T-STING-HA) for use in the gain of function screen. Each of the 20 candidates genes was subcloned into a lentiviral-based expression vector (pLenti6.2-V5) and co-transfected with cGAS and an IFN-Stimulated Response Element (ISRE) promoter driving expression of luciferase (ISRE-luc) into 293T-STING-HA cells (Figure 1; of the 20 candidates, 17 were successfully cloned and expressed by the pLenti6.2-V5 vector). Our targeted screen indicated that five of the candidates suppressed cGAS-mediated ISRE promoter activation by 40% or more (TRIM42, TGIF2LX, C4orf42, ZPLD1, and NLRP14). Of these, we focused on NLRP14, a member of the inflammasome family of proteins (Schattgen and Fitzgerald, 2011), which achieved the highest suppression of cGAS mediated signaling (Figure 1, right panel). Indeed, we also confirmed that NLRP14 expression was rapidly down-regulated following fertilization (Supplemental Figure 1a). In short, our combined unbiased computational and experimental approach resulted in the identification of NLRP14 as a putative inhibitor of NAS during fertilization.
NLRP14 negatively regulates pattern recognition receptor (PRR)-mediated signaling
The cytosolic sensing of DNA is regulated by cGAS in collaboration with the endoplasmic reticulum (ER)-associated multiple transmembrane protein STING (Barber, 2015; Cai et al., 2014). Downstream signaling activates IRF3- and Nuclear Factor Kappa-Light-Chain-Enhancer of Activated B Cells (NFκB)- dependent transcription that induces the production and secretion of type I interferons (such as IFNβ) and other pro-inflammatory cytokines (Figure 2a). A key member of this pathway is TBK1 which phosphorylates IRF3 and, to a lesser extent, IRF7. To examine the effect of NLRP14 on cytosolic DNA-sensing pathway in more detail, HA-tagged-STING, or FLAG-tagged-TRIF, TBK1, IRF3, and IRF7 were co-transfected with N-terminal Myc-tagged-NLRP14 (Myc-NLRP14) together with ISRE or NFκB reporter plasmids into 293T cells (Figure 2b–c). We found that transduction of Myc-NLRP14 significantly reduced ISRE- and NFκB-mediated promoter activation in the context of STING, TRIF, TBK1, and IRF3, but not IRF7 (Figure 2b–c), suggesting that NLRP14 preferentially targets IRF3 dependent signaling. The effect on TBK1 (the kinase that is critical for signaling downstream of cytosolic sensing of both DNA and RNA) suggested a more general role for NLRP14 in dampening PRR signaling. Consistent with this, we found that NLRP14 reduced RIG-I and MAVS induced activation of ISRE- and NFκB-mediated promoters (Figure 2d). Indeed, TBK1-mediated induction of endogenous C-X-C Motif Chemokine Ligand 10 (IP10) and IFNβ mRNA expression was suppressed by NLRP14 in a dose-dependent manner (Figure 2e).
Figure 2. NLRP14 suppresses TBK1-dependent innate immunity.
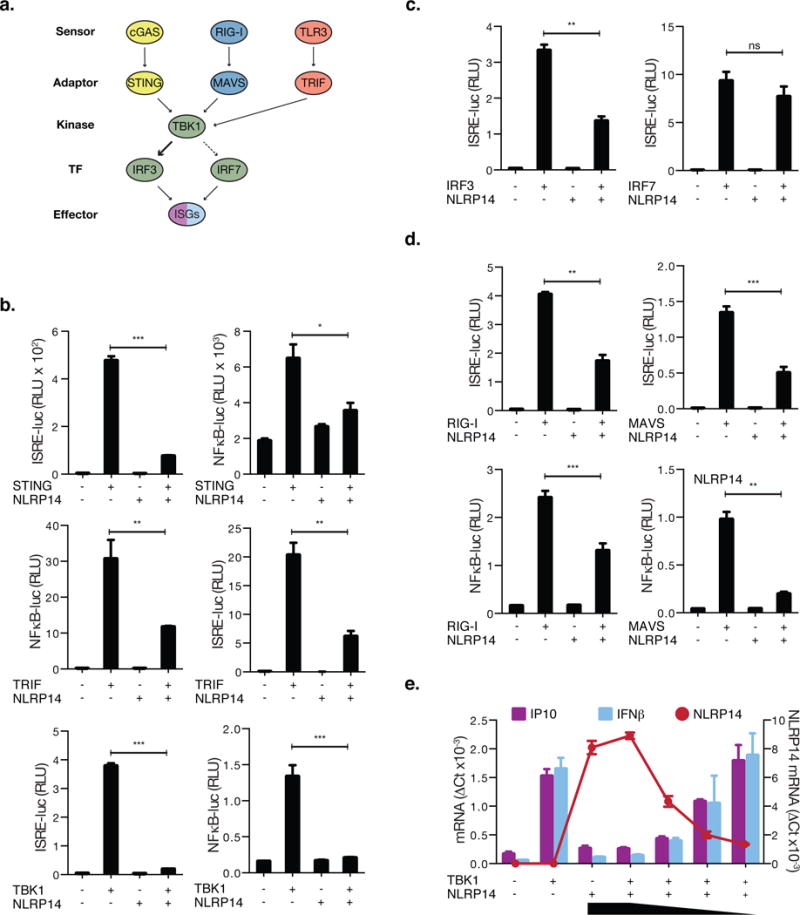
(A) Schematic diagram outlining signaling pathway components involved in cytosolic nucleic acid sensing. (B–D) 293T cells were cotransfected with Myc-NLRP14 and RIG-I-Flag, MAVS-Flag, STING-HA, TBK1-Flag, TRIF-Flag, IRF3-Flag, or untagged-IRF7 along with the luciferase gene under control of the ISRE or NFκB promoter. Shown are relative luciferase units (RLU) normalized to Renilla luciferase expression. (E) 293T cells were cotransfected with TBK1-Flag and decreasing amounts of Myc-NLRP14. IP10, IFNβ, and NLRP14 mRNA levels were determined by real-time PCR 24h post transfection. RT-PCR data were normalized to GAPDH mRNA. **P<0.01, Student’s T-test. Error bars indicate SD. Data are representative of at least three independent experiments. (See also Supplemental Figure S1 and S3)
While not normally expressed physiologically in somatic tissues, we found that cancer and immortalized cell lines including 293T cells and primary human bronchial epithelial cells (HBECs) express modest levels of NLRP14 (Supplemental Figure 1b). Therefore, to further confirm NLRP14’s involvement in modulating cGAS-STING-triggered signaling, we targeted NLRP14 via CRISPR-Cas9-mediated gene silencing in 293T cells stably expressing HA-tagged STING (293T-STING-HA-NLRP14-CRISPR; Supplemental Figure 2b). Transfection of cGAS-Flag or cGAMP (Cyclic guanosine monophosphate-edenosine monophosphate) into 293T-STING-HA-NLRP14-CRISPR cells resulted in enhanced IFNβ and ISRE-mediated promoter activation as compared to parental cells in a dose-dependent manner (Figure 3A–B) confirming that NLRP14 indeed participates in dampening immune responses via the cGAS-STING axis. Consistent with these results, siRNA (Short Interfering RNA) knockdown of NLRP14 in primary HBECs resulted in enhanced responses to transfected viral RNA as measured by secreted IFNβ and a concomitant decrease in influenza virus replication (Supplemental Figure 1c–d).
Figure 3. Loss of NLRP14 results in augmentation of immune responses via the cGAS/RIG-I axis.
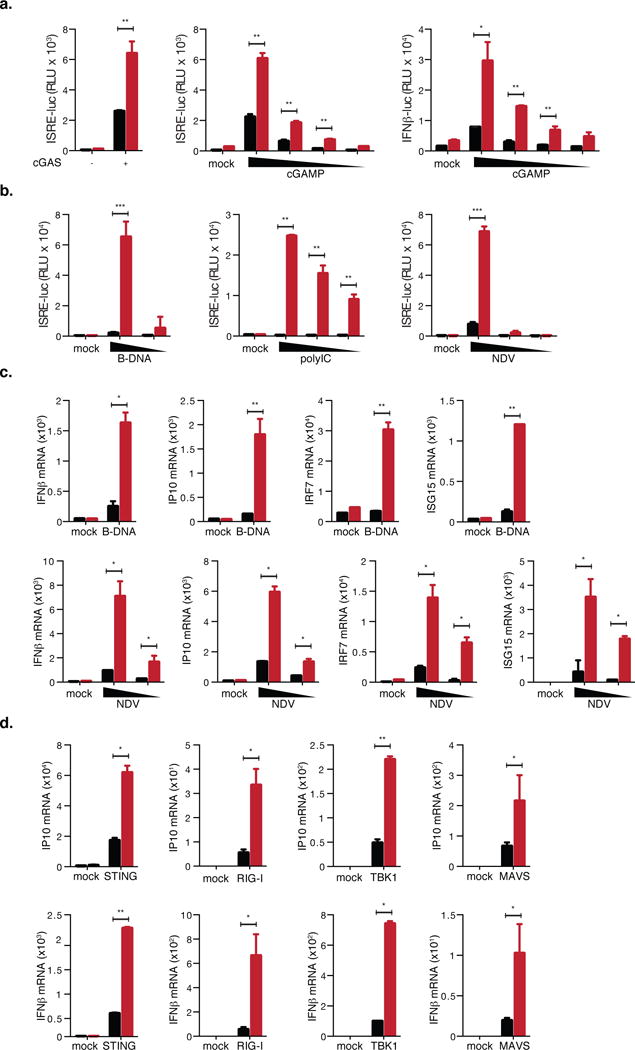
(A) 293T-STING-HA-NLRP14-CRISPR cells were transfected with cGAS-Flag, or decreasing doses of 2′3′-cGAMP (30, 15, 7.5 and 1.5 μg/ml) along with ISRE or IFNβ promoters. Shown are relative luciferase units (RLU) normalized to Renilla luciferase expression. (B) Parental and 293T-NLRP14-CRISPR cells were stimulated with B-DNA (0.06 or 0.03 μg/ml), PolyI:C (25, 12.5, and 2.5 μg/ml), or infected with NDV-GFP (moi of 20, 4, or 0.8). 24h after treatment, levels of ISRE inducing cytokines in supernatants were determined using 293T-ISRE-B8 cells. (C) Rhodamine-labeled B-DNA (indicated as B-DNA Rho) was transfected into parental (WT) and 293T-NLRP14-CRISPR (KO) cells, and observed by fluorescence microscopy. (D) Parental and 293T-NLRP14-CRISPR cells were stimulated with indicated ligands (0.06 μg/ml of B-DNA and NDV-GFP at the m.o.i of 10), and levels of IP10, IFNβ, IRF7 and ISG15 mRNA were determined by RT-PCR 6h post transfection of B-DNA and 15h post NDV inoculation. (E) Parental and 293T-NLRP14-CRISPR cells were transfected with RIG-I-Flag, MAVS-Flag, TBK1-Flag, or STING-HA, and IP10 and IFNβ mRNA levels were determined by RT-PCR at 24h post transfection. RT-PCR data were normalized to GAPDH mRNA. **P<0.01, ***P<0.001, Student’s T-test. Error bars indicate SD. Data are representative of at least three independent experiments. Black and red bars indicate parental and CRISPR-targeted cells, respectively. (See also Supplemental Figures S2 and S4)
To further explore the role of NLRP14 as a general dampener of NAS, we again took advantage of CRISPR-Cas9 mediated genome editing to establish NLRP14 deficient 293T cells (which do not express STING). Sequencing of genomic DNA derived from 293T-NLRP14-CRISPR cells revealed a 23-nucleotide deletion within the targeted exon (Supplemental Figure 2).
We first monitored innate immune activation through the indirect measurement of type I interferons in supernatants from cells stimulated with ligands of the RNA nucleic sensing pathway: B-DNA, polyI:C (Polyinosinic:polycytidylic acid), and the RNA virus Newcastle disease virus (NDV). We found that cells lacking NLRP14 were more responsive to ligand stimulation and RNA virus infection as compared to WT controls (Figure 3b, d). Of note, transfection efficiency of Rhodamine-labeled B-DNA in 293T-NLRP14-CRISPR cells was equivalent to that of parental cells (Supplemental Figure 2c), and thus the hyper-responsive phenotype observed in 293T-NLRP14-CRISPR cells was not simply due to differences in ligand uptake. Stimulation with B-DNA or NDV infection resulted in significant enhancement of endogenous mRNA gene expression, including IP10, IFNβ, IRF7, and ISG15 (Interferon Stimulated Gene 15), (Figure 3d). Furthermore, exogenous expression of STING, RIG-I, TBK1, and MAVS resulted in enhanced transcription of IP10 and IFNβ in 293T-NLRP14-CRISPR cells (Figure 3e). Collectively, these results demonstrate that while overexpression of NLRP14 results in dampening of NAS, deletion of endogenous NLRP14 expression results in augmented NAS-mediated signaling and anti-viral immunity. Thus, NLRP14 functionally serves as a potent negative regulator of cytosolic sensing of nucleic acids.
NLRP14 associates physically with components of NAS
Activation of cGAS (through interaction with DNA) leads to the production of cGAMP, a second messenger molecule that in turn triggers STING-mediated activation of TBK1. To investigate how NLRP14 counteracts cGAS/STING and RIG-I mediated signal transduction, we explored if NLRP14 interacts with components of the cytosolic NAS machinery (Figure 4). We first interrogated whether NLRP14 associates with exogenously expressed cGAS and/or STING independently of downstream signaling and found that it interacted with STING but not cGAS in WT 293T cells (Figure 4a). Next, Myc-NLRP14 was expressed in the presence or absence of HA-cGAS in 293T-STING-HA cells followed by immunoprecipitation with anti-HA and immunoblotting with an anti-Myc antibody (Figure 4). While we observed slight precipitation of NLRP14 with STING, it was enhanced in the presence of cGAS (Figure 4a and b), suggesting that NLRP14 may participate in the DNA sensing pathway through interaction with activated STING. While cGAS-mediated STING activation, such as phosphorylation and degradation, were intact, STING-TBK1 interaction as well as phosphorylation of IRF3 was reduced in the presence of NLRP14 (Figure 4b–c). Together, these results suggested that NLRP14 does not inhibit STING-mediated signaling via the cGAS-STING axis. Rather, NLRP14 suppresses downstream activation of IRF3 by interfering with STING-TBK1 signaling complexes.
Figure 4. NLRP14 interacts with components of cytosolic nucleic acid sensing machinery.
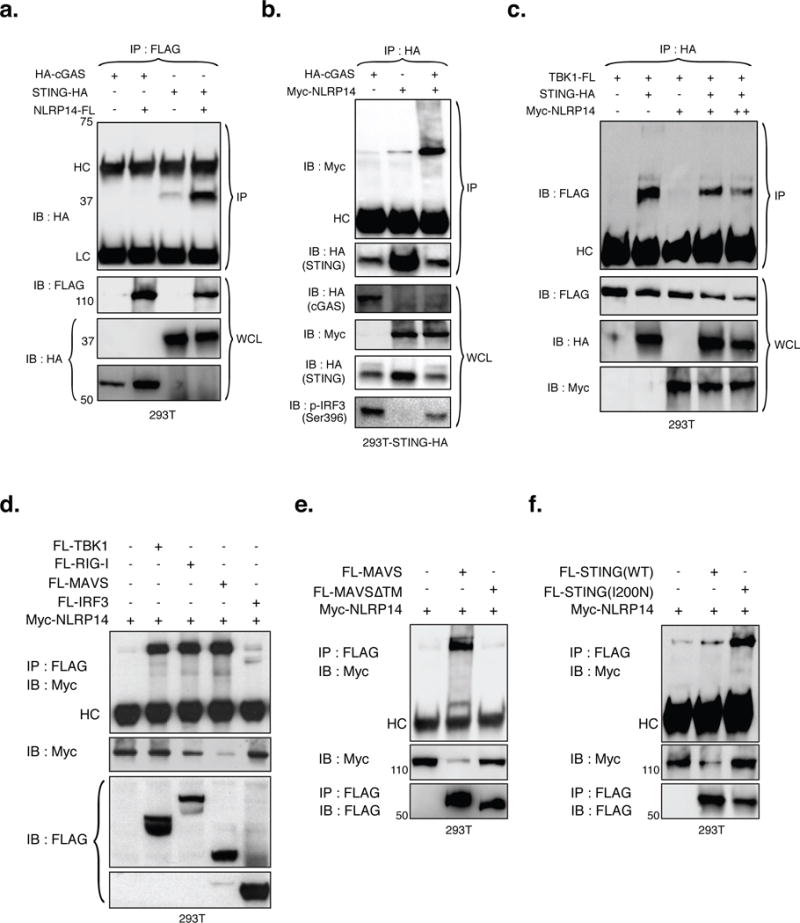
(A) NLRP14-Flag was coexpressed with HA-cGAS or STING-HA in 293T cells and subjected to immunoprecipitation (IP) and immunoblotting (IB) using appropriate antibodies. (B) Myc-NLRP14 was coexpressed with or without HA-cGAS in 293T-STING-HA cells and subjected to IP and IB using the appropriate antibodies. (C) The effect of NLRP14 on TBK1-STING interaction was examined in the context of 1μg (+) or 2μg (++) Myc-NLRP14 expression plasmid. Myc-NLRP14 was coexpressed with (D) RIG-I-Flag, MAVS-Flag, TBK1-Flag, or IRF3-Flag (E) FL-MAVS or FL-MAVSΔTM (F) FL-STING or FL-STING(I200N) in 293T cells and monitored for interactions by IP followed by IB using appropriate antibodies. WCL, HC and LC indicate whole cell lysates, Heavy-Chain, and Light-Chain, respectively. (See also Supplemental Figure S6)
We next sought to determine if NLRP14 interacts with molecules that participate in RIG-I-mediated signaling. To examine this possibility, Myc-NLRP14 was co-expressed with Flag-tagged versions of RIG-I, MAVS, TBK1, or IRF3 followed by immunoprecipitation with anti-Flag and immunoblotting with anti-Myc antibody (Figure 4d–f). We found that NLRP14 interacts not only with TBK1 but each of the other signaling molecules tested (RIG-I, MAVS and IRF3), although its interaction with IRF3 was modest (Figure 4d). In addition, cells transduced with NLRP14 exhibited suppression of IRF3 phosphorylation both in the presence and absence of NDV infection (which triggers TBK1-mediated IRF3 activation; Supplemental Figure 3). Similarly, NLRP14 expression suppressed Ser172 phosphorylation of TBK1 and Ser536 phosphorylation of NFκB p65 (Supplemental Figure 3a, indicated panels). We observed significant increase in TBK1 and IRF3 phosphorylation in 293T-NLRP14-CRISPR cells in the context of TBK1 overexpression as well as in response to NDV infection (Figure 6a and Supplemental Figure 3a–c). In contrast, similar to the results shown in Figure 2, infection-induced phosphorylation of IRF7 was not inhibited by NLRP14, suggesting possible specificity in NLRP14 function (Supplemental Figure 3b). Together with the observation that NLRP14 influences formation of STING-TBK1 complexes, these results indicated that NLRP14 neutralizes cytosolic sensing of nucleic acids at the level of TBK1 through direct interaction with complexes critical for IRF3 phosphorylation and immune activation.
Figure 6. NLRP14 dampens TBK1-mediated signaling through interactions with TBK1-kinase domain (KD) and induction of TBK1 ubiquitination.
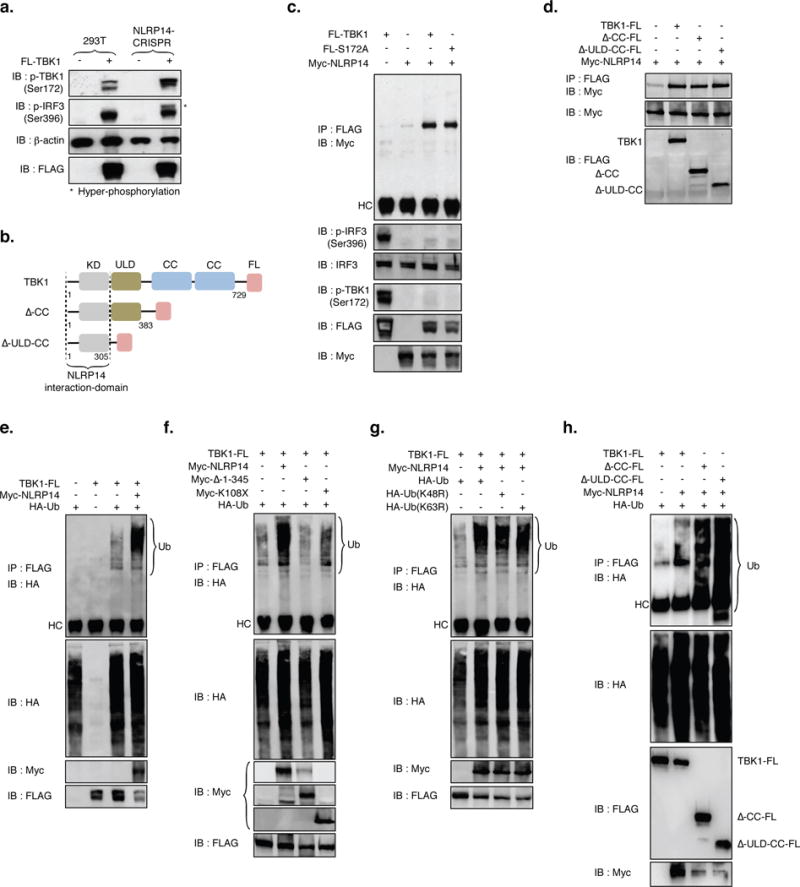
(A) Parental and 293T-NLRP14-CRISPR cells were transfected with TBK1-Flag and 24h later monitored for TBK1 expression and phosphorylation (S172), phosphorylation of IRF3 (S396), and β-actin using appropriate antibodies. Asterisks indicates hyper-phosphorylation of IRF3. (B) Schematic of human TBK1 and deletion mutants generated. Kinase domain (KD); ubiquitin-like domain (ULD); coiled-coil domain (CC). (C, D) Immunoprecipitation and immunoblot of 293T cell extracts transfected with various combinations (noted above the lanes) of NLRP14 and TBK1 mutants using the appropriate antibodies. (E–H) Immunoprecipitation and immunoblot of 293T cell extracts transfected with various combinations (noted above lanes) of NLRP14 variants, TBK1 variants, and linked ubiquitin. (I) Schematic of human TBK1 K670R (constitutively active) variant generated. (J) ISRE-luc and NFκB-luc activity in cells transfected with combinations of TBK1-FL, myc-NLRP14, and K670R TBK1 mutant. Shown are relative luciferase units (RLU) normalized to Renilla luciferase expression. **P<0.01, Student’s T-test. Error bars indicate SD. Data are representative of at least two independent experiments. (See also Supplemental Figure S6)
N-terminal domains of NLRP14 are required for suppression of TBK1-mediated IFNβ production
We sought to determine the functional protein elements in NLRP14 responsible for modulating TBK1-mediated signaling. We constructed N-terminal deletion mutants of NLRP14 lacking amino acids 1 to 100 (Δ1-100) and 1 to 345 (Δ1-345), as well as a C-terminal deletion mutant lacking amino acids 531 to 1093 (Δ531-1093) and monitored their capacity to affect NAS signaling (Figure 5). Consistent with our previously described observations (Figure 3), neither full-length nor any of the NLRP14 variants tested suppressed IRF7-mediated ISRE promoter activation (Supplemental Figure 5f). However, while Δ1-100 and Δ531-1093 significantly inhibited TBK1-mediated IFNβ, and NFκB promoter activation, deletion of amino acids 1-345 (Δ1-345) resulted in abolishment of the inhibitory function (Figure 5b). This correlated with the observation that Δ1-345 failed to inhibit endogenous expression of IP10 and IFNβ mRNA normally induced by TBK1 (Figure 5e). Phosphorylation of IRF3 and TBK1 in cells transduced with full-length or NLRP14 variants correlated with downstream promoter activation (Figure 5b–c). On the other hand, the NACHT-domain of NLRP14 (consisting of amino acid residues 176-345) was not sufficient to suppress TBK1-mediated promoter activation (Supplemental Figure 4), suggesting redundant role for this domain in modulating NAS signaling.
Figure 5. N-terminal PYD and NACHT domains mediate NLRP14 function.
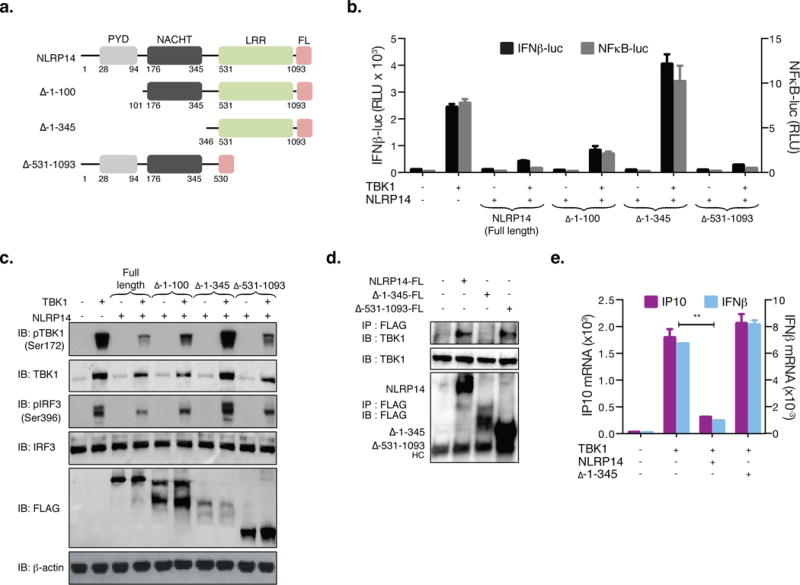
(A) Schematic of human NLRP14 and deletion mutants generated. Pyrin domain (PYD); NAIP, CIITA, HET-E, and TP1 domain (NACHT); Leucine rich repeat (LRR). 293T cells were cotransfected with TBK1 and NLRP14 variants along with IFNβ-luc or NFκB-luc and monitored for promoter activity (RLU; B) or subjected to immunoblotting with appropriate antibodies (C). (D) 293T cells were transfected with full length, Δ1-345 or Δ531-1093 NLRP14 and interaction with endogenous TBK1 monitored by immunoprecipitation 48h post transfection. (E) 293T cells were cotransfected with TBK1 and NLRP14 variants and IP10 and IFNβ mRNA levels were determined by RT-PCR 24h post transfection. **P<0.01, Student’s T-test. Error bars indicate SD. Data are representative of at least two independent experiments. (See also Supplemental Figure S6)
To further investigate how the N-terminal region of NLRP14 – which includes both PYD and NACHT domains – contributes to inhibiting signaling downstream of STING-cGAS and RIG-I, we examined their requirement for NLRP14-TBK1 interaction. We found that loss of amino acids 1-345 (Δ1-345; containing both PYD and NACHT domains) resulted in complete abolishment of TBK1 co-immunoprecipitation, indicating that this region is required for NLRP14 interaction and suppression of TBK1 signaling (Figure 5d, e). Similarly, we sought to determine domains in TBK1 responsible for its interaction with NLRP14. To this end, Flag-tagged TBK1 variants (1-383 and 1-305, lacking the Coiled-Coil (CC) domain and the Ubiquitin-like domain (ULD) as well as the CC domain, respectively) were co-transfected along with Myc-NLRP14 into 293T cells and monitored for interaction through immunoprecipitation (using anti-Flag antibody) followed by immunoblotting (with anti-Myc antibody; Figure 6b, d). As shown in Figure 6d, aa1-305 were sufficient for TBK1-NLRP14 binding, suggesting that the N-terminal kinase domain (KD) of TBK1 participates in this interaction. We also found that a kinase-dead mutant of TBK1 (S172A) did not abrogate interaction with NLRP14 (Figure 6c), implying that kinase activity was not required for its association with NLRP14 and suggested that the suppression of TBK1-mediated signaling by NLRP14 was not likely to be associated with the modulation of TBK1 kinase activity.
NLRP14 induces ubiquitination and degradation of TBK1
Given the physical interaction between NLRP14 and TBK1, we next sought to determine how NLRP14 abrogates TBK1-dependent signaling. Exogenous expression of NLRP14 resulted in a decrease in TBK1 protein both in the presence and absence of NDV infection (Figure 5c and Supplemental Figure 3). We therefore hypothesized that the ubiquitination pathway may provide clues to the molecular mechanism through which NLRP14 targets TBK1 for degradation; Flag-TBK1, Myc-NLRP14 and HA-tagged ubiquitin (HA-Ub) were co-transduced in 293T cells followed by immunoprecipitation by anti-Flag and immunoblotting with anti-HA antibody. We found that NLRP14 expression lead to poly-ubiquitination of TBK1 (Figure 6e) and resulted in a reduction in TBK1 levels. Importantly, NLRP14 loss of function mutants (Δ1-345; which do not interfere with TBK1-mediated signaling; Figure 5), failed to induce poly-ubiquitination of TBK1 (Figure 6f, lane 3). Previous studies have indicated that TBK1 may be subject Lysine-48 (K48) and Lysine-63 (K63) poly-ubiquitination (which control degradation and activation of TBK1, respectively (Zhao, 2013)). We observed that K48R and K63R mutations in ubiquitin did not alter NLRP14-induced poly-ubiquitination of TBK1 (Figure 6g), suggesting that TBK1 may be targeted for degradation in a K48-ubiquitin independent manner. Moreover, we observed that similar to full-length TBK1, ΔCC and ΔULD-CC variants (which did not alter interactions with NLRP14; Figure 6d) still underwent NLRP14-dependent poly-ubiquitination (Figure 6h). Together, these results indicated the kinase domain of TBK1 was sufficient for its interaction with NLRP14 and subsequent targeting for degradation.
Recently, Cui and colleagues proposed that NLRP4 (a member of the inflammasome family of proteins that shares 38% homology with NLRP14) negatively regulates RNA sensing by targeting TBK1 for ubiquitination and proteasomal degradation(Cui et al., 2012). We explored whether NLRP4 and NLRP14 have overlapping or redundant functions. Indeed, similarly to the observations previously reported, we generated NLRP4-CRISPR-293T cells and found that they exhibited enhanced responses to NDV infection and PolyI:C transfection (Supplemental Figure 5a, b), suggesting a possible similarity between NLRP4 and NLRP14-mediated immunosuppression. However, exogenous expression of NLRP14 in NLRP4-CRISPR-293T cells resulted in induction of TBK1 ubiquitination, suppression of TBK1 phosphorylation, as well as inhibition of TBK1-mediated ISRE and NFκB promoter activation (Supplemental Figure 5c–d), indicating an NLRP4-independent, NLRP14-dependent, inhibition of this pathway. Furthermore, we found that NLRP14 suppressed a mutant version of TBK1 where lysine-670 (which is associated with DTX4-dependent ubiquitination and degradation of TBK1) was replaced with an arginine (K670R; Supplemental Figure 6c–e) suggesting a non-redundant role for NLRP14 in modulating TBK1-dependent signaling through an as of yet unknown E3 ligase in a K670-independent manner. So, while further studies are needed to clarify the molecular mechanism by which NLRP14 induces ubiquitination and degradation of TBK1, these data indicated that it may serve as a safeguard against inappropriate sensing of nucleic acids in germ cells.
A Negative feedback loop regulates NLRP14 action on NAS
The mitochondrially localized scaffold protein MAVS is a critical adaptor molecule for signal transduction downstream of RIG-I mediated sensing of RNA (Kawai and Akira, 2006). As shown in Figure 4D, we found that NLRP14 interacts with MAVS (along with STING, RIG-I, TBK1, and IRF3). However, we also observed that expression of these signaling molecules resulted in reduction of NLRP14 protein levels. In particular, MAVS and STING expression lead to marked decreases in NLRP14 protein (Figure 4a, and 4d). Previous reports have demonstrated that the transmembrane region of MAVS (consisting of amino acids 514-535 is required for proper mitochondrial localization and signaling (Baril et al., 2009; Seth et al., 2005). We found that expression of MAVS lacking the transmembrane domain (MAVSΔTM) resulted in complete abrogation of the NLRP14 interaction and MAVS-induced loss of NLRP14 expression (Figure 4e). Similarly, STING, but not a loss of function mutant (I200N), interacted with NLRP14 and induced loss of expression (Figure 4f), suggesting that functional signaling was required. To determine the pathway responsible for controlling NLRP14 levels, cells were treated with the proteasome inhibitor, MG132, or the autophagy inhibitor, Bafilomycin A1. While MAVS-induced loss of NLRP14 levels was resistant to Bafilomycin A1 treatment, we found that MG132 treatment resulted in recovery of NLRP14 protein (Supplemental Figure 6f). Together, these results suggested that proper MAVS localization and signaling were required for inducing a proteasome-dependent process that lead to the degradation of NLRP14. Finally, to determine the element responsible for controlling the degradation NLRP14, Myc-tagged variants of NLRP14, lacking PYD, NACHT, and LRR were transfected into 293T cells in presence or absence of MAVS or STING (Supplemental Figure 6a–b). While deleting PYD (ΔPYD) and NACHT (ΔNACHT) domains in NLRP14 did not affect MAVS or STING induced depletion, deletion of the LRR domain (ΔLRR) resulted in resistance to degradation, suggesting that the LRR controls NLRP14 protein stability (Supplemental Figure 6a–b). Furthermore, we found that increased stability of ΔLRR-NLRP14 resulted in enhanced capacity to suppress TBK1 and STING-mediated IFNβ promoter activation in a dose dependent manner (Supplemental Figure 6a–b). As with other NLRP proteins (Martinon et al., 2002) we suspect that NLRP14 exists in two conformations that differentially regulate protein function and deletion of the LRR results in exposure of the functional PYD and NACHT domains that participate in inhibiting the cytosolic nucleic acid sensing pathways. The existence of such tight on/off regulation may ensure that persistent immunosuppression by NLRP14 does not occur (Figure 7e).
Figure 7. Naturally occurring SNP associated with male sterility results in loss of NLRP14 function.
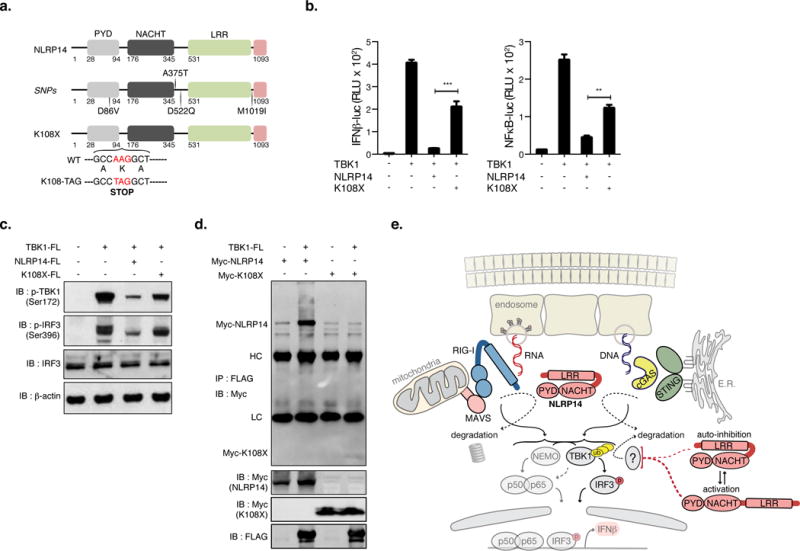
(A) Schematic of human NLRP14 and location of SNPs identified in men with spermatogenic failure. (B) 293T cells were transfected with combinations (noted above lanes) of NLRP14 (WT and K108X mutant), TBK1 and (where noted) IFNβ-luc or NFκB-luc and monitored for luciferase activity (B; shown are RLU normalized to Renilla luciferase expression), phosphorylation of TBK1 and IRF3 (C), and TBK1-NLRP14 interactions by immunoprecipitation and immunoblotting (D). (E) Summary model of NLRP14-mediated suppression of cytosolic nucleic acid sensing. **P<0.01, ***P<0.001, Student’s T-test. Error bars indicate SD. Data are representative of at least two independent experiments. (See also Supplemental Figure S7)
Naturally occurring SNPs in NLRP14 result in reduced inhibition of the nucleic acid sensing pathway
Previous reports have suggested a role for NLRP14 in embryo development (Yurttas et al., 2010) and one study identified SNPs within the several genes including NLRP14 in sterile men with spermatogenic failure (Westerveld et al., 2006). These included missense mutations D86V, A375T, D522Q, M1019I, as well as K108X that introduces a stop codon at aa108 (Westerveld et al., 2006). With the exception of the K108X allele, these SNPs are extremely rare in the human population (≤0.0001%; 1000 Genome Project). Though a functional relationship between these mutations and spermatogenic failure and/or sterility remains elusive, we hypothesized that they may regulate NLRP14 immunosuppressive function. We generated full-length NLRP14 expression constructs containing each of the reported mutations and functionally interrogated their capacity to inhibit TBK1-mediated signaling (Figure 7 and Supplemental Figure 7a). We found that NLRP14 variants harboring missense mutations inhibited TBK1-mediated activation of IFNβ and NFκB promoters, impaired TBK1 and IRF3 phosphorylation and resulted in the loss of TBK1 expression similarly to wild-type NLRP14 (Supplemental Figure 7a). In contrast, we observed that the K108X mutation, which introduces a stop codon at aa108, resulted in impaired inhibition of TBK1-mediated activation of both IFNβ and NFκB promoter activation (Figure 7b). Furthermore, this corresponded to significant recovery of TBK1 levels, TBK1 and IRF3 phosphorylation, and IFNβ and IP10 mRNA expression (Figure 7c and Supplemental Figure 7b). Moreover, co-immunoprecipitation analysis revealed that K108X-NLRP14 did not associate with TBK1 (Figure 7d) and induced markedly lower polyubiquitination of TBK1 (Figure 6f), further demonstrating a loss of normal NLRP14-mediated function. Since the possibility exists for transcriptional read-through in the K108X-NLRP14 construct (resulting in full-length protein), we sought to confirm our findings by constructing a Flag-tagged C-terminal deletion mutant possessing exclusively amino acid residues 1-108 of NLRP14 (referred to as 1-108-FL; Supplemental Figure 7c–f). Indeed, 1-108-FL failed to inhibit TBK1 induced IFNβ and NFκB promoter activation as well as endogenous IFNβ and IP10 mRNA expression. However, we observed that signaling was partially enhanced as compared to full-length NLRP14 (Supplemental Figure 7c–f), consistent with our observation that both PYD and NACHT domains are required for NLRP14’s inhibitory function. Together, these data suggested that loss of NLRP14 function could result in inappropriate induction of PRR-mediated immune responses that may lead to spermatogenic failure and infertility. Further studies, including small animal models, will be critical in further delineating the molecular mechanism and developmental role of NLRP14 and naturally occurring SNPs in modulating cytosolic sensing of nucleic acids.
Discussion
Recent advances have revealed that some members of the inflammasome family of proteins may be involved not only in pathogen-recognition but also in the regulation of tissue homeostasis (Kufer and Sansonetti, 2011; Rathinam and Fitzgerald, 2016). Though several NLRPs (like NLRP2, 4, 5, 7, 9, and 14) were shown to be expressed specifically in mammalian oocytes, little was known about their function (Tian et al., 2009). Here, we demonstrated that human NLRP14 (expression of which has been confirmed through an unbiased proteomics approach in oocytes (Wang et al., 2010)) negatively regulates cytosolic sensing of DNA and RNA. Additionally, loss and gain of function experiments revealed that NLRP14 targets TBK1 for ubiquitination and degradation, albeit through an as of yet unknown pathway. Similarly, there is growing evidence that the inflammasome family of proteins have non-inflammasome functions that are critical in maintaining tissue homeostasis and negatively regulate innate immune responses though none target cytosolic sensing of both DNA and RNA (Chen, 2014; Cui et al., 2012; Cui et al., 2010; Guo et al., 2016; Kumar et al., 2011b; Lupfer and Kanneganti, 2013; Moore et al., 2008; Rathinam and Fitzgerald, 2016; Tattoli et al., 2008; Wang et al., 2015; Zhang et al., 2014). One possibility is that members of this family operate to fulfill tissue specific requirements to control aberrant responses to self and foreign ligands. Indeed, the expression of NLRP14 is limited to gonadal tissues and immortalized cell lines such as 293T cells used in the present study. Here, we demonstrated that NLRP14 associates with both STING and MAVS and prevents downstream signal transduction (see model in Figure 7E). In turn, these adaptor molecules induce the proteasomal degradation of NLRP14, a feedback loop that may be critical in preventing persistent immunosuppression and proper induction of innate immune responses under appropriate conditions (for example, post fertilization of oocytes).
We found that ectopic expression of the K108X allele (rs76274604; coding for a nonsense mutation that introduces an early STOP codon immediately after the Pyrin domain and reduced expression of full-length NLRP14) resulted in reduced suppression of TBK1-mediated signaling. This allele has an average frequency of 1.7% in the human population, and a minor allele frequency of 3% in East Asian and Ad Mixed American populations (Genomes Project et al., 2015), suggesting that infertility associated with homozygosity of this gene may affect 3 in 10,000 individuals. While NLRP14 mutations associated with female infertility have yet to be reported, the possibility cannot be ruled out and is the subject of further investigation. However, we found that a sub-cloned version of the allele, containing only sequence for aa1-108 and lacking the coding sequence for aa109-1093 exhibited further enhancement of TBK1-mediated IFNβ and NFκB activation. Recent work suggests that PYRIN domains can modulate inflammatory responses in human monocytes, macrophages, and dendritic cells through interactions with other inflammasome components (de Almeida et al., 2015). Therefore, the enhancement in signaling we observe in the context of K108X-FL expression may be due to similar mechanisms, though further work will be necessary to explore this possibility.
Satie et al. reported that the excess amount of type I IFN signaling disrupts seminiferous tubules in mice, leading to a loss of germ cells and infertility (Satie et al., 2011). Additionally, intra-peritoneal injection of IFNα results in impaired spermatogenesis in rodents (Ulusoy et al., 2004), and in infertile men, high levels of IFNα were detected in semen (Fujisawa et al., 1998). These reports support the notion that some NLRP14 mutations may cause spermatogenesis failure through inappropriate of innate immune responses in testis. While the in vivo function of NLRP14 remains to be explored, microinjection of siRNA targeting NLRP14 into mouse oocytes results in aborted fertilization and early embryo development (Hamatani et al., 2004) and anecdotal evidence indicates that mice require a functional nlrp14 for sterility (data not shown). Furthermore, NLRP14 transcript levels appear to decrease with age, coinciding with reduced fertility (Hamatani et al., 2004). Though these observations may be related to additional functions, such as polarization and maturation of fertilized oocytes, this is the first report that attributes any function to NLRP14 and describes an operative role for this protein in modulating innate recognition of nucleic acids.
Our combined findings highlight the importance of controlling innate immune responses to foreign and endogenous ligands and suggest that tight regulation of these processes is critical in maintaining proper immunologic homeostasis in germline. In line with this idea, a recent report illustrates that NLRP3 inflammasome signaling and mitosis are mutually exclusive events that are controlled through interactions with NEK7, which mediates inflammasome assembly and activation (He et al., 2016; Shi et al., 2016). Similarly, as an immunological rheostat, NLRP14 safeguards against inappropriate cytosolic responses to nucleic acids and the acquisition of such a function may have been a prerequisite to sexual reproduction. Homologues of the inflammasome family of proteins have been found widely across life – arguing in favor of their involvement in immunity and broader cellular processes, as is observed with other expanded gene families. Indeed, we found that mouse Nlrp14 inhibits NAS pathway similarly to human NLRP14. In addition, knock-down of Ced-3, the C. elegans homolog of mammalian NLR-proteins, results in reduced fertility and increased resistance to certain pathogens (Green et al., 2011; Jia et al., 2009; Maeda et al., 2001) — a finding that further suggests evolutionary conservation of NLR proteins across life. Speculation aside however, further studies will be necessary to clarify the molecular determinants that control NLRP14 function, including i) identification of the ubiquitin ligase that mediates TBK1-targeting for degradation; ii) resolving the conformational dynamics that mediate NLRP14 protein stability and activity; and iii) the use of genetically engineered mice to determine if manipulating NLRP14 expression could serve as a viable target of clinical importance.
Experimental procedures
GTEx Data analysis
Data preparation
To determine the set of genes with preferential expression in testis or ovary, we utilized tissue specific gene expression data from the GTEx portal (Lonsdale et al. 2013). For this particular analysis, we used the version 3 data (GTEx_Analysis_RNA-seq_RNA-SeQCv1.1.8_gene_reads__Pilot_V3_patch1.gct.gz) from http://www.gtexportal.org/home/datasets2. Since the number of available samples was different for individual tissues, we randomly sampled 20 samples from 23 different tissues. Samples selected are listed in Supporting Table 1. Tissues with less than 20 replicates (Salivary Gland, Fallopian Tube, Kidney, Cervix Uteri, Bladder, Small Intestine, Bone Marrow, Blood) were excluded for downstream analysis. To minimize the potential batch effect of platform, we restricted our analysis to the samples generated from PAXgene Tissue miRNA Kit. We performed the multidimensional scaling with correlation distance to confirm that there were no batch effects in our selected samples.
Identification of genes preferentially expressed in testes and/or ovary
Differences in library size within the GTEx data set were normalized using the Bioconductor package DESeq 1.20.0 (Anders and Huber 2010). To identify gonad specific genes (expressed in gonads and not expressed in any other tissues), the following criteria had to be met:
1. No duplicate entries for any given gene.
2. Mean of normalized expression in testis or ovary had to be greater than 20 (this threshold was chosen to reflect the fact that there are 20 replicates, and on average each replicate should have at least one read to be considered as being expressed).
3. Within non-gonad tissues, normalized expression had to be less than 20.
4. Within non-gonad tissues, fold difference (FD) between testis (or ovary) and non-gonad tissue had to be greater than 2.
To quantify the expression specificity for testis and ovary, we used EdgeR (where expression is modeled as a linear combination of different tissue factors; (Robinson et al., 2010)) to obtain confidence scores (p-values) for each gene. We used log likelihood ratio test to determine if the mean expression of a given tissue is equal to that of the rest of the tissues. We selected those genes with multiple testing adjusted p-value of less than 1%. This resulted in identifying 1180 genes as being preferentially expressed in testes and/or ovary (Supporting Table 2).
Using all GTEX data and redefining the filter
As an alternative approach from the one described above (where the complete GTEx data set was subsampled to yield results with lower computational power), we also used the full data set to identify gonad specific genes, with similar results. For this analysis, we downloaded “GTEx_Analysis_V4_RNA-seq_RNA-SeQCv1.1.8_gene_rpkm.gct.gz” from the GTEx portal (Consortium, 2013). Similarly to the original analysis, we used the “RNA isolation_PAXgene Tissue miRNA” samples to minimize inherent batch effects that could be induced by different RNA preparations. In total, all 2126 libraries, spanning 29 tissues were used. (Supporting Table 1).
In order to determine the threshold to use for presence or absence calls, we used the mean logRPKM as the expression unit in each tissue. The meanLogRPKM for GTEx data clearly follows a bimodal distribution indicating artifacts and real gene expressions. Based on this result, we used a value of -3 log RPKM as a threshold to call present or absent genes. We then used a pairwise one-tailed t-test between gonadal and non-gonadal tissues to determine how significant the tissue specificity of a given gene was. We adjusted p-values for multiple hypothesis testing, and used a 0.01 cutoff.
Selection of genes regulated during early development
To screen for the set of genes only expressed in oocyte and not in the tissues of early embryos such as trophectoderm and inner cell mass, we download the expression (measured in RPKM) of known RefSeq genes in 124 single cells from mature oocytes, preimplantation embryos and embryonic stem cells from Yan et al 2013 (Yan et al., 2013). We filtered for the genes whose RPKM expression values are greater than 1 in oocyte and equal to 0 in trophectoderm, endoderm, and epiblast. This resulted in 571 genes.
Experimental Materials and Methods
Cells
293T and A549 cells were obtained from ATCC and Vero cells were kindly provided by Dr. Vincent Racaniello (Columbia University Medical Center, NY, USA). The stable cell lines, 293-ISRE-B8 cells containing a luciferase reporter gene (luc) under the control of ISRE (IFN-Stimulated Response Element) were described previously(Shapira et al., 2009). Cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Life Technology) or Eagle’s minimum essential medium (EMEM) supplemented with 10% fetal bovine serum (FBS; Hyclone Laboratories, Inc.) and 1% penicillin and streptomycin (Life technology). Single-cell 293T cell clones stably expressing STING-HA (293T-STING-HA) were generated by transduction of pLX302-STING-HA/BSD using LT-1 transfection reagent (Mirus) followed by selection with 10 g/ml blasticidin (BSD). CRISPR/Cas9 was used to target NLRP4 and NLRP14 in 293T cells and 293T-STING-HA cells and generate single-cell clones which were maintained in DMEM supplemented with 10% FBS containing of puromycin (Life technology) at a final concentration of 1ug/ml, or blasticidin at a final concentration of 10ug/ml. All cells were cultured at 37 °C in a humidified atmosphere with 5% CO2.
Antibodies, Reagents and Plasmids
Antibodies were purchased from the indicated sources: antibodies specific to phosphorylated I0RF3 (Ser396; 4D4G), phosphorylated IRF7 (Ser471/472), phosphorylated TBK1 (Ser172; D52C2), IκBα (L35A5), phosphorylated NF Bp65 (Ser536), and mouse monoclonal HA; 6E2) were purchased from Cell Signaling Technology (CST). TBK1 (EP611Y; ab40676), and β-actin antibody (AC-15; ab62760) were purchased from Abcam. Antibodies specific to IRF7 (H-246), IRF3 (FL-425), STAT1 (C-24), and Myc (9E10) were purchased from Santa Cruz Biotechnology. Mouse monoclonal anti-FLAG M2 was purchased from Sigma and mouse-α–V5 was purchased from Life Technology. Polyinosinic-polycytidylic acid (polyI:C) was purchased from Sigma and 2′3′-cGAMP (STING ligand) and Poly(dA:dT) were purchased from Invivogen. Vesicular stomatitis virus (VSV), and Newcastle disease virus (NDV) carrying green fluorescein protein (GFP) were kindly provided by Dr. Vincent Racaniello (Columbia University Medical Center, NY, USA). Proteasome inhibitor (MG132) and Bafilomycin A1 were purchased from Sigma and Invivogen, respectively. Recombinant human IFNβ was purchased from PBL Biomedical Laboratories (New Brunswick, N.J.).
The plasmids encoding N-terminal Myc-tagged human NLRP14, NLRP4, IL17C, C4orf22, or TRIM42, RIG-I, MAVS, TBK1, IRF3 or HA-tagged Ubiquitin (HA-Ub) were purchased from TransOMIC Technologies, and Addgene. ACTL7B, DMRTC2, CCDC27, ASZ1a, ZPLD1, C10orf120, FAM154A, SPERT, ZPBP2, TGIF2LX, INSL6, and LYPD4 were cherry picked from the human ORFeome Collection 8.1. cDNA encoding C5orf47 and IRF7 was generated reverse transcription-PCR from total RNA from human testis (included the Human Total RNA Master Panel II; Clontec Laboratories, Inc) and 293T cells, respectively, and subcloned into pcDNA3.1(−) (Life Technology). N- and C-terminal deletion mutants of FLAG-tagged NLRP14 (Δ-1-100, Δ-1-345, Δ-531-1093 and Δ-109-1093), and deletion mutants of TBK1 (Δ-CC, and Δ-ULD-CC) were amplified by PCR using Phusion High Fidelity DNA polymerase (Thermo Fisher) and subcloned into p3xFLAG-CMV-14 (Sigma). N-terminal Myc-tagged NLRP14 lacking the PYD, NACHT, or LRR domains, Δ-1-345, K108X, or C-terminal FLAG-tagged NLRP14 SNP-containing mutants (D86V, A375T, D522Q, M1019I, K108X, transmembrane deleted version of MAVS Δ-514-540, as well as S172A and K670R mutants of TBK1 were generated by PCR based site-directed mutagenesis methods (Agilent Technologies). HA-tagged human cGAS and STING were kindly provided by Dr. Glen Barber (University of Miami School of Medicine, FL, USA)(Ishikawa and Barber, 2008). IFNβ, NFκB, and ISRE reporter constructs were kindly provided by Dr. Stephen Goff (Columbia University Medical Center, NY, USA). All constructions were sequence validated by GENEWIZ, Inc., or ETON Bioscience.
Establishment of CRISPR/Cas9-mediated gene editing cell clones
293T cells were transfected with pLenti-CRISPR/Cas9/puro plasmid encoding appropriate gRNA sequence (designed by using the GenScript gRNA design tool; http://www.genscript.com/CRISPR-gRNA-constructs.html) targeting exon 1 or 4 of human NLRP14 or exon 2 of human NLRP4. 5h following transfection, cells were seeded in 100mm dishes and selected with puromycin at a final concentration of 1ug/ml. Single cell clones selected and gene editing was confirmed by DNA sequencing of purified genomic DNA (QIAamp DNA Mini Kit, Qiagen). The primer pairs used for PCR amplification were as follows:
NLRP14 exon 1: 5′-GTCCCATTTATTCCATGTGCT-3′ and 5′-TCACAGTTGATCTCTTCTTTC-3′
NLRP14 exon 4: 5′-ACCTGCTATATTTCTAGCTTG-3′ and 5′-TCGATCTTCATTCAAAAGGCC-3′
NLRP4 exon 2: 5′-GTCAAGCAAGAAGAATGTGACCAT-3′ and 5′-TTCGAAGCTGTCGATGACGAACAA-3′
Reporter Analysis
Cells were stimulated with ligands at indicated concentrations and multiplicity of infection (moi; NDV-GFP) or transfected with polyI:C or B-DNA using LT-1 (Mirus). After 24 h of incubation, supernatant was collected, and 20ul were transferred into 96-well microplates seeded with 293-ISRE-B8 cells. ISRE promoter-mediated luciferase activity was determined after 24h of incubation. Similarly, cells were cotransfected with reporter constructs encoding the luciferase gene under control of the IFNβ, NFκB, or ISRE promoter. Luciferase activity was determined with dual-luciferase reporter assay system (Promega) at 24h post-transfection. All values shown are relative to Renilla luciferase reporter gene that was simultaneously monitored and used as an internal control.
Immunoprecipitation and immunoblotting
293T cells were seeded into 6-well tissue culture plates for 24h and cotransfected with corresponding plasmids as indicated in each figure. Cells were harvested 48h post-transfection, and suspended in 0.4 ml RIPA lysis buffer (50mM Tris-HCl; pH 8.0, 150 mM NaCl, 0.1% SDS, 1% NP-40, 0.5% DOC, and Complete, EDTA-free protease inhibitor cocktail; Roche) at 4°C for 2h. Cell lysates were centrifuged at 14,000g for 30 min at 4°C. The supernatant was immunoprecipitated with 1ug of indicated antibodies, and 30ul of Dynabeads (Protein G; Life Technology) at 4°C overnight. The immunocomplex was then washed five times with 0.5ml of RIPA lysis buffer. Proteins bound to the beads were boiled in 20ul of Laemmli sample buffer (BIORAD) containing 5% β-mercaptoethanol and then subjected to NuPAGE 10% Bis-Tris Gel (Life technology) and transferred to nitrocellulose membranes (AmershamTM protranTM 0.45 m NC; GE Healthcare Life Science). Membranes were blocked with TBST containing 5% non-fat milk at room temperature for 2h and incubated with corresponding antibodies, followed by incubation with horseradish peroxidase (HRP)-conjugated goat α-mouse or α-rabbit polyclonal IgG (DAKO) at room temperature for 2h. Membranes were visualized with Supersignal West Femto Maximum Sensitivity Substrate (Thermo Scientific) and detected using ChemDoc Touch Imaging System (BIORAD).
Cell Fractionation
Cells were seeded into 6-well tissue culture plates for 24h and cotransfected with indicated plasmids. 24h later, transfected cells were inoculated with NDV-GFP at an moi of 10 and fractionated after a 24h incubation. Briefly, cytoplasmic fractions were isolated with hypotonic lysis buffer (10mM Hepes; pH 7.75, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, 0.1% NP-40, and protease inhibitors) at 4°C for 10 min. Nuclear fractions were isolated with high salt nuclei extraction buffer (20mM Hepes; pH 7.5, 420 mM NaCl, 1 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 0.4% NP-40, and protease inhibitors) at 4°C for 60 min.
Real-time PCR
Total cellular RNA was prepared by using RNeasy mini kit (QIAGEN). First-strand cDNA was synthesized by using ProtoScript II First Strand cDNA Synthesis Kit (NEB). Real-time PCR was performed by using FastStart Universal SYBR green master (Rox; Roche) according to the manufacturer’s protocol. Fluorescent signals were analyzed by an CFX96TM Real-Time System (BIORAD). Human IP10, IFNβ, IRF7, ISG15, and GAPDH genes were amplified using the following primer pairs:
IP10: 5′-GGCCATCAAGAATTTACTGAAAGCA-3′ and 5′-TCTGTGTGGTCCATCCTTGGAA-3′
IFNβ: 5′-TCTGGCACAACAGGTAGTAGGC-3′ and 5′-GAGAAGCACAACAGGAGAGCAA-3′
IRF7: 5′-TGGTCCTGGTGAAGCTGGAA-3′ and 5′-GATGTCGTCATAGAGGCTGTTGG-3′
ISG15: 5′-GAGAGGCAGCGAACTCATCT-3′ and 5′-CTTCAGCTCTGACACCGACA-3′
GAPDH: 5′- ACCACAGTCCATGCCATCAC -3′ and 5′- TCCACCACCCTGTTGCTGTA -3′
The qRT-PCR primers for NLRP14 and NLRP4 were purchased from Origene.
In vitro cytopathic effect assay
Cells were stimulated with polyI:C or B-DNA. 24h later supernatant was collected 30ul was transferred onto the 96 well microplates seeded with Vero cells. After 24 h of incubation, VSV-GFP was inoculated at a moi of 1 and cell viability and VSV infectivity was determined by crystal violet staining on a fluorescein microplate reader (absorbance 570nm; SynergyH1, BioTek) or by microscopic observation, respectively.
Statistics
Results are expressed as mean ± standard deviations. A Student’s t test was used to determine p values; p-val ≤ 0.05 was considered as significant.
Supplementary Material
Highlights.
Data-driven approach identifies NLRP14 as a rheostat of cytosolic nucleic acid sensing in germ cells
NLRP14 interacts with signaling components regulating immune responses via the cGAS/RIG-I axis
NLRP14 targets TBK1 for ubiquitination and degradation thereby inhibiting both DNA and RNA sensing
Naturally occurring SNP associated with male sterility results in loss of NLRP14 function
Acknowledgments
The work was supported by the following NIH grants: S.D.S. acknowledges funding from NIH R01 (GM109018-04) and NIH U54 (CA121852-07). R.R. acknowledges NIH R01 (GM117591-01), NIH R01 (R01 GM109018), and HDTRA1-14-1-0016. We also would like to thank Stephen P. Goff, Vincent Racaniello, and Saul Silverstein at Columbia University for discussions and critical reading of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions:
T.A. designed and conducted all of the experiments. A.L. performed all of the computational analysis. R.S. and J.K. assisted with of the cloning of some of the 20 candidates screened. S.D.D. and R.R. oversaw the computational analysis. S.D.S. conceived of and directed the research project. R.R. and A.L. wrote the methods section pertaining to the computational analysis. T.A. and S.D.S. wrote the manuscript.
References
- Barber GN. STING: infection, inflammation and cancer. Nature reviews. Immunology. 2015;15:760–770. doi: 10.1038/nri3921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baril M, Racine ME, Penin F, Lamarre D. MAVS dimer is a crucial signaling component of innate immunity and the target of hepatitis C virus NS3/4A protease. Journal of virology. 2009;83:1299–1311. doi: 10.1128/JVI.01659-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X, Chiu YH, Chen ZJ. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Molecular cell. 2014;54:289–296. doi: 10.1016/j.molcel.2014.03.040. [DOI] [PubMed] [Google Scholar]
- Chen GY. Role of Nlrp6 and Nlrp12 in the maintenance of intestinal homeostasis. European journal of immunology. 2014;44:321–327. doi: 10.1002/eji.201344135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium GT. The Genotype-Tissue Expression (GTEx) project. Nature genetics. 2013;45:580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Li Y, Zhu L, Liu D, Songyang Z, Wang HY, Wang RF. NLRP4 negatively regulates type I interferon signaling by targeting the kinase TBK1 for degradation via the ubiquitin ligase DTX4. Nature immunology. 2012;13:387–395. doi: 10.1038/ni.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J, Zhu L, Xia X, Wang HY, Legras X, Hong J, Ji J, Shen P, Zheng S, Chen ZJ, Wang RF. NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell. 2010;141:483–496. doi: 10.1016/j.cell.2010.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida L, Khare S, Misharin AV, Patel R, Ratsimandresy RA, Wallin MC, Perlman H, Greaves DR, Hoffman HM, Dorfleutner A, Stehlik C. The PYRIN Domain-only Protein POP1 Inhibits Inflammasome Assembly and Ameliorates Inflammatory Disease. Immunity. 2015;43:264–276. doi: 10.1016/j.immuni.2015.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujisawa M, Fujioka H, Tatsumi N, Inaba Y, Okada H, Arakawa S, Kamidono S. Levels of interferon alpha and gamma in seminal plasma of normozoospermic, oligozoospermic,and azoospermic men. Arch Androl. 1998;40:211–214. doi: 10.3109/01485019808987944. [DOI] [PubMed] [Google Scholar]
- Genomes Project C. Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RA, Kao HL, Audhya A, Arur S, Mayers JR, Fridolfsson HN, Schulman M, Schloissnig S, Niessen S, Laband K, et al. A high-resolution C. elegans essential gene network based on phenotypic profiling of a complex tissue. Cell. 2011;145:470–482. doi: 10.1016/j.cell.2011.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Konig R, Deng M, Riess M, Mo J, Zhang L, Petrucelli A, Yoh SM, Barefoot B, Samo M, et al. NLRX1 Sequesters STING to Negatively Regulate the Interferon Response, Thereby Facilitating the Replication of HIV-1 and DNA Viruses. Cell host & microbe. 2016;19:515–528. doi: 10.1016/j.chom.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamatani T, Falco G, Carter MG, Akutsu H, Stagg CA, Sharov AA, Dudekula DB, VanBuren V, Ko MS. Age-associated alteration of gene expression patterns in mouse oocytes. Human molecular genetics. 2004;13:2263–2278. doi: 10.1093/hmg/ddh241. [DOI] [PubMed] [Google Scholar]
- He Y, Zeng MY, Yang D, Motro B, Nunez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 2016;530:354–357. doi: 10.1038/nature16959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia K, Thomas C, Akbar M, Sun Q, Adams-Huet B, Gilpin C, Levine B. Autophagy genes protect against Salmonella typhimurium infection and mediate insulin signaling-regulated pathogen resistance. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:14564–14569. doi: 10.1073/pnas.0813319106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S. Innate immune recognition of viral infection. Nature immunology. 2006;7:131–137. doi: 10.1038/ni1303. [DOI] [PubMed] [Google Scholar]
- Kufer TA, Sansonetti PJ. NLR functions beyond pathogen recognition. Nature immunology. 2011;12:121–128. doi: 10.1038/ni.1985. [DOI] [PubMed] [Google Scholar]
- Kumar H, Kawai T, Akira S. Pathogen recognition by the innate immune system. International reviews of immunology. 2011a;30:16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- Kumar H, Pandey S, Zou J, Kumagai Y, Takahashi K, Akira S, Kawai T. NLRC5 deficiency does not influence cytokine induction by virus and bacteria infections. J Immunol. 2011b;186:994–1000. doi: 10.4049/jimmunol.1002094. [DOI] [PubMed] [Google Scholar]
- Lupfer C, Kanneganti TD. Unsolved Mysteries in NLR Biology. Front Immunol. 2013;4:285. doi: 10.3389/fimmu.2013.00285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda I, Kohara Y, Yamamoto M, Sugimoto A. Large-scale analysis of gene function in Caenorhabditis elegans by high-throughput RNAi. Current biology: CB. 2001;11:171–176. doi: 10.1016/s0960-9822(01)00052-5. [DOI] [PubMed] [Google Scholar]
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Molecular cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, Accavitti-Loper MA, Madden VJ, Sun L, Ye Z, et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature. 2008;451:573–577. doi: 10.1038/nature06501. [DOI] [PubMed] [Google Scholar]
- Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell. 2010;140:619–630. doi: 10.1016/j.cell.2010.02.014. [DOI] [PubMed] [Google Scholar]
- Rathinam VA, Fitzgerald KA. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell. 2016;165:792–800. doi: 10.1016/j.cell.2016.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satie AP, Mazaud-Guittot S, Seif I, Mahe D, He Z, Jouve G, Jegou B, Dejucq-Rainsford N. Excess type I interferon signaling in the mouse seminiferous tubules leads to germ cell loss and sterility. The Journal of biological chemistry. 2011;286:23280–23295. doi: 10.1074/jbc.M111.229120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schattgen SA, Fitzgerald KA. The PYHIN protein family as mediators of host defenses. Immunological reviews. 2011;243:109–118. doi: 10.1111/j.1600-065X.2011.01053.x. [DOI] [PubMed] [Google Scholar]
- Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Shapira SD, Gat-Viks I, Shum BO, Dricot A, de Grace MM, Wu L, Gupta PB, Hao T, Silver SJ, Root DE, et al. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell. 2009;139:1255–1267. doi: 10.1016/j.cell.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, Su L, Pratt D, Bu CH, Hildebrand S, et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nature immunology. 2016;17:250–258. doi: 10.1038/ni.3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tattoli I, Carneiro LA, Jehanno M, Magalhaes JG, Shu Y, Philpott DJ, Arnoult D, Girardin SE. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-kappaB and JNK pathways by inducing reactive oxygen species production. EMBO reports. 2008;9:293–300. doi: 10.1038/sj.embor.7401161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian X, Pascal G, Monget P. Evolution and functional divergence of NLRP genes in mammalian reproductive systems. BMC Evol Biol. 2009;9:202. doi: 10.1186/1471-2148-9-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulusoy E, Cayan S, Yilmaz N, Aktas S, Acar D, Doruk E. Interferon alpha-2b may impair testicular histology including spermatogenesis in a rat model. Arch Androl. 2004;50:379–385. doi: 10.1080/01485010490474823. [DOI] [PubMed] [Google Scholar]
- Wang P, Zhu S, Yang L, Cui S, Pan W, Jackson R, Zheng Y, Rongvaux A, Sun Q, Yang G, et al. Nlrp6 regulates intestinal antiviral innate immunity. Science. 2015;350:826–830. doi: 10.1126/science.aab3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Kou Z, Jing Z, Zhang Y, Guo X, Dong M, Wilmut I, Gao S. Proteome of mouse oocytes at different developmental stages. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:17639–17644. doi: 10.1073/pnas.1013185107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerveld GH, Korver CM, van Pelt AM, Leschot NJ, van der Veen F, Repping S, Lombardi MP. Mutations in the testis-specific NALP14 gene in men suffering from spermatogenic failure. Hum Reprod. 2006;21:3178–3184. doi: 10.1093/humrep/del293. [DOI] [PubMed] [Google Scholar]
- Wu J, Chen ZJ. Innate immune sensing and signaling of cytosolic nucleic acids. Annual review of immunology. 2014;32:461–488. doi: 10.1146/annurev-immunol-032713-120156. [DOI] [PubMed] [Google Scholar]
- Yan L, Yang M, Guo H, Yang L, Wu J, Li R, Liu P, Lian Y, Zheng X, Yan J, et al. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nature structural & molecular biology. 2013;20:1131–1139. doi: 10.1038/nsmb.2660. [DOI] [PubMed] [Google Scholar]
- Yurttas P, Morency E, Coonrod SA. Use of proteomics to identify highly abundant maternal factors that drive the egg-to-embryo transition. Reproduction. 2010;139:809–823. doi: 10.1530/REP-09-0538. [DOI] [PubMed] [Google Scholar]
- Zhang L, Mo J, Swanson KV, Wen H, Petrucelli A, Gregory SM, Zhang Z, Schneider M, Jiang Y, Fitzgerald KA, et al. NLRC3, a member of the NLR family of proteins, is a negative regulator of innate immune signaling induced by the DNA sensor STING. Immunity. 2014;40:329–341. doi: 10.1016/j.immuni.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W. Negative regulation of TBK1-mediated antiviral immunity. FEBS letters. 2013;587:542–548. doi: 10.1016/j.febslet.2013.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.