

Abstract
Human methionine S-adenosyltransferase (MAT2A) catalyzes the formation of S-adenosylmethionine (SAM) from ATP and methionine. Synthetic lethal genetic analysis has identified MAT2A as an anticancer target in tumor cells lacking expression of 5′-methylthioadenosine phosphorylase (MTAP). Approximately 15% of human cancers are MTAP−/−. The remainder can be rendered MTAP− through MTAP inhibitors. We used kinetic isotope effect (KIE), commitment factor (Cf), and binding isotope effect (BIE) measurements combined with quantum mechanical (QM) calculations to solve the transition state structure of human MAT2A. The reaction is characterized by an advanced SN2 transition state. The bond forming from the nucleophilic methionine sulfur to the 5′-C of ATP is 2.03 Å at the transition state (bond order of 0.67). Departure of the leaving group triphosphate of ATP is well advanced and forms a 2.32 Å bond between the 5′-C of ATP and the oxygen of the triphosphate (bond order of 0.23). Interaction of MAT2A with its MAT2B regulatory subunit causes no change in the intrinsic KIEs, indicating the same transition state structure. The transition state for MAT2A is more advanced along the reaction coordinate (more product-like) than that from the near-symmetrical transition state of methionine adenosyltransferase from E. coli.
Graphical abstract

INTRODUCTION
S-Adenosyl-L-methionine (SAM) is an essential metabolite often referred to as the universal methyl donor. It is synthesized in mammals by the methionine S-adenosyltransferases (MATs).1,2 These enzymes catalyze an atypical reaction that uses adenosine 5′-triphosphate (ATP) and L-methionine (Met) as substrates to form SAM. The triphosphate leaving-group is hydrolyzed to phosphate and pyrophosphate before loss from the catalytic site.3,4 In humans, MAT I/III is encoded by the MAT1A gene and is expressed in liver tissue, while MAT2A is encoded by the MAT2A gene and is widely distributed. MAT2A is the primary source of SAM in most cell types, including cancer cells, in cell development, and in the developing fetal liver.5,6
SAM is used as a substrate for epigenetic regulation by methyltransferases for histones, DNA, regulatory proteins and ribonucleic acids.7,8 Methyl transfer from SAM is also critical for neurotransmitters, as ethanolamine N-methyltransferase and catechol-O-methyltransferase are necessary for catecholamine synthesis.9 SAM donates its propylamino group to the synthesis of polyamines, and interacts with the folate pathway at the level of methyl group transfer for folate-mediated thymidine synthesis.10,11
Extensive synthetic lethal genome analysis of human cancer cells with 5′-methylthioadenosine phosphorylase (MTAP) deletion at 9p21, together with the nearby CDK2, has demonstrated cell sensitivity to inhibition of PRMT5 and to MAT2A.12–14 Thus, MAT2A represents a target for novel anticancer therapeutics. In addition to its specific effects on the MTAP/PRMT5/MAT2A regulatory axis, disruption of SAM synthesis limits substrate availability for metabolic processes associated with rapid cellular growth, including polyamine synthesis.15,16 Thus, a deeper understanding of the chemical mechanism of MAT2A is expected to aid in developing anticancer agents by targeting this enzyme. Genetic information indicates that MTAP-negative cancer cells or MTAP-positive cancer cells concurrently treated with inhibitors of MTAP to induce a MTAP-negative phenotype would exhibit increased anticancer effects in response to MAT2A inhibition.12–14,17–20
We measured chemically intrinsic kinetic isotope effects (KIEs) at five positions relevant to the reaction chemistry of SAM synthesis and combined that data with QM calculations to determine the TS structure for human MAT2A. The MAT2A TS structure provides new insights into the mechanism of catalysis for this developing target in human cancer cell growth. Transition state information is informative for future design of agents that mimic transition state structure and thus provide transition state analogues.21,22
MATERIALS AND METHODS
Expression and Purification of MAT2A and MAT2B
A synthetic plasmid encoding human MAT2A was designed in a modified pET vector (now deposited at AddGene). The N-terminus of the MAT2A gene was modified with the addition of 22 amino acids including a His6 tag and a TEV cleavage site. E. coli BL21(DE3) cells harboring the plasmid were grown for 14 h at 37 °C in 25 mL of LB broth containing 50 μg/mL ampicillin. The starter culture was inoculated into 1 L of the same media and incubated at 37 °C to an OD600 of 0.6 to 0.8. MAT2A expression was induced with 1 mM IPTG for 4 h at 37 °C. The resulting cells were harvested via centrifugation at 4000g for 25 min.
Cells were suspended in cell lysis buffer containing 20 mM Tris-HCl, 300 mM NaCl, 10 mM imidazole and 1 mM dithiothreitol (DTT) at a pH of 8.0, disrupted by sonication at 30% amplitude for 10 min and clarified by three rounds of centrifugation at 25 000g for 20 min, each time discarding the pellets. Clarified lysate was purified using 5 mL of Ni-NTA resin previously equilibrated with four column volumes of lysis buffer. Protein was eluted from the column with 6 mL volumes of elution buffers containing 20 mM Tris-HCl, 1 mM DTT and imidazole concentrations increasing at 10, 30, 50, 100, and 250 mM, pH 8.0. Protein purity of elution fractions was assessed by SDS-PAGE. Pure samples were pooled and dialyzed twice for 14 h against 20 mM Tris-HCl and 1 mM DTT at a pH of 8.0. Protein was concentrated and stored at −80 °C. MAT2A concentration was assessed with spectrophotometry using an ε280 = 44.35 mM−1 cm−1.
Human MAT2B was expressed and purified using a similar expression and purification protocol as used for MAT2A. A modified pET vector encoding the MAT2B protein and an N-terminal His6 tag was expressed in E. coli.
Isotopically Labeled ATP and Methionine
Isotopically labeled ATP was synthesized by coupled enzymatic reactions. Syntheses using glucose and adenine as starting material contained 2 mM D-glucose, 2 mM adenine, 20 μM ATP, 200 μM NADP+, 20 mM phosphoenolpyruvate, 20 mM α-ketoglutaric acid, 6 mM ammonium chloride, 5 mM magnesium chloride, 50 mM potassium chloride, and 100 mM potassium phosphate at pH = 8.0 in a volume of 1 mL. Reactions were initiated by the addition of 1 unit each of hexokinase, adenylate kinase, glucose 6-phosphate dehydrogenase, 6-phosphogluconate dehydrogenase, phosphoriboisomerase, pyruvate kinase, 5-phosphoribosyl 1-pyrophosphate synthetase, adenine phosphoribosyl transferase, and α-ketoglutarate dehydrogenase. Reactions were incubated under ambient conditions (20 °C) overnight and were terminated by the addition of sulfuric acid to a final concentration of 0.01 M. Reactions with ribose as the starting material were similar to that above except that 2 mM D-ribose was used in the place of D-glucose, and 1 unit of ribokinase was used in the place of hexokinase, glucose-6-phosphate dehydrogenase, 6-phosphogluconate dehydrogenase, and phosphoriboisomerase (see Figure S1). All enzymes used in this synthesis were purchased from Sigma-Aldrich with the exception of ribokinase, 5-phosphoribosyl 1-pyrophosphate synthetase and adenine phosphoribosyl transferase, which were expressed and purified as previously described.23
ATP products were purified from reaction mixtures by reverse phase HPLC using a Kinetex C18 250 mm × 4.6 mm column eluted with 50 mM potassium phosphate and 8 mM tetrabutylammonium bisulfate, pH of 6.0, as previously described.7 Heavy atoms were incorporated into ATP from appropriately labeled commercially available glucose, ribose or adenine as starting materials. Thus, [6-14C]glucose, [6-3H2]glucose, [1-3H]ribose and [8-14C]adenine produced [5′-14C]ATP, [5′-3H2]ATP, [1′-3H]ATP, and [8-14C]ATP, respectively. Likewise, dual labeled [5′-18O, 8-14C]ATP was produced from both [6-18O]glucose and [8-14C]adenine as starting materials (see Table 1).
Table 1.
Summary of V/K Kinetic Isotope Effects, Intrinsic Kinetic Isotope Effects, Binding Isotope Effects and Calculated Kinetic Isotope Effects from QM Calculations for Human MAT2A at 5 Atomic Positions
| ATP
| ||||||
|---|---|---|---|---|---|---|
| heavy ATP substrate | starting material | light ATP substrate | KIEV/K | intrinsic KIE | calc KIE | BIE |
| [5′-14C] | 6-14C Glc | [1′-3H] | 1.063 ± 0.001 | 1.091 ± 0.003 | 1.092 | N/A |
| [5′-3H2] | 6-3H Glc | [8-14C] | 0.998 ± 0.001 | 0.997 ± 0.003 | 0.997 | 1.001 ± 0.013a |
| [5′-18O, 8-14C] | 6-18O Glc, 8-14C Adenine | [8-14C] | 1.013 ± 0.003 | 1.019 ± 0.004 | 1.019 | N/A |
|
Met | ||||||
| heavy Met substrate | starting material | light Met substrate | KIEV/K | intrinsic KIE | calc KIE | BIE |
|
| ||||||
| [Me-14C] | N/A | [Me-3H] | 1.0012 ± 0.003 | 1.0012 ± 0.003 | 1.0014 | N/A |
| [Me-3H3] | N/A | [1-14C] | 1.042 ± 0.008 | 1.024 ± 0.009 | 1.024 | 1.018 ± 0.007b |
BIE was measured for the formation of the MAT2A-ATP binary complex.
BIE was measured for the formation of that MAT2A-αβATP-Met ternary complex.
Radiolabeled methionine substrates were purchased from commercially available sources. [Me-14C]Methionine and [Me-3H3]methionine were purchased from PerkinElmer while [1-14C]methionine was purchased from American Radiolabeled Chemicals.
Measurement of Kinetic Isotope Effects
Kinetic isotope effects (KIEs) were measured by the competitive radiolabel method.24 For ATP KIEs, the appropriately labeled “heavy” substrate was mixed with “light” substrate labeled with a remote [1′-3H]ATP or [8-14C]ATP to indicate the reaction rate of the light substrate. For methionine, light isotopes were labeled with a remote [1-14C]- or [Me-3H3]methionine. The heavy and light substrates were mixed at approximately equal radioactivity count values prior to KIE experiments. Reactions contained 50 mM Tris-HCl, 10 mM MgCl2, 50 mM KCl, 50 μM ATP, 50 μM Met, and 1 μM MAT2A at pH = 8.0 and under ambient temperature (20 °C). Portions of the reactions were quenched at four reaction fractions of conversion, (f = 0.00, 0.33, 0.66, 1.00) and reactants and products resolved via reverse phase HPLC as described above. Separation of ATP and SAM for ATP-based KIE measurements used 50 mM potassium phosphate and 8 mM tetrabutylammonium bisulfate at pH = 6.0 to give retention times of 2.5 and 24 min for SAM and ATP respectively (Figure S8A). Methionine and SAM resolution for methionine based KIE measurements used 50 mM potassium phosphate and 4 mM 1-octanesulfonic acid at pH = 6.0 to increase the retention time of SAM. Under these conditions, methionine and SAM eluted at 4 and 20 min respectively (Figure S8C). HPLC-resolved samples were dried under vacuum-centrifugation, dissolved in 0.5 mL H2O and mixed with 10 mL of PerkinElmer Ultima Gold scintillation fluid. The radioactivity of samples was measured in a PerkinElmer Tricarb 2910 TR scintillation counter in dual channel analysis where 3H signals appear exclusively in channel 1 while 14C signals appear in channels 1 and 2. The ratio of 14C signal appearing in channel 1 versus channel 2 (r) was determined using standards containing only 14C. Counts per minute (CPM) were used as a measure of radioactivity and were measured by the average of ten 10 min counts for each sample. The 3H and 14C CPM were calculated according to eqs 1 and 2.
| (1) |
| (2) |
Experimental KIEs (KIEV/K) were calculated from the heavy to light isotope ratio (Rf) for reaction samples at each fraction of conversion (f) and comparing those ratios to the starting heavy to light isotope ratio (R0) according to eq 3.
| (3) |
Measurement of Forward Commitment Factor and Intrinsic KIEs
Forward commitment factors (Cf) for ATP and methionine were measured via the isotope trapping method at ambient temperature (20 °C).25 For measurement of the ATP Cf, a 24 μL mixture containing 42 μM MAT2A and 158 μM [8-14C]ATP in 50 mM Tris-HCl, 10 mM MgCl2 and 50 mM KCl at a pH = 8.0 was prepared and allowed to incubate for 1 h at 4 °C. No ATP hydrolysis was observed following this 1 h incubation (Figure S5). The incubation mixture was diluted with 976 μL of a chase solution containing 2 mM unlabeled ATP and 50 μM methionine in 50 mM Tris-HCl, 10 mM MgCl2 and 50 mM KCl at a pH = 8.0. Aliquots of 100 μL were taken at 30, 60, 90, and 120 s and quenched with sulfuric acid as described above. Samples were purified by the HPLC protocol described above to determine the amounts of [14C]SAM at each time interval. The ratio of radiolabeled SAM to the starting MAT2A-ATP complex was plotted as a function of time to determine the amount of radiolabeled SAM produced when extrapolated to time zero (Y), the y-intercept of this function (Figure S2). The forward commitment factor was calculated according to eq 4.
| (4) |
The Cf for methionine was determined in a similar experiment but with 143 μM [Me-14C]Met and 48 μM MAT2A in the preincubation mixture and 2 mM unlabeled methionine and 50 μM ATP in the chase solution. Concentrations of MAT2A complexes were estimated from the kinetic constants (Figure S7).
Intrinsic KIE values (k) were then calculated using the experimental KIEV/K values and the forward commitment factor according to eq 5.
| (5) |
Computational Methods
The transition state structure for human MAT2A was determined by using the experimentally measured intrinsic KIE values as constraints for theoretical transition state structures determined via density functional theory in Gaussian 09 using the B3LYP level of theory and the 6-31g(d) basis set. Calculated KIE values for theoretical transition states were calculated from the scaled vibrational frequencies of optimized structures of methionine and ATP at the ground state and at the transition state. The transition state calculated KIE values were iteratively refined to match the experimentally measured values.26,27
The starting coordinates for the transition state structure were determined from the crystal structure of MAT2A in complex with S-adenosylethionine and (disphosphono)aminophosphonic acid (imido-triphosphoric acid; PDB ID: 5A1G). The structures of ATP and methionine were truncated to form computational cutoff models (see Figure 2). Ground state structures for truncated ATP and methionine were taken from the same coordinates. All calculations were carried out with water as an implicit solvent and by assigning a multiplicity of 0 and a formal charge of 0 to all TS structures. Transition state calculations were carried out by fixing the S–5′C and the 5′C–5′O bonds and varying their lengths from 1.8 to 2.5 Å in 0.1 Å increments. TS structures were then refined by optimizing structures with the S–5′C bond varied from 2.00 to 2.10 Å in 0.01 Å increments while the 5′C–5′O bond was varied from 2.30 to 2.40 Å in 0.01 Å increments. A final refinement step then involved varying the S–5′C bond length from 2.03 to 2.04 Å in 0.005 Å increments while keeping the 5′C–5′O bond length fixed at 2.32 Å.
Figure 2.
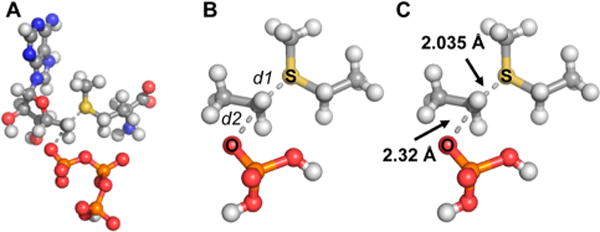
Full structures of ATP and methionine (A) were simplified in an atomic cutoff model as shown (B) for QM transition state modeling calculations. Calculations were made by iterating the lengths of the C–S (d1) bond and the C–O (d2) bond lengths, and calculating kinetic isotope effects for each theoretical transition state. The best fit of KIE values to the transition state is shown (C).
Calculated KIEs for each theoretical transition state were determined using ISOEFF98 from the scaled vibrational frequencies (SCFACT = 0.964) of each theoretical transition state and the ground state structures for ATP and methionine.27 All KIEs were calculated at 25 °C (TKELV = 298.15). Calculated KIEs at each theoretical transition state were then compared to the experimental intrinsic KIEs to determine the final transition state structure (see Figure 3 and S4). The final transition state structure had a single imaginary vibrational frequency.
Figure 3.
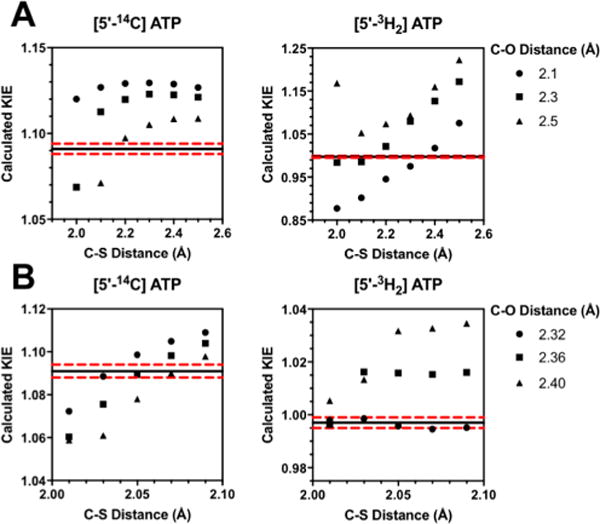
(A) KIEs predicted for the 5′-14C and 5′-3H2 as a function of C–O and C–S bond lengths. The horizontal black line indicates the experimentally measured intrinsic KIE which is flanked by the two dashed red lines indicated the upper and lower error margins for the measured KIE values. Bond lengths are iterated by units of 0.1 Å (B) Calculated KIEs for a refined set of C–S and C–O bond lengths iterated by units of 0.01 Å.
Bond orders for the final transition state were determined from the Pauling bond distance/bond order correlation.28 Reference C–S and C–O bond lengths in SAM and ATP respectively were taken from bond lengths from the Coot database29 and correlated to TS bond distances determined from the above-mentioned calculations according to eq 6 where nx is the transition state bond order, n0 is the reference bond order (assumed to be 1.0), r0 is the reference bond length, rx is the TS bond length and c is the proportionality constant. A proportionality constant of c = 0.6 was used as it is reported to be more appropriate for transition state structures where total bond order may vary at the reaction center.30
| (6) |
Measurement of Binding Isotope Effects
Binding isotope effects (BIEs) were measured by the competitive radiolabeled approach using a rapid equilibrium dialysis (RED) apparatus.31,32 The RED apparatus contained multiple chambers encased in 8 K MWCO dialysis membranes (sample wells) surrounded by a larger well (buffer well).
BIEs were measured for formation of the MAT2A-ATP binary complex with 100 μL of 20 μM radiolabeled ATP (containing a ∼ 1:1 CPM ratio of [8-14C]ATP and [5′-3H2]ATP, 5 μM MAT2A in 50 mM Tris-HCl, 10 mM MgCl2 and 50 mM KCl at a pH = 8.0 in the sample well with 300 μL of 50 mM Tris-HCl, 10 mM MgCl2 and 50 mM KCl with 20 μM radiolabeled ATP added to the buffer well. The apparatus was sealed and incubated for 4 h in a 120 rpm shaker at ambient temperature (20 °C) to allow the contents of the sample well and buffer well to equilibrate. Following incubation 50 μL aliquots of sample and buffer wells were added to 450 μL H2O before being mixed with 10 mL of PerkinElmer Ultima Gold scintillation fluid. The radioactivity of each sample was determined by scintillation counting in a PerkinElmer Tricarb 2910 TR scintillation counter. The 14C and 3H contents of each sample were determined (eqs 1 and 2). Binding isotope effects were calculated (eq 7). In this equation 14CSW and 14CBW refer to the 14C contents of the sample well and buffer well, respectively. Similarly 3HSW and 3HBW refer to the 3H contents of the sample and buffer wells, respectively.
| (7) |
The Met BIE for formation of the MAT2A-ATP-methionine ternary complex used α,β-methyleneadenosine 5′-triphosphate (αβATP) as a catalytically inert ATP analogue to allow the formation of the ternary complex without reaction. Radiolabeled methionine (100 μL of 20 μM containing a ∼ 1:1 CPM ratio of [Me-14C]methionine and [Me-3H3]methionine), 5 μM MAT2A, and 2 mM αβATP in 50 mM Tris-HCl, 10 mM MgCl2 and 50 mM KCl at a pH = 8.0 was added to the sample well while 300 μL of 50 mM Tris-HCl, 10 mM MgCl2 and 50 mM KCl with 20 μM radiolabeled methionine and 2 mM αβATP added to the buffer well. The dialysis apparatus was equilibrated and analyzed as above. The BIE for formation of the ternary complex was calculated (eq 7). The Met BIE was used to determine the intrinsic KIE value for [Me-3H3]methionine according to eq 8.
| (8) |
KIEs of Human MAT2A in Complex with MAT2B
Intrinsic kinetic isotope effects were measured for the MAT2A/MAT2B complex, using the same reaction conditions for the measurement of MAT2A KIEs. The complex was formed by 4 μM MAT2B to give a [MAT2A]:[MAT2B] of 1:4. The MAT2A-MAT2B complex has a stoichiometry of 1 MAT2A dimer for every single MAT2B monomer. The KD for MAT2B in solution has been demonstrated to be 4.5 nM.33 Under these conditions >99% of MAT2A would be complexed MAT2B. Protein mixtures were incubated for 1 h prior to the initiation of the KIE reaction.5 KIEs were measured using the same protocol described above for MAT2A alone.
RESULTS AND DISCUSSION
Measurement of Kinetic Isotope Effects
Kinetic isotope effects were measured at five atomic positions relevant to the transition state of human MAT2A (Figure 1, S3).23,24 KIEs measured by competitive isotope labeling (KIEV/K) report on all steps in the reaction mechanism up to and including the first irreversible step, here the loss of triphosphate from ATP. KIE experiments measure the MAT2A reaction rate with a “heavy” ATP or methionine substrate, containing an isotopic label near the reaction center together with the same reactant containing a “light” isotope at the reaction center, but containing a 14C or 3H label at a remote position unaffected by bond changes at the reaction center.
Figure 1.
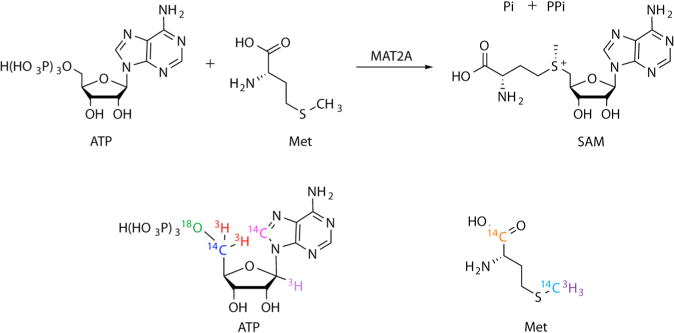
MAT2A catalyzes the formation of SAM from ATP and methionine. KIE measurements used isotopically labeled substrates with the labels indicated. Each color corresponds to labels found on an individual substrate molecule.
Experimental KIEs can be altered or obscured by BIEs or by forward commitment factors (Cf). We used isotope trapping and BIE methods,25,26 to correct experimental KIEV/K values to intrinsic KIE values. The Cf values for MAT2A were measured for both ATP and methionine, and were found to be 0.44 ± 0.05 and 0.008 ± 0.008 respectively under the conditions used for the KIE experiments (Figure S3). The relatively large Cf for ATP indicates that conversion of the ternary complex to SAM occurs at a rate similar to ATP release from this complex for equilibration with unbound ATP. The negligible Cf for methionine is consistent with an ordered reaction mechanism with ATP binding first and being trapped by subsequent methionine binding. This observation is contradictory to literature reports of the kinetic mechanism of MAT2A, which support an ordered binding mechanism where methionine binds the enzyme first.4 However, literature reports also detail the closure of an active site loop following the formation of the MAT2A-ATP-Met ternary complex prior to catalysis, a structural mechanism which supports the high observed Cf for ATP. The relatively slow catalytic turnover rate of MAT2A (0.07 s−1)3 in the absence of the activating MAT2B subunits is consistent with methionine equilibration with unbound methionine prior to catalysis.
The intrinsic KIE measurements provide information about the transition state structure of MAT2A (Table 1). The primary [5′-14C]ATP KIE at the reaction center is an indication of the nucleophilic character of this displacement. A relatively large intrinsic 14C KIE of 1.091 ± 0.003 was measured at this position, indicating that the reaction proceeds by an SN2 mechanism.27 The KIE at this position is the largest when the transition state is a symmetrical nucleophilic displacement. Nucleophilic displacements at carbon have an intrinsic 14C KIE limit near 1.14.34 Thus, some asymmetric character is present in this transition state. The E. coli methionine S-adenosyl transferase has a nearly symmetrical transition state and a reaction center 14C KIE of 1.13.35 The [5′-3H2]ATP position reports on the hybridization state of the reaction center carbon. The insignificant KIE of 0.997 ± 0.003 for the 5′-3H2 position indicates minimal changes to the vibrational modes of the C–H bonds between reactant and transition state at the reaction center carbon. This, together with the 14C KIE supports an asymmetric but well-developed SN2 reaction mechanism. The primary [5′-18O]ATP KIE reports on the degree of participation of the leaving group triphosphate oxygen in the transition state. A KIE of 1.019 ± 0.004 at the 5′-18O position indicates substantial loss of the bond order to the 5′-18O at the transition state and provides information about the C–O bond to the leaving triphosphate at the transition state. Finally the [Me-14C]methionine and [Me-3H3]methionine KIEs report on the hybridization state of the methionine sulfur nucleophile and vibrational freedom of the methyl group at the transition state. KIEs of 1.001 ± 0.003 and 1.042 ± 0.008 for [Me-14C]-methionine and [Me-3H3]methionine, respectively indicate a small change toward looser methyl C–H bonds at the transition state.
Finding a Transition State
The transition state for MAT2A was modeled by using the experimental intrinsic KIEs as constraints for computational chemistry. The initial coordinates for the ATP and methionine substrates (reactant states) were taken from a crystal structure of MAT2A bound to S-adenosylethionine and (disphosphono)aminophosphonic acid (PDB ID: 5A1G). A simplified cutoff model containing 26 atoms related to the transition state was used in the calculations (Figure 2). Calculations were performed in Gaussian 09 using the B3LYP level of theory and the 6-31g(d) basis set.36 Theoretical transition states were generated by varying the lengths of bonds marked d1 and d2 in Figure 2, corresponding to the C–S and C–O bonds, respectively. Theoretical KIEs for each potential transition state were calculated from scaled vibrational frequencies in ISOEFF98.37 The C–S and C–O bond lengths were varied from 1.8–2.5 Å (Figure 3) to find a theoretical transition state that would generate calculated KIEs best matching those measured experimentally (Table 1). The theoretical transition state structure with C–S and C–O bond lengths of 2.03–2.04 and 2.32 respectively gave calculated KIEs within experimental error to the measured intrinsic KIEs. More precisely, 2.035 and 2.320, respectively, gave the closest match of calculated KIEs to intrinsic KIEs. The only calculated KIE outside the error of the experimental intrinsic KIE measurements was the [Me-3H3]-methionine KIE which was 0.02 lower than the experimental intrinsic KIE. We considered it possible that binding isotope effects might contribute to this measurement. BIEs were measured for [Me-3H3]methionine and for [5′-3H2]ATP, as differences in binding between the light and heavy substrates lie between free reactants and the first irreversible step in the MAT2A mechanism and may therefore contribute to the experimentally determined intrinsic kinetic isotope effects.
Binding Isotope Effect Measurement
BIEs were measured using the competitive radiolabeled approach in a Rapid Equilibrium Dialysis (RED) device.31,32,38 ATP BIEs were measured for the formation of the MAT2A-ATP binary complex while the methionine BIE was measured for the formation of the MAT2A-ATP-methionine ternary complex, using α,β-methylene-adenosine 5′-triphosphate as an inert ATP analogue. The BIE for [5′-3H2]ATP position was determined to be 1.001 ± 0.013, indicating an insignificant difference in binding character between the heavy and light ATPs. A BIE of 1.018 ± 0.007 was measured for [Me-3H3]methionine, indicating that the light substrate binds preferentially to MAT2A. This BIE influences the experimental intrinsic KIE at this position by decreasing the amount of [Me-3H3]-methionine in the Michaelis complex. The [Me-3H3]Met KIE of 1.042 ± 0.008 is obtained from the 3H-depleted ternary complex. Correcting the apparent KIE for the BIE gives an intrinsic [Me-3H3]Met KIE of 1.024 ± 0.009, which closely matches the computational estimate for this position in the calculated transition state. In the ternary Michaelis complex the methyl tritiums are less constrained in their vibrational modes relative to solution. At the transition state increased vibrational freedom occurs as the sulfonium ion develops. One potential caveat for this interpretation is the assumption that the α,β-methyleneadenosine 5′-triphosphate analogue of ATP stabilizes methionine the same as the ATP complex, information which is not experimentally accessible from these BIE experiments.
MAT2A Transition State Structure
Bond orders for the forming 5′C–S and breaking 5′C–O bonds in the MAT2A transition state were estimated from the Pauling bond distance/bond order correlation. A proportionality constant of 0.6 was used as recommended for transition state structures.28,30 The transition state is late, with 0.668 bond order to the attacking sulfur nucleophile and a weak bond order of 0.227 to the oxygen of the departing triphosphate. The asymmetry of this transition state is consistent with the 5′-14C KIE measured for ATP. The total bond order calculated for the reaction coordinate at the 5′-carbon is 0.895, and indicates a loose SN2 character for the reaction.27 In contrast to the transition state for MAT2A, the E. coli methionine adenosyl transferase has a tighter transition state with a net bond order of 0.96 and a reaction center 14C KIE of 1.13.35 For human MAT2A, C–O bond breaking is ahead of C–S bond formation at the transition state, whereas these are more balanced in the E. coli enzyme. The catalytic turnover number for the E. coli enzyme is also 21-fold greater than for the human enzyme and is 1.5 s−1 versus 0.07 s−1, suggesting the stabilization of a reaction coordinate with a lower energetic barrier.39 A caveat to this analysis is that steady-state parameters may represent rate-limiting product release, rather than a strict measure of transition state barrier height. Transition state comparison establishes that, while catalyzing the same reaction, the human and E. coli enzymes differ in their kinetic properties and in the chemical properties of their transition state structures.
An electrostatic potential surface (EPS) map for the full atomic model of the MAT2A transition state reactants was generated assuming a net charge of 0 (fully protonated ATP and developing sulfonium ion neutralized by leaving group charge) for this transition state (Figure 4). The EPS structure was compared to ATP and methionine, as well as to SAM, to distinguish features unique to the transition state.
Figure 4.
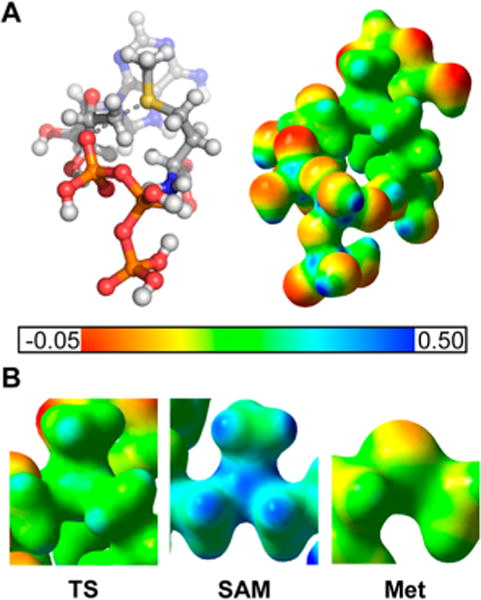
(A) Geometric and electrostatic potential surface (EPS) maps for the MAT2A transition state. Red color designates partial negative charge while blue color designates partial positive charge. (B) Zoomed in EPS maps of the central sulfur atom for the MAT2A transition state (TS), S-adenosyl-L-methionine (SAM) and methionine (Met) are shown for comparison. The partial positive charge on the sulfonium group is significantly reduced in the transition state structure when compared to the SAM structure.
The key features of the MAT2A transition state to distinguish it from reactants and products center on the charge distribution around the sulfur atom and the geometry surrounding the reaction center 5′-carbon. The SAM sulfonium charge is greater than the charge on the sulfur in the transition state. Additionally, the distance between the reaction center 5′-carbon and the leaving group triphosphate is significantly larger than a typical C–O bond length. In this late transition state, the sulfonium ion is more developed than in a symmetric SN2 transition state.
KIE Analysis of Human MAT2A in Complex with MAT2B
The intrinsic KIE values for the MAT2A/MAT2B complex were measured for [5′-14C]- and the [5′-3H2]ATPs and were found to be 1.089 ± 0.003 and 0.995 ± 0.002 respectively. Within experimental error, the intrinsic KIEs measured with MAT2A alone are identical to those determined for the complex of MAT2A with MAT2B. As intrinsic KIE values report on the nature of the transition state, the transition state structures of MAT2A are the same with and without the MAT2B regulatory protein.
CONCLUSIONS
Human MAT2A catalyzes the synthesis of S-adenosylmethionine from ATP and methionine. As the donor for methyl groups for epigenetic control of both histones and DNA, it has been identified as a potential anticancer drug target. Despite its significance, its catalytic mechanism has not been extensively explored. In human cells, MAT2A is reported to exist in a corepressor complex with MarK, Swi/Snf, BAF53a, CHD4, PARP1 and MAT2B to position the production of SAM near the sites of histone methyltransferases.40 Existence as a multiprotein nuclear complex is likely to alter the functional catalytic properties of the complex as catalysis by MAT2A alone is slow. Here we define the transition state structure of MAT2A, and demonstrate the reaction to be a late, loose, SN2 displacement mechanism. The transition state structure has the incoming nucleophilic sulfur forming a 2.03–2.04 Å partial bond to the 5′C of ATP and the leaving triphosphate group 5′C–O bond having a length of 2.32 Å at the transition state. Additionally, a complex of MAT2A with its regulatory protein, MAT2B, does not significantly affect the transition state structure of the enzyme. This information may be useful in the design of transition state analogues, as inhibition of MAT2A may be important in altering epigenetic control as an anticancer approach.
Supplementary Material
Acknowledgments
We thank Drs. Scott A. Cameron, Zhen Wang, Myles B. Poulin, and Christopher F. Stratton for helpful discussions. This work was supported by research grant CA135405, training grant GM007288 and F30 fellowship CA210372 from the National Institutes of Health.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b05803.
Figures S1–S8; Methods for kinetic analysis and chromatography information for purifications; Atomic coordinates for the QM transition state analysis (PDF)
ORCID
Vern L. Schramm: 0000-0002-8056-1929
Notes
The authors declare no competing financial interest.
References
- 1.Shafqat N, Muniz JR, Pilka ES, Papagrigoriou E, von Delft F, Oppermann U, Yue WW. Biochem J. 2013;452:27. doi: 10.1042/BJ20121580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roje S. Phytochemistry. 2006;67:1686. doi: 10.1016/j.phytochem.2006.04.019. [DOI] [PubMed] [Google Scholar]
- 3.Murray B, Antonyuk SV, Marina A, Van Liempd SM, Lu SC, Mato JM, Hasnain SS, Rojas AL. IUCrJ. 2014;1(4):240. doi: 10.1107/S2052252514012585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murray B, Antonyuk SV, Marina A, Lu SC, Mato JM, Hasnain SS, Rojas AL. Proc Natl Acad Sci U S A. 2016;113:2104. doi: 10.1073/pnas.1510959113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.LeGros HL, Jr, Halim AB, Geller AM, Kotb M. J Biol Chem. 1999;275:2359. doi: 10.1074/jbc.275.4.2359. [DOI] [PubMed] [Google Scholar]
- 6.Lu SC, Mato JM. Physiol Rev. 2012;94:1515. doi: 10.1152/physrev.00047.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Du Q, Wang Z, Schramm VL. Proc Natl Acad Sci U S A. 2016;113:2916. doi: 10.1073/pnas.1522491113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Poulin MB, Schneck JL, Matico RE, McDevitt PJ, Huddleston MJ, Wangfang H, Johnson NW, Thrall SH, Meek TD, Schramm VL. Proc Natl Acad Sci U S A. 2016;113:1197. doi: 10.1073/pnas.1521036113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stratton CF, Poulin MB, Du Q, Schramm VL. ACS Chem Biol. 2017;12:342. doi: 10.1021/acschembio.6b00922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu SC, Mato JM. J Gastroenterol Hepatol. 2008;23I(Suppl 1I):S73. doi: 10.1111/j.1440-1746.2007.05289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wallace HM, Fraser AV, Hughes A. Biochem J. 2003;576:1. doi: 10.1042/BJ20031327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marjon K, Cameron MJ, Quang P, Clasquin MF, Mandley E, Kunii K, McVay M, Choe S, Kernytsky A, Gross S, Konteatis Z, Murtie J, Blake ML, Travins J, Dorsch M, Biller SA, Marks KM. Cell Rep. 2016;15:574. doi: 10.1016/j.celrep.2016.03.043. [DOI] [PubMed] [Google Scholar]
- 13.Kryukov GV, Wilson FH, Ruth JR, Paulk J, Tsherniak A, Marlow SE, Vazquez F, Weir BA, Fitzgerald ME, Tanaka M, Bielski CM, Scott JM, Dennis C, Cowly GS, Boehm JS, Root DE, Dolub TR, Clish CB, Bradner JE, Hahn WC, Garraway L. A cience. 2016;351:1214. doi: 10.1126/science.aad5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mavrakis KJ, McDonald ER, 3rd, Schlabach MR, Billy E, Hofman GR, deWeck A, Ruddy DA, Venkatesan K, Yu J, McAllister G, Stump M, deBeaumont R, Ho S, Yue Y, Liu Y, Yan-Neale Y, Yang G, Lin F, Yin H, Gao H, Kipp DR, Zhao S, McNamara JT, Sprague ER, Zheng B, Lin Y, Cho YS, Gu J, Crawford K, Ciccone D, Vitari AC, Lai A, Capka V, Hurov K, Porter JA, Tallarico J, Mickanin C, Lees E, Pagliarini R, Keen N, Schmelzie T, Hofmann F, Stegmeier F, Sellers WR. Science. 2016;351:1208. doi: 10.1126/science.aad5944. [DOI] [PubMed] [Google Scholar]
- 15.Chen H, Xia M, Lin M, Yang H, Kuhlehkamp J, Li T, Sodir NM, Chen YH, Josef-Lenz H, Laird PW, Clarke S, Mato JM, Lu SC. Gastroenterology. 2006;133:207. doi: 10.1053/j.gastro.2007.03.114. [DOI] [PubMed] [Google Scholar]
- 16.Jani TS, Gobejishvili L, Hote PT, Barve AS, Joshi-Barve S, Kharebava G, Suttles J, Chen T, McClain CJ, Barve S. Cell Res. 2009;19:358. doi: 10.1038/cr.2008.314. [DOI] [PubMed] [Google Scholar]
- 17.Singh V, Schramm VL. J Am Chem Soc. 2006;128:14691. doi: 10.1021/ja065419p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Evans GB, Furneaux RH, Lenz DH, Painter GF, Schramm VL, Singh V, Tyler PC. J Med Chem. 2005;48:4679. doi: 10.1021/jm050269z. [DOI] [PubMed] [Google Scholar]
- 19.Basu I, Cordovano G, Das I, Belbin TJ, Guha C, Schramm VL. J Biol Chem. 2007;282:21477. doi: 10.1074/jbc.M702287200. [DOI] [PubMed] [Google Scholar]
- 20.Basu I, Locker J, Cassera MB, Belbin TJ, Merino EF, Dong X, Hemeon I, Evans GB, Guha C, Schramm VL. J Biol Chem. 2011;286:4902. doi: 10.1074/jbc.M110.198374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schramm VL. J Biol Chem. 2007;282:28297. doi: 10.1074/jbc.R700018200. [DOI] [PubMed] [Google Scholar]
- 22.Schramm VL. Annu Rev Biochem. 2011;80:703. doi: 10.1146/annurev-biochem-061809-100742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parkin DW, Leung HB, Schramm VL. J Biol Chem. 1984;259:9411. [PubMed] [Google Scholar]
- 24.Cleland WW. Methods Enzymol. 1995;249:341. doi: 10.1016/0076-6879(95)49041-8. [DOI] [PubMed] [Google Scholar]
- 25.Rose IA. Methods Enzymol. 1980;64:47. doi: 10.1016/s0076-6879(80)64004-x. [DOI] [PubMed] [Google Scholar]
- 26.Lewis BE, Schramm VL. J Am Chem Soc. 2003;125:4785. doi: 10.1021/ja0298242. [DOI] [PubMed] [Google Scholar]
- 27.Westaway KC, Fang YR, MacMillar S, Matsson O, Poirier RA, Islam SM. J Phys Chem A. 2008;112:10361. doi: 10.1021/jp804237g. [DOI] [PubMed] [Google Scholar]
- 28.Pauling L. J Am Chem Soc. 1947;69:542. [Google Scholar]
- 29.Emsley P, Lohkamp B, Scott WG, Cowtan K. Acta Crystallogr, Sect D: Biol Crystallogr. 2010;66(Pt 4):486. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Houk KN, Gustafson SM, Black KA. J Am Chem Soc. 1992;114:8565. [Google Scholar]
- 31.Stratton CF, Namanja-Magliano HA, Cameron SA, Schramm VL. ACS Chem Biol. 2015;10:2182. doi: 10.1021/acschembio.5b00490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Poulin MB, Schneck JL, Matico RE, Hou W, McDevitt PJ, Holbert M, Schramm VL. J Am Chem Soc. 2016;138:6699. doi: 10.1021/jacs.6b01612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quinlan CL, Kaiser SE, Bolaños B, Nowlin D, Grantner R, Karlicek-Bryant S, Feng JL, Jenkinson S, Freeman-Cook K, Dann SG, Wang X, Wells PA, Fantin VR, Stewart AE, Grant SK. Nat Chem Biol. 2017;13:785. doi: 10.1038/nchembio.2384. [DOI] [PubMed] [Google Scholar]
- 34.Schramm VL. Annu Rev Biochem. 1998;67:693. doi: 10.1146/annurev.biochem.67.1.693. [DOI] [PubMed] [Google Scholar]
- 35.Markham GD, Parkin DW, Mentch F, Schramm VL. J Biol Chem. 1987;262:5609. [PubMed] [Google Scholar]
- 36.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima TE, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramilo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09. Gaussian, Inc; Wallingford, CT: 2009. [Google Scholar]
- 37.Anisimov V, Paneth P. J Math Chem. 1999;26:75. [Google Scholar]
- 38.Schramm VL. Curr Opin Chem Biol. 2007;11:529. doi: 10.1016/j.cbpa.2007.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taylor JC, Takusagawa F, Markham GD. Biochemistry. 2002;41:9358. doi: 10.1021/bi025851t. [DOI] [PubMed] [Google Scholar]
- 40.Katoh Y, Ikura T, Hoshikawa Y, Tashiro S, Ito T, Ohta M, Kera Y, Noda T, Igarashi K. Mol Cell. 2011;41:554. doi: 10.1016/j.molcel.2011.02.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.