

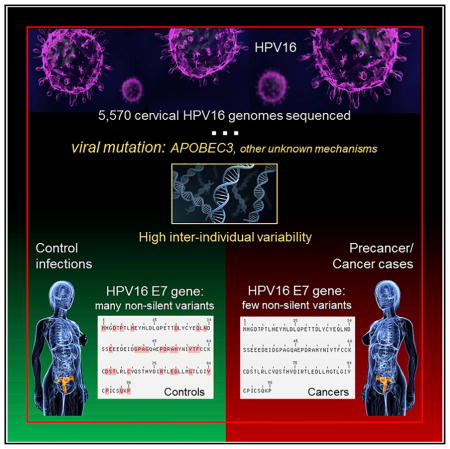
SUMMARY
Although most cervical human papillomavirus type 16 (HPV16) infections become undetectable within 1–2 years, persistent HPV16 causes half of all cervical cancers. We used a novel HPV whole-genome sequencing technique to evaluate an exceptionally large collection of 5,570 HPV16-infected case-control samples to determine whether viral genetic variation influences risk of cervical precancer and cancer. We observed thousands of unique HPV16 genomes; very few women shared the identical HPV16 sequence, which should stimulate a careful re-evaluation of the clinical implications of HPV mutation rates, transmission, clearance, and persistence. In case-control analyses, HPV16 in the controls had significantly more amino acid changing variants throughout the genome. Strikingly, E7 was devoid of variants in precancers/cancers compared to higher levels in the controls; we confirmed this in cancers from around the world. Strict conservation of the 98 amino acids of E7, which disrupts Rb function, is critical for HPV16 carcinogenesis, presenting a highly specific target for etiologic and therapeutic research.
In Brief
A genomic survey of thousands of cases of HPV infection in women around the world identifies a conserved sequence in the viral genome that is critical for carcinogenesis.
INTRODUCTION
A persistent infection with one of a dozen high-risk human papillomaviruses (HR-HPV) is the cause of virtually all cases of cervical cancer and its precursors (Schiffman et al., 2016a). More than half a million women are diagnosed with cervical cancer and more than 200,000 deaths occur each year (Fitzmaurice et al., 2015). However, most cervical HR-HPV infections are benign and resolve (become undetectable) spontaneously (Ho et al., 1998). Only a small fraction (<5%) of women infected with one of the HR-HPV types will, in fact, develop cervical precancer (cervical intraepithelial neoplasia grade 3 [CIN3] or adenocarcinoma in situ [AIS]) (Rodríguez et al., 2008), and only the minority of precancerous lesions will invade (McCredie et al., 2008). Host and viral factors clearly influence risk of progression of infected cells to precancer and invasive cancer (Kulasingam et al., 2002; Schiffman et al., 2007). Given the small size and relative simplicity of the HPV genome (double-stranded DNA genome of ~8,000 bp encoding 8 genes) and advances in HPV whole-genome sequencing (Cullen et al., 2015), it is now technically feasible to search comprehensively for viral genetic variation linked strongly to risk of cancer, thereby providing new clues into viral carcinogenic mechanisms.
It is already well established that although all the HR-HPV types are genetically related, they differ profoundly in prevalence, a measure of evolutionary fitness, and risk of causing precancer and cancer (Burk et al., 2013; Guan et al., 2012). By definition, each type differs from all others genetically by at least 10% in the conserved L1 region coding for the major capsid protein (Bernard et al., 2010). HPV genetic variation represents slow evolutionary drift; the HR-HPV types all belong to one phylogenetic clade within the Alpha genus (Schiffman et al., 2005). Nonetheless, HPV type 16 (HPV16) is uniquely carcinogenic, accounting for half of all cervical cancer cases and even higher proportions of HPV-induced cancers at other sites (de Sanjose et al., 2010; Guan et al., 2012; Serrano et al., 2015; Taylor et al., 2016). Nearly half of the small subset of women with HPV16 infections persisting for at least 2 years develop precancer within the subsequent 5 years (Kjær et al., 2010), making this small virus one of the most powerful known carcinogenic exposures. The mechanisms underlying risk differences between the other HR types and the unique carcinogenicity of HPV16 are currently unknown, although some aspect of variation in the two major HPV oncogenes, E6 and E7, is suspected based on laboratory experiments (Harden et al., 2017; Moody and Laimins, 2010).
HPV types including HPV16 are comprised, in turn, of phylogenetic variant lineage and sublineage evolutionary clades that differ from each other by ~1%–9% of the 8,000 bp HPV genome (Burk et al., 2013). HPV16 can be divided into four main variant lineages (A, B, C, D) and at least ten sublineages (A1, A2, A3, A4, B1, C1, D1, D2, D3, D4) (Burk et al., 2013). Recently, we used high-throughput HPV16 whole-genome sequencing (Cullen et al., 2015) to conduct the largest case-control study to date of variant sublineage risk of precancer and cancer among 3,215 HPV16-infected women drawn from approximately a million screened women from northern California (Mirabello et al., 2016). We observed that specific sublineages, A4, C, D2, and D3, had a significantly increased risk of precancer and cancer compared to the most common A1/A2 sublineages (Mirabello et al., 2016). For example, D2 had the strongest risk of cancer, predominantly for glandular lesions (adenocarcinomas) with an odds ratio (OR) compared to A1/A2 of >100 (Mirabello et al., 2016).
The current question is whether even finer variations demonstrate risk differences that could pinpoint molecular mechanisms. Using a novel whole-HPV-genome sequencing method (Cullen et al., 2015), we are conducting HPV genetic studies to find specific genotype-phenotype associations that can be discerned by large epidemiologic studies to generate clues to HPV carcinogenicity. We address this question by focusing on HPV16 and by sequencing thousands of HPV16-infected specimens, ranging from benign infections to cancers. To our knowledge, there have been no previous large studies evaluating finer details of HPV genetic variation and carcinogenicity. Here, we sequenced whole-genomes of a total of 5,570 HPV16 specimens to evaluate genetic variants within HPV16 and determine the associations of HPV16 single nucleotide polymorphisms (SNPs) and risk of cervical precancer/cancer.
RESULTS
A summary of the study populations is shown in Table 1. To evaluate HPV16 genome variability, we evaluated variation among cervical specimens from 3,215 HPV16-infected women (Mirabello et al., 2016) from California (PaP cohort) and paired multiple anatomical site HPV16 samples (from cervical scrape, vulvar swab, anal swab, and/or oral cavity rinse) from 58 women in Costa Rica (CVT). We utilized a case-control study design to evaluate agnostically the impact on carcinogenicity of each individual SNP and the rare variant distribution within an HPV16 lineage. We then confirmed the generalizability of the major genetic risk associations found by testing 688 HPV16-infected cervical cell specimens from CIN grade 2 or higher (CIN2+) cases from Oklahoma (SUCCEED) and a case series of 1,609 HPV16-infected cervical cell or tissue specimens from invasive cervical cancers collected internationally by the International Agency for Research on Cancer (IARC).
Table 1.
Summary of the 5,570 HPV16-Infected Women Analyzed from Four Studies by Age Group and Infection Outcome
| Cohort | Characteristics | Total Women | ||
|---|---|---|---|---|
| N | Col % | |||
| PAP | age (years) | 21–29 | 942 | 29.3 |
| 30–39 | 1,326 | 41.2 | ||
| 40–49 | 544 | 16.9 | ||
| 50–59 | 268 | 8.3 | ||
| 60+ | 135 | 4.2 | ||
| infection outcome | control | 1,107 | 34.4 | |
| CIN2 | 906 | 28.2 | ||
| CIN3/AIS | 1,093 | 34.0 | ||
| cancer | 109 | 3.4 | ||
| CVT | age (years) | 22–25 | 29 | 50.0 |
| 26–29 | 25 | 43.1 | ||
| 30–32 | 4 | 6.9 | ||
| infection outcome | control | 58 | 100 | |
| SUCCEED | age (years) | 18–20 | 60 | 9.1 |
| 21–29 | 346 | 52.4 | ||
| 30–39 | 115 | 17.4 | ||
| 40–49 | 79 | 12.0 | ||
| 50–59 | 29 | 4.4 | ||
| 60+ | 31 | 4.7 | ||
| infection outcome | CIN2 | 244 | 35.5 | |
| CIN3/AIS | 314 | 45.6 | ||
| cancer | 130 | 18.9 | ||
| IARC | age (years) | 21–29 | 42 | 2.7 |
| 30–39 | 341 | 21.9 | ||
| 40–49 | 486 | 31.2 | ||
| 50–59 | 375 | 24.1 | ||
| 60+ | 313 | 20.1 | ||
| infection outcome | cancer | 1,609 | 100 | |
| Total | infection outcome | controls | 1,165 | 20.9 |
| CIN2 | 1,150 | 20.6 | ||
| CIN3/AIS | 1,407 | 25.3 | ||
| cancer | 1,848 | 33.2 | ||
| total | 5,570 | |||
CIN2, cervical intraepithelial neoplasia grade 2; CIN3, cervical intraepithelial neoplasia grade 3; AIS, adenocarcinoma in situ; cancer includes adenocarcinoma and squamous cell carcinoma.
The majority of women in all four studies had an HPV16 A1 or A2 variant sublineage infection: 84.7% of the 3,215 women in the PaP cohort (Table S1) (Mirabello et al., 2016), 91.4% of the 58 women in CVT, 85.8% of the 688 women in the SUCCEED study, and 53.7% of 1,609 women in the internationally collected IARC study. We initially focused our analyses on variation occurring within the A1 and A2 sublineages, because these included the largest number of women and thus provided the most statistical power to detect an effect; we then evaluated if the significant SNPs and/or regions replicated across the other less common lineages (A3–A4, B1, C1–C4, D1–D4).
HPV16 Isolate Diversity between Women and within a Woman at Multiple Body Sites
Between women in the PaP cohort, we determined that 2,445 (76%) of the 3,215 HPV16 genome sequences evaluated were unique isolates (differing by ≥2 nucleotides); only 24% of the isolate genomes were shared between two or more women in this population (Table 2). This high isolate diversity was consistent across variant lineages (data not shown). To replicate and extend this finding, we compared cervical sample isolates between women and paired multiple-site sample isolates within women in the CVT study. Between these 58 women’s cervical samples, we also found a high level of HPV16 isolate diversity, 84.5% of the viral isolates were unique (n = 49) (Table 2).
Table 2.
HPV16 Isolate Diversity between Women and within a Woman at Multiple Sites
| Between Women at the Cervix | N | % of Total |
|---|---|---|
| PAP cohort | 3,215 | |
| shared HPV16 isolatesa | 770 | 24.0 |
| unique HPV16 isolatesb | 2,445 | 76.0 |
| CVT study | 58 | |
| shared HPV16 isolates | 9 | 15.5 |
| unique HPV16 isolates | 49 | 84.5 |
| Within a Woman: 54 Multi-site Pairs | N | % of Pairs |
| cervical-anal | 41 | 75.9 |
| cervical-vulvar | 11 | 20.4 |
| cervical-oral | 2 | 3.7 |
| Shared HPV16 isolatesa | 44 | 81.5 |
| cervical-anal | 34 | 82.9c |
| cervical-vulvar | 8 | 72.7d |
| cervical-oral | 2 | 100.0e |
| Unique HPV16 isolatesb | 10 | 18.5 |
| cervical-anal | 7 | 17.1c |
| cervical-vulvar | 3 | 27.3d |
| cervical-oral | 0 | 0.0 |
Shared isolate is defined as the same HPV16 genome sequence (<2 bp different) present in more than one women in the study population or shared within a woman at multiple sites.
Unique isolate is defined as an HPV16 genome sequence differing by 2 or more bp from all other sequences in the population or differing from the paired sample within a woman.
Percent of cervical-anal pairs.
Percent of cervical-vulvar pairs.
Percent of cervical-oral pairs.
In contrast, the evaluation of 54 paired multiple-site samples from 52 women (two women had 3 sites sampled) showed that within a woman, the viral isolates were the same (shared) in 81.5% of paired samples (n = 44; Table 2). Of these shared isolates, 72.2% (n = 39) were exactly the same HPV16 genome sequence. There were five pairs that differed by one nucleotide. Paired cervical-anal, cervical-vulvar, and cervical-oral samples all showed this high level of HPV16 isolate sharing among the pairs within a woman.
This confirms that there is high HPV16 isolate diversity between women, but not within a woman at multiple body sites; and importantly, the paired sample analysis supports the notion that the high isolate diversity we observe is not due to sequencing errors because similar diversity was not seen within women.
SNP Associations with Precancer and Cancer
There were 1,985 total site-specific variable positions (SNPs), most were rare SNPs, and only 103 had a minor allele frequency (MAF) ≥1.0% within the HPV16 A1/A2 sublineages. 48 SNPs were associated with risk of CIN3+ (p < 0.05), and the minor alleles for the top ten SNPs with the smallest p values were associated with a reduced risk of CIN3+ (Figure 1; Table S2). Two SNPs located in the URR region were strongly associated with a reduced risk of CIN3+ and remained significant after adjustment for multiple tests, at positions 7387 (OR 0.04, p = 1.6 × 10−9) and 7359 (OR 0.06, p = 7.2 × 10−5; Table 3).
Figure 1. Individual SNP Associations with CIN3+ Compared to the Controls within the A1/A2 Lineages in Women in the PaP Cohort.

Logistic regression models were used to obtain the odds ratio and 95% confidence intervals for CIN3+ risk for eachSNP minor alleleusing the controls asthe referent group. The x axis indicates the viral genome nucleotide position; The y axis indicates the p-value. The vertical arrows indicate the most significant SNPs identified. SNPs are colored by gene region in the legend. URR, upstream regulatory region; E6, early gene 6; E7, early gene 7; E1, early gene 1; E2, early gene 2; E4, early gene 4; E5, early gene 5; L2, late gene 2; L1, late gene 1; NC, non-coding region.
See also Table S2.
Table 3.
Summary of the Top SNPs Associated with CIN3+ within the Specified Sublineages
| Position | Viral Gene | Allelesa | MAF (%) | p Valueb | OR | 95% CI Lower | 95% CI Upper |
|---|---|---|---|---|---|---|---|
| A1/A2 Sublineages | |||||||
| 7387 | URR | C/G | 1.9 | 1.57 × 10−09 | 0.044 | ∞ | ∞ |
| 7359 | URR | A/T/G | 2.0 | 7.19 × 10−05 | 0.055 | 0.01 | 0.23 |
| A2 Sublineage | |||||||
| 5032 | L2 | G/T | 21.9 | 0.002 | 2.50 | 1.39 | 4.49 |
| 4247 | L2 | G/A | 24.2 | 0.010 | 2.14 | 1.20 | 3.82 |
| 7507 | URR | C/A | 15.8 | 0.014 | 2.34 | 1.19 | 4.61 |
| 5121 | L2 | T/C | 4.2 | 0.026 | 10.46 | 1.33 | 82.04 |
| 7868 | URR | A/G | 7.1 | 0.031 | 3.46 | 1.12 | 10.72 |
MAF, minor allele frequency; OR, odds ratio; 95% CI, 95% confidence interval; CIN3+, cervical intraepithelial neoplasia grade 3 and cancer; URR, upstream regulatory region; L2, late gene 2.
Minor allele(s)/major allele.
Logistic regression models were used to obtain the p value, OR and 95% CI for CIN3+ risk using the controls as the referent group.
Within the A2 sublineage, there were five SNPs associated with CIN3+ (Table 3). A SNP at position 5121 in L2 was associated with an increased risk of CIN3+ (OR 10.5, 95% CI 1.3–82.0, p = 0.02). This SNP was part of a haplotype including variants at positions 5032, 4247, and 7507 (Table 3), which represents a sub-branch of the main A2 phylogenetic branch. Considering just the A1 sublineage, the URR SNPs, 7387 and 7359, were the top SNPs associated with a reduced risk of CIN3+ (data not shown). Within the D sublineages separately, there were no individual SNPs significantly associated with CIN3+, likely due to the much smaller numbers.
Cumulative Variant Analysis by Viral Gene Region
We evaluated genetic variants by the type of substitution (i.e., nonsynonymous, nonsense, synonymous) for each viral gene region or open reading frame (ORF) within the HPV16 A1/A2 sublineages. A burden test was used to determine if the variant distribution was different between the cases and controls by viral region within the A1/A2 sublineages. Despite nearly equal numbers of cases and controls, the controls overall had a significantly higher number of variants compared to the precancer/cancer cases (10,290 total variants versus 9,485; summarized in Table S3). In particular, there were more nonsynonymous and nonsense variants in the controls. Strikingly, the E7 ORF had statistically significantly fewer nonsynonymous and nonsense variants in the cases compared to the controls (OR 0.16, p = 1.1 × 10−7).
A large amount of the variation across the HPV16 genome was comprised of rare nucleotide variants, and controls had a much higher number of rare variants (2,574 rare variants versus 1,811 for CIN3+; beta of −0.13 per mutation, p = 6.5 × 10−7). Therefore, we also conducted burden analyses focusing on rare variants (Table 4). Figure 2 illustrates the cumulative nonsynonymous and nonsense rare variant counts observed in the cases and controls; and, clearly shows that E7 has significantly fewer variants in the cases compared to the controls (OR 0.16, p = 1.1 × 10−7; all E7 non-silent variants were rare). There were statistically significantly more nonsynonymous and nonsense rare variants also in the E1 (OR 0.64, p = 0.001) and L1 (OR 0.55, p = 7.9 × 10−5) gene regions (Figure 2; Table 4). The nonsynonymous/nonsense rare variant differences appear to be spread fairly evenly throughout the L1 ORF for the cases and controls, however for E7 and E1 more rare variants occurred in the 3′ portion of the gene (Figure S1). On closer evaluation of E7 variants, there were a few invariant regions whereas, the majority of the variants observed in the controls occurred in the central or C terminus regions (Figure 3). We further determined that the majority of rare nonsynonymous/nonsense DNA changes observed within the E7 ORF (56.8%), particularly in the controls, matched an APOBEC3-associated motif (p < 0.01; Figure 3). Thus, these specific DNA changes are potentially due to the antiviral activity of human APOBEC3 (hA3) cytidine deaminases. The E7 protein is one of two main HPV oncoproteins (along with E6); however, in contrast, there were nearly equal numbers of nonsynonymous/nonsense rare variants within the E6 ORF in the controls and CIN3+ cases (Figure S2).
Table 4.
Rare Variant Burden Analysis for Nonsynonymous and Nonsense Variants within the A1 and A2 Sublineages for Controls (N = 972) and CIN3+ Cases (N = 936) in the PaP Cohort
| Viral Gene | N Variants, Controls | N Controls with Variants | % of Controls | N CIN3 with Variants | % of CIN3 | N Cancer with Variants | % of Cancer | N Variants, CIN3+ | N CIN3+ with Variants | % of CIN3+ | p Valuea |
|---|---|---|---|---|---|---|---|---|---|---|---|
| URR | 303 | 228 | 23.5 | 201 | 23.0 | 12 | 19.4 | 267 | 213 | 22.8 | 0.745 |
| E6 | 104 | 86 | 8.8 | 69 | 7.9 | 3 | 4.8 | 77 | 72 | 7.7 | 0.362 |
| E7 | 48 | 45 | 4.6 | 7 | 0.8 | 0 | 0.0 | 7 | 7 | 0.7 | 1.1 × 10−07b |
| E1 | 241 | 161 | 16.6 | 106 | 12.1 | 0 | 0.0 | 136 | 106 | 11.3 | 0.001b |
| E2 | 238 | 167 | 17.2 | 127 | 14.5 | 11 | 17.7 | 154 | 138 | 14.7 | 0.151 |
| E4 | 57 | 43 | 4.4 | 34 | 3.9 | 2 | 3.2 | 39 | 36 | 3.8 | 0.566 |
| E5 | 56 | 54 | 5.6 | 44 | 5.0 | 3 | 4.8 | 50 | 47 | 5.0 | 0.611 |
| L2 | 354 | 256 | 26.3 | 197 | 22.5 | 18 | 29.0 | 271 | 215 | 23.0 | 0.090 |
| L1 | 163 | 130 | 13.4 | 63 | 7.2 | 10 | 16.1 | 90 | 73 | 7.8 | 7.9 × 10−05b |
| Total | 1,564 | 620 | 63.8 | 521 | 59.5 | 36 | 58.1 | 1,091 | 557 | 59.5 | 6.5 × 10−07b |
CIN3, cervical intraepithelial neoplasia grade 3; CIN3+, includes CIN3 and cancer; URR, upstream regulatory region; E6, early gene 6; E7, early gene 7; E1, early gene 1; E2, early gene 2; E4, early gene 4; E5, early gene 5; L2, late gene 2; L1, late gene 1. URR is a non-coding regulatory region; all rare variation is included for this region.
p value for the rare variant burden comparing the controls with CIN3+.
p values remain significant after Bonferroni adjustment for multiple tests.
Figure 2. Rare Non-silent Variant Distributions across the Viral Genomes of A1/A2 Lineages for the CIN3+ Cases and Controls in the PaP Cohort.

Each viral gene region is represented by a wedge in the plots and the size of the wedge corresponds to the nucleotide size of the region. The nucleotide positions of each viral gene region are shown in the center. The two inner rings illustrate the genome positions of the rare variants within each of the viral gene regions for cases in red and controls in blue. Each vertical line represents a variant and the height of the line corresponds to how many individuals had this variant. The total variant counts for each viral region are given in Table 4. The outermost ring illustrates the cumulative rare variant counts by viral gene region. The density of the variants and the variant increase across each region are represented by the slope of the line for the cases and controls. URR, upstream regulatory region; E6, early gene 6; E7, early gene 7; E1, early gene 1; E2, early gene 2; E4, early gene 4; E5, early gene 5; L2, late gene 2; L1, late gene 1.
See also Figure S1 and Tables S3 and S4–S6.
Figure 3. HPV16 Rare Nonsynonymous and Nonsense Nucleotide Variants Observed in the E7 ORF.

Women in the PAP Cohort with HPV16 A1/A2 lineage infections were included in this analysis. Rare nonsynomous and nonsensense variants are shown as black and red sticks, respectively. Controls are shown as blue lollipops and CIN3+ cases as either red or grey lollipops corresponding to CIN3 or AIS, respectively. Amino acid changes are indicated with each lollipop, and number of lollipop circles indicates the number of individuals with that variant. The domains of E7 are colored, see legend, and the stars indicate changes consistent with an APOBEC3-induced change. CR1, conserved region 1; CR2, conserved region 2; RB1, retinoblastoma (Rb) protein interacting region. CIN3, cervical intraepithelial neoplasia grade 3; AIS, adenocarcinoma in situ.
See also Figure S2.
In the other HPV16 sublineages (A3–A4, B1, C1–C4, D1–D4), there were smaller numbers of controls to evaluate because these sublineages are very rare in this population and/or are more carcinogenic (A4, C1–C4, D2, D3) (Mirabello et al., 2016). Nevertheless we observed hypovariation in E7 in all of the CIN3+ cases (Table S4). In the SUCCEED study, there was also hypovariation in E7 in the cases, and the nonsynonymous/nonsense variants decreased with severity of the lesion from CIN2 to CIN3+, supporting the pattern seen in the PaP cohort (Table S4). E7 had the lowest number of rare nonsynonymous/nonsense variants in the CIN3+ cases (n = 1, 0.2%), and pairwise comparisons between each viral gene region indicated that the E7 gene was significantly less variable than all other HPV16 regions (each p < 0.01; data not shown).
Given the strong signal we observed within the E7 gene region, we additionally sequenced the HPV16 genomes from 1,609 cervical cancers cases collected internationally to evaluate the generalizability of reduced genetic variants within E7. We confirmed hypovariation at E7 in this set of cancers, with only 0.8% of cancers showing any rare variants within the E7 gene region (Table S4). Further, pairwise comparisons of the genetic variants among all the viral gene regions within cancers determined that the E7 gene was significantly less variable than all other gene regions (each p < 0.001; Table S5). Importantly, compared to E6, E7 had significantly fewer, rare non-silent genetic variants in cancers (p = 6.1 × 10−5).
Nonsynonymous and Synonymous Sequence Divergence
We estimated the dN/dS ratios as a measure of nonsynonymous and synonymous sequence divergence to better understand the increased genetic variation we observed in the controls by viral ORF. In the PaP cohort, within all ORFs, dN < dS (Table S6), which is evidence of widespread purifying selection and has been observed for nearly all viruses (Holmes, 2009).
For all individuals, E7 was the most constrained ORF, having the smallest overall dN/dS ratio of 0.054 (Table S6). For the A1/A2 lineages, E7 had a dN/dS ratio 5.6 times higher in controls (dN/dS ratio 0.28) than in cases (dN/dS ratio 0.05; p = 1.3 × 10−4). This suggests that the controls have substantially relaxed constraint in E7 compared to very high constraint in the cases. Both dN and dS were lower in the cases, but dN decreased by a factor of 6.5 and dS decreased by a factor of 1.2. Therefore, the lower dN/dS ratio in E7 in the cases is likely due to purifying selection against nonsynonymous changes (i.e., carcinogenicity depends on a highly conserved E7 protein). Interestingly, in contrast, E6 showed the highest overall dN/dS ratio near 1.0 in cases and controls, consistent with neutrality. In the IARC A1 cancers, patterns were consistent with the PaP cases. The E7 ORF had the smallest overall dN/dS ratio of 0.087 (i.e., high constraint) and E6 had the highest ratio of 1.12 (i.e., the least constraint).
DISCUSSION
This intensive whole-genome sequencing study of HPV16 and cervical cancer risk has increased the published number of full HPV16 sequences by an order of magnitude and revealed several fundamental discoveries. First, we have uncovered tremendous variability below the level of HPV16 variant lineages. HPV16 actually includes thousands of viral isolates (each a unique HPV16 genome). This suggests a paradigm shift from thinking of HPV16 as a single viral entity undergoing slow genetic drift to considering each HPV16 isolate to be a separate virus, with possibly different carcinogenic potential, which will necessarily lead to a re-interpretation of HPV natural history and carcinogenesis. For example, re-appearance of HPV16 after initial clearance can now be subdivided into (1) a new infection with different isolate(s) versus (2) possible loss of immunologic control of a single variant.
We have previously related variation within a HPV16 variant lineage to carcinogenic “strength.” Realizing that HPV16 is one of the most important human carcinogens, conferring much greater risks of precancer and cancer over many years compared to other high-risk HPV types, understanding the mechanism of HPV16’s unique carcinogenicity would be useful for etiologists, prevention researchers and designers of individualized therapy.
We have found that HPV16’s unique risk might be due in part to a particular structure of the E7 protein; specifically, we report here the results of a very large whole-genome study of HPV16, which identified E7 conservation significantly associated with cervical carcinogenesis. The agnostic SNP analyses and rare variant burden tests indicate that HPV16 isolates within controls have a significantly increased burden of genetic variants compared with the precancer and cancer cases. But, most importantly, we have determined that there is a much higher rate of rare nucleotide variation within E7 in the infections that did not progress to precancer/cancer during our multi-year period of observation (median follow-up time = 4.3 years; data not shown).
We observed that the HPV16 E7 protein leading to cervical cancer is virtually invariant. In general, E7 has several cellular targets but its inhibition of RB1 seems most important for transformation, and during progression to precancer, E7 becomes overexpressed (Roman and Munger, 2013). Previous studies had shown that the HPV16 E7 gene is generally conserved (Eschle et al., 1992; Roman and Munger, 2013; Safaeian et al., 2010; Song et al., 1997). Further, HPV16 E7 has been suggested in a very small study to be more hypovariable than HPV31 E7 and HPV73 E7, and this was thought to be a possible clue to HPV16’s greater carcinogenicity (Safaeian et al., 2010). Our findings strongly corroborate the earlier report of E7 hypovariability, and we further show that in a large case-control analysis E7 is less constrained in benign infections, and genetic variation in E7 reduces HPV16 carcinogenicity. Furthermore, analysis of cervical cancer cases from around the world indicates that E7 hypovariation is consistent in different geographic locations and racial groups and suggests, in summary, that E7 variation greatly decreases the risk of invasive cancer.
The genetic variants that do occur in HPV16, particularly in the controls, may be induced at least in part by the antiviral activity of human APOBEC3 (hA3) cytidine deaminases (removes an amino group from cytosine, leading to a C-to-T base change at specific motifs) (Mangeat et al., 2003). It has been shown that hA3A-mediated cytidine deaminase activity induces HPV16 mutations (Vartanian et al., 2008) and can inhibit HPV infectivity (Warren et al., 2015). The latter paper further suggested that these induced mutations, if not lethal, may also be responsible for the long-term accumulation of genomic changes that contribute to HPV-associated cancers (Warren et al., 2015). We have determined that a large portion of the non-silent variants we observed specifically in E7 were consistent with APOBEC3-induced mutations. It is possible that a significant portion of the genetic variations across the HPV genome can be explained by APOBEC3-induced mutations, a hypothesis we are now pursuing.
The control HPV16 genomes with greater variation were from benign HPV16 infections presumed to be sampled during the productive, virion-producing, stage of the viral life cycle (Doorbar, 2005; Doorbar et al., 2012). To evaluate whether the greater variation we are observing in these genomes is related to variation produced during a productive infection and viral genome amplification, we evaluated the rare variants among controls stratified by cytology results. HPV16-infected women with normal (NILM) cytology had significantly more variation than those with cytomorphologic manifestations of an HPV infection (LSIL, p = 0.01). Those with equivocal cytologic changes (ASC-US) had intermediate levels. This does not suggest that variability is introduced by high levels of viral replication; on the contrary, variability might be associated with decreased fitness. However, a longitudinal series of samples from women with HPV16 infections would allow a more accurate estimation of HPV16 variation accumulating within a woman over time and would also allow a more precise estimate of the HPV16 mutation rate.
The limited repertoire of E7 proteins has features of purifying selection. As an interesting contrast with E7, the other oncogene, E6, was the most unconstrained ORF in the viral genome in both A1/A2 cases and controls. In cancers, E6 was even less constrained, with a dN/dS ratio over 1, suggesting an excess of non-silent variants in this region. It is unknown why there would be a relaxation of purifying selection against the accumulation of mutations in this important oncogene, while E7 is the opposite. It is possible that there could be positive selection for immune escape in certain E6 regions or APOBEC3-induced mutations that balances out the background tendency toward purifying selection.
Earlier studies have shown that transgenic mice that express E6 and E7 from HPV16 develop cancer (Lambert et al., 1993). HPV16 E6 was shown to have a modest transforming function dependent on its interaction with PDZ domain partners and inactivation of p53 (reviewed in Vande Pol and Klingelhutz, 2013), and it was HPV16 E7 that was the more potent driver for cervical cancer (Jabbar et al., 2009, 2012; Riley et al., 2003; Roman and Munger, 2013). This was more recently supported by the observation that more HPV16 E7 was required to sustain a malignant phenotype compared to E6 in an extensive characterization of primary cervical cancer HPV transcripts (The Cancer Genome Atlas Research, 2017). HPV16 E7 is also critical in earlier phases of the viral life cycle, particularly for genome amplification when the episomal DNA of the virus is undergoing replication (Doorbar, 2005; Doorbar et al., 2012). It has been directly shown that the E7 protein is required for viral replication in human keratinocytes (Flores et al., 2000).
Because the crystal structure of the ternary complex of HPV16 E6, E6AP (the cellular ubiquitin ligase), and p53 has been solved, it is possible to speculate on the contributions of particular residues of E6 (Figure S2) to p53 degradation (Martinez-Zapien et al., 2016). The E6 protein must bind first to the leucine-rich LxxLL motif of E6AP, this interaction exposes a structured p53-binding cleft on E6 allowing it to interact and target p53 for degradation; none of the E6 residues with a pivotal role in the E6-E6AP complex formation show variants predicted to influence the E6 protein phenotype leading to decreased biological activity. The amino acid changes at R15Q, R17I, P20L, Q21H, and Q21E likely render E6 unable to support p53 binding and degradation (Cooper et al., 2003; Liu et al., 1999; Martinez-Zapien et al., 2016; Zanier et al., 2013), and except for the single R15Q change in a CIN3, these variations occurred in controls (n = 8). D51 and F54 are highly conserved positions in high-risk HPV E6 proteins and establish critical contacts to p53, here a D51N was observed in two controls and a CIN3 and F54S in a control, which could disrupt p53 degradation for these E6 variants. Residues from L107 to K122 in the carboxy-terminal zinc-binding domain of E6 are highly conserved in low- and high-risk HPV E6 proteins, this region shows minimal non-synonymous variation with only one control with a L107S change in this region. The amino acid changes at D11N, K18T, I59V, and between residues H31 and E36 were observed in multiple cases and controls and likely do not impair the binding and p53 degradation activities of E6 (Cooper et al., 2003; Dalal et al., 1996; Martinez-Zapien et al., 2016; Zanier et al., 2013). Thus, based on review of the predicted impact of variants found in isolates from cases and controls on the structure and function of the E6 protein, we did not identify a clear pattern explaining the amino acid changes in cases or controls.
We also saw a significant increase in rare variants in L1 and E1 in the controls. The L1 protein is the major component of the HPV16 capsid. L1 is expressed in the upper differentiated layers of the epithelium during productive infection after genome amplification and it participates in packaging of the amplified viral genomes (Doorbar, 2005; Doorbar et al., 2012). E1 is an ATP-dependent DNA helicase that is critical for replication and amplification of the viral episome in infected cells, particularly during the initial phase of viral genome amplification (Bergvall et al., 2013; Egawa et al., 2012; Kim and Lambert, 2002). Interestingly, both E7 and E1 are essential mediators of viral genome amplification (Doorbar, 2005; Doorbar et al., 2012). Perhaps maintenance of these functions not for viral replication, but incidentally driving host cell DNA replication in cells headed toward quiescence is a mechanism involved in mutation and selection of cervical epithelial cells advancing toward cancer. Interestingly, E1 has been considered one of the most highly conserved proteins encoded by papillomaviruses infecting different animal species, reflecting its essential role in the life cycle (Bergvall et al., 2013). Here, we did observe constraint in E1, slightly more in the cases; however, it was not the most constrained ORF. It is unclear how the rare variation we observe in these regions may affect HPV16 carcinogenicity.
In our sequence divergence analyses, we additionally observed that overall the E2 ORF exhibited little constraint while the overlapping E4 ORF showed strong constraint. The hinge region of E2 contains both CD4+ T and B cell epitopes that overlap a portion of E4 (Dillner, 1990; Lehtinen et al., 1995), and this region has been previously shown to exhibit simultaneous positive selection in E2 and purifying selection in E4 (Hughes and Hughes, 2005; Narechania et al., 2005). This is consistent with our observation of elevated dN in E2 concurrent with elevated dS in E4, which may reflect selection of nonsynonymous changes in E2 that are disproportionately synonymous in the overlapping E4. Therefore, changes observed in E2 can at least in part be explained by positive selection for immune escape acting on epitopes which overlap portions of E4.
The paired samples within a woman at multiple body sites showed that there is limited HPV16 isolate diversity within a woman. This also supports the robustness and reproducibility of our sequencing assay. There were a smaller number of pairs that only differed from each other by 1 to 2 nucleotides, and although we cannot rule out sequencing error, more interestingly, this could be evidence of within-host viral evolution, especially considering the large number of unique isolates observed between women. We are planning a more detailed longitudinal analysis to explore the possibility of within-host viral mutations. Given the high level of diversity we identified in HPV16 isolates, we can now use unique HPV isolates as an endpoint in epidemiologic studies to re-evaluate previous ideas of transmission, sexual networks, persistence, clearance, and reappearance in longitudinal studies.
In conclusion, our findings suggest that within-lineage hypovariation in specific regions of the viral genome is important for HPV16 carcinogenicity. It will be important to follow-up our E7 findings, and the other implicated regions, in subsequent studies to characterize genetic variation in these regions in more depth. A priority should be to conduct functional studies to understand how the particular E7 genetic sequence apparently common to virtually all cervical cancers caused by HPV16 worldwide is linked to this type’s unique carcinogenicity and might be an appropriate target for therapeutic intervention.
STAR★METHODS
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical Commercial Assays | ||
| ION AMPLISEQ LIB 2.0-384LV | ThermoFisher Scientific | 4480442 |
| Ion 540 Kit-Chef (2/init) | ThermoFisher Scientific | A30011 |
| Ion 540 Chip Kit | ThermoFisher Scientific | A27766 |
| Deposited Data | ||
| HPV16 sequence data | This study | GEO: |
| Oligonucleotides | ||
| Ampliseq HPV16 | ThermoFisher Scientific Ampliseq Custom White Glove assay; Cullen et al., 2015 |
WG00038_HPV16_2 |
| Software and Algorithms | ||
| Torrent Mapping Alignment Program v5.0.13 | ThermoFisher Scientific | https://github.com/iontorrent/TS/tree/master/Analysis/TMAP |
| Torrent Variant Caller v.5.0.3 | ThermoFisher Scientific | https://github.com/domibel/IonTorrent-VariantCaller |
| GATK LeftAlignIndels module v.3.3 | Broad Institute | https://software.broadinstitute.org/gatk/ |
| snpEff v.3.6c | Pablo Cingotani | https://github.com/cgrlab/cgrHPV16 |
| Integrative Genomics Viewer (IGV) | Broad Institute | http://software.broadinstitute.org/software/igv/ |
| SNPGenie | Nelson et al., 2015 | https://github.com/chasewnelson/snpgenie |
| SPSS version 21.0 | IBM Corp. | Released 2013. IBM SPSS Statistics for Windows, Version 22.0. Armonk, NY: IBM Corp. |
| R statistical package version 3.3.1 | R Core Team | https://www.r-project.org/ |
CONTACT FOR RESOURCE AND REAGENT SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by Lead Contact, Lisa Mirabello (mirabellol@mail.nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Study populations
PaP cohort
The cases and controls for the large discovery phase of our study were chosen from the Kaiser Permanente Northern California (KPNC)-NCI HPV Persistence and Progression (PaP) cohort study (Mirabello et al., 2016). This study population has been previously described (Schiffman et al., 2015, 2016b). Briefly, KPNC is a very large integrated health care system. The PaP cohort includes approximately 55,000 women out of approximately 1 million who underwent routine cervical cancer screening using Hybrid Capture 2 (HC2) (QIAGEN, Germantown, MD) and cytology between January 2007 and January 2011. Participants could opt-out of having their residual cervical specimens retained, which are normally discarded. We obtained coded information on subsequent cervical cancer screening test results and histology results from women in the cohort from electronic health records. All personal identifying information was kept strictly at KPNC.
The specimen storage was focused on women found to be HPV-positive by HC2; the collection includes more than three-quarters of HC2-positive women during the enrollment time period. Only 8% of women with collected specimens opted out from having their specimen banked and tested for HPV-related biomarkers including HPV genotypes. Our study included 3,579 exfoliated cervical cell specimens known to contain HPV16 DNA (Castle et al., 2011), including 1,032 women diagnosed with CIN2, 1,079 CIN3 and 71 squamous cell carcinoma (SCC) cases; 91 adenocarcinoma in situ (AIS) and 41 adenocarcinoma; and 1,265 women with < CIN2 in follow-up through 2013. The cases were diagnosed at baseline or during the study follow-up period after the baseline specimens were collected. The controls were defined as women having baseline specimens with HPV16 DNA and no histologic evidence of equivocal precancer or worse (CIN2+) during the follow-up study period according to the coded data obtained from electronic health records. Women included in our study population were followed for a median of 4.3 years (interquartile range = 3.4 years; range = 7.8 years). Women were censored if they received treatment for a CIN2+ lesion, or until the last documented follow-up cytology or histology. The study protocol was reviewed and approved yearly by Kaiser Permanente and the National Cancer Institute Institutional Review Boards.
CVT
Sixty-one women with paired (N = 56) or triplet (N = 5) multiple-site HPV16-positive infections from the control arm of the Costa Rica Vaccine Trial (CVT) were evaluated for viral isolate diversity within a woman at multiple anatomical sites and between the women at the cervix. All women chosen had HPV16 at the cervix and also at the anus, vulva, and/or oral region, including 49 cervical-anal, 13 cervical-vulvar and 4 cervical-oral pairs (127 total specimens).
CVT is a NCI–sponsored community-based, double-blind, randomized clinical trial of Cervarix™(GlaxoSmithKline Biologicals, Rixensart, Belgium) in Guanacaste, Costa Rica. This study was a nested analysis within the previously reported CVT (clinical trials registration NCT00128661). Written informed consent was obtained from all participants in CVT. Institutional review board approval was obtained at both the NCI and in Costa Rica. CVT study design and main outcomes have been previously described (Herrero et al., 2008, 2011; Hildesheim et al., 2014). The study enrolled female residents initially aged 18–25 years living in Guanacaste and Puntarenas, Costa Rica, with community-based census recruitment occurring during 2004–2005.
At the 4-year study visit (when women were 22 to 29 years), all women were asked to provide oral rinse samples and sexually-experienced women were asked to provide a vulvar swab, cervical sample and anal swab specimens, which were collected by study clinicians. The anal specimen was collected prior to the pelvic exam among sexually active women (defined by a history of vaginal intercourse) using a dry swab (Kreimer et al., 2011). A vulvar sample was then collected by swabbing the mucosal surface of the labia minora (using a single Dacron swab), and cervical cells were obtained using a Cervex-Brush (Rovers Medical Devices, Oss, the Netherlands) (Herrero et al., 2011). The oral rinse specimen was collected by use of a 30 s oral rinse and gargle with 15mL of Scope mouthwash (Procter and Gamble Company, Cincinnati, OH) (Herrero et al., 2013).
SUCCEED
To confirm the findings from the PaP Cohort, we studied a set of HPV16-positive women from the Study to Understand Cervical Cancer Early Endpoints and Determinants (SUCCEED). The details of the study design and specimen collection have been previously described (Wang et al., 2009; Wentzensen et al., 2009a, 2009b). Briefly, a total of 1,899 women were enrolled into SUCCEED between November 2003 and September 2007. We recruited women that were referred to colposcopy or treatment at the University of Oklahoma Dysplasia Clinic based at the University of Oklahoma Health Sciences Centre (OUHSC), with a recent abnormal Pap smear diagnosis or a biopsy diagnosis of CIN/cancer. Women were excluded if they were less than 18 years of age, pregnant at the time of their visit, previously treated with chemotherapy or radiation for any cancer, or scheduled for vaginal colposcopy. Here, we included all CIN2+ exfoliated cervical cell specimens previously found to contain HPV16 DNA, including 716 women: 252 women diagnosed with CIN2, 316 CIN3 and 116 SCC cases; 12 AIS and 20 adenocarcinoma. Written informed consent was obtained from all women enrolled in the study and Institutional Review Board approval was provided by OUHSC and the US National Cancer Institute.
IARC
1,701 additional HPV16-positive cervical cell or tissue (frozen biopsy or formalin-fixed paraffin-embedded [FFPE]) specimens from cervical cancer cases were studied to assess the worldwide generalizability of our main finding, from the biobank at IARC. These samples were part of the IARC-coordinated cervical cancer case series, cervical cancer case–control studies and population-based HPV prevalence surveys from 39 countries worldwide (Bosch et al., 1995; Clifford et al., 2005; Cornet et al., 2012; Crosbie et al., 2013; Muñoz et al., 2003). Both local and IARC ethical committees approved all studies. We sequenced all HPV16-positive histologically confirmed cervical cancers with adequate DNA left in the IARC biobank.
METHOD DETAILS
HPV16 detection and DNA isolation
PaP cohort
DNA was extracted from the banked STM specimens as previously described (Burk et al., 1996). Typing methods varied for different subsets of the cohort. To test many of the enrollment PaP samples, the Burk laboratory (Bronx, NY) used MY09/M11 L1 degenerate primer PCR (MY09/11 PCR) and type-specific dot-blot hybridization methods (Burk et al., 1996; Castle et al., 2002). Another large group of enrollment specimens was tested using the Linear Array® HPV Genotyping System (Roche Molecular Diagnostics, Pleasanton, CA). A third group of specimens was typed by BD using Onclarity (BD, Sparks, MD).
CVT
DNA was extracted from the cervical, oral, and anal samples using the MagNAPure LC DNA isolation procedure (Roche Diagnostics, Indianapolis, IN). DNA specimens were then tested for 25 HPV DNA types utilizing the SPF10 PCR-DEIA (DNA enzyme immunoassay)-LiPA25 version 1 method (Labo Biomedical Products, Rijswijk, the Netherlands) (Kleter et al., 1998, 1999). To increase the sensitivity of HPV16/18 detection, all positive cervical, anal and vulvar specimens on SPF10 PCR/DEIA that were negative for HPV16 or HPV18 by LiPA25 were also tested using HPV16 and 18 type-specific primers; all oral samples were tested using HPV16 and 18 type-specific primers (van Doorn et al., 2006).
SUCCEED
Details of DNA isolation and HPV detection have been previously described (Dunn et al., 2007; Wentzensen et al., 2009a). Briefly, DNA was isolated from 1 mL aliquots of PreservCyt-fixed cells using the QIAamp DNA Blood Mini Kit (QIAGEN) following a rinse in Hanks’ Balanced Salt Solution (HBSS). The Linear Array® HPV Genotyping System (Roche Molecular Diagnostics) was used to detect HPV genotypes. Hybridization of PCR products to linear arrays and subsequent signal detection were performed using the Auto-LiPA automated staining system (Innogenetics N.V., Belgium). Hybridization to both β-globin probes was required to report genotyping results. A hybridization signal was called “positive” when an unambiguous, continuous band was observed on the array.
IARC
DNA was extracted from frozen biopsy specimens, cervical cells, or FFPE at IARC, as previously described (Cornet et al., 2012). Samples were genotyped for 37 HPV types using a GP5+/6+based PCR system (Jacobs et al., 2000) in one centralized laboratory (Department of Molecular Pathology, Vrije University, Amsterdam, the Netherlands).
Ion Torrent library preparation and sequencing
We used a custom Thermo Fisher Ion Torrent AmpliSeq HPV16 panel approach to amplify the entire 7906 bp HPV16 genome (Cullen et al., 2015). The next-generation sequencing (NGS) assay used the Thermo Fisher Life Sciences’ Ion Torrent Proton and a custom HPV16 Ion Ampliseq panel of 47 multiplexed primer sets. Custom overlapping degenerate primers were designed to cover the entire viral genomes for all HPV16 variant lineages. After amplification, an Ion Torrent adaptor-ligated library was generated following the manufacturer’s Ion AmpliSeq Library Preparation kit 2.0-96LV protocol with slight modifications (Life Technologies, Part #4480441). Raw sequencing reads were quality and adaptor trimmed using the Torrent Suite Software and aligned to the HPV16 reference sequence (7906bp) from GenBank (NC_001526.4) using the Torrent Mapping Alignment Program v5.0.13. BAM files were left-aligned using the GATK LeftAlignIndels module v.3.3 (McKenna et al., 2010). SNP calls were made using the Torrent Variant Caller v.5.0.3, and variants were annotated with HPV gene/region using snpEff v.3.6c (Cingolani et al., 2012). Pipeline settings and parameters can be found at https://github.com/cgrlab/cgrHPV16.
QUANTIFICATION AND STATISTICAL ANALYSIS
HPV16 variant lineage classification
HPV16 variant lineage assignment was based on the maximum likelihood (ML) tree topology constructed using RAxML MPI v7.2.8.27 (Stamatakis, 2006) that included sixteen HPV16 A, B, C, and D variant lineage reference sequences. In case of multiple HPV16 lineages present in a specimen (coinfection with multiple isolates of HPV16 was observed in more than 20% of HPV16-infected women), a ‘predominant’ variant lineage was assigned if there was a low level coinfection. The predominant lineage variable sites were called based on presence in at least 60% of the sequence reads to avoid calling variants from multiple HPV16 genomes; therefore, variable sites are presumed from a single ‘predominant’ genome. Women without a clear ‘predominant’ HPV16 isolate, i.e., with equal levels of multiple isolates that preclude identification of a predominant genome, were excluded from the analysis.
We previously showed that dichotomized dot blot low viral load (PCR signal strength index of 1–3, compared to high PCR signal strength index of 4–5) was directly correlated with sequence completion rates (Cullen et al., 2015), thus samples with a low viral load had fewer reads. Here, we excluded the samples with overall poor coverage across the genome (< 2000 total reads per sample or < 2000 nucleotide positions callable), and individual nucleotide sites per sample with low reads (< 5). A total of 364 PaP cohort specimens (10.2%) were removed from the discovery stage due to poor read depth (Cullen et al., 2015), poor or spotty coverage across the genome, within-HPV16 coinfection for which a predominant lineage could not be assigned, or inability to assign lineage. 12 specimens were similarly excluded from the CVT dataset (9.4%), 28 specimens were excluded from the SUCCEED dataset (3.9%), and 92 from the IARC dataset (5.4%). 95.1% of all samples included in the study had at least 10x coverage across each viral gene region (99% of samples had at least 10x coverage of E7), with a median of 3,120 reads per gene and sample (IQR 605-8,695). We used this stringent quality control threshold to minimize sequencing errors in the dataset.
Statistical analyses
We first evaluated HPV16 genome diversity by estimating the number of variable positions and annotation by gene region. We used hierarchical pairwise comparisons of all the PaP HPV16 genome sequences to determine the number of shared and unique HPV16 sequences present in each population. A unique HPV16 ‘isolate’ was defined as differing by 2 or more nucleotide positions (to minimize potential error) from all other sequences. Missing sequence data at a variable position was ignored for pairwise comparison, which could result in underestimation of the true number of unique HPV16 sequences. For the paired analysis, all pairs were additionally examined manually to verify homogeneity or number of differences using the Integrative Genomics Viewer (IGV) (Robinson et al., 2011). Sample pairs that were ≥2bp different at the nucleotide level were determined to be different or unique viruses.
We evaluated the association of all individual single nucleotide polymorphisms (SNP) with risk of precancer/cancer. Squamous and glandular lesions were combined for analysis of precancer and cancer cases. We assessed each SNP occurring in each lineage and/or sublineage for those lineages with enough numbers (> 100 case and control specimens) to analyze individually (A1, A2, D), and for A1/A2 combined and all non-A1/A2 sublineages combined. A logistic regression model was used to obtain the odds ratio (OR) and 95% confidence intervals (CI) for precancer/cancer risk for each SNP using the controls (i.e., women with HPV16 and < CIN2) as the referent group. We chose CIN3/AIS as the histopathologic definition of precancer; we opted not to include as precancer less severe cases (CIN2) because of the mediocre reproducibility and histopathologic ambiguity of this diagnosis (i.e., it is a mixture of benign infections and precancers).
We then conducted aggregation tests to evaluate the cumulative effects of multiple genetic variants within an HPV16 lineage in each viral gene region (open reading frame [ORF]) and the URR region using a custom ‘burden’ test (Ionita-Laza et al., 2013). We evaluated if there was an association between all genetic variants, and specifically rare genetic variants, by each viral region in the precancer and cancer (CIN3+) cases compared to the controls using the Fishers Exact test and burden statistical analyses (Ionita-Laza et al., 2013). The burden tests for rare variants included nucleotide variants that had a rare allele frequency of < 1% (based on prevalence within sublineages A1/A2 in PaP) or < 3% (based on prevalence within other rare sublineages in PaP, SUCCEED, and IARC specimens). Burden tests were used to evaluate all coding region variants (i.e., synonymous and nonsynonymous), and also separately the nonsynonymous rare variants (missense and nonsense, excluding synonymous) since these potentially alter protein function. For the non-coding URR, all rare variants were included in the burden test. Evaluating variation within the specified sublineages enabled us to exclude the lineage defining variants from this analysis and focus on the non-lineage defining variants. We also determined the proportion of rare variants that matched an APOBEC3-induced mutation signature for specific regions of the viral genome by estimating the DNA changes that occurred at one of the eight possible APOBEC3-associated motifs (5′ [C/T]•C > T•N 3′) out of the 96 possible DNA motifs in a 3 base-pair residue defined by the variant in the center as well as neighboring nucleotides in its 5′ side and 3′ side (Vartanian et al., 2008).
Statistical analyses were performed with SPSS version 21.0 and R version 3.3.1; all statistical tests were two-sided.
Mean sequence divergence was estimated as , where Di is the mean number of pairwise differences at each of s polymorphic sites over a sequence alignment L nucleotides in length (Nelson and Hughes, 2015). Aligned sequences were analyzed to determine mean numbers of nonsynonymous and synonymous pairwise differences and sites for each ORF using SNPGenie (Nelson et al., 2015) (https://github.com/chasewnelson/snpgenie), yielding estimates of mean nonsynonymous (dN) and mean synonymous (dS) nucleotide distance. Codons containing indeterminate nucleotides (N’s) were excluded from analysis. The hypotheses of neutrality (dN = dS) and equivalent nucleotide distance ratios in cases and controls (dN/dS-cases = dN/dS-controls) were evaluated using a Z-test, with the variance of the differences estimated using a bootstrap method (100 replicates) (Nei, 2000).
DATA AND SOFTWARE AVAILABILITY
Data and software are available as described in the Key Resources Table.
Supplementary Material
Figure S1. Rare Nonsynonymous Variation within the E7, E1, and L1 Gene Regions for the CIN3+ Cases and Controls with an A1/A2 HPV16, Related to Figure 2 and Table 4
Cases and controls have a significantly different rare variant distribution in E7, E1, and L1. Cumulative rare variant counts (y axis) are shown for the CIN3+ cases in red and controls in blue by viral genome position for the specified gene (x axis). CIN3+, cervical intraepithelial neoplasia grade 3 and cancer.
Figure S2. HPV16 Rare Nonsynonymous and Nonsense Nucleotide Variants Observed in the E6 ORF. Related to Figure 3 and Table 4
Women in the PAP Cohort with HPV16 A1/A2 lineage infections were included in this analysis. Rare nonsynomous and nonsensense variants are shown as black and red sticks, respectively. Controls are shown as blue lollipops and CIN3+ cases as either red, dark red, grey or black lollipops corresponding to CIN3, SCC, AIS, or Adeno, respectively. Amino acid changes are indicated with each lollipop, and number of circles indicates the number of individuals with that variant. Specific domains of E6 are colored, see legend, and the stars indicate changes consistent with an APOBEC3-induced change. CIN3, cervical intraepithelial neoplasia grade 3; SCC, squamous cell carcinoma; AIS, adenocarcinoma in situ; Adeno, adenocarcinoma.
Highlights.
5,570 cervical HPV16 genomes revealed high inter-individual variability
Higher number of non-silent variants in benign HPV16 infections compared with cases
E7 gene was strikingly devoid of genetic variants in precancer and cancer cases
HPV16 rare variants potentially induced by the antiviral activity of human APOBEC3
Acknowledgments
We thank Ayax Perez Gallegos, Albert Einstein College of Medicine, for helpful input on HPV16 E6 protein structure. C.W.N. was supported by a Gerstner Scholars Fellowship from the Gerstner Family Foundation at the American Museum of Natural History. This study was funded by the intramural research program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute, NIH. This project has been funded in whole or in part with federal funds from the National Cancer Institute, NIH (HHSN261200800001E); and, the National Cancer Institute (CA78527) and the Einstein Cancer Research Center (P30CA013330) from the National Cancer Institute (to R.D.B.). Work at IARC was supported by a grant from the Institut National du Cancer (INCa), France (SHSESP 16-006). The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Supplemental Information includes two figures and six tables and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2017.08.001.
AUTHOR CONTRIBUTIONS
Study conceptualization and supervision was carried out by L.M., M.S, and R.D.B. Sample collection, resources, and/or clinical characterization was performed by G.C., N.W., T.R.-B., T.L., S.F., P.E.C., J.W., R.Z., A.R.K., A.H., P.G., C.P., and D.C.B. HPV sequencing and data curation was performed by M.Y., M.C., J.F.B., S.B., Q.Y., M.S., L.B., D.R., J.M., and M.D. Sequence bioinformatics and assessment was performed by M.Y., L.M., Q.Y., M.S., and Z.C. Formal statistical analyses were performed by L.M., K.Y., B.Z., L.S., Y.X., and C.W.N. The manuscript was drafted by L.M., R.B. and M.S. and reviewed by all co-authors.
References
- Bergvall M, Melendy T, Archambault J. The E1 proteins. Virology. 2013;445:35–56. doi: 10.1016/j.virol.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard HU, Burk RD, Chen Z, van Doorslaer K, Hausen H, de Villiers EM. Classification of papillomaviruses (PVs) based on 189 PV types and proposal of taxonomic amendments. Virology. 2010;401:70–79. doi: 10.1016/j.virol.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch FX, Manos MM, Muñoz N, Sherman M, Jansen AM, Peto J, Schiffman MH, Moreno V, Kurman R, Shah KV. Prevalence of human papillomavirus in cervical cancer: a worldwide perspective. International biological study on cervical cancer (IBSCC) Study Group. J Natl Cancer Inst. 1995;87:796–802. doi: 10.1093/jnci/87.11.796. [DOI] [PubMed] [Google Scholar]
- Burk RD, Ho GYF, Beardsley L, Lempa M, Peters M, Bierman R. Sexual behavior and partner characteristics are the predominant risk factors for genital human papillomavirus infection in young women. J Infect Dis. 1996;174:679–689. doi: 10.1093/infdis/174.4.679. [DOI] [PubMed] [Google Scholar]
- Burk RD, Harari A, Chen Z. Human papillomavirus genome variants. Virology. 2013;445:232–243. doi: 10.1016/j.virol.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castle PE, Schiffman M, Gravitt PE, Kendall H, Fishman S, Dong H, Hildesheim A, Herrero R, Bratti MC, Sherman ME, et al. Comparisons of HPV DNA detection by MY09/11 PCR methods. J Med Virol. 2002;68:417–423. doi: 10.1002/jmv.10220. [DOI] [PubMed] [Google Scholar]
- Castle PE, Shaber R, LaMere BJ, Kinney W, Fetterma B, Poitras N, Lorey T, Schiffman M, Dunne A, Ostolaza JM, et al. Human papillomavirus (HPV) genotypes in women with cervical precancer and cancer at Kaiser Permanente Northern California. Cancer Epidemiol Biomarkers Prev. 2011;20:946–953. doi: 10.1158/1055-9965.EPI-10-1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, Platts A, Wang L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clifford GM, Gallus S, Herrero R, Muñoz N, Snijders PJF, Vaccarella S, Anh PTH, Ferreccio C, Hieu NT, Matos E, et al. IARC HPV Prevalence Surveys Study Group. Worldwide distribution of human papillomavirus types in cytologically normal women in the International Agency for Research on Cancer HPV prevalence surveys: a pooled analysis. Lancet. 2005;366:991–998. doi: 10.1016/S0140-6736(05)67069-9. [DOI] [PubMed] [Google Scholar]
- Cooper B, Schneider S, Bohl J, Jiang Yh, Beaudet A, Vande Pol S. Requirement of E6AP and the features of human papillomavirus E6 necessary to support degradation of p53. Virology. 2003;306:87–99. doi: 10.1016/s0042-6822(02)00012-0. [DOI] [PubMed] [Google Scholar]
- Cornet I, Gheit T, Franceschi S, Vignat J, Burk RD, Sylla BS, Tommasino M, Clifford GM IARC HPV Variant Study Group. Human papillomavirus type 16 genetic variants: phylogeny and classification based on E6 and LCR. J Virol. 2012;86:6855–6861. doi: 10.1128/JVI.00483-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosbie EJ, Einstein MH, Franceschi S, Kitchener HC. Human papillomavirus and cervical cancer. Lancet. 2013;382:889–899. doi: 10.1016/S0140-6736(13)60022-7. [DOI] [PubMed] [Google Scholar]
- Cullen M, Boland JF, Schiffman M, Zhang X, Wentzensen N, Yang Q, Chen Z, Yu K, Mitchell J, Roberson D, et al. Deep sequencing of HPV16 genomes: A new high-throughput tool for exploring the carcinogenicity and natural history of HPV16 infection. Papillomavirus Res. 2015;1:3–11. doi: 10.1016/j.pvr.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalal S, Gao Q, Androphy EJ, Band V. Mutational analysis of human papillomavirus type 16 E6 demonstrates that p53 degradation is necessary for immortalization of mammary epithelial cells. J Virol. 1996;70:683–688. doi: 10.1128/jvi.70.2.683-688.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Sanjose S, Quint WGV, Alemany L, Geraets DT, Klaustermeier JE, Lloveras B, Tous S, Felix A, Bravo LE, Shin H-R, et al. Retrospective International Survey, HPV Time Trends Study Group. Human papillomavirus genotype attribution in invasive cervical cancer: a retrospective cross-sectional worldwide study. Lancet Oncol. 2010;11:1048–1056. doi: 10.1016/S1470-2045(10)70230-8. [DOI] [PubMed] [Google Scholar]
- Dillner J. Mapping of linear epitopes of human papillomavirus type 16: the E1, E2, E4, E5, E6 and E7 open reading frames. Int J Cancer. 1990;46:703–711. doi: 10.1002/ijc.2910460426. [DOI] [PubMed] [Google Scholar]
- Doorbar J. The papillomavirus life cycle. J Clin Virol. 2005;32(Suppl 1):S7–S15. doi: 10.1016/j.jcv.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Doorbar J, Quint W, Banks L, Bravo IG, Stoler M, Broker TR, Stanley MA. The biology and life-cycle of human papillomaviruses. Vaccine. 2012;30(Suppl 5):F55–F70. doi: 10.1016/j.vaccine.2012.06.083. [DOI] [PubMed] [Google Scholar]
- Dunn ST, Allen RA, Wang S, Walker J, Schiffman M. DNA extraction: an understudied and important aspect of HPV genotyping using PCR-based methods. J Virol Methods. 2007;143:45–54. doi: 10.1016/j.jviromet.2007.02.006. [DOI] [PubMed] [Google Scholar]
- Egawa N, Nakahara T, Ohno S, Narisawa-Saito M, Yugawa T, Fujita M, Yamato K, Natori Y, Kiyono T. The E1 protein of human papillomavirus type 16 is dispensable for maintenance replication of the viral genome. J Virol. 2012;86:3276–3283. doi: 10.1128/JVI.06450-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eschle D, Dürst M, ter Meulen J, Luande J, Eberhardt HC, Pawlita M, Gissmann L. Geographical dependence of sequence variation in the E7 gene of human papillomavirus type 16. J Gen Virol. 1992;73:1829–1832. doi: 10.1099/0022-1317-73-7-1829. [DOI] [PubMed] [Google Scholar]
- Fitzmaurice C, Dicker D, Pain A, Hamavid H, Moradi-Lakeh M, MacIntyre MF, Allen C, Hansen G, Woodbrook R, Wolfe C, et al. Global Burden of Disease Cancer Collaboration. The global burden of cancer 2013. JAMA Oncol. 2015;1:505–527. doi: 10.1001/jamaoncol.2015.0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores ER, Allen-Hoffmann BL, Lee D, Lambert PF. The human papillomavirus type 16 E7 oncogene is required for the productive stage of the viral life cycle. J Virol. 2000;74:6622–6631. doi: 10.1128/jvi.74.14.6622-6631.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan P, Howell-Jones R, Li N, Bruni L, de Sanjosé S, Franceschi S, Clifford GM. Human papillomavirus types in 115,789 HPV-positive women: a meta-analysis from cervical infection to cancer. Int J Cancer. 2012;131:2349–2359. doi: 10.1002/ijc.27485. [DOI] [PubMed] [Google Scholar]
- Harden ME, Prasad N, Griffiths A, Munger K. Modulation of microRNA-mRNA target pairs by human papillomavirus 16 oncoproteins. MBio. 2017;8:e02170–16. doi: 10.1128/mBio.02170-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero R, Hildesheim A, Rodríguez AC, Wacholder S, Bratti C, Solomon D, González P, Porras C, Jiménez S, Guillen D, et al. Costa Rica Vaccine Trial (CVT) Group. Rationale and design of a community-based double-blind randomized clinical trial of an HPV 16 and 18 vaccine in Guanacaste, Costa Rica. Vaccine. 2008;26:4795–4808. doi: 10.1016/j.vaccine.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero R, Wacholder S, Rodríguez AC, Solomon D, González P, Kreimer AR, Porras C, Schussler J, Jiménez S, Sherman ME, et al. Costa Rica Vaccine Trial Group. Prevention of persistent human papillomavirus infection by an HPV16/18 vaccine: a community-based randomized clinical trial in Guanacaste, Costa Rica. Cancer Discov. 2011;1:408–419. doi: 10.1158/2159-8290.CD-11-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrero R, Quint W, Hildesheim A, Gonzalez P, Struijk L, Katki HA, Porras C, Schiffman M, Rodriguez AC, Solomon D, et al. CVT Vaccine Group. Reduced prevalence of oral human papillomavirus (HPV) 4 years after bivalent HPV vaccination in a randomized clinical trial in Costa Rica. PLoS ONE. 2013;8:e68329. doi: 10.1371/journal.pone.0068329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildesheim A, Wacholder S, Catteau G, Struyf F, Dubin G, Herrero R CVT Group. Efficacy of the HPV-16/18 vaccine: final according to protocol results from the blinded phase of the randomized Costa Rica HPV-16/18 vaccine trial. Vaccine. 2014;32:5087–5097. doi: 10.1016/j.vaccine.2014.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho GY, Bierman R, Beardsley L, Chang CJ, Burk RD. Natural history of cervicovaginal papillomavirus infection in young women. N Engl J Med. 1998;338:423–428. doi: 10.1056/NEJM199802123380703. [DOI] [PubMed] [Google Scholar]
- Holmes E. The Evolution and Emergence of RNA Viruse. New York, NY: Oxford University Press; 2009. [Google Scholar]
- Hughes AL, Hughes MAK. Patterns of nucleotide difference in overlapping and non-overlapping reading frames of papillomavirus genomes. Virus Res. 2005;113:81–88. doi: 10.1016/j.virusres.2005.03.030. [DOI] [PubMed] [Google Scholar]
- Ionita-Laza I, Lee S, Makarov V, Buxbaum JD, Lin X. Sequence kernel association tests for the combined effect of rare and common variants. Am J Hum Genet. 2013;92:841–853. doi: 10.1016/j.ajhg.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabbar SF, Abrams L, Glick A, Lambert PF. Persistence of high-grade cervical dysplasia and cervical cancer requires the continuous expression of the human papillomavirus type 16 E7 oncogene. Cancer Res. 2009;69:4407–4414. doi: 10.1158/0008-5472.CAN-09-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabbar SF, Park S, Schweizer J, Berard-Bergery M, Pitot HC, Lee D, Lambert PF. Cervical cancers require the continuous expression of the human papillomavirus type 16 E7 oncoprotein even in the presence of the viral E6 oncoprotein. Cancer Res. 2012;72:4008–4016. doi: 10.1158/0008-5472.CAN-11-3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs MV, Walboomers JMM, Snijders PJF, Voorhorst FJ, Verheijen RHM, Fransen-Daalmeijer N, Meijer CJLM. Distribution of 37 mucosotropic HPV types in women with cytologically normal cervical smears: the age-related patterns for high-risk and low-risk types. Int J Cancer. 2000;87:221–227. [PubMed] [Google Scholar]
- Kim K, Lambert PF. E1 protein of bovine papillomavirus 1 is not required for the maintenance of viral plasmid DNA replication. Virology. 2002;293:10–14. doi: 10.1006/viro.2001.1305. [DOI] [PubMed] [Google Scholar]
- Kjær SK, Frederiksen K, Munk C, Iftner T. Long-term absolute risk of cervical intraepithelial neoplasia grade 3 or worse following human papillomavirus infection: role of persistence. J Natl Cancer Inst. 2010;102:1478–1488. doi: 10.1093/jnci/djq356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleter B, van Doorn L-J, ter Schegget J, Schrauwen L, van Krimpen K, Burger M, ter Harmsel B, Quint W. Novel short-fragment PCR assay for highly sensitive broad-spectrum detection of anogenital human papillomaviruses. Am J Pathol. 1998;153:1731–1739. doi: 10.1016/S0002-9440(10)65688-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleter B, van Doorn L-J, Schrauwen L, Molijn A, Sastrowijoto S, ter Schegget J, Lindeman J, ter Harmsel B, Burger M, Quint W. Development and clinical evaluation of a highly sensitive PCR-reverse hybridization line probe assay for detection and identification of anogenital human papillomavirus. J Clin Microbiol. 1999;37:2508–2517. doi: 10.1128/jcm.37.8.2508-2517.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreimer AR, González P, Katki HA, Porras C, Schiffman M, Rodriguez AC, Solomon D, Jiménez S, Schiller JT, Lowy DR, et al. CVT Vaccine Group. Efficacy of a bivalent HPV 16/18 vaccine against anal HPV 16/18 infection among young women: a nested analysis within the Costa Rica Vaccine Trial. Lancet Oncol. 2011;12:862–870. doi: 10.1016/S1470-2045(11)70213-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulasingam SL, Hughes JP, Kiviat NB, Mao C, Weiss NS, Kuypers JM, Koutsky LA. Evaluation of human papillomavirus testing in primary screening for cervical abnormalities: comparison of sensitivity, specificity, and frequency of referral. JAMA. 2002;288:1749–1757. doi: 10.1001/jama.288.14.1749. [DOI] [PubMed] [Google Scholar]
- Lambert PF, Pan H, Pitot HC, Liem A, Jackson M, Griep AE. Epidermal cancer associated with expression of human papillomavirus type 16 E6 and E7 oncogenes in the skin of transgenic mice. Proc Natl Acad Sci USA. 1993;90:5583–5587. doi: 10.1073/pnas.90.12.5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen M, Hibma MH, Stellato G, Kuoppala T, Paavonen J. Human T helper cell epitopes overlap B cell and putative cytotoxic T cell epitopes in the E2 protein of human papillomavirus type 16. Biochem Biophys Res Commun. 1995;209:541–546. doi: 10.1006/bbrc.1995.1535. [DOI] [PubMed] [Google Scholar]
- Liu Y, Chen JJ, Gao Q, Dalal S, Hong Y, Mansur CP, Band V, Androphy EJ. Multiple functions of human papillomavirus type 16 E6 contribute to the immortalization of mammary epithelial cells. J Virol. 1999;73:7297–7307. doi: 10.1128/jvi.73.9.7297-7307.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424:99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- Martinez-Zapien D, Ruiz FX, Poirson J, Mitschler A, Ramirez J, Forster A, Cousido-Siah A, Masson M, Vande Pol S, Podjarny A, et al. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature. 2016;529:541–545. doi: 10.1038/nature16481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCredie MR, Sharples KJ, Paul C, Baranyai J, Medley G, Jones RW, Skegg DC. Natural history of cervical neoplasia and risk of invasive cancer in women with cervical intraepithelial neoplasia 3: a retrospective cohort study. Lancet Oncol. 2008;9:425–434. doi: 10.1016/S1470-2045(08)70103-7. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirabello L, Yeager M, Cullen M, Boland JF, Chen Z, Wentzensen N, Zhang X, Yu K, Yang Q, Mitchell J, et al. HPV16 sublineage associations with histology-specific cancer risk using HPV whole-genome sequences in 3200 women. J Natl Cancer Inst. 2016;108:djw100. doi: 10.1093/jnci/djw100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10:550–560. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- Muñoz N, Bosch FX, de Sanjosé S, Herrero R, Castellsagué X, Shah KV, Snijders PJF, Meijer CJLM International Agency for Research on Cancer Multicenter Cervical Cancer Study Group. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N Engl J Med. 2003;348:518–527. doi: 10.1056/NEJMoa021641. [DOI] [PubMed] [Google Scholar]
- Narechania A, Terai M, Burk RD. Overlapping reading frames in closely related human papillomaviruses result in modular rates of selection within E2. J Gen Virol. 2005;86:1307–1313. doi: 10.1099/vir.0.80747-0. [DOI] [PubMed] [Google Scholar]
- Nei MKS. Molecular Evolution and Phylogenetics. New York, NY: Oxford University Press; 2000. [Google Scholar]
- Nelson CW, Hughes AL. Within-host nucleotide diversity of virus populations: insights from next-generation sequencing. Infect Genet Evol. 2015;30:1–7. doi: 10.1016/j.meegid.2014.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CW, Moncla LH, Hughes AL. SNPGenie: estimating evolutionary parameters to detect natural selection using pooled next-generation sequencing data. Bioinformatics. 2015;31:3709–3711. doi: 10.1093/bioinformatics/btv449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley RR, Duensing S, Brake T, Münger K, Lambert PF, Arbeit JM. Dissection of human papillomavirus E6 and E7 function in transgenic mouse models of cervical carcinogenesis. Cancer Res. 2003;63:4862–4871. [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez AC, Schiffman M, Herrero R, Wacholder S, Hildesheim A, Castle PE, Solomon D, Burk R Proyecto Epidemiológico Guanacaste Group. Rapid clearance of human papillomavirus and implications for clinical focus on persistent infections. J Natl Cancer Inst. 2008;100:513–517. doi: 10.1093/jnci/djn044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman A, Munger K. The papillomavirus E7 proteins. Virology. 2013;445:138–168. doi: 10.1016/j.virol.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safaeian M, van Doorslaer K, Schiffman M, Chen Z, Rodriguez AC, Herrero R, Hildesheim A, Burk RD. Lack of heterogeneity of HPV16 E7 sequence compared with HPV31 and HPV73 may be related to its unique carcinogenic properties. Arch Virol. 2010;155:367–370. doi: 10.1007/s00705-009-0579-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffman M, Herrero R, Desalle R, Hildesheim A, Wacholder S, Rodriguez AC, Bratti MC, Sherman ME, Morales J, Guillen D, et al. The carcinogenicity of human papillomavirus types reflects viral evolution. Virology. 2005;337:76–84. doi: 10.1016/j.virol.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Schiffman M, Castle PE, Jeronimo J, Rodriguez AC, Wacholder S. Human papillomavirus and cervical cancer. Lancet. 2007;370:890–907. doi: 10.1016/S0140-6736(07)61416-0. [DOI] [PubMed] [Google Scholar]
- Schiffman M, Boyle S, Raine-Bennett T, Katki HA, Gage JC, Wentzensen N, Kornegay JR, Apple R, Aldrich C, Erlich HA, et al. The role of human papillomavirus genotyping in cervical cancer screening: a large-scale evaluation of the cobas HPV test. Cancer Epidemiol Biomarkers Prev. 2015;24:1304–1310. doi: 10.1158/1055-9965.EPI-14-1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffman M, Doorbar J, Wentzensen N, de Sanjosé S, Fakhry C, Monk BJ, Stanley MA, Franceschi S. Carcinogenic human papillomavirus infection. Nat Rev Dis Primers. 2016a;2:16086. doi: 10.1038/nrdp.2016.86. [DOI] [PubMed] [Google Scholar]
- Schiffman M, Hyun N, Raine-Bennett TR, Katki H, Fetterman B, Gage JC, Cheung LC, Befano B, Poitras N, Lorey T, et al. A cohort study of cervical screening using partial HPV typing and cytology triage. Int J Cancer. 2016b;139:2606–2615. doi: 10.1002/ijc.30375. [DOI] [PubMed] [Google Scholar]
- Serrano B, de Sanjosé S, Tous S, Quiros B, Muñoz N, Bosch X, Alemany L. Human papillomavirus genotype attribution for HPVs 6, 11, 16, 18, 31, 33, 45, 52 and 58 infemale anogenitallesions. Eur J Cancer. 2015;51:1732–1741. doi: 10.1016/j.ejca.2015.06.001. [DOI] [PubMed] [Google Scholar]
- Song YS, Kee SH, Kim JW, Park NH, Kang SB, Chang WH, Lee HP. Major sequence variants in E7 gene of human papillomavirus type 16 from cervical cancerous and noncancerous lesions of Korean women. Gynecol Oncol. 1997;66:275–281. doi: 10.1006/gyno.1997.4756. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Taylor S, Bunge E, Bakker M, Castellsagué X. The incidence, clearance and persistence of non-cervical human papillomavirus infections: a systematic review of the literature. BMC Infect Dis. 2016;16:293. doi: 10.1186/s12879-016-1633-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas Research. Integrated genomic and molecular characterization of cervical cancer. Nature. 2017;543:378–384. doi: 10.1038/nature21386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Doorn L-J, Molijn A, Kleter B, Quint W, Colau B. Highly effective detection of human papillomavirus 16 and 18 DNA by a testing algorithm combining broad-spectrum and type-specific PCR. J Clin Microbiol. 2006;44:3292–3298. doi: 10.1128/JCM.00539-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Pol SB, Klingelhutz AJ. Papillomavirus E6 oncoproteins. Virology. 2013;445:115–137. doi: 10.1016/j.virol.2013.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartanian JP, Guétard D, Henry M, Wain-Hobson S. Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science. 2008;320:230–233. doi: 10.1126/science.1153201. [DOI] [PubMed] [Google Scholar]
- Wang SS, Zuna RE, Wentzensen N, Dunn ST, Sherman ME, Gold MA, Schiffman M, Wacholder S, Allen RA, Block I, et al. Human papillomavirus cofactors by disease progression and human papillomavirus types in the study to understand cervical cancer early endpoints and determinants. Cancer Epidemiol Biomarkers Prev. 2009;18:113–120. doi: 10.1158/1055-9965.EPI-08-0591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren CJ, Xu T, Guo K, Griffin LM, Westrich JA, Lee D, Lambert PF, Santiago ML, Pyeon D. APOBEC3A functions as a restriction factor of human papillomavirus. J Virol. 2015;89:688–702. doi: 10.1128/JVI.02383-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentzensen N, Schiffman M, Dunn ST, Zuna RE, Gold MA, Allen RA, Zhang R, Sherman ME, Wacholder S, Walker J, et al. Multiple HPV genotype infections in cervical cancer progression in the Study to Understand Cervical Cancer Early Endpoints and Determinants (SUCCEED) Int J Cancer. 2009a;125:2151–2158. doi: 10.1002/ijc.24528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wentzensen N, Schiffman M, Dunn ST, Zuna RE, Walker J, Allen RA, Zhang R, Sherman ME, Wacholder S, Jeronimo J, et al. Grading the severity of cervical neoplasia based on combined histopathology, cytopathology, and HPV genotype distribution among 1,700 women referred to colposcopy in Oklahoma. Int J Cancer. 2009b;124:964–969. doi: 10.1002/ijc.23969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanier K, Charbonnier S, Sidi AOMO, McEwen AG, Ferrario MG, Poussin-Courmontagne P, Cura V, Brimer N, Babah KO, Ansari T, et al. Structural basis for hijacking of cellular LxxLL motifs by papillomavirus E6 oncoproteins. Science. 2013;339:694–698. doi: 10.1126/science.1229934. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Rare Nonsynonymous Variation within the E7, E1, and L1 Gene Regions for the CIN3+ Cases and Controls with an A1/A2 HPV16, Related to Figure 2 and Table 4
Cases and controls have a significantly different rare variant distribution in E7, E1, and L1. Cumulative rare variant counts (y axis) are shown for the CIN3+ cases in red and controls in blue by viral genome position for the specified gene (x axis). CIN3+, cervical intraepithelial neoplasia grade 3 and cancer.
Figure S2. HPV16 Rare Nonsynonymous and Nonsense Nucleotide Variants Observed in the E6 ORF. Related to Figure 3 and Table 4
Women in the PAP Cohort with HPV16 A1/A2 lineage infections were included in this analysis. Rare nonsynomous and nonsensense variants are shown as black and red sticks, respectively. Controls are shown as blue lollipops and CIN3+ cases as either red, dark red, grey or black lollipops corresponding to CIN3, SCC, AIS, or Adeno, respectively. Amino acid changes are indicated with each lollipop, and number of circles indicates the number of individuals with that variant. Specific domains of E6 are colored, see legend, and the stars indicate changes consistent with an APOBEC3-induced change. CIN3, cervical intraepithelial neoplasia grade 3; SCC, squamous cell carcinoma; AIS, adenocarcinoma in situ; Adeno, adenocarcinoma.