

Abstract
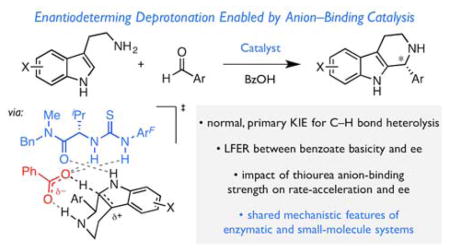
An investigation of the mechanism of benzoic acid/thiourea co-catalysis in the asymmetric Pictet–Spengler reaction is reported. Kinetic, computational, and structure–activity relationship studies provide evidence that rearomatization via deprotonation of the pentahydro-β-carbolinium ion intermediate by a chiral thiourea•carboxylate complex is both rate- and enantioselectivity-determining. The thiourea catalyst induces rate acceleration over the background reaction mediated by benzoic acid alone by stabilizing every intermediate and transition state leading to up to and including the final selectivity-determining step. Distortion–interaction analyses of the transition structures for deprotonation predicted using density functional theory indicate that differential π–π and C–H···π interactions within a scaffold organized by multiple hydrogen-bonds dictate stereoselectivity. The principles underlying rate acceleration and enantiocontrol described herein are expected to have general implications for the design of selective transformations involving deprotonation of high-energy intermediates.
Keywords: Brønsted acid co-catalysis, anion-binding catalysis, kinetic isotope effects, reaction mechanism, tetrahydro-β-carboline
SYNOPSIS
Mechanistic study of a small-molecule catalyst system for the Pictet–Spengler reaction reveals an unusual enantioselectivity-determining step for the classic transformation.
INTRODUCTION
The intramolecular Friedel–Crafts alkylation of β-arylethylimines has attracted intense research interest since it was first reported by Pictet and Spengler over a century ago.1,2 Synthetic chemists and biological systems routinely employ this transformation for the efficient construction of bioactive heterocycles including tetrahydro-β-carbolines and tetrahydroisoquinolines.3–15 The products arising from Pictet–Spengler reactions of tryptamine derivatives alone account for the cores of all monoterpene indole alkaloids16,17 and numerous other secondary metabolites (Figure 1A), and similar motifs are of continual interest as pharmaceutical candidates.18–20 The enzymes catalyzing these transformations have been scrutinized by the structural biology and enzymology communities,3,16,17,21–23 and parallel attention has been devoted to the development of small molecule catalysts for stereocontrolled Pictet–Spengler reactions.24–35
Figure 1.

The Pictet–Spengler Reaction of Tryptamines. Key catalytic residues (silver) in the enzyme active site of strictosidine synthase (B) depicted in the overlay of structures bound to tryptamine (light pink, PDB 2FPB) and secolognin (slate blue, PDB 2FPC); see ref 46. Glc = glucose. Glu = glutamic acid. His = histidine. Phe = phenylalanine. TMB = 3,4,5-trimethoxybenzoyl. Tyr = tyrosine.
Interest in the synthetic applications of the Pictet–Spengler reaction has long been intimately linked to speculation and investigation regarding the details of its molecular mechanism. Following condensation of a β-arylethylamine with an aldehyde, addition of the pendant arene to the resultant iminium ion and subsequent rearomatization (via deprotonation) are necessary to generate the final product. In the case of tryptamine derivatives, the C–C bond-forming step may, in principle, occur by direct electrophilic aromatic substitution at the C2 position or by alkylation at the more nucleophilic C3 position followed by C–C migration (Figure 1B). System-dependent evidence has been provided for the viability of both paths to the requisite pentahydro-β-carbolinium ion intermediate.36–45 The important questions surrounding the mechanism of C–C bond formation notwithstanding, kinetic isotope effect (KIE) and pH–rate dependence studies of the biosynthesis of strictosidine provide evidence that rearomatization, mediated by an active site glutamate residue, is in fact rate-limiting (Figure 1C).21–23,46 The analogous, nonenzymatic reaction in aqueous acetic acid buffer displays similar behavior.21 However, little is known about the rearomatization process in the nonpolar media commonly employed for stereoselective synthetic methods.
In 2009, we reported an enantioselective protocol for the Pictet–Spengler reaction of substituted tryptamines based on the cooperative action of chiral thiourea (1a) and benzoic acid co-catalysts (Figure 1D).47 This method exhibits broad substrate scope: tryptamine nucleophiles bearing diverse substitution (2) as well as both aryl and aliphatic aldehyde electrophiles (3) undergo reaction to afford chiral tetrahydro-β-carboline products (4) in high enantioselectivity. The cooperative catalysis between weak achiral Brønsted acids and chiral thioureas revealed in this work has subsequently proven to be broadly applicable to a variety of enantioselective transformations of interest.34,48–52 Motivated by the synthetic utility of the Pictet–Spengler method and the generality of the catalytic approach,53 we undertook a thorough analysis of this reaction system with the goal of elucidating its mechanism.
Herein we describe experimental and computational studies toward this aim. On the basis of kinetic characterization, kinetic isotope effects, structure–enantioselectivity relationships, and computational analyses, we advance that the thiourea catalyst stabilizes every intermediate and transition state leading to up to and including the rate- and enantioselectivity-determining deprotonation of the final pentahydro-β-carbolinium ion intermediate. The factors controlling rate acceleration and enantioselectivity in this transformation may guide the design of other stereoselective methods requiring the deprotonation of high-energy intermediates.
RESULTS AND DISCUSSION
Thiourea and Benzoic Acid Both Contribute to Rate Acceleration
We initiated our investigation with the goal of identifying the rate- and stereoselectivity-determining step(s) of the transformation. The originally reported conditions (Figure 1D) produced heterogeneous reaction mixtures and long reaction times that were not conducive to rigorous kinetic analysis. The use of pre-formed imine 5a as a substrate along with binary mixtures of toluene and 1,2-dichloroethane (DCE) as solvent resulted in homogenous reaction media and formation of tetrahydro-β-carboline 4a in good yield and with only a modest reduction in observed enantioselectivity (88% ee under homogeneous conditions compared with 94% ee under those reported initially; Scheme 1).54 The distinctive absorption spectrum of cyano-substitued imine 5a enabled facile reaction progress kinetic analysis by in situ attenuated total reflectance Fourier-transform infrared (ATR FTIR) spectroscopy.55,56 Through such an analysis, the reaction was observed to exhibit a first-order dependence on substrate [5a].57
Scheme 1.
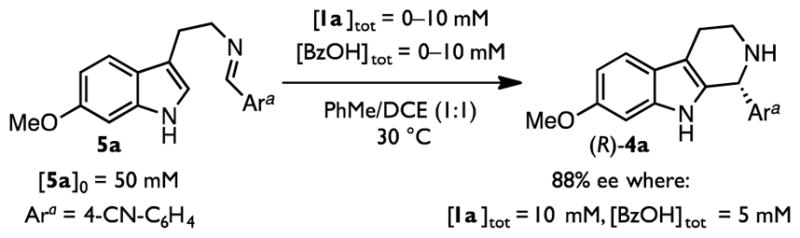
Reaction Conditions for Kinetic Analysis.
The kinetic order of each of the co-catalysts was also determined by varying either [1a]tot or [BzOH]tot while holding the other constant. A competitive racemic background reaction is observed when [1a]tot ≪ [BzOH]tot, but the rate is too slow to measure accurately under the conditions used to probe the catalytic reaction. However, the rate constant for this racemic, benzoic acid-mediated process (krac) can be extrapolated from the non-zero y-intercept in Figure 2A. The kinetic dependence on [1a]tot is further complicated by the propensity for amido-thioureas to aggregate in solution. Catalysts similar to 1a are known to exist as resting-state inactive homodimeric complexes in related anion-abstraction reactions,58–61 and 1H NMR titration experiments in toluene-d8 and CD2Cl2 reveal that 1a is partially aggregated at the concentrations used in the catalytic reaction (see Supporting Information). Taking this behavior into account, the non-linear dependence of reaction rate on [1a]tot can be fit readily to a model involving a single molecule of 1a in the transition state and a change in the stoichiometry of the resting-state from a monomer at low [1a]tot to a dimer at high [1a]tot (Figure 2A).
Figure 2.

Kinetic Evidence for Thiourea and Benzoic Acid Co-Catalysis. (A) The rate dependence for consumption of imine 5a (see Scheme 1) on [1a]tot. (B) The rate dependence for consumption of imine 5a on [BzOH]tot. Rates determined at 20% conversion. Blue dashed lines represent the fit to a model accounting for non-productive catalyst self-aggregation, using independently determined self-dimerization constants (Kdim). See Supporting Information for derivation. (C) Job plot of the association of thiourea 1a and a guest, either tetrabutylammonium benzoate (purple diamonds) or 4-fluorobenzoic acid (green squares), generated by monitoring the CF3 resonance in the 19F NMR spectrum of catalyst 1a in toluene-d8 at 25 °C. [1a + guest]tot = 0.01 M
Similar analysis over a range of [BzOH]tot reveals that no product formation occurs in the absence of the Brønsted acid. The reaction exhibits a first-order dependence on [BzOH]tot over much of the concentration range examined (Figure 2B), but a slight deviation from linearity at high [BzOH]tot reflects ground-state aggregation between two molecules of the carboxylic acid.62 While the propensity for benzoic acid self-dimerization is weaker than that observed with the amido-thiourea catalyst, the self-dimerization equilibrium constant for 4-fluorobenzoic acid, measured independently through 19F NMR titration experiments in toluene-d8 and CD2Cl2, could be used to fit the kinetic data.
The combined kinetic data thus indicate that the rate-limiting transition state for the enantioselective Pictet–Spengler reaction involves a ternary 1a•BzOH•5a complex. The cooperative association of the two co-catalysts in the mechanism of catalysis is further supported by the observation of 1:1 complexation between thiourea 1a and 4-fluorobenzoic acid by 19F NMR analysis in toluene-d8 (Figure 2C).63,64 We hypothesize that the resulting complex exhibits increased acidity relative to free benzoic acid and thus contributes to substrate activation en route to product formation, as proposed in the catalytic cycle outlined in Scheme 2. Consistent with this analysis, added tetrabutylammonium benzoate (NBu4OBz), which forms a much stronger complex with thiourea 1a (Figure 2C), 34,49,51,52,65–80 inhibits the reaction. This inhibition is also of significance given that the secondary amine product of the Pictet–Spengler reaction is substantially more basic than the imine substrate,81,82 thereby introducing the potential for formation of a similarly counterproductive ammonium benzoate complex during the course of the reaction (Scheme 2, step V).34
Scheme 2.
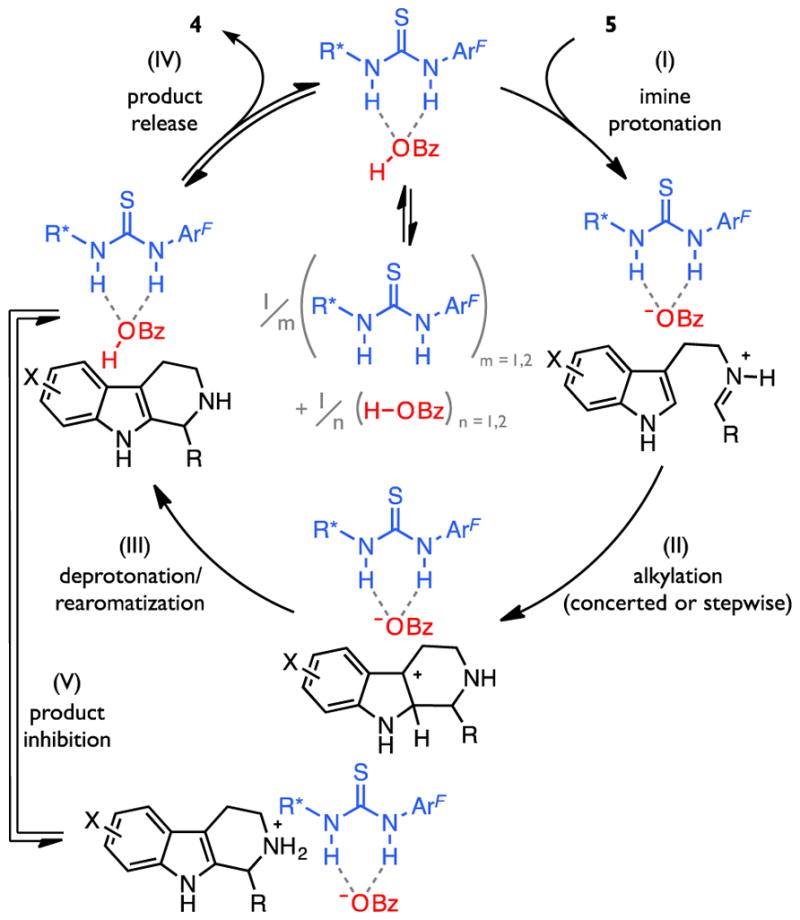
General Catalytic Cycle.a
a ArF = 3,5-bis(trifluoromethyl)phenyl
To probe whether product inhibition is operative, the absolute rates of two reactions with the same catalyst concentrations, [1a]tot and [BzOH]tot, but different initial imine concentrations, [5a]0, were compared over the entire reaction course.83 The resultant rate vs. concentration curves do not overlay (Figure 3A, blue dots and green squares); this behavior is a signature of diminishing catalytic activity during the course of the reaction as a result of either product inhibition or catalyst decomposition. Much better overlay of the rate vs. concentration plots is observed when the reaction is conducted with initial imine and product concentrations selected to mimic a reaction at partial conversion (Figure 3A, blue dots and purple diamonds). This result provides unequivocal evidence that product inhibition, rather than catalyst decomposition, is responsible for the significant catalyst deactivation occurring over the course of the reaction.
Figure 3.

Kinetic Evidence for Product Inhibition and New Reaction Conditions for Minimizing Product Inhibition. See Scheme 1 for reaction conditions used for kinetic analysis; results from multiple independent runs are overlaid. Ara = 4-cyanophenyl. Boc = tert-butoxycarbonyl.
This insight sheds light on a likely cause for the poor catalytic efficiency observed under the conditions optimized originally, where 20 mol % loading of each of the two co-catalysts was required to achieve good product yields within 1–5 day reaction times. To overcome the deleterious effects of product inhibition on reaction rate, modified conditions were devised to trap the product amine in situ (Figure 3B). With the addition of a slight excess of Boc2O,84 the enantioselective Pictet–Spengler reaction can be conducted over the same reaction period with a 10-fold reduction in the loading of each co-catalyst. The lower catalyst concentrations carry the added benefit of disfavoring nonproductive catalyst self-aggregation, thereby amplifying the increase in catalytic efficiency. These modified conditions overcome an important limitation of the original procedure, thereby enhancing the practical value of this method for the preparation of enantioenriched tetrahydro-β-carbolines.
Rate- and Enantiodetermining Rearomatization is Promoted through the Cooperative Action of the Thiourea and Benzoic Acid Co-Catalysts
As noted above, the kinetic data provide evidence that the rate-determining step in the thiourea/benzoic acid co-catalyzed Pictet–Spengler reaction involves a ternary 1a•BzOH•5a complex. However, the kinetic analysis is consistent with any of the three fundamental steps in the proposed mechanism (Scheme 2, steps I–III) being rate- and enantioselectivity-determining, since they all involve a complex with the same stoichiometry. In order to understand the basis for catalysis and enantioselectivity, a set of isotope effect experiments was designed to distinguish which step is rate-limiting. Namely, analogs of 5a bearing deuterium at either the Cϕ or the C2 positions were prepared and their cyclizations under standard catalytic conditions were analyzed (Figure 4). If imine protonation were rate-determining (Scheme 2, step I), no significant kinetic isotope effect (KIE) would be expected for either the 5a-d1(Cϕ) or 5a-d1(C2) isotopomers, since the deuterium-bearing carbons are essentially unaffected in that step. If the alkylation step (Scheme 2, step II), either via C2- or C3-addition, were rate-limiting, deuteration of the Cϕ position of imine 5a would be expected to result in an inverse, secondary kinetic isotope effect arising from an sp2 to sp3 hybridization change at the carbon bearing the isotope. In contrast, the C2-labelled isotopomer would be expected to display an inverse, secondary KIE in the case of rate-limiting C2-addition or C–C migration but not C3-addition. Finally, only rate-limiting deprotonation/rearomatization (Scheme 2, step III) should result in a relatively large primary KIE with 5a-d1(C2).
Figure 4.
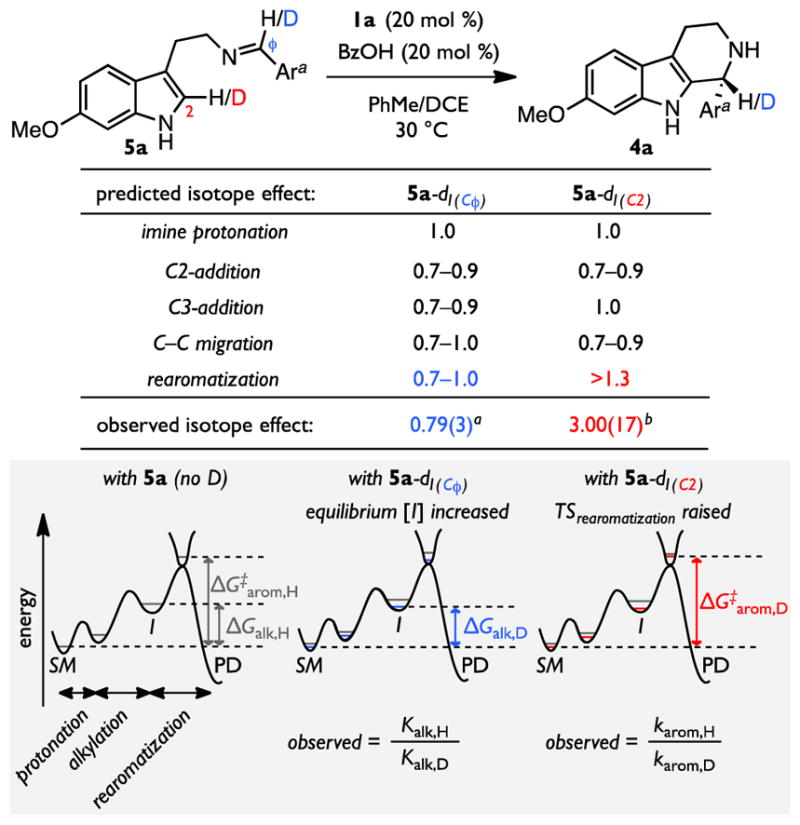
Isotope Effect Measurements. a Competition isotope effect determined from the ratio of recovered 5a and 5a-d1(Cϕ) at low conversion. b Isotope effect determined from the relative first-order rate constants for formation of 4a measured independently with 5a and 5a-d1(C2). Rates are uncorrected for incomplete deuterium incorporation (94% D for 5a-d1(C2)). See ref 86. Ara = 4-cyanophenyl.
Competition experiments between imine isotopologues 5a and 5a-d1(Cϕ) under standard catalytic conditions afforded a secondary inverse isotope effect of 0.79(3) at the imine position. Independent rate measurements with imine isotopologues 5a and 5a-d1(C2) revealed a normal isotope effect of 3.0(2) at C2.85,86 These results are consistent only with a scenario involving rate-limiting deprotonation/rearomatization. The increased rate from Cϕ-deuteration can be attributed to an increased equilibrium concentration of the high-energy pentahydro-β-carbolinium ion intermediate, I, (i.e. an equilibrium isotope effect, EIE).87 Normal, primary KIEs of similar magnitude to that observed with 5a-d1(C2) were measured through intermolecular competition experiments with imines bearing different aryl substituents (see Supporting Information),85 providing evidence that rate-limiting rearomatization is general for this catalyst system. As noted in the introduction, rearomatization has also been identified as the rate-determining step in Pictet–Spengler reactions catalyzed by enzymes including strictosidine synthase21 (Figure 1B) and norcoclaurine synthase,22 which play central roles in alkaloid biosynthesis.88
The observation that rearomatization is rate-determining in the thiourea- and benzoic acid co-catalyzed Pictet–Spengler reaction implies that the preceding protonation and alkylation steps are reversible; therefore, rearomatization must also be enantioselectivity-determining.89 Thus, while two stereogenic centers are formed during reversible C2- or C3-addition steps, only diastereomer-selective deprotonation of the resultant chiral pentahydro-β-carbolinium ion intermediate dictates the final stereochemistry observed in the product.90 Given the non-intuitive nature of a rate- and enantioselectivity-determining deprotonation event from such a high-energy intermediate, we became interested in elucidating the features of the reaction system responsible for substrate activation and enantiocontrol. As a first step in this effort, we sought to determine the identity of the active Brønsted base, given that the dual catalyst system consists of multiple potentially basic sites.
In order to determine whether the thiourea S or amide O of catalyst 1a may be responsible for deprotonation of the pentahydro-β-carbolinium ion intermediate, the electronic properties of each of the two motifs were varied systematically (Figure 5A). Ureas are generally stronger Brønsted bases and H-bond acceptors than analogous thioureas by several orders of magnitude.91–93 On this basis, urea 1b would be expected to demonstrate improved reactivity and/or enantioselectivity if thiourea 1a serves as a Brønsted base in the rate- and enantioselectivity-determining transition state. However, urea 1b is a slightly less active and enantioselective catalyst than thiourea 1a. Likewise, a more electron-rich thiourea, 1c, also exhibits reduced activity and enantioselectivity.94 Taken together, these results provide evidence that the thiourea moiety of catalyst 1a does not serve as a base in the rate and selectivity-determining step. Instead, the modestly reduced activity and enantioselectivity observed with 1b and 1c most likely reflect their diminished H-bonding ability.95,96
Figure 5.

Co-Catalyst Structure–Reactivity–Enantioselectivity Relationships. Enantiomeric excess reported for the formation of 4b under the conditions shown. Relative rate constants (krel) determined from the first-order rate constants for formation of 4a under the conditions in Scheme 1. Arb = 4-chlorophenyl. ArF = 3,5-bis(trifluoromethyl)phenyl.
Reactivity and enantioselectivity are similarly insensitive to the basicity of the amide moiety. Ester 1e,97 which is predicted to be at least an order of magnitude less basic than an analogous amide,93 is only slightly less enantioselective than 1b and a sterically similar amido-thiourea, 1d, and it affords modestly improved reaction rates. This relationship suggests that any interaction with the carbonyl O must, therefore, be similar in strength in the major and minor transition structures and must lead to a net increase in the activation barrier. Thus, while the amide moiety of catalyst 1a may be involved in substrate binding, it must not be responsible for deprotonation of the pentahydro-β-carbolinium ion intermediate in the rate- and selectivity-determining step.
Given the evidence that amido-thiourea 1a does not act as a Brønsted base, we hypothesized that the conjugate base of the benzoic acid co-catalyst may serve this role instead. A series of 4-substituted benzoic acid co-catalysts spanning acidities over more than an order of magnitude were therefore evaluated in the enantioselective Pictet–Spengler reaction of imine 5b (Figure 5B).98 No linear relationship is observed between the electronic properties of the benzoic acid and the reaction rate (see Supporting Information), but this may simply reflect the competing demands of equilibrium imine protonation prior to the rate-determining step. Nonetheless, the reaction exhibits a strong correlation between the benzoic acid Hammett σp value and enantioselectivity, wherein the most basic benzoates (or least acidic benzoic acids) afford tetrahydro-β-carboline 4b with the highest level of enantioselectivity.99,100 However, despite the excellent correlation, the overall electronic effect is in fact quite small (ρ = −0.33). Taken together, these observations implicate the benzoate as the Brønsted base responsible for enantioselectivity-determining deprotonation but suggest that it plays only a minor role in enantiodiscrimination.
The Thiourea Catalyst Influences Multiple Steps by Anion-Binding
The thiourea/benzoic acid co-catalyzed Pictet–Spengler reaction is observed to be approximately four-fold faster than the process mediated by benzoic-acid alone.101 The basis for this rate acceleration was probed through construction of a reaction coordinate energy diagram based on computed models of energy-minimized intermediates and transition states using density functional theory (DFT).102–107 To ascertain an appropriate starting point for this analysis,108 the relative acidities of acetic acid and a model iminium ion were assessed computationally using an implicit solvent model,109,110 and imine protonation by acetic acid was calculated to be highly unfavorable in the nonpolar medium relevant to this study. The degree of interaction between model imine 7111 and benzoic acid was assessed experimentally by 1H NMR spectroscopy (Scheme 3). The chemical shift of an imine’s formyl hydrogen is highly sensitive to electrostatic perturbation,112 and changes in imine chemical shift have previously been taken as evidence of imine protonation by stronger Brønsted acids.113 No change in the signal was observed upon addition of a full equivalent of benzoic acid, thus providing further support for the prediction that imine protonation is highly endergonic in the reaction solvent. As such, the neutral, H-bonded encounter complex between 5b and acetic acid114 (rather than the free iminium ion) was employed as the starting point for subsequent evaluation. Furthermore, while only (E)-5b is detected under the standard conditions employed for the imine preparation, interconversion with the (Z)-isomer may, in principle, occur in situ. Accordingly, the full reaction coordinate en route to (R)-5b accessed through either of the isomers was examined. In both cases, the results obtained with the inclusion of N,N′-dimethylthiourea (1f) were compared with the background process mediated by acetic acid alone.
Scheme 3.
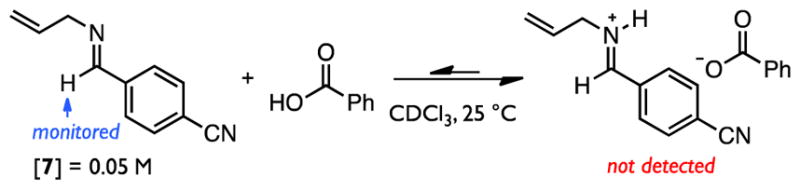
Imine Protonation is Endergonic in Nonpolar Media.
Consistent with experimental observations, no discrete iminium ion intermediate could be located in the reaction coordinate analysis of the uncatalyzed reaction (Figure 5A, red). Instead, imine protonation and C2-addition to form high-energy pentahydro-β-carbolinium ion I2 occur in a single, highly asynchronous step. In contrast, the inclusion of 1f results in a reduced penalty to imine protonation such that the stationary point for the resultant ion pair is slightly lower in energy than the neutral encounter complex (Figure 6A, blue). These observations suggest that anion-binding interactions between 1f and the conjugate base of acetic acid enhance the acidity of the weak Brønsted acid, thereby enabling formation of the protioiminium ion intermediate under the non-polar reaction conditions.
Figure 6.

Computational Evaluation of the Basis for Rate-Acceleration by Thiourea and Brønsted Acid Co-Catalysis. With the exception of (R,R)-TS2,chair, only structures arising from the major, (E)-imine isomer are represented. Calculations performed with B3LYP-D3(BJ)/6-31+G(d,p)/CPCM(toluene)//B3LYP/6-31G(d)/CPCM(toluene). Electronic energies reported in kcal/mol. Corrected free energies listed in parentheses in kcal/mol. For additional information and discussion, including analyses of structures arising from the (Z)-imine isomer, see Supporting Information. Arb = 4-chlorophenyl
While the impact of the H-bond donor on preequilbrium protonation is pronounced, the calculations reveal that it is the persistence of anion-binding interactions throughout subsequent steps involving cationic transition states and intermediates that results in a net reduction of the energetic span of the reaction.115,116 Specifically, the acetate anion engages in attractive C2–H···O interactions to guide C2-addition (TS2) and facilitate deprotonation (TS3). While the pentahydro-β-carbolinium ion I2 is stabilized to an even greater extent than either of these transition states, the overall barrier to initial addition (TS2) and subsequent deprotonation (TS3) are reduced by 4–6 kcal/mol and ~3 kcal/mol, respectively, relative to the lowest energy ground state.117 Furthermore, the significant stabilization of the chair-like transition structure for C2-addition (TS2,chair) renders it the only accessible path for cyclization en route to pentahydro-β-carbolinium ion (I2). Alternative modes of cyclization, such as those involving a boat-like conformation (TS2,boat) or C3-addition (TS2,spiro) are predicted to be at least 3 kcal/mol higher in energy than either TS2,chair or TS3 (Figure 6B).118 Nonetheless, in the presence of the thiourea co-catalyst, C2-addition through both the (E)- and (Z)-imine isomers is energetically accessible. While (R,R)-TS2,chair is higher in energy than (S,R)-TS2,chair, it is energetically comparable with (S,R)-TS3,chair and (R,R)-TS3,chair (see Supporting Information). As such, the intermediacy of all possible diastereomers of the pentahydro-β-carbolinium ion intermediate—namely, (S,R)-I2 and (R,R)-I2 en route to the major enantiomer of product, (R)-4b, as well as (R,S)-I2 and (S,S)-I2 en route to (S)-4b—must be considered in examination the rate-determining deprotonation event. Nonetheless, it is clear that the thiourea serves as an anion-binding catalyst throughout the reaction. While anion-binding interactions attenuate the kinetic basicity of the benzoate and therefore decelerate the step with the highest overall transition state energy (i.e. I2 → I3), reduction in the net energetic span of the reaction arising from the stabilization of all charged intermediates and transition states results in an overall rate-acceleration.115,116
Enantiodetermination Arises from Attractive π–π and C–H···π Interactions Enabled by a Cooperative Network of Conserved H-Bonds
In an effort to elucidate the specific interactions between the amido-thiourea catalyst, the benzoic acid co-catalyst, and the reactive pentahydro-β-carbolinium ion responsible for enantiodetermination, we focused subsequent computational work on the deprotonation step. Chiral thiourea 1g (Figure 7) was employed in this analysis, as truncation of the amide benzyl substituent and aryl CF3 groups has only a modest impact on the enantioselectivity observed experimentally (Figure 5B), but affords a substantial reduction in the size of the system and therefore in the computational expense. Through this analysis, several conserved catalyst–substrate interactions were observed across the lowest energy transition structures derived from each of the four possible diastereomers of the pentahydro-β-carbolinium ion intermediate (Figure 7). These include: (a) two N–H···O H-bonding interactions between the thiourea and carboxylate base, (b) an H-bonding interaction between the amine N–H and the distal O of the carboxylate, (c) an H-bonding interaction between the amide O and the indole N–H, and (d) highly symmetric breaking and forming C···H···O bonds (1.30–1.36 Å), wherein the proximal O of the carboxylate base effects the deprotonation.
Figure 7.

Enantiodetermining Rearomatization Occurs within a Conserved Network of Cooperative Hydrogen-Bonding Interactions. Calculations performed with B3LYP-D3(BJ)/6-31+G(d,p)/CPCM(toluene)//B3LYP/6-31G(d)/CPCM(toluene). Electronic energies reported in kcal/mol. Corrected free energies listed in parentheses in kcal/mol. Select bond lengths listed in Å. Dotted lines represent H-bonding or C–H···π interactions; dashed lines represent breaking and forming bonds. H = white, C = silver, N = blue, O = red, S = yellow, Cl = magenta.
These features account for the observation that decreasing the amide Lewis basicity does not impact the observed enantioselectivity (Figure 5A), as the N–H···O lengths are comparable for all of the structures examined. However the importance of this interaction for enabling well-defined organization of the reactive complex is underscored by the observation that masking the indole N–H leads to a dramatic reduction in the enantioselectivity observed experimentally (Scheme 4). Furthermore, while the conformations of the amido-thiourea and carboxylate are predicted to be fairly constant across the computed structures, the orientations of the reacting pentahydro-β-carbolinium ion with respect to the catalyst differ substantially, with the lowest-energy structure, (S,R)-TS3•−OAc•1g, accurately predicting formation of the major enantiomer of product, (R)-4b. Analogous transition structures optimized for the full experimental system (i.e. catalyst 1d and substrate 5b) with a functional designed to account for dispersive interactions (M062X/6-31+g(d,p)/SMD(toluene)) are qualitatively similar to those depicted in Figure 7,104,107,119–121 and the predicted enantioselectivity (ΔΔG‡ = 1.6 kcal/mol) is in excellent agreement with experimental values.122
Scheme 4.
The Indole N–H is Necessary for Enantiocontrol.
To better understand the factors contributing to stereochemical discrimination, a modified distortion–interaction analysis of each of the four energetically accessible, diastereomeric transition structures was conducted (Table 1).123–125 Through this analysis, the differential electronic energy of activation was decomposed into terms describing the relative energy required to distort amido-thiourea catalyst 1g into the conformation assumed in the transition structure (ΔΔE‡dist,1g), the relative energy required to distort the (E)-5b•HOAc complex126 into the conformation assumed in the catalyzed transition structure (ΔΔE‡dist,5b•HOAc), and the relative interaction energy between the distorted 5b•HOAc complex and catalyst 1g (ΔΔE‡int). These differential distortion and interaction terms reveal that the degree of catalyst distortion has only a small impact on enantioselectivity, consistent with the qualitative assessment that its conformation changes little across the structures examined. The degree of substrate distortion is modestly more important, particularly in contributing to the destabilization of (R,S)-TS3•−OAc•1g, but the overall, discrimination between the diastereomeric complexes is overwhelmingly dictated by the interactions between the catalyst and the reactive species.
Table 1.
Stereodiscrimination Arises from Stabilizing π–π and C–H···π Interactions
| Entry | Transition Structure | Imine | Product | ΔΔE‡tot (kcal/mol) | ΔΔE‡dist,1g(kcal/mol) | ΔΔE‡dist,5b•HOAc(kcal/mol) |
ΔΔE‡int (kcal/mol) with:
|
||
|---|---|---|---|---|---|---|---|---|---|
| 1g | 1h | 1i | |||||||
| 1 | (S,R)-TS3•−OAc•1g | (E)-5b | (R)-4b | +0.0 | +0.0 | +0.0 | +0.0 | +0.0 | +0.0 |
| 2 | (S,S)-TS3•−OAc•1g | (Z)-5b | (S)-4b | +3.1 | +0.3 | +0.5 | +2.3 | +2.0 | +0.2 |
| 3 | (R,S)-TS3•−OAc•1g | (E)-5b | (S)-4b | +5.1 | −0.4 | +2.6 | +2.9 | +3.1 | +1.8 |
| 4 | (R,R)-TS3•−OAc•1g | (Z)-5b | (R)-4b | +5.7 | +0.1 | +0.8 | +4.8 | +3.5 | +3.5 |
Numerous noncovalent interactions contribute to the total interaction energy for each of the predicted transition structures. While very little variation is observed in the H-bond lengths between acetate and the pentahydro-β-carbolinium ion, we noted sizable differences (up to 0.14 Å) in the lengths of the H-bonds between acetate and the thiourea catalyst. These differences are consistent with the observation that enantioselectivity is correlated to electronic properties of both the H-bond donor and the benzoate anion (Figure 5). However, as noted above, even significant changes in benzoate basicity have a relatively small net impact on the overall enantioselectivity;98 we were thus compelled to consider other interactions that could potentially contribute to stereodiscrimination to a greater degree.
Visual inspection of the computed structures in Figure 7 reveals that the catalyst isopropyl side-chain and phenyl thiourea moieties are positioned in close proximity to other components of the cationic assembly in the lowest energy transition state, (R,S)-TS3•−OAc•1g. We hypothesized that these groups might be engaged in stabilizing π···π and C–H···π interactions that are absent or diminished in the competing transition structures. To evaluate the relative contributions of these interactions, the differential interaction energies were recomputed with truncated catalysts (1h and 1i; Figure 7) that lack the substituents in question. Ablation of the phenyl group (as in 1h) is predicted to have only modest impact on enantioselectivity. The lowest-energy transition structure (R,S)-TS3•−OAc•1 as well as the transition structures leading to the minor enantiomer of product all position either the substrate indole or 4-chlorophenyl moieties directly over the ablated phenyl group and are, thus, similarly impacted by the loss of a π···π interaction. In contrast, ablation of the isopropyl group (as in 1i) has a substantial impact on the relative interaction energies of all four transition structures. Of particular note, the relative interaction energy of (S,S)-TS3•−OAc•1 is decreased by 2.1 kcal/mol such that ΔΔE‡int is almost identical for (S,R)-TS3•−OAc•1i and (S,S)-TS3•−OAc•1i. Only (S,R)-TS3•−OAc•1g exhibits a C–H···π interaction with the isopropyl group of the catalyst, and this pronounced change in the relative interaction energies indicates that, along with the H-bonding interactions between the thiourea and benzoate anion, this attractive interaction is a major controlling feature in the observed enantioselectivity.
CONCLUSION
The experimental and computational analyses described herein provide insight into the cooperative mechanism of thiourea and Brønsted acid co-catalysis in the enantioselective protio-Pictet–Spengler reaction. The combined application of kinetic studies, isotope effects, structure-enantioselectivity relationships, and computational analyses enable identification of rearomatization as the rate- and enantioselectivity-determining step, wherein the co-catalysts play key roles in multiple steps leading up to and including the rate-determining deprotonation event. Acidification of the weak benzoic acid co-catalyst upon coordination with the thiourea promotes pre-equilibrium substrate protonation, and anion-binding interactions with the conjugate base further stabilize the pentahydro-β-carbolinium ion intermediate. These interactions increase the concentrations of high-energy intermediates en route to the rate-determining step and thereby contribute to rate acceleration, while enantioinduction is mediated by differential π···π and C–H···π interactions within a scaffold organized by multiple hydrogen-bonding interactions. This remarkable small-molecule catalyst system recapitulates many features of enzymatic Pictet–Spengler catalysts, and we posit that the principles underlying rate acceleration and enantiocontrol in this system may prove to be general tools for the design of other stereoselective transformations relying on the deprotonation of high-energy intermediates.
Supplementary Material
Acknowledgments
This paper is dedicated to Professor Robert Bergman on the occasion of his 75th birthday. This work was supported by the NIH (GM-43214). The authors thank Stephan Zuend and Chris Uyeda for helpful suggestions and Professor Theodore Betley and Evan King for access to UV/Vis instrumentation. The computations in this paper were run on the Odyssey cluster supported by the FAS Sciences Division Research Computing Group at Harvard University.
ABBREVIATIONS
- Ara
4-cyanophenyl
- Arb
4-chlorophenyl
- ArF
3,5-bis(trifluoromethyl)phenyl
- Boc
tert-butoxycarbonyl
- DCE
1,2-dichloroethane
- Glc
glucose
- TMB
3,4,5-trimethoxybenzoyl
Footnotes
The Supporting Information is available free of charge on the ACS Publications website.
Complete experimental procedures, characterization data, kinetic data, derivation of the rate law, 1H NMR titration data, further computational data, geometries and energies of all calculated stationary points (PDF)
Notes
The authors declare no competing financial interest.
References
- 1.Pictet A, Spengler T. Berichte Der Deutschen Chem Ges. 1911;44:2030–2036. [Google Scholar]
- 2.Tatsui G. Yakugaku Zasshi. 1928;48:453–459. [Google Scholar]
- 3.Stöckigt J, Antonchick AP, Wu F, Waldmann H. Angew Chem Int Ed. 2011;50:8538–8564. doi: 10.1002/anie.201008071. [DOI] [PubMed] [Google Scholar]
- 4.Whaley WM, Govindachari TR. Org React. 2011;6:151–190. doi: 10.1002/0471264180.or006.03. [DOI] [Google Scholar]
- 5.Cox ED, Cook JM. Chem Rev. 1995;95:1797–1842. doi: 10.1021/cr00038a004. [DOI] [Google Scholar]
- 6.Chrzanowska M, Rozwadowska MD. Chem Rev. 2004;104:3341–3370. doi: 10.1021/cr030692k. [DOI] [PubMed] [Google Scholar]
- 7.Lorenz M, van Linn ML, Cook JM. Curr Org Synth. 2010;7:189–223. doi: 10.2174/157017910791163011. [DOI] [Google Scholar]
- 8.Mailvan AK, Eickhoff JA, Minakova AS, Gu Z, Lu P, Zakarian A. Chem Rev. 2016;116:4441–4557. doi: 10.1021/acs.chemrev.5b00712. [DOI] [PubMed] [Google Scholar]
- 9.Chrzanowska M, Grajewska A, Rozwadowska MD. Chem Rev. 2016;116:12369–12465. doi: 10.1021/acs.chemrev.6b00315. [DOI] [PubMed] [Google Scholar]
- 10.For select examples from natural product and pharmaceutical synthesis, see: van Tamelen EE, Shamma M, Burgstahler AW, Wolinsky J, Tamm R, Aldrich PE. J Am Chem Soc. 1958;80:5006–5007. doi: 10.1021/ja01551a062.
- 11.Fu X, Cook JM. J Am Chem Soc. 1992;114:6910–6912. doi: 10.1021/ja00043a043. [DOI] [Google Scholar]
- 12.Bailey PD, Morgan KM. J Chem Soc, Perkin Trans 1. 2000:3578–3583. doi: 10.1039/B005695M. [DOI] [Google Scholar]
- 13.Yamashita T, Kawai N, Tokuyama H, Fukuyama T. J Am Chem Soc. 2005;127:15038–15039. doi: 10.1021/ja055832h. [DOI] [PubMed] [Google Scholar]
- 14.Stork G, Tang PC, Casey M, Goodman B, Toyota M. J Am Chem Soc. 2005;127:16255–16262. doi: 10.1021/ja055744x. [DOI] [PubMed] [Google Scholar]
- 15.Mergott DJ, Zuend SJ, Jacobsen EN. Org Lett. 2008;10:745–748. doi: 10.1021/ol702781q. [DOI] [PubMed] [Google Scholar]
- 16.O’Connor SE, Maresh JJ. Nat Prod Rep. 2006;23:532–547. doi: 10.1039/B512615K. [DOI] [PubMed] [Google Scholar]
- 17.Salim V, De Luca V. Towards Complete Elucidation of Monoterpene Indole Alkaloid Biosynthesis Pathway: Catharanhus roseus as a Pioneer System. In: Giglioli-Guivarch N, editor. New Light on Alkaloid Biosynthesis and Future Prospects. Elsevier; Oxford: 2013. pp. 1–38. [Google Scholar]
- 18.Pulka K. Curr Opin Drug Discov Devel. 2010;13:669–684. [PubMed] [Google Scholar]
- 19.Rottmann M, McNamara C, Yeung BKS, Lee MCS, Zou B, Russell B, Seitz P, Plouffe DM, Dharia NV, Tan J, Cohen SB, Spencer KR, González-Páez GE, Lakshminarayana SB, Goh A, Suwanarusk R, Jegla T, Schmitt EK, Beck H-P, Brun R, Nosten F, Renia L, Dartois V, Keller TH, Fidock DA, Winzeler EA, Diagana TT. Spiroindolones. Science. 2010;329:1175–1180. doi: 10.1126/science.1193225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laine AE, Lood C, Koskinen AMP. Molecules. 2014;19:1544–1567. doi: 10.3390/molecules19021544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maresh JJ, Giddings LA, Friedrich A, Loris EA, Panjikar S, Trout BL, Stockigt J, Peters B, O’Connor SE. J Am Chem Soc. 2008;130:710–723. doi: 10.1021/ja077190z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Similar conclusions were reached following analysis of norcoclaurine synthase: Louis YP, Luk SB, Liscombe DK, Facchini PJ, Tanner ME. Biochemistry. 2007;46:10153–10161. doi: 10.1021/bi700752n.
- 23.Bonamore A, Barba M, Botta B, Boffi A, Macone A. Molecules. 2010;15:2070–2078. doi: 10.3390/molecules15042070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.For select examples of catalytic, enantioselective methods, see: Taylor MS, Jacobsen EN. J Am Chem Soc. 2004;126:10558–10559. doi: 10.1021/ja046259p.
- 25.Seayad J, Seayad AM, List B. J Am Chem Soc. 2006;128:1086–1087. doi: 10.1021/ja057444l. [DOI] [PubMed] [Google Scholar]
- 26.Raheem IT, Thiara PS, Peterson EA, Jacobsen EN. J Am Chem Soc. 2007;129:13404–13405. doi: 10.1021/ja076179w. [DOI] [PubMed] [Google Scholar]
- 27.Wanner MJ, van der Haas RNS, de Cuba KR, van Maarseveen JH, Hiemstra H. Angew Chem Int Ed. 2007;46:7485–7487. doi: 10.1002/anie.200701808. [DOI] [PubMed] [Google Scholar]
- 28.Sewgobind NV, Wanner MJ, Igemann S, de Gelder R, van Maarseveen JH, Hiemstra H. J Org Chem. 2008;73:6405–6408. doi: 10.1021/jo8010478. [DOI] [PubMed] [Google Scholar]
- 29.Raheem IT, Thiara PS, Jacobsen EN. Org Lett. 2008;10:1577–1580. doi: 10.1021/ol800256j. [DOI] [PubMed] [Google Scholar]
- 30.Muratore ME, Holloway CA, Pilling AW, Storer RI, Trevitt G, Dixon DJ. J Am Chem Soc. 2009;131:10796–10797. doi: 10.1021/ja9024885. [DOI] [PubMed] [Google Scholar]
- 31.Holloway CA, Muratore ME, Storer RI, Dixon DJ. Org Lett. 2010;12:4720–4723. doi: 10.1021/ol101651t. [DOI] [PubMed] [Google Scholar]
- 32.Wanner MJ, Claveau E, van Maarseveen JH, Hiemstra H. Chem Eur J. 2011;17:13680–13683. doi: 10.1002/chem.201103150. [DOI] [PubMed] [Google Scholar]
- 33.Huang D, Xu F, Lin X, Wang Y. Chem Eur J. 2012;18:3148–3152. doi: 10.1002/chem.201103207. [DOI] [PubMed] [Google Scholar]
- 34.Mittal N, Sun DX, Seidel D. Org Lett. 2014;16:1012–1015. doi: 10.1021/ol403773a. [DOI] [PubMed] [Google Scholar]
- 35.Ruiz-Ollalla A, Würdemann MA, Wanner MJ, Ingemann S, van Maarseveen JH, Hiemstra H. J Org Chem. 2015;80:5125–5132. doi: 10.1021/acs.joc.5b00509. [DOI] [PubMed] [Google Scholar]
- 36.Jackson AH, Smith P. Chem Commun. 1967:264–266. doi: 10.1039/C19670000264. [DOI] [Google Scholar]
- 37.Jackson AH, Smith P. Tetrahedron. 1968;24:403–413. doi: 10.1016/0040-4020(68)89038-6. [DOI] [Google Scholar]
- 38.Jackson AH, Smith AE. Tetrahedron. 1968;24:2227–2239. doi: 10.1016/0040-4020(68)88125-6. [DOI] [Google Scholar]
- 39.Woodward RB, Cava MP, Hunger A, Ollis WD, Daeniker HU, Schenker K. Tetrahedron. 1963;19:247–288. doi: 10.1016/S0040-4020(01)98529-1. [DOI] [Google Scholar]
- 40.Ungemach F, Cook JM. Heterocycles. 1978;9:1089–1119. doi: 10.3987/R-1978-08-1089. [DOI] [Google Scholar]
- 41.Bailey PD. Tetrahedron Lett. 1987;28:5181–5184. doi: 10.1016/S0040-4039(00)95623-5. [DOI] [Google Scholar]
- 42.Bailey PD, Hollinshead SP, McLay NR, Morgan K, Palmer SJ, Prince SN, Reynolds CD, Wood SD. J Chem Soc, Perkin Trans 1. 1993:431–439. doi: 10.1039/P19930000431. [DOI] [Google Scholar]
- 43.Trzupek JD, Li CM, Chan C, Crowley BM, Heimann AC, Danishefsky SJ. Pure Appl Chem. 2010;82:1735–1748. doi: 10.1351/PAC-CON-09-11-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kowalski P, Bojarski AJ, Mokrosz JL. Tetrahedron. 1995;51:2737–2742. doi: 10.1016/0040-4020(95)00022-Z. [DOI] [Google Scholar]
- 45.Overvoorde LM, Grayson MN, Luo Y, Goodman JG. J Org Chem. 2015;80:2634–2640. doi: 10.1021/jo5028134. [DOI] [PubMed] [Google Scholar]
- 46.For crystal structures of Strictosidine Synthase bound to each its substrates (PDB 2FPB, 2FPC) see: Ma X, Panjikar S, Koepke J, Loris E, Stockigt J. Plant Cell. 2006;18:907–920. doi: 10.1105/tpc.105.038018.
- 47.Klausen RS, Jacobsen EN. Org Lett. 2009;11:887–890. doi: 10.1021/ol802887h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lee Y, Klausen RS, Jacobsen EN. Org Lett. 2011;13:5564–5567. doi: 10.1021/ol202300t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Min C, Mittal N, Sun DX, Seidel D. Angew Chem Int Ed. 2013;52:14084–14088. doi: 10.1002/anie.201308196. See also: ref. 31. [DOI] [PubMed] [Google Scholar]
- 50.Lalonde MP, McGowan MA, Rajapaksa NS, Jacobsen EN. J Am Chem Soc. 2013;135:1891–1894. doi: 10.1021/ja310718f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Min C, Lin CT, Seidel D. Angew Chem Int Ed. 2015;54:6608–6612. doi: 10.1002/anie.201501536. [DOI] [PubMed] [Google Scholar]
- 52.Zhao C, Chen SB, Seidel D. J Am Chem Soc. 2016;138:9053–9056. doi: 10.1021/jacs.6b05225. [DOI] [PubMed] [Google Scholar]
- 53.For example, the catalyst system has been applied in total synthesis: Piemontesi C, Wang Q, Zhu J. J Am Chem Soc. 2016;138:11148–11151. doi: 10.1021/jacs.6b07846.
- 54.The high enantioselectivity observed under these conditions underlies our assumption that product formation occurs predominantly through a single mechanistic manifold.
- 55.Blackmond DG. Angew Chem Int Ed. 2005;44:4302–4320. doi: 10.1002/anie.200462544. [DOI] [PubMed] [Google Scholar]
- 56.Blackmond DG. J Am Chem Soc. 2015;137:10852–10866. doi: 10.1021/jacs.5b05841. [DOI] [PubMed] [Google Scholar]
- 57.The reaction was observed to exhibit a first-order dependence on 5a over much of the reaction (up to 60–80% conversion, fit to the integrated rate law), although the reaction rate slows at high conversion. While the order in reactant is generally determined over >4 half-lives, the order in substrate is distorted at high conversion due to substantial product inhibition. See below and Supporting Information for additional discussion.
- 58.Ford DD, Lehnherr D, Kennedy CR, Jacobsen EN. J Am Chem Soc. 2016;138:7860–7863. doi: 10.1021/jacs.6b04686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ford DD, Lehnherr D, Kennedy CR, Jacobsen EN. ACS Catal. 2016;6:4616–4620. doi: 10.1021/acscatal.6b01384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lehnherr D, Ford DD, Bendelsmith AJ, Kennedy CR, Jacobsen EN. Org Lett. 2016;18:3214–3217. doi: 10.1021/acs.orglett.6b01435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kennedy CR, Lehnherr D, Rajapaksa NS, Ford DD, Park Y, Jacobsen EN. J Am Chem Soc. 2016;138:13525–13528. doi: 10.1021/jacs.6b09205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Novak P, Vikić-Topić D, Meić Z, Sekušak S, Sabljić A. J Mol Struct. 1995;356:131–141. doi: 10.1016/0022-2860(95)08939-S. [DOI] [Google Scholar]
- 63.Renny JS, Tomasevich LL, Tallmadge EH, Collum DB. Angew Chem Int Ed. 2013;52:11998–12013. doi: 10.1002/anie.201304157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.H-bonded complexes between specialized ureas or thioureas and carboxylate anions have been observed with binding constants on the order of 103–104 in dimethyl sulfoxide and as high as 106–107 in acetonitrile. See: Smith PJ, Reddington MV, Wilcox CS. Tetrahedron Lett. 1992;33:6085–6088. doi: 10.1016/S0040-4039(00)60012-6.
- 65.Fan E, Vanarman SA, Kincaid S, Hamilton AD. J Am Chem Soc. 1993;115:369–370. doi: 10.1021/ja00054a066. [DOI] [Google Scholar]
- 66.Gomez DE, Fabbrizzi L, Licchelli M, Monzani E. Org Biomol Chem. 2005;3:1495–1500. doi: 10.1039/B500123D. [DOI] [PubMed] [Google Scholar]
- 67.Gunnlaugsson T, Davis AP, O’Brien JE, Glynn M. Org Biomol Chem. 2005;3:48–56. doi: 10.1039/B409018G. [DOI] [PubMed] [Google Scholar]
- 68.Amendola V, Esteban-Gómez D, Fabbrizzi L, Licchelli M. Acc Chem Res. 2006;39:343–353. doi: 10.1021/ar050195l. [DOI] [PubMed] [Google Scholar]
- 69.Jose DA, Singh A, Das A, Ganguly BA. Tetrahedron Lett. 2007;48:3695–3698. doi: 10.1016/j.tetlet.2007.03.120. [DOI] [Google Scholar]
- 70.Pérez-Casas C, Yatsimirsky AK. J Org Chem. 2008;73:2275–2284. doi: 10.1021/jo702458f. [DOI] [PubMed] [Google Scholar]
- 71.Li AF, Wang JH, Wang F, Jiang YB. Chem Soc Rev. 2010;39:3729–3745. doi: 10.1039/B926160P. [DOI] [PubMed] [Google Scholar]
- 72.This propensity for carboxylate-binding has been exploited in the design of thiourea catalysts for enantioselective acyl transfer reactions and related transformations. See: Seidel D. Synlett. 2014;25:783–794. doi: 10.1055/s-0033-1340618.
- 73.De CK, Klauber EG, Seidel D. J Am Chem Soc. 2009;131:17060–17061. doi: 10.1021/ja9079435. [DOI] [PubMed] [Google Scholar]
- 74.Klauber EG, De CK, Shah TK, Seidel D. J Am Chem Soc. 2010;132:13624–13626. doi: 10.1021/ja105337h. [DOI] [PubMed] [Google Scholar]
- 75.Klauber EG, Mittal N, Shah TK, Seidel D. Org Lett. 2011;13:2464–2467. doi: 10.1021/ol200712b. [DOI] [PubMed] [Google Scholar]
- 76.De CK, Seidel D. J Am Chem Soc. 2011;133:14538–14541. doi: 10.1021/ja2060462. [DOI] [PubMed] [Google Scholar]
- 77.De CK, Mittal N, Seidel D. J Am Chem Soc. 2011;133:16802–16805. doi: 10.1021/ja208156z. [DOI] [PubMed] [Google Scholar]
- 78.Mittal N, Sun DX, Seidel D. Org Lett. 2012;14:3084–3087. doi: 10.1021/ol301155b. [DOI] [PubMed] [Google Scholar]
- 79.Min C, Mittal N, De CK, Seidel D. Chem Commun. 2012;48:10853–10855. doi: 10.1039/C2CC36361E. [DOI] [PubMed] [Google Scholar]
- 80.Mittal N, Lippert KM, De CK, Klauber EG, Emge TJ, Schreiner PR, Seidel D. J Am Chem Soc. 2015;137:5748–5758. doi: 10.1021/jacs.5b00190. [DOI] [PubMed] [Google Scholar]
- 81.Consistent with the proposed inhibition of BzOH by amines, reactions conducted with in situ imine formation are significantly slower when an excess of tryptamine is employed. For reference pKa values in dimethyl sulfoxide, pKa(BzOH) = 11.1, pKa(piperidinium) = 10.9. See: Olmstead WN, Bordwell FG. J Org Chem. 1980;45:3299–3305. doi: 10.1021/jo01304a033.
- 82.Bordwell FG, Algrim DJ. J Am Chem Soc. 1988;110:2964–2968. doi: 10.1021/ja00217a045. [DOI] [Google Scholar]
- 83.This experiment is conceptually analogous to the “same-excess” experiment introduced by Blackmond for the analysis of catalytic reactions involving two or more substrates. See refs. 55 and 56.
- 84.A similar strategy was employed by Seidel and coworkers. See ref. 34.
- 85.Simmons EM, Hartwig JF. Angew Chem Int Ed. 2012;51:3066–3072. doi: 10.1002/anie.201107334. [DOI] [PubMed] [Google Scholar]
- 86.Observed isotope effects are determined from the relative first-order rate constants for formation of 4a and are uncorrected for incomplete deuterium incorporation in the starting material (94% D). The uncorrected values may, thus, lead to an underestimation of the magnitude of the isotope effect.
- 87.The isotope effect data are also consistent with a mechanism involving concerted C2-addition and rearomatization. These two possibilities cannot be distinguished on the basis of isotope effects alone, but computational predictions favor a stepwise process, vide infra.
- 88.The isotope effect observed with 5a-d1(C2) for the racemic, benzoic acid-mediated reaction in CHCl3 (1.8 ± 0.2) is also consistent with rate-limiting rearomatization.
- 89.The product-determining step or selectivity-determining step is “[t]he step of a stepwise reaction in which the product distribution is determined. The product-determining step may be identical to, or occur later than, the rate-controlling step on the reaction coordinate.” See: McNaught AD, Wilkinson A, compilers. IUPAC. In: Compendium of Chemical Terminology, 2nd ed (the “Gold Book”) Blackwell Scientific Publications; Oxford: 1997. XML on-line corrected version: http://goldbook.iupac.org. (2006-) created by M. Nic, J. Jirat, B. Kosata, updates compiled by A. Jenkins. ISBN 0-9678550-9-8. DOI:10.1351/goldbook.P04862. Last update: 2014-02-24; version: 2.3.3.
- 90.Isotopic substitution has only a small effect on the enantiomeric excess of product 4a or 4a-d1 (83% ee with 5a; 85% ee with 5a-d1(Cϕ); 81% ee with 5a-d1(C2)) indicating that the isotope effects for the formation of the major and minor enantiomer of product are similar in magnitude. As such, deprotonation/rearomatization can be assigned definitively as the rate- and selectivity-determining step following rapid and reversible cyclization.
- 91.Lequestel JY, Laurence C, Lachkar A, Helbert M, Berthelot M. J Chem Soc, Perkin Trans 2. 1992:2091–2094. doi: 10.1039/P29920002091. [DOI] [Google Scholar]
- 92.Laurence C, Berthelot M, Lequestel JY, Elghomari MJ. J Chem Soc, Perkin Trans 2. 1995:2075–2079. doi: 10.1039/P29950002075. [DOI] [Google Scholar]
- 93.Laurence C, Brameld KA, Graton J, Le Questel JY, Renault E. J Med Chem. 2009;52:4073–4086. doi: 10.1021/jm801331y. [DOI] [PubMed] [Google Scholar]
- 94.A related N-phenylthiourea catalyst was found to be superior to the analogous N-(3,5-bis(trifluoromethyl)phenyl)thiourea for a transformation in which the Lewis basic S atom participates directly in substrate activation. See: Park Y, Schindler CS, Jacobsen EN. J Am Chem Soc. 2016;138:14848–14851. doi: 10.1021/jacs.6b09736.
- 95.Jakab G, Tancon C, Zhang Z, Lippert KM, Schreiner PR. Org Lett. 2012;14:1724–1727. doi: 10.1021/ol300307c. [DOI] [PubMed] [Google Scholar]
- 96.Lippert KM, Hof K, Gerbig D, Ley D, Hausmann H, Guenther S, Schreiner PR. Eur J Org Chem. 2012:5919–5927. doi: 10.1002/ejoc.201200739. [DOI] [Google Scholar]
- 97.The thiourea analogue of urea 1e decomposes rapidly in the presence of acid, precluding a direct comparison.
- 98.Due to the poor solubility of substituted benzoic acids in toluene, this analysis was conducted in chloroform, instead. Under these conditions all of the substituted benzoic acids tested afforded product 4b in relatively high enantioselectivity (86–94% ee), thereby suggesting that competing background reactivity is minimal.
- 99.Hansch C, Leo A, Taft RW. Chem Rev. 1991;91:165–195. doi: 10.1021/cr00002a004. [DOI] [Google Scholar]
- 100.Bess EN, Sigman MS. Linear Free Energy Relationships (LFERs) in Asymmetric Catalysis. In: Christmann M, Bräse S, editors. Asymmetric Synthesis II: More Methods and Applications. Wiley-VCH Verlag GmbH & Co. KGaA; Weinheim: 2012. pp. 363–370. [Google Scholar]
- 101.Relative rates determined at the standard catalyst concentration [1a]tot = [BzOH]tot = 5 mM, such that νrel = νobs/krac, where νobs is the rate observed experimentally and krac is determined from the fit to νobs vs. [1a]tot at a given [5a] (Figure 2). Values for νrel range from 3.2 to 5.9 over the course of the reaction.
- 102.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA, Jr, Peralta JE, Ogliaro F, Bearpark MJ, Heyd J, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision D.01. Gaussian Inc; Wallingford, CT: 2009. [Google Scholar]
- 103.For a discussion of methods in the computational analysis of catalytic, enantioselective reactions: Lam Y-h, Grayson MN, Holland MC, Simon A, Houk KN. Acc Chem Res. 2016;49:750–762. doi: 10.1021/acs.accounts.6b00006.
- 104.Becke AD. J Chem Phys. 1993;98:5648–5652. doi: 10.1063/1.464913. [DOI] [Google Scholar]
- 105.Lee C, Yang W, Parr RG. Phys Rev B: Condens Matter Mater Phys. 1988;37:785–789. doi: 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- 106.Miehlich B, Savin A, Stoll H, Preuss H. Chem Phys Lett. 1989;157:200–206. doi: 10.1016/0009-2614(89)87234-3. [DOI] [Google Scholar]
- 107.Grimme S, Antony J, Ehrlich S, Krieg H. J Chem Phys. 2010;132:154104. doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- 108.Computational studies reported previously begin with a fully protonated iminium ion intermediate, which should form readily in aqueous media. (See refs. 21 and 44.). However, the iminium carboxylate protonation state is expected to be substantially less favored in a nonpolar medium.
- 109.Calculations were performed using the CPCM implicit solvent model for toluene. See Supporting Information for details.
- 110.Scalmani G, Frisch MJ. J Chem Phys. 2010;132:114110. doi: 10.1063/1.3359469. [DOI] [PubMed] [Google Scholar]
- 111.Imine 7 was employed to avoid the potential for confounding cyclization during the analysis.
- 112.Mayr H, Ofial AR, Würthwein EU, Aust NC. J Am Chem Soc. 1997;119:12727–12733. doi: 10.1021/ja972860u. [DOI] [Google Scholar]
- 113.Xu H, Zuend SJ, Woll MG, Tao Y, Jacobsen EN. Science. 2010;327:986–990. doi: 10.1126/science.1182826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Under the conditions shown in Figure 1D, use of the benzoic acid co-catalyst affords tetrahydro-β-carboline 4b in 94% ee. Use of acetic acid instead affords 4b in 92% ee with modestly reduced yield. The minimal change in enantioselectivity lends credence to the assumption that the features of the path en route to the major enantiomer of product are highly conserved even upon this simplification.
- 115.Kozuch S, Shaik S. Acc Chem Res. 2011;44:101–110. doi: 10.1021/ar1000956. [DOI] [PubMed] [Google Scholar]
- 116.Kozuch S. WIREs Comput Mol Sci. 2012;2:795–815. doi: 10.1002/wcms.1100. [DOI] [Google Scholar]
- 117.Consistent with experimental observations, the turnover-limiting intermediate is predicted to be the product-bound complex (R)-4b•HOAc•1f.
- 118.A strong energetic preference for C2 addition over C3 addition was also predicted in other computational studies. See refs. 21, 44, and 45.
- 119.While the relative energies of TS2,chair and TS2,spiro varied across the functionals evaluated, the transition state for rearomatization (TS3) was predicted to be higher in energy than that for cyclization in all cases.
- 120.Zhao Y, Truhlar DG. Theor Chem Acc. 2008;120:215–241. doi: 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- 121.Marenich AV, Cramer CJ, Truhlar DG. J Phys Chem B. 2009;113:6378–6396. doi: 10.1021/jp810292npmid:19366259. [DOI] [PubMed] [Google Scholar]
- 122.The free energies predicted at this high level of theory provide a superior quantitative description of the degree of enantioselectivity but little additional qualitative information relative to the calculations performed with B3LYP-D3(BJ)/6-31+G(d,p)/CPCM(toluene)//B3LYP/6-31G(d)/CPCM(toluene). Given the significant additional computational expense with little added benefit, the bulk of the calculations described here were performed at the lower level of theory. See Supporting Information.
- 123.Wheeler and coworkers have employed this strategy in the analysis of transformations catalyzed by chiral phosphoric acids; see: Seguin TJ, Lu T, Wheeler SE. Org Lett. 2015;17:3066–3069. doi: 10.1021/acs.orglett.5b01349.
- 124.Seguin TJ, Wheeler SE. ACS Catal. 2016;6:2681–2688. doi: 10.1021/acscatal.6b00538. [DOI] [Google Scholar]
- 125.Seguin TJ, Wheeler SE. Angew Chem Int Ed. 2016;55:15889–15893. doi: 10.1002/anie.201609095. [DOI] [PubMed] [Google Scholar]
- 126.For this analysis, we assume that E/Z isomerization is rapid under the reaction conditions. The (E)-5b•HOAc complex is 3.6 kcal/mol lower in energy than (Z)-5b•HOAc and thus represents the minimum starting point for this analysis.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.