

Abstract
Introduction
Despite substantial improvements in standards of care, the most common aggressive pediatric and adult high-grade gliomas (HGG) carry uniformly fatal diagnoses due to unique treatment limitations, high recurrence rates and the absence of effective treatments following recurrence. Recent advancements in our understanding of the pathophysiology, genetics and epigenetics as well as mechanisms of immune surveillance during gliomagenesis have created new knowledge to design more effective and target-directed therapies to improve patient outcomes.
Areas covered
In this review, the authors discuss the critical genetic, epigenetic and immunologic aberrations found in gliomas that appear rational and promising for therapeutic developments in the presence and future. The current state of the latest therapeutic developments including tumor-specific targeted drug therapies, metabolic targeting, epigenetic modulation and immunotherapy are summarized and suggestions for future directions are offered. Furthermore, they highlight contemporary issues related to the clinical development, such as challenges in clinical trials and toxicities.
Expert opinion
The commitment to understanding the process of gliomagenesis has created a catalogue of aberrations that depict multiple mechanisms underlying this disease, many of which are suitable to therapeutic inhibition and are currently tested in clinical trials. Thus, future treatment endeavors will employ multiple treatment modalities that target disparate tumor characteristics personalized to the patient’s individual tumor.
1. The Diverse Spectrum of Malignant Gliomas
In the United States, approximately 23,000 people are diagnosed with a malignant brain tumor each year [1], of which approximately 80% belong to the heterogeneous group of diffuse gliomas [1]. Histologically, gliomas are graded into individual classes as defined by the World Health Organization (WHO) classification of central nervous system (CNS) tumors based on architectural and cytological features, such as cellular atypia, mitotic activity, necrosis and vascular proliferation [2]. Clinically, we frequently distinguish between low-grade (LGG, grade II) and high-grade (HGG, grades III and IV) tumors to reflect their anticipated biologic behavior and clinical course. Over time, LGGs frequently progress to higher-grade projecting the natural course of this disease rather than a new entity. Three main glioma subtypes exist, which are based on the morphological similarities of the predominant tumor cell population; the most frequent subtypes are astrocytoma (~75%, WHO grade I–IV), followed by oligodendroglioma (~6%, WHO grade II–III) and ependymoma (~7%, WHO grade II–III), which resemble astrocytes, oligodendrocytes and ependymal cells, respectively [1].
Glioblastoma (GBM, grade IV astrocytoma) is the most malignant variant of diffuse astrocytoma in adulthood accounting for 55% of all gliomas [1]. The majority (95%) of GBM arise as de novo lesions (i.e. primary GBM). Less common are secondary GBM (~5%) that arise from the progression of grade II or III glioma. Secondary GBM is a genetically and clinically distinct entity that typically occurs in younger patients (mean age 45 years versus 60 years for primary GBM) [1 3].
Oligodendroglioma comprises the second most common glioma in adults [2]. The hallmark molecular abnormality that is now increasingly used to define oligodendroglioma is co-deletion of chromosomes 1p and 19q, which also predicts a better prognosis and responsiveness to radiation therapy and chemotherapy [4].
Brain cancer has superseded leukemia as the most common cause of cancer-related death in children [1,5]. As in adults, gliomas are the most common CNS neoplasms in children accounting for ~53% of all tumors although the predilection sites differ. The majority of childhood gliomas (~60%) are grade I or II lesions and of astrocytic lineage, while oligodendroglioma and ependymoma are rare [5]. Childhood HGG traditionally include GBM, anaplastic astrocytoma, anaplastic oligodendroglioma and diffuse intrinsic pontine glioma (DIPG) [5,6]. DIPG is a childhood specific brain cancer that presents most commonly between ages 6–8. Despite varying histological grades DIPGs share a universally lethal outcome [7,8]. In contrast to other HGGs, the diagnosis of DIPG has relied solely upon radiological and clinical findings, and not on histopathological features. However, a role for image-guided stereotactic biopsy has recently been re-introduced toward the goals of sample collection for biological studies and molecular characterization required for experimental personalized therapeutics [9].
Aside from radiation exposure and uncommon inherited genetic syndromes, there are few proven causes of primary brain tumors beyond the role of random spontaneous mutation associated with physiologic DNA replication. About 5% of gliomas are due to inherited germ-line mutations associated with known syndromes such as Cowden’s disease (PTEN mutation), tuberous sclerosis (TSC1 9q34, TSC2 16p13), Li–Fraumeni syndrome (p53 mutation), neurofibromatosis types 1 and 2 (neurofibromin and merlin mutations) and Lynch Syndrome (miss-match DNA repair defect) [10].
2. Current standards of treatment and outcomes
Tumor grade, histology and their effect on prognosis are the key factors influencing therapeutic decision-making. Non-infiltrative grade I gliomas have the most favorable prognosis and chance for cure due to their indolent nature and potential for complete surgical resection with 5- and 10-year survival rates of 94% and 92%, respectively [1]. Well-differentiated oligodendroglioma (WHO grade II) and anaplastic oligodendroglioma (WHO grade III) are known to be particularly responsive to cytotoxic therapies including radiation, temozolomide (TMZ), and the combination chemotherapy regimen PCV (Procarbazine, CCNU, Vincristine). Two recent randomized Phase III trials, initiated prior to the introduction of TMZ, definitively demonstrated the benefit of aggressive combination radiation + chemotherapy for anaplastic oligodendroglioma [11,12]. Patients treated with PCV + radiation lived remarkably longer than patients treated with radiation therapy alone (~15 vs. 7 years, median survival). These studies have all but established a new standard of care that combines radiation and PCV chemotherapy for patients with anaplastic 1p19q co-deleted oligodendroglioma. An informative sub-analysis in these studies revealed no statistically significant benefit from combination PCV + radiation compared to radiation alone in the non-codeleted tumors that represent anaplastic astrocytoma. A randomized trial comparing combination radiation + TMZ with combination radiation + PCV is currently underway (CODEL study, NCT00887146). Results are anxiously awaited since anaplastic oligodendroglioma is known to be sensitive to TMZ, which is less toxic, more easily tolerated and more easily administered than PCV.
In contrast, HGGs of the astrocytic lineage are less sensitive to therapy and have a drastically lower median survival. Although complete surgical resection of these infiltrative tumors is virtually impossible, retrospective studies increasingly point to a probable survival benefit from > 70% tumor resection with clear survival benefit following gross total resection [13,14]. Surgery is followed by fractionated radiotherapy with concomitant and adjuvant treatment with the alkylating agent temozolomide (TMZ), a regimen that was established in GBM as the gold standard in 2005 [15]. In this study, the 2-year survival of GBM patients increased from ~8% to 28% [1]. Many neuro-oncologists also recommend the use of TMZ for the treatment of anaplastic astrocytomas; however, a clear benefit has not been convincingly demonstrated and is subject of an ongoing Phase III trial by the European Organisation for Research and Treatment of Cancer (EORTC, CATNON intergroup trial (NCT00626990). The strongest predictive and prognostic factor for a therapeutic response and a longer survival is the epigenetic silencing of the DNA repair enzyme O-6-methylguanine-DNA methyltransferase (MGMT) [14–17]. Nonetheless, almost all GBMs ultimately relapse, and there are no treatments proven to prolong survival in patients with recurrent GBM. This is exemplified by recent disappointing results showing no significant survival benefit from the neutralizing anti-VEGF monoclonal antibody bevacizumab (Avastin®), administered either alone or in combination with existing cytotoxic regimens, which was granted accelerated approval in 2009 based on its ability to improve MRI abnormalities and reverse patient symptoms in patients with recurrent GBM [18–20].
For pediatric DIPG patients, treatment options are even more limited with no conventional chemotherapeutic agents proven effective, and radiation therapy alone representing the standard of care resulting in a survival rate of less than 10% 2 years after diagnosis [8,21].
In spite of extensive efforts to optimize existing therapies, we have likely reached the limits of what can be achieved with traditional cytotoxic regimens. Drawing from the advancements in understanding the underlying biology, new nodes of vulnerabilities have been identified that have paved the way for novel target-directed therapies, onco-metabolic and epigenetic focused strategies, as well as new immunotherapeutic approaches (Fig. 1, Tab. 1). Especially exciting have been the advances made in next-generation immunotherapies, which, in principle, can affect every single tumor cell by simultaneously or sequentially evoking specific immune responses against a theoretically unlimited number of tumor-associated antigens. In this review, we summarize the current state of these newly emerging therapeutic developments and highlight major remaining obstacles and challenges for these approaches.
Figure 1. Schematic overview of current targeted therapies in HGGs.
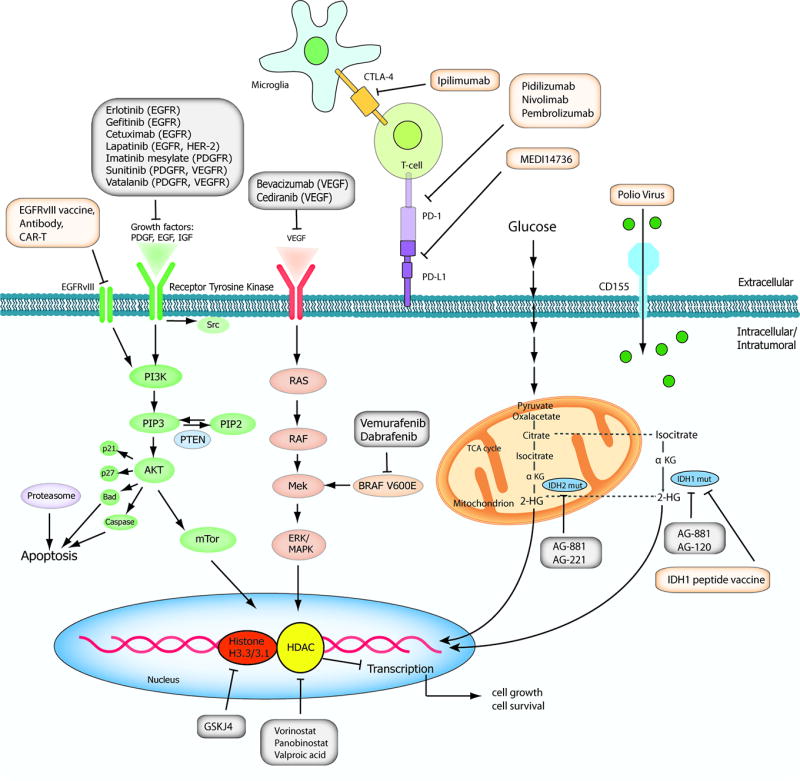
Aberrant oncogenic RTK pathways including the PI3K-AKT (green) and RAS (pink) oncogenic pathways are targeted with a variety of small molecule inhibitors (grey boxes) and monoclonal antibodies. Oncometabolites produced by IDH1/2-mutated glioma cells can be intracellularly targeted by small molecule inhibitors and a IDH1 mutant specific vaccine, which is currently being tested HGG patients. Epigenetic modifiers targeting on HDAC and histone 3.3/3.1 are shown as well. Notably, various immunotherapeutic strategies that are currently undergoing evaluations in clinical trials are pictured including tumor vaccines, immune checkpoint inhibitors, CAR-T cell therapies, and oncolytic viruses (yellow box). Abbreviations: mut: mutant.
Table 1.
Overview of selected completed and ongoing clinical trials for malignant gliomas in adults and children.

|

|

|

|
A search was carried out on http://www.clinicaltrials.gov. Currently active trials are highlighted in bold.
Abbreviations: Bev: bevacizumab; CEA: carcinoembryonic antigen; DIPG: diffuse intrinsic pontine glioma; FSRT: fractionated stereotactic radiation therapy; GBM: glioblastomas; HGG: high-grade glioma; HDACi: HDAC inhibitor; HU: hydroxyurea; IA: intra-arterial; mut: mutant; Ped: pediatric; rec: recurrent; Prog: progressive; RDX: radiation; subc: subcutaneous; TMZ: temozolomide.
3. Emerging Targeted Therapy for Malignant Glioma
3.1 Receptor tyrosine kinase (RTK) Pathway Inhibitors
Nearly all HGGs show genetic alterations in the receptor tyrosine kinase (RTK)–PI3K–RAS pathway, although specific aberrations and frequencies greatly differ between adults and children [22,23]. RTKs are a family of cell surface receptors that include platelet-derived growth factor receptors (PDGFR-α and PDGFR-β), epidermal growth factor receptors (ErbB1/EGFR, ErbB2, ErbB3, ErbB4), vascular endothelial growth factor receptors (VEGFR1–4), fibroblast growth factor receptors (FGFR1, FGFR2) and c-MET. Oncogenic RTK hyperactivation is driven by multiple mechanisms such as RTK gene amplification and overexpression resulting in ligand-independent receptor oligomerization, receptor mutation resulting in constitutive activation and ligand over-expression. Dysregulated RTK hyperactivation drives multiple oncogenic processes such as cell proliferation, aberrant survival and therapeutic resistance, migration/invasion, and tumor cell stemness that is closely linked to tumor propagating capacity [24–26].
In adult HGGs, epidermal growth factor receptor (EGFR) EGFR is the most frequently amplified gene, with approximately one-third of GBMs also having gene rearrangements [27]. The most common is EGFR variant III (EGFRvIII), a truncated 801 base pair in-frame deletion of the wild-type EGFR, which is tumor-specific and absent in healthy tissue. Despite this frequency and that fact that EGFR inhibitors are efficacious in other molecularly defined malignancies (e.g. EGFR mutated non-small cell lung cancer) [28], multiple EGFR inhibitors (erlotinib, gefitinib, lapatinib and the chimeric EGFR monoclonal antibody cetuximab), alone or in combination, have been largely ineffective against HGG in multiple clinical trials [29–39], attributed to the absence of the kinase domain mutations required for durable therapeutic responses, insufficient CNS drug penetration or toxicity, particularly when used in combination [23,40,41]. For example, lapatinib, a dual RTK inhibitor that also targets erbB2, was investigated as a single agent for recurrent GBM and discontinued prematurely due to lack of clinical effect, owing to sub-therapeutic tissue concentrations [37]. Likewise, the combined use of lapatinib with pazopanib, a multiple kinase inhibitor against VEGFR1–3, PDGFRα/β and c-Kit, failed to show any effectiveness in a Phase I/II study [42], while a follow-up Phase I study of lapatinib + TMZ in recurrent HGGs showed a slight effect on progression-free survival, albeit with moderate toxicity [43]. This gave rise to the current Phase II trial evaluating high-dose lapatinib in conjuncture with standard chemo-radiation in patients with newly diagnosed GBM (NCT01591577).
Despite such disappointing results to date, the development of new EGFR inhibitors continues to be of high interest due to its critical role in gliomagenesis, which is also underscored by the fact that the majority of agents evaluated in clinical trials for glioma patients have targeted one or multiple components of this signaling pathway. The novel agent Dacomitinib (PF-299804), an irreversible pan-HER inhibitor, was found to have promising activity in preclinical EGFR mutated glioma models [44] and is currently being evaluated in two Phase II studies for recurrent GBM patients with EGFR amplification or EGFRvIII expression (NCT01520870, NCT01112527). Afatinib (BIBW 2992) is another novel dual EGFR/Her2 inhibitor that had no meaningful single-agent activity in non-selected recurrent GBMs, but perhaps a modest response in patients with EGFRvIII mutations [45]. Although standard daily dosing was inactive, alternative dosing schedules such as pulsatile-increased dosing may be evaluated in the future, as observed in other cancer types.
Conjugated and unconjugated antibodies have also been developed to target both wildtype EGFR and EGFRvIII, including cetuximab, panitumumab, and nimotuzumab. Although none of them have been able to demonstrate a significant survival advantage in patients with recurrent GBM, several new agents are currently undergoing clinical trial evaluations: AMG 595, a first-in-human mAb for patients with EGFRvIII recurrent HGG (NCT01475006); Sym004, a recombinant antibody that specifically binds to EGFR in patients with EGFR amplified GBM (NCT02540161); and ABT-414, an antibody-drug conjugate, with a toxic payload (monomethylauristatin F) targeted to EGFR or EGFRvIII amplified GBMs (Intellance 1; NCT02573324). ABT-414, which is currently undergoing evaluation in randomized, placebo-controlled Phase IIb/III with concomitant radiotherapy and temozolomide (TMZ), demonstrated preliminary efficacy in Phase I, however toxicities affecting the eye and liver were observed as well [46].
Small molecule tyrosine kinase inhibitors with anti-PDGFR activity, which is amplified in 10% of HGGs [47], include imatinib mesylate, the dual VEGFR/PDGFR inhibitors (sunitinib, sorafenib, pazopanib, and vatalanib), and dasatinib that targets BCR-ABL, the Src family, c-Kit, and PDGFR-a/β. PDGFR inhibitors have also largely disappointed in the treatment of HGGs despite encouraging preclinical results. For example, imatinib, which was investigated as monotherapy and in combination with hydroxyurea in newly diagnosed and recurrent GBM did not improve outcome [48–50]. Likewise, a Phase II clinical trial of pazopanib in patients with recurrent GBM failed, reporting a median progression-free survival and overall survival of 12 and 35 weeks, respectively [51]. Furthermore, dasatinib was ineffective in multiple Phase II clinical trials as a single agent and in combination with radiation, TMZ, erlotinib, lomustine, as well as crizotinib [52–57]; the results of an ongoing randomized Phase II trial adding dasatinib to bevacizumab in recurrent disease (Alliance N0872, NCT00892177) are anticipated shortly. Dose-limiting toxicities, particularly when used in combination with other cytotoxic agents (e.g. lomustine), and poor CNS bioavailability due to the fact that dasatinib is a substrate for P-glycoprotein and breast cancer resistance protein (BCRP) have contributed to its poor efficacy [58 59]. The novel PDGFRα/β and Flt3 inhibitor crenolanib is currently in Phase II for adult HGG (NCT01229644) and in a Phase I study for recurrent pediatric HGG (NCT01393912).
Other inhibitors target c-MET or its ligand hepatocyte growth factor (HGF), which, when overexpressed promotes angiogenesis, proliferation and invasion of cancer cells and correlates with poor prognosis [60–62]. Preclinical studies have conferred sensitivity to c-Met inhibitors in glioblastomas xenograft models [60,63]. Cabozantinib, which targets Met and other kinases, failed in the treatment of primary and recurrent GBM, although patients were not pre-selected for c-Met amplification [64]. Further studies with AMG 102 (rilotumumab), a humanized monoclonal antibody against HGF, and SGX523, a direct inhibitor of c-MET, evaluated in patients with recurrent molecularly undefined GBMs were also unsuccessful in a Phase II and two Phase I trials (NCT00606879, NCT00607399) respectively [65]. Met-specific inhibitors are in clinical development, including onartuzumab, a monoclonal antibody targeting c-Met that is currently being evaluated in Phase II combination with bevacizumab in recurrent GBM (NCT01632228).
In contrast to the RTKs discussed above that are primarily expressed by glioma cells, VEGFRs are primarily expressed by glioma vascular endothelial cells and ligand-dependent activation drives tumor-associated vascular proliferation (i.e. angiogenesis) and BBB dysfunction. Bevacizumab, a mAb against the VEGF-A ligand, is the most extensively studied and successful VEGFR pathway inhibitor. Bevacizumab was FDA-approved for recurrent GBM based on objective imaging response data with no evidence of survival benefit (see Section 2, above). Two large Phase III trials have since failed to demonstrate any survival benefit for patients with newly diagnosed GBM [18,20,66]. Furthermore, the Phase III EORTC-26101 trial, investigating the therapeutic efficacy of the combination bevacizumab and lomustine versus lomustine alone in GBM patients at first recurrence, did not improve the neurological deterioration-free survival or the overall survival [67]. Apart from mAbs, various small molecule inhibitors directed against VEGFRs have been developed including cabozanitinib, cediranib, pazopanib, sorafenib, sunitinib, vandetanib and vatalanib; however, clinical trials have been uniformly disappointing. Several Phase II trials of sorafenib and sunitinib in combination with other therapies in GBM have failed to meet primary endpoints [68–75]. Cediranib, which targets all VEGFRs, c-kit and PDGFR, initially showed modest potential efficacy as monotherapy in a Phase II trial in recurrent GBM patients [76]. However, these results were not substantiated in a subsequent randomized Phase III trial comparing cediranib monotherapy, lomustine monotherapy and combination cediranib and lomustine [77]. Lenvatinib (E7080), a novel multitargeted tyrosine kinase inhibitor for all VEGFRs and PDGFR, did not warrant further testing in recurrent GBM patients beyond Phase II because the therapeutic effect was very limited with no objective responses (NCT01137604) [78]. Finally, axitinib, a TKI with high affinity and specificity for the VEGF-receptors that approved for the treatment of metastatic renal cell carcinoma, did not result in any meaningful treatment response in patients with recurrent GBM who had failed surgery, radiation, and TMZ [79].
The failures of RTK pathway inhibitors in HGG have been especially frustrating in light of strong evidence supporting the molecular basis for RTK pathway targeting and mounting successes in other malignancies. Several potential explanations exist for these treatment failures in GBM. Most clinical trials have not focused sufficiently on patient cohorts with tumors enriched for target hyperactivation. Both parallel signaling pathways and bypass feedback loops can contribute to intrinsic resistance to target inhibition. Small molecule RTKs are typically hydrophilic resulting in poor drug bioavailability in the CNS due to the blood-brain barrier and insufficient target inhibition. Moreover, multiple surveys have shown substantial intratumoral cellular heterogeneity for target expression and hyperactivation, which adds another layer of complexity [80,81]. As such, future clinical trials will have to focus on stringent patient selection criteria (genotype-enriched clinical trials), customized drug design based on tumor characteristics, and drug dosing regimens rigorously proven to sufficiently inhibit target activation in patients. Combinations instead of single tyrosine kinase inhibitors may be required to sufficiently inhibit parallel bypass and feedback pathways.
3.2 PI3K/AKT and mTOR Pathway Inhibitors
Almost 90% of patients with GBM show at least one alteration in the phosphatidylinositol 3-kinase (PI3K) signaling, which can either result from activating mutations in PI3K itself (25%), loss of the tumor suppressor gene Phosphatase and Tensin Homolog (PTEN) (41%) or downstream activation of RTKs [47]. The downstream effectors of PI3K include AKT and mammalian target of rapamycin (mTOR) consisting of mTOR complexes mTORC1 and 2, which play key roles in cell metabolism, survival, and protein translation. Hence, the addition of pharmacological inhibitors should theoretically lead to therapeutic benefits; however, the majority of clinical trials have failed to fulfill this expectation.
Several first-generation mTOR inhibitors, including rapamycin, temsirolimus (CCI-779), and everolimus (RAD001) posses antineoplastic activity as single agents in vitro and in vivo, and have been studied in various Phase I/II trials for newly diagnosed and recurrent HGGs. Although radiographic responses were observed in a subset of HGG patients receiving everolimus or temsirolimus, no significant effect on progression-free survival and overall survival was reported for any of these inhibitors when used alone, in combination with standard chemoradiation or with bevacizumab in recurrent GBM [82–89]. Notably, the combined use of temsirolimus with TMZ and radiation therapy led to an increased infection risk, which prohibited the analysis of the efficacy of this combination [87]. Currently ongoing clinical trials focus on the evaluation of everolimus in the treatment of progressive LGGs in combination with TMZ (NCT02023905), as single agent in children with progressive LGGs (NCT01734512) and ependymoma (NCT02155920). In addition, dual mTORC1/2 inhibitor sapanisertib (INK1280) is in early clinical investigation to assess CNS penetration and response in patients with recurrent GBM (NCT02133183).
Among PI3K-targeting drugs, buparlisib (BKM120), a pan-PI3K inhibitor, has been studied in the treatment of recurrent GBM based on encouraging preclinical experiments in U87 glioma cells [90]. An initial Phase II trial achieved intratumoral drug concentrations sufficient to inhibit the PI3K pathway although efficacy was not reported. Ongoing studies with buparlisib include a Phase I/II dose-escalation trial with concurrent standard chemoradiation in newly diagnosed GBM (NCT01473901), a Phase II trial in recurrent GBM (NCT01339052) and combination trials for recurrent GBM with bevacizumab (NCT01349660), and carboplatin or lomustine (NCT01934361). PX-886 (Oncothyreon) is a semi-synthetic derivative of wortmannin and irreversibly inhibits PI3K. In glioma cells, PX-866 dramatically inhibited proliferation, autophagy and angiogenic potential in a variety of cell lines and U87 mouse xenograft models [91]. A Phase II trial evaluating the efficacy and safety of PX-866 in patients with recurrent GBM was recently completed and results are soon to be published (NCT01259869). Other PI3K inhibitors currently undergoing clinical evaluation include XL-147 (NCT01240460).
GBM with mutant PI3K alleles may also benefit from inhibition of AKT. Among the various agents that have entered the clinical trial Phase perifosine (KRX-0401) is the most advanced. A Phase II trial of single agent perifosine failed in recurrent GBM (NCT00590954). As data suggesting that the combined inhibition of AKT and mTOR may be more effective in combination with temsirolimus, two Phase II trials have been launched (NCT01051557, NCT02238496).
3.3 RAF–MEK–ERK pathway inhibitors
The presence of activating mutations in constituents of the mitogen activate protein kinase (MAPK) pathway has raised considerable interest in inhibiting this pathway in patients with HGGs and also in patients with unresectable or recurrent LGGs, which typically harbor oncogenic activation of BRAF resulting from a BRAFV600E point mutation or a KIAA1549:BRAF fusion. Evidence for the effectiveness of BRAF and MEK inhibitors or combinations thereof comes from multiple pre-clinical data in murine intracranial xenografts of pediatric gliomas and a few anecdotal clinical case reports [92,93].
There are in fact several BRAF inhibitors available, but only few have been evaluated in CNS tumors due to poor BBB penetration. Vemurafenib (PLX4032), which has demonstrated remarkable activity against metastatic BRAFV600E mutated melanoma [94], is currently undergoing Phase I/II testing in patients with BRAFV600E positive recurrent or refractory pediatric glioma (NCT01748149). Additional agents currently in clinical trial for pediatric BRAFV600E mutated glioma are the BRAF inhibitor dabrafenib (GSK436) (NCT01677741) and the two MEK1/2 inhibitors trametanib (GSK212) (NCT02034110, NCT02124772) and MEK162 (NCT02285439). Notably, the BBB penetrance has not been sufficiently studied in the majority of these agents except for dabrafenib, which appears to achieve adequate CNS drug levels [95]. Isolated case reports detailing complete clinical responses to dabrafenib in children with BRAFV600E-mutant GBMs have sparked excitement [96]; however, responses are rarely durable due to MAPK reactivation [97,98]. Therefore, multiple research groups have proposed the co-administration with other MAPK pathway inhibitors, such as the MEK inhibitor trametinib, in an effort to delay the development of resistance and to minimize the toxicities, particularly cutaneous, associated with BRAF inhibition [97].
Current BRAF inhibitors are mutation specific explaining why BRAFV600E inhibitors are ineffective against other mutations or oncogenic BRAF fusions [93,99]. This limitation can potentially be overcome in appropriate patient subsets using MEK inhibitors that target wild-type MEK1/2 kinase that is hyperactivated downstream of oncogenic BRAF driver mutations [99]. In addition to trametinib and MEK162, sorafenib (NCT02450149) and selumetinib (AZD6244) are under clinical investigation for this purpose. The latter, selumetinib, a second-generation MEK1/2 inhibitor, is undergoing Phase II evaluation in LGGs and neurofibromatosis 1 (NF1)-associated tumors, including optic pathway gliomas as well as plexiform neurofibromas (NCT01386450, NCT01089101) that has resulted in some encouraging therapeutic responses, particularly in patients with NF1 [100].
3.5 Inhibitors of Onco-Metabolism
Activation of oncogenes and loss of tumor suppressors promote metabolic reprogramming to enhance nutrient uptake and energy supply in order to sustain growth and survival. In diffuse gliomas, the premier oncogenic mutations that result in metabolic reprogramming are within the genes coding for isocitrate dehydrogenases, IDH1 and IDH2. Originally detected in primary GBMs (5–10%) [23], IDH1 mutations occur in 50–80% of grade II and III astrocytoma and oligodendroglioma, as well as secondary GBM, while IDH2 mutations are less common and are mutually exclusive with mutations in IDH1 [101,102]. The most common mutation in IDH1 is arginine 132 to histidine (R132H) that results in the ability to convert α-ketoglutarate (α-KG) to the R(−)-2-hydroxyglutarate (2-HG), a potential onco-metabolite, with the coincident conversion of NADPH to NADP+. Current evidence suggests that 2-HG alters the epigenetic machinery in glioma, affecting histone demethylases and DNA hydroxylases, leading to chromatin modifications and gene expression dysregulation [103].
There are currently no therapies approved targeting mutant IDH; however, multiple therapeutic approaches targeting mutant IDH1/2 and the altered metabolic pathway are currently in clinical and preclinical development stages. Because IDH1 mutation is involved in the inhibition of histone lysine demethylases, DNA methyltransferase (DNMT) inhibitors have been suggested as a potential therapeutic approach [104], including decitabine and 5-azacytidine that reverse cancer-promoting hypermethylation restoring cell differentiation and tumor suppressing functions [105,106]. Specific inhibitors of IDH1 R132H are AGI-5198 and ML309, which reduced 2-HG levels and significantly decreased the growth of IDH1R132H –expressing glioma cells in vitro and human glioma xenografts [107,108]. Similar compounds of mutant-selective inhibitors of IDH1 (AG-120), IDH2 (AG-221) or both IDH1/2 (AG-881) have entered the clinical trial Phase (NCT02073994, NCT02273739, NCT02481154) (Tab. 1). While we are awaiting the results for glioma patients, an ongoing Phase I trial of AG-120 in 62 patients with advanced IDH1-mutant solid tumors reported a partial response in 1/55, stable disease in 52.7% (29/55), and progressive disease in 38.2% (21/55) [109].
Dichloroacetate (DCA) represents another strategy for targeting the oncogenic effects of IDH mutation. DCA is an inhibitor of mitochondrial pyruvate dehydrogenase kinase and thus, increases intracellular levels of pyruvate dehydrogenase, which is reduced in IDH1 mutated tumors. DCA has been shown to reduce GBM cell proliferation and clonogenicity in preclinical animal models [110]. A small case series of 5 patients treated with oral DCA confirmed the inhibition of pyruvate dehydrogenase kinase along with p53 activation inducing apoptosis in human GBMs [111] and gave rise to a Phase II trial for primary and recurrent GBM, albeit unselected for IDH1 status, which was recently completed (NCT00540176).
In contrast to IDH-mutated tumors, IDH wild-type HGGs adapt to the increased metabolic demands of tumor growth through alternative metabolic pathways dependent on branched-chain amino acids (BCAAs), which generate nitrogen for the synthesis of neurotransmitter glutamate and macromolecule precursors for mitochondrial ATP synthesis. The key enzyme involved in this process is branched-chain amino acid transaminase 1 (BCAT1) that is restricted to a small number of tissues, including the brain. Interestingly, expression of BCAT1 is dependent on intracellular levels of α-KG, which is produced by wild-type IDH1 [112]. IDH1 expression inhibition was found to impair BCAA metabolism and reduce tumor cell proliferation and invasiveness in a GBM xenograft model [112]. Gabapentin, a leucin analog that specifically inhibits BCAT1 significantly reduces glutamate concentrations in U-87MG cells in-vitro [112]. As BCAAs are nutritionally essential in that humans cannot synthesize them endogenously, a diet deficient in BCAAs (e.g. ketogenic diet) might provide a unique way to downregulate BCAT1 and target these neoplastic metabolic processes [113]. Currently, there are multiple clinical trials under way investigating the feasibility of ketogenic dietary therapy as an adjunct to radiation and chemotherapy in adult patients with HGGs (NCT02046187, NCT02302235, NCT01754350, NCT01865162). Historically, the success of the ketogenic diet in brain tumor patients, reported as case reports or small case series, have been mixed [114,115]. A recently reported Phase I trial examined the feasibility of a ketogenic diet in 20 patients with recurrent GBM (ERGO trial; NCT00575146). Although well tolerated, no clinical activity was found when used as single agent [116]. Regardless, these data highlight that energy metabolism reprogramming drugs or diets might be an attractive treatment option for GBM patients with mutated or wildtype IDH1.
Acetate is another nutrient of increasing interest for its possible role in HGGs and other malignancies. Acetate ligation to coenzyme A (CoA) by acetyl-CoA synthetases (ACSSs) to form acetyl-CoA is critical for the synthesis of fatty acids (lipogenesis), nucleotides, amino acids, and histone acetylation. Cancer cells increase their dependence on acetate under conditions of hypoxia and nutrient stress. Mashimo and Comerford et al. found that primary brain tumors utilize exogenous acetate to meet their high metabolic demands and facilitate growth [117,118]. The incorporation of exogenous acetate is achieved by acetyl-CoA synthetase 2 (ACSS2) and its expression inversely correlates with survival of patients with HGGs. Knockdown of ACSS2 expression was found to dramatically impair the incorporation of exogenously supplied acetate into lipids and histone protein and increased animal survival. These findings have been validated in patients with HGGs and brain metastasis [117]. Selective ACSS2 inhibitors are currently under development and could offer a highly promising new approach to metabolic therapy since ACSS2 is dispensable in normal cells.
3.6 Targeting epigenetic modulators
Epigenetic alterations, i.e. the acetylation and deacetylation of histones allowing the regulation of gene expression patterns, play a crucial role in gliomagenesis. Histone acetylation leads to the unfolding of chromatin (euchromatin) and promotes increased gene transcription while deacetylation induces chromatin condensation (heterochromatin) and mediates suppression of transcription [119,120]. Histone deacetylases (HDAC), responsible for maintaining this balance are frequently overactivated in cancers and inhibition of HDAC in glioma cell lines and GBM animal models demonstrated anti-neoplastic activity [121,122]. Nonetheless, the role of HDAC inhibitors in the treatment of adult HGG remains questionable because most of the HDAC genes are, in fact, downregulated in GBM [123], and clinical trials with the HDAC inhibitors vorinostat and panobinostat failed [124,125]. Although a Phase I/II study of vorinostat in comination with TMZ or radiotherapy + TMZ demonstrated good tolerability in recurrent and newly diagnosed GBM, respectively, it did not result in a significant survival improvement. Likewise, Phase II studies evaluating the combinations of bevacizumab and vorinostat or bevacizumab and panobinostat in recurrent GBM revealed no efficacy. In the context of these results, it is somewhat surprising that valproic acid, an anticonvulsant with relatively weaker HDAC inhibitory activity, showed modestly improved outcome in newly diagnosed GBM when used in combination with radiotherapy (Tab. 1) [126]. A possible explanation for the failure to translate the pre-clinical results is that only specific patient subgroups selected based on molecular biomarkers such as HDAC expression and/or histone acetylation patterns have the potential to benefit from HDAC inhibition. To date, clinical trial designs have not selected for such populations.
In contrast to adult HGG, chromatin-remodeling defects appear to be central to the pathogenesis of pediatric HGG. Of particular significance are mutations in histone H3.3, with 78% of DIPG carrying the amino acid substitution lysine 27 to methionine (K27M) and up to 31% of non-brainstem pediatric HGGs harboring glycine 34 to valine or arginine (G34V/R) or K27M mutations [127,128]. These mutations are thought to sequester polycomb repressive complex 2 (PRC2), which in turn represses gene expression through histone methylation resulting in broad epigenetic dysregulation [8,129–131]. It is thus not surprising that epigenetic modifier panobinostat demonstrated significant anti-tumor efficacy in a preclinical DIPG animal model, further substantiating the underlying mechanism, and has rapidly emerged as a promising therapeutic strategy for this devastating disease [132]. However, panobinostat resistant DIPG cells exist, particularly following chronic drug exposure highlighting the need to optimize regimens by utilizing combination approaches. As H3K27M DIPG cells are transcriptionally more active with reduced di- and trimethylation, one potential strategy has focused on inhibiting the Jumonji-domain demethylase JMJD3, a key enzyme responsible for the demethylation [130]. The use of the GSKJ4, an inhibitor of JMJD3 demethylase, resulted in complete growth inhibition of H3K27M -expressing DIPG cells and brainstem glioma xenografts along with increased K27 methylation and a subsequent decrease in gene expression [133], an effect that could be further enhanced by panobinostat [132,134]. With these results as a foundation, panobinostat that is FDA approved for other indications could rapidly be translated into clinic use and first clinical trials are currently being planned, as single therapy or in combination to increase clinical benefit and reduce the development of resistance.
4. Immunotherapy in malignant glioma
4.1 Vaccines in Malignant Glioma
Several vaccine studies against HGG have been completed or are underway (Tab. 1). Particularly exciting has been the development of vaccines based on GBM-specific antigens, such as EGFRvIII or IDHR132H. Rindopepimut (PEPvIII vaccine), the most extensively studied vaccine for CNS neoplasms, is targeted against the EGFRvIII peptide (H-Leu-Glu-Glu-Lys-Lys-Gln-Asn-Tyr-Val-Val-Thr-Asp-His-Cys-OH) conjugated to keyhole limpet hemocyanin (KLH). ACTIVATE (A Complementary Trial of an Immunotherapy Vaccine against Tumor Specific EGRFvIII) trial was the first Phase II study to evaluate the efficacy of rindopepimut in newly diagnosed EGFRvIII-positive GBM patients, who had undergone gross total resection and standard chemoradiation therapy [135]. 19 patients were enrolled and compared favorably with a control group that received the standard therapy with temozolomide and radiation. Not surprisingly, patients who developed immune sensitization to EGFRvIII had a longer median overall survival compared to patients that lacked this response (47.7 months vs. 22.8 months) [135]. Notably, in cases of recurrence, most tumors were found to have lost EGFRvIII expressing cells. A follow-up Phase II trial (ACT II) investigated rindopepimut/GM-CSF concurrently with standard or dose-intensive adjuvant TMZ and enrolled 22 patients with newly diagnosed EGFRvIII-positive GBM [136]. All patients were found to have an immune response to EGFRvIII, with the greatest serum response seen in the dose-intensive cohort. Most importantly, overall survival was significantly improved (23.6 months) compated to historical case-matched controls [136]. The ACT III trial was the third and largest Phase II trial that evaluated the efficacy of the peptide vaccine CDX-110 in 65 patients with newly diagnosed EGFRvIII-positive GBM following gross total resection and the successful completion of combination radiation and TMZ therapy [137]. The median overall survival was 24.6 months compared to 15.2 months for matched EGFRvIII-positive controls [137,138]. A further sub-analyzes revealed that MGMT methylated patients receiving rindopepimut were found to have a significantly longer PFS (17.5 months vs. 11.2 months) and OS (32.3 months vs. 20.9 months) [137]. Although these trials rigorously confirm that rindopepimut vaccine can be safe and suggested a possible benefit in the treatment of EGFRvIII positive GBMs, these results are unlikely to be confirmed in the ongoing Phase III trial for newly diagnosed EGFRvIII positive GBM (ACT IV, NCT01480479) [139]. However, the limited clinical responses observed in Phase III and recurrence of EGFRvIII-negative tumors highlight a potential limitation of single antigen vaccines in GBMs that display considerable intratumoral cellular heterogeneity and require additive therapies to maximize the therapeutic response and control tumor growth at recurrence.
Apart from newly diagnosed GBM, rindopepimut is currently also being evaluated in a Phase II trial in patients with EGFR-positive recurrent GBM in combination with the anti-VEGF mAb bevacizumab in an attempt to optimize the EGFRvIII specific immune response through reversing VEGF mediated immunsuppression (ReACT, NCT01498328) [140]. Preliminary results presented at the 2015 ASCO Annual Meeting demonstrated that 27% of patients reached a 6-month progression free survival (vs. 11% in contemporary controls) with a median overall survival of 12 months (vs. 8.8 months in contemporary controls) [141]. The primary data collection of the ReAct trial was completed in April 2015 and results are anxiously awaited. Also, a Phase I trial is underway utilizing the EGFRvIII vaccine in the treatment of children with DIPG, which show EGFR expression in ~50% [142].
Another promising tumor-specific neoantigen with high uniformity and penetrance is IDH1R132H. The immunogenic epitope encircling this mutation is found on major histocompatibility complexes (MHC) class II and induces mutation-specific CD4+ T-helper-1 (TH1) responses [143]. Notably, peptide vaccination of mice that are transgenic for human MHC class I and II with IDH1R132H resulted in an effective MHC class II-restricted mutation-specific antitumor immune response [143]. Based on these results two Phase I trials were initiated for patients with IDH1R132H–positive grade III and IV gliomas (NCT02454634) and for IDH1R132H–positive grade II gliomas (RESIST, NCT02193347). Other notable contemporary clinical trials target the chaperone heat shock protein (HSP) 96-kD, which is highly expressed in GBM and when carrying a tumor antigenic peptide can be delivered to dendritic cells resulting in presentation of tumor peptides [144]. By purifying HSP-96 protein complexes from each patient's tumor, a personalized polyvalent vaccine, known as HSP protein complex-96 vaccine (HSPPC-96; Prophage), was developed that has been clinically tested in Phase I and II studies in recurrent and newly diagnosed HGG suggesting good tolerability and safety [145]. Although the vaccine resulted in a measurable systemic immune response to the patient's specific tumor antigens, the therapeutic benefit is unclear and requires further clinical studies [145]. In addition, HSPPC-96 vaccine + bevacizumab is currently undergoing Phase II testing in patients with recurrent GBM (NCT01814813).
ICT-107 is a patient derived DC vaccine, which targets six GBM associated antigens, AIM-2, TRP-2, HER2/neu, IL-13Ra2, gp100, MAGE1. The vaccine has been evaluated in newly diagnosed HLA-A1+ and/or HLA-A2+ GBM patients following resection and concomitant chemo-radiotherapy. Preliminary results from the Phase I study were highly exciting, demonstrating a median a median overall survival of 38.4 months and 5-year overall survival rate was 50% [146]. A follow-up randomized, double blind, placebo-controlled Phase II trial suggested potential therapeutic benefits, particularly in HLA-A2+/MGMT-methylated subgroup with a median progression free survival of 24 months (vs. 8.5 months in the historic controls), although the efficacy was less than what was observed in Phase I [147]. In contrast, the prognostically poorer MGMT-unmethylated subgroup did not show any therapeutic response when compared to historic controls [147]. A multi-center randomized, double blind Phase III trial has been launched to study about 400 HLA-A2+ patients with newly diagnosed GBMs (NCT02546102).
In the future, vaccine therapies may be combined with other means of immune augmentation to optimize their efficacy and escape the tumor-induced immunosupression [148–150]. Potential strategies include the use of cytokines, that have been studied in combination with anti-tumor vaccines, and site preconditioning with the tetanus/dipheria (Td) toxoid, which significantly improved the tumor-antigen specific therapeutic response [151].
4.2 Immune checkpoint inhibitors
Historically, the CNS was classified as an immunologically privileged site. Recent work demonstrated the presence of a lymphatic system within the CNS and new insights in the key mechanisms of tumor escape from the immune system [152,153]. Those include the tumor’s ability to produce specific inhibitors of antigen-presenting cell (APC) maturation and T-cell mediated cytotoxicity, as well as other immunosuppressive factors such PD-L1, prostaglandin E2, TGFβ, indoleamine 2,3-dioxygenase (IDO), IL-10 and STAT3 and the recruitment of immunosuppressive host cells such as regulatory T cells, or myeloid-derived suppressor cells to the tumor microenvironment [153].
Immune checkpoint inhibitors, aimed at overcoming these obstacles, are humanized antibodies, which block various cell surface receptors expressed on the tumor or the host’s T-cells that negatively regulate the anti-tumor immune response, e.g. cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4), the programmed death-1 (PD-1) receptor and the PD-L1 ligand (Fig. 2) [153]. Clinical trials in other cancers have already convincingly demonstrated that inhibition of CTLA4, PD1, and PDL1 can activate anti-tumor immune responses, with significant and durable therapeutic benefits. In human GBMs, PD-1 ligands are also expressed on the tumor surface, however, whether this can predict a clinical response to PD-1 blockade is questionable based on the data from other cancers [154,155]. Despite lacking evidence for clinical, preclinical studies in murine syngeneic glioma models convincingly demonstrated that blocking specific co-inhibitory receptors, such as PD1 and CTLA4, alone or in combination with chemotherapy or radiation, inhibited T- cell activation and resulted in dramatic tumor regressions and long-term survival [156–158].
Figure 2. Immune checkpoint interactions on T cells and cancer.

Antigens released from tumor cells are taken up by antigen-presenting cells. The T-cell receptor (TCR) interacts with a presented antigen to activate the immune system. PD-1, which is expressed on T-cells, has an inhibitory role (red arrow) through its interaction with PD-L1 on the tumor cells resulting in tolerance and inhibition of tumor destruction. Similarly, CTLA-4, which is also expressed on T-cells, mediates inhibitory signals after binding with its ligands CD80 or CD86 dampening the immune response and preventing activation. Blocking both interactions, with monoclonal antibodies to PD-1, PD-L1 or CTLA-4 (for example, nivolumab, pembrolizumab, ipilimumab) precludes inhibition allowing for T-cell activation (green arrow) with subsequent tumor cell lysis.
Five immune checkpoint inhibitors are currently undergoing clinical evaluations in HGGs, both as monotherapy or in combination with other agents (eg. bevacizumab, TMZ, radiation and vaccines): nivolumab, pidilizumab, and pembrolizumab, which inhibit the interaction of the PD1 receptor with its PDL1 ligand; MEDI4736 and MPDL3280A, which neutralize the PDL1-ligand; and ipilimumab, that targets CTLA4. While preliminary data for nivolumab and nivolumab plus ipilimumab in the treatment of recurrent GBMs are cautiously encouraging with an overall survival of 60% at 9 months, two randomized open-label Phase III studies investigating the efficacy and safety of nivolumab compared to bevacizumab and nivolumab with or without ipilimumab are underway for newly diagnosed and recurrent GBM (CheckMate 143; NCT02017717), as well as nivolumab + radiation in newly diagnosed GBM (CheckMate 498; NCT02617589). In addition, the safety of ipilimumab, nivolumab, and the combination of ipilimumab and nivolumab will be evaluated in newly diagnosed GBM patients undergoing temozolomide therapy (Phase I, NCT02311920). Furthermore, pidilizumab is being tested in Phase I/II to assess safety and efficacy in children with DIPG (NCT01952769). For pembrolizumab, four clinical trials have recently opened or are ongoing: a Phase I study investigating pembrolizumab in combination with bevacizumab and hypofractionated stereotactic irradiation in recurrent HGGs (NCT02313272); a Phase I/II study investigating the safety and efficacy of pembrolizumab in combination with MRI-guided laser ablation in recurrent HGG (NCT02311582); a Phase II study of pembrolizumab with and without bevacizumab in recurrent GBM (NCT02337491); and a Phase I/II study evaluating the safety and efficacy of pembrolizumab in combination with standard chemoradiation in newly diagnosed GBM (NCT02530502).
Despite the large number of ongoing studies, the evaluation of the immune-checkpoint inhibitors must be systematic and proceed with caution as evidenced by the recent suspension of the Phase I/II trial evaluating pembrolizumab in children with recurrent HGGs and DIPG due to severe adverse reactions (NCT02359565). This highlights one of the major challenges associated with immunotherapies: uncontrolled immune activation. Hence, there is a crucial need to identify tools to accurately assess and control the intensity of immune response to avoid potentially life-threatening conditions, such as toxic autoimmunity directed at brain antigens (allergic encephalomyelitis), elevated intracranial pressure and cerebral edema. Similar immune-mediated toxicities have also been observed with other immunotherapies, such as T-cell based immunotherapies, particularly chimeric antigen receptor T cell (CAR T-cell) therapy targeting EGFRvIII (Tab.1) [159]. Apart from toxicities, we will face unique challenges in the assessment of the radiographic response because the inflammatory responses might mimic radiological features of tumor progression with increased enhancement and edema. This has led to the development of the immunotherapy Response Assessment for Neuro-Oncology (iRANO) criteria, in which patients with imaging findings suggestive of progressive disease within 6 months of starting immunotherapy including the development of new lesions, should undergo follow-up imaging in a 3 months’ time before defining the patient as non-responsive to treatment or progressive disease [160]. In addition, insights yielded from other clinical trials evaluating the immune checkpoint inhibitors in brain metastasis will hopefully provide further guidance towards addressing these unresolved questions. Finally, biomarkers that identify patients who will likely benefit from specific agents are needed. Recent data suggests that tumors with mismatch repair deficiency, which is only present in a small fraction of brain cancers leading to a higher mutational load, may be more susceptible to immune checkpoint blockade and thus, providing a potential screening tool [161]. Its role in CNS neoplasms, however, has yet to be defined although early data are encouraging [162].
4.4 Oncolytic viruses
Oncolytic viruses represent a promising class of therapeutic agents that promote antineoplastic responses through a combination of two mechanisms: cytotoxicity via selective replication within neoplastic cells, resulting in direct tumor cell killing and induction of a systemic antitumour immunity. The concept of oncolytic virotherapy has a long history of nearly 100 years and is based on the observation that viruses have a preferential, although nonexclusive, tropism for tumors [163]. Virotherapy became a popular tool in cancer therapy in the 1990s, when advances in recombinant technology met an improved molecular understanding of virology and cancer genetics [163,164]. Many viruses have been investigated as agents for oncolytic immunotherapy in gliomas, and considerable work has been done to optimize viral vectors by attenuating pathogenicity and enhancing immunogenicity. As of 2015, approximately 15 viruses of six different species have advanced to clinical trials: adenoviruses, herpes simples virus-1 (HSV-1), coxsackie virus, poliovirus, measles virus, poxvirus, Newcastle disease virus (NDV) and reovirus (Tab. 1) [163]. The majority of these oncolytic viruses have a natural tropism for cell surface proteins that are expressed by glioma cells. For example, poliovirus uses CD155, a widely expressed receptor on GBM cells, for cell entry. Coxsackievirus can enter cells via intercellular adhesion molecule 1 and decay accelerating factor, and HSV-1 utilizes the herpesvirus entry mediator (HVEM) and selected nectins for cell entry. Adenovirus DNX-2401 was developed by genetically modifying adenovirus type 5 to bind integrins that are highly expressed on cancer cells. In addition, some of these viruses have also been engineered to selectively replicate exclusively in GBM-specific conditions, such as hypoxia or oncogenic mutation (e.g. p53 mutation, Retinoblastoma (Rb) mutation) to increase the tumor specificity and spare normal cells. For instance, adenovirus ONYX-015 (dl1520) is deleted in the E1B gene, which encodes a 55 kDa protein that inactivates p53 protein and thus, exclusively replicates in and lyses p53-deficient tumor cells [165,166]. Likewise, tumor-selective adenovirus Delta24 carries a deletion in the E1A region responsible for binding Rb protein and can only replicate in Rb deficient glioma cells [167].
There are more than 20 clinical trials of viral oncolytics completed or active in CNS tumors. The majority of them have used direct intratumoral injections to bypass the architectural barriers of the tumor and surrounding tissue as well as increase infectivity. Two oncolytic HSV-1 strains (G207 and 1716) have completed Phase I and II testing in adult recurrent HGGs, alone and in combination with radiation. For G207, no serious adverse events were observed and at best, 38% of patients had a clinical response, and one patient had long-term survival (>5.5 years) [168]. However, a follow-up Phase Ib study, was not able to reproduce these results [169]. A third study in combination with a single dose of 5 Gy radiation (NCT00157703) resulted in a probable clinical response and further studies are necessary to define its role in the therapy of HGGs [170]. G207 is also currently undergoing Phase I testing in children with progressive and recurrent HGGs (NCT02457845). Three Phase I clinical trials with HSV-1716 have been conducted in recurrent HGGs, demonstrating safety and paved the way for a Phase I trial in children with recurrent HGGs (NCT02031965). Furthermore G47 delta, derived from G207 with an additional deletion of nonessential α47 gene to enhance replication efficacy, and M032, an HSV-1 strain equipped with a human IL-12 transgene, have entered early clinical testing for recurrent or progressive HGGs (NCT02062827).
DNX-2401 (DNATrix) is a conditionally replicative oncolytic adenovirus designed to target tumor cells that are defective in the Rb pathway. It is currently being evaluated in a Phase I trial for the treatment of recurrent HGGs (NCT00805376). Preliminary results presented at the Society of Neuro-Oncology (SNO) meeting in 2014 were promising showing complete and durable responses in up to 12% of the patients [171]. Interestingly, responders also demonstrated interleukin responses, particularly IL-12 elevations, re-emphasizing the concept that intratumoral administration of oncolytic viruses does not only act to directly kill tumor cells though viral replication but also induces anticancer immunity.
Another promising oncolytic is the TOCA 511 virus that belongs to the family of non-lytic retroviruses, and carries the cytosine deaminase (CD) gene to enhance the direct cancer cell killing via local conversion of systemically administered prodrug 5-fluorocytosine (5-FC) to the active 5-fluorouracil (5-FU). It is currently undergoing Phase I clinical trial in adults with newly diagnosed HGGs co-administered with standard chemoradiation (NCT02598011), and recurrent HGGs (NCT01156584), which is soon to be completed. A Phase II/III trial just initiated patient enrollment for recurrent HGGs in November 2015 (NCT02414165).
Poliovirus, termed PVS-RIPO in its therapeutic engineered form, demonstrated potent lytic effects and dramatic tumor regressions in preclinical HTB-15 glioma xenografts [172]. PVS-RIPO has advanced to a Phase I clinical trial for patients with recurrent HGGs (NCT01491893). Although the results are not published yet, early data suggest moderate therapeutic efficacy in selected patient subsets (40% survival reported at 20 months) [173] and Phase II/III trials as well as an expansion to pediatric HGGs are currently planned. Other oncolytic viruses in early stages of clinical development include parvovirus H1 (NCT01301430), and measles virus (NCT00390299).
Although the replicating nature of oncolytic viruses poses unique and potentially life-threatening toxicities, clinical trials have shown that the majority of oncolytic therapies results in few adverse events, even in patients with brain tumors [164]. However, clinical responses in HGG clinical trials have fallen short of the promise initially generated from preclinical animal testing, in part because dosing and application regimens were chosen conservatively and the inability of current animal model systems to sufficiently replicate human viral pathogenesis. Human HGG xenograft models lack immune competency and immune-competent mouse models are frequently misleading because the viruses behave differently in rodents and humans. The BBB likely limits oncolytic virus entry into the CNS, and except for Parvovirus that naturally crosses the BBB, there have been few studies to determine the viral penetrance and distribution in the CNS. CNS bioavailability can be overcome by direct infusion of virus into the brain tumor but the viral distribution can still be limited due to patient-specific differences in tumor anatomy (e.g. cysts, necrosis). Furthermore, viral efficacy may be hampered by genetic modifications intended to limit toxicity that inadvertently affect the viral replication and spread [164]. In summary, it remains to be determined if oncolytic virotherapy can be designed in a way to manage toxicities at clinically effective doses. Perhaps more importantly, anti-tumor immune responses generated during oncolytic virus infections could be used to stimulate the systemic anti-tumor immunity and provide a strong rationale for the clinical exploration of combinations of immunoregulatory antibodies with oncolytic viruses [174,175]. Clinical trials using oncolytic viruses, such as HSV-1, in combination with immune checkpoint inhibitors (ipilimumab) are currently under way for advanced solid cancers and will provide a clinical proof of concept in humans (NCT02272855).
5. Conclusion
Unlike other solid tumors, the complexity and diversity of malignant gliomas poses unique challenges to the therapeutic approaches including intratumoral heterogeneity, divergent signaling mechanisms, insufficient tumor targeting strategies and limited drug delivery through the BBB. These obstacles have made this cancer one of the most difficult ones to treat despite an unprecedentedly high number of new investigational agents that have evolved over the last years. Though the therapeutic success has been limited so far, multiple promising strategies are still untested such as the concurrent use of multiple modalities that target disparate tumor features, the use of local cytotoxic therapies to overcome limitations from the BBB and minimize the systemic immune-suppression, particularly when combined with immunotherapeutics, and the use of specific patient subgroups selected based on molecular biomarkers or mutations such as HDAC or RTK expression to evaluate realistically potential benefits of candidate agents. We need to be aware that novel cancer therapeutics may introduce new hurdles, as observed with immunotherapeutics and their immune-related response patterns, which pose an increasing clinical challenge for practitioners and patients. Finally, research has yet to discover a single definitive target or event that deserves the major research focus of the neuro-oncological community to successfully battle this devastating disease.
6. Expert Opinion
This is a time of progress and guarded optimism in Neuro-Oncology. Remarkable progress in survival has been achieved for subsets of patients with GBM and anaplastic oligodendroglioma by combining chemotherapy with radiation therapy highlighting the fact that significant advances can be made by using traditional therapeutics in creative ways. The guarded optimism while fully warranted is counterbalanced by the complexities and heterogeneity of brain cancer biology and the unique obstacles associated with diagnosing, treating, and monitoring tumors within the central nervous system. We are likely reaching the limits of what can be achieved by combining existing cytotoxic regimens and it remains frustrating that chemotherapies proven to be effective against malignant glioma remain woefully limited to the class of alkylating agents. There remains no meaningful therapy for many of our patients including but not limited to those with recurrent GBM and aggressive pediatric HGGs. Brain cancer patients have not yet benefited from the promise of personalized targeted therapy despite the fact that GBM, the most common brain malignancy, is one the most extensively characterized cancers at the genetic and epigenetic levels.
Optimism regarding the prospect for totally new therapeutic approaches is warranted and driven by research advances in multiple directions. Progress in brain cancer genomics and epigenomics has identified multiple oncogenic drivers amenable to therapeutic inhibition many of which are just entering clinical trial. However, single cell analyses of GBM are revealing an unprecedented degree of intratumoral cellular heterogeneity, findings that most certainly reflect GBM’s resilience and highlight the need to more precisely define the evolutionary biology of these malignancies at the cellular and molecular levels, especially their evolution during treatment and the emergence of therapeutic resistance. The expectation is that this information will provide a roadmap to guide emerging sequential and combinatorial treatment regimens. The original focus of targeted therapy on oncogenic kinase inhibitors (e.g. EGFR, PDGFR inhibitors) has now expanded to include strategies for targeting oncometabolite production, chromatin dysregulation, and their effects on gene expression regulation. The brain is no longer misconceived as an immune-privileged site opening the prospect of harnessing the immune system to overcome brain cancer’s heterogeneity and ability to evade cytotoxic and targeted therapies. This, in conjunction with advances in our understanding of the immune-suppressive strategies employed by cancer, has stimulated the development of innumerable exciting immune-based clinical trials using vaccines and immune checkpoint inhibition. Vigilance is needed to not let current immune-suppressive standards of care (i.e. chemo-radiation) block the efficacy of immune-based therapy.
Significant challenges to the rapid and successful development of these promising approaches remain. The path from the preclinical laboratory to FDA approval is time consuming and increasingly expensive taking decades and hundreds of millions of dollars for new agents. New and more efficient clinical trial designs that minimize patient numbers and maximize the rapid detection of therapeutic signal are critical. Employing the personalized medicine approaches of tumor subtyping and patient selection for therapeutic target expression is vital to achieving these goals. Limited CNS bioavailability of systemically administered drugs remains a serious obstacle to brain tumor therapy and should no longer be relegated to the background. Toward overcoming this obstacle direct intratumoral therapies delivered by increasingly advanced forms of convection enhanced delivery, implanted biomaterials, or replication competent biological agents are becoming more sophisticated and practical. Effectively addressing current challenges will require increased financial and programmatic commitments from national funding agencies, the pharmaceutical industry and philanthropy. With this support, the growing critical mass of brain cancer scientists and clinicians, attributable in great part to the founding of The Society for Neuro-Oncology two decades ago, in conjunction with the critically important clinical trials consortia are prepared to overcome these challenges and accelerate the discovery and implementation of more effective therapies for our highly motivated brain cancer patients for whom time is of the essence.
Highlight Box.
Pediatric and adult high-grade gliomas (HGG) carry a uniformly fatal diagnosis due to high recurrence rates and the absence of effective treatments.
Progress in brain cancer genomics and epigenomics have identified multiple oncogenic drivers targetable by therapeutic inhibition, many of which are currently undergoing evaluations in clinical trial but have yet to show improvements in survival.
Promising new developments focusing on immunotherapies such as immune-checkpoint inhibitors or oncolytic viruses are underway to overcome the genetic drift and could lead to durable anti-tumor immune responses against a theoretically unlimited number of tumor-associated antigens.
Future successful therapies will concurrently use multiple modalities that target disparate tumor characteristics personalized to the particular tumor of a given patient.
Acknowledgments
V Staedtke is supported by the Francis S Collins Scholars Program and RY Bai is supported by National Cancer Institute grant 1R03CA178118-01A1.
Footnotes
Declaration of Interest
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Bibliography
Papers of special note have been highlighted as either of interest • or •• of considerable interest to readers.
- 1.Ostrom QT, Gittleman H, Fulop J, et al. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2008–2012. Neuro-oncology. 2015;17(Suppl 4):iv1–iv62. doi: 10.1093/neuonc/nov189. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta neuropathologica. 2007;114(2):97–109. doi: 10.1007/s00401-007-0243-4. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ohgaki H, Kleihues P. Genetic pathways to primary and secondary glioblastoma. The American journal of pathology. 2007;170(5):1445–53. doi: 10.2353/ajpath.2007.070011. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jenkins RB, Blair H, Ballman KV, et al. A t(1;19)(q10;p10) mediates the combined deletions of 1p and 19q and predicts a better prognosis of patients with oligodendroglioma. Cancer research. 2006;66(20):9852–61. doi: 10.1158/0008-5472.CAN-06-1796. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 5.Ostrom QT, de Blank PM, Kruchko C, et al. Alex's Lemonade Stand Foundation Infant and Childhood Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007–2011. Neuro-oncology. 2015;16(Suppl 10):x1–x36. doi: 10.1093/neuonc/nou327. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fangusaro J. Pediatric high grade glioma: a review and update on tumor clinical characteristics and biology. Frontiers in oncology. 2012;2:105. doi: 10.3389/fonc.2012.00105. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Warren KE. Diffuse intrinsic pontine glioma: poised for progress. Frontiers in oncology. 2012;2:205. doi: 10.3389/fonc.2012.00205. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jones C, Baker SJ. Unique genetic and epigenetic mechanisms driving paediatric diffuse high-grade glioma. Nature reviews. Cancer. 2014;14(10) doi: 10.1038/nrc3811. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cage TA, Samagh SP, Mueller S, et al. Feasibility, safety, and indications for surgical biopsy of intrinsic brainstem tumors in children. Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery. 2013;29(8):1313–9. doi: 10.1007/s00381-013-2101-0. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 10.Farrell CJ, Plotkin SR. Genetic causes of brain tumors: neurofibromatosis, tuberous sclerosis, von Hippel-Lindau, and other syndromes. Neurologic clinics. 2007;25(4):925–46. viii. doi: 10.1016/j.ncl.2007.07.008. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 11•.Cairncross JG, Wang M, Jenkins RB, et al. Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of IDH. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2014;32(8):783–90. doi: 10.1200/JCO.2013.49.3726. [published Online First: Epub Date]|. first to show that non-codeleted IDH-mutated tumors also benefit from chemoradiation, but to a lesser degree. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van den Bent MJ, Brandes AA, Taphoorn MJ, et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31(3):344–50. doi: 10.1200/JCO.2012.43.2229. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 13.Chaichana KL, Jusue-Torres I, Navarro-Ramirez R, et al. Establishing percent resection and residual volume thresholds affecting survival and recurrence for patients with newly diagnosed intracranial glioblastoma. Neuro-oncology. 2014;16(1):113–22. doi: 10.1093/neuonc/not137. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sanai N, Polley MY, McDermott MW, et al. An extent of resection threshold for newly diagnosed glioblastomas. Journal of neurosurgery. 2011;115(1):3–8. doi: 10.3171/2011.2.JNS10998. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 15••.Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England journal of medicine. 2005;352(10):987–96. doi: 10.1056/NEJMoa043330. [published Online First: Epub Date]|. first to show the added benefit of TMZ to radiation in GBM, which subsequently became standard of care. [DOI] [PubMed] [Google Scholar]
- 16.Esteller M, Hamilton SR, Burger PC, et al. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer research. 1999;59(4):793–7. [PubMed] [Google Scholar]
- 17••.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. The New England journal of medicine. 2005;352(10):997–1003. doi: 10.1056/NEJMoa043331. [published Online First: Epub Date]|. demonstrated the correlation of MGMT silencing and response to TMZ in GBM patients. [DOI] [PubMed] [Google Scholar]
- 18••.Gilbert MR, Dignam JJ, Armstrong TS, et al. A randomized trial of bevacizumab for newly diagnosed glioblastoma. The New England journal of medicine. 2014;370(8):699–708. doi: 10.1056/NEJMoa1308573. [published Online First: Epub Date]|. first-line use of bevacizumab did not improve overall survival in patients with newly diagnosed glioblastoma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cohen MH, Shen YL, Keegan P, et al. FDA drug approval summary: bevacizumab (Avastin) as treatment of recurrent glioblastoma multiforme. The oncologist. 2009;14(11):1131–8. doi: 10.1634/theoncologist.2009-0121. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 20••.Chinot OL, Wick W, Mason W, et al. Bevacizumab plus radiotherapy-temozolomide for newly diagnosed glioblastoma. The New England journal of medicine. 2014;370(8):709–22. doi: 10.1056/NEJMoa1308345. [published Online First: Epub Date]|. Adding bevacizumab to radiotherapy–TMZ did not improve survival in GBM patients. [DOI] [PubMed] [Google Scholar]
- 21•.Cohen KJ, Pollack IF, Zhou T, et al. Temozolomide in the treatment of high-grade gliomas in children: a report from the Children's Oncology Group. Neuro-oncology. 2011;13(3):317–23. doi: 10.1093/neuonc/noq191. [published Online First: Epub Date]|. first to indicate that TMZ may be less effective in pediatric HGGs compared to adults. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cancer Genome Atlas Research N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–8. doi: 10.1038/nature07385. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23••.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321(5897):1807–12. doi: 10.1126/science.1164382. [published Online First: Epub Date]|. Discovered the IDH1 mutation in gliomas. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2000;103(2):211–25. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 25.Carrasco-Garcia E, Saceda M, Martinez-Lacaci I. Role of receptor tyrosine kinases and their ligands in glioblastoma. Cells. 2014;3(2):199–235. doi: 10.3390/cells3020199. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Y, Li A, Glas M, et al. c-Met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(24):9951–6. doi: 10.1073/pnas.1016912108. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heimberger AB, Hlatky R, Suki D, et al. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11(4):1462–6. doi: 10.1158/1078-0432.CCR-04-1737. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 28.Siegelin MD, Borczuk AC. Epidermal growth factor receptor mutations in lung adenocarcinoma. Laboratory investigation; a journal of technical methods and pathology. 2014;94(2):129–37. doi: 10.1038/labinvest.2013.147. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 29.De Witt Hamer PC. Small molecule kinase inhibitors in glioblastoma: a systematic review of clinical studies. Neuro-oncology. 2010;12(3):304–16. doi: 10.1093/neuonc/nop068. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rich JN, Reardon DA, Peery T, et al. Phase II trial of gefitinib in recurrent glioblastoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2004;22(1):133–42. doi: 10.1200/JCO.2004.08.110. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 31.van den Bent MJ, Brandes AA, Rampling R, et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27(8):1268–74. doi: 10.1200/JCO.2008.17.5984. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reardon DA, Desjardins A, Vredenburgh JJ, et al. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. Journal of neuro-oncology. 2010;96(2):219–30. doi: 10.1007/s11060-009-9950-0. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Prados MD, Chang SM, Butowski N, et al. Phase II study of erlotinib plus temozolomide during and after radiation therapy in patients with newly diagnosed glioblastoma multiforme or gliosarcoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27(4):579–84. doi: 10.1200/JCO.2008.18.9639. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uhm JH, Ballman KV, Wu W, et al. Phase II evaluation of gefitinib in patients with newly diagnosed Grade 4 astrocytoma: Mayo/North Central Cancer Treatment Group Study N0074. International journal of radiation oncology, biology, physics. 2011;80(2):347–53. doi: 10.1016/j.ijrobp.2010.01.070. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Prados MD, Yung WK, Wen PY, et al. Phase-1 trial of gefitinib and temozolomide in patients with malignant glioma: a North American brain tumor consortium study. Cancer chemotherapy and pharmacology. 2008;61(6):1059–67. doi: 10.1007/s00280-007-0556-y. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chakravarti A, Wang M, Robins HI, et al. RTOG 0211: a phase 1/2 study of radiation therapy with concurrent gefitinib for newly diagnosed glioblastoma patients. International journal of radiation oncology, biology, physics. 2013;85(5):1206–11. doi: 10.1016/j.ijrobp.2012.10.008. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thiessen B, Stewart C, Tsao M, et al. A phase I/II trial of GW572016 (lapatinib) in recurrent glioblastoma multiforme: clinical outcomes, pharmacokinetics and molecular correlation. Cancer chemotherapy and pharmacology. 2010;65(2):353–61. doi: 10.1007/s00280-009-1041-6. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 38.Fouladi M, Stewart CF, Blaney SM, et al. A molecular biology and phase II trial of lapatinib in children with refractory CNS malignancies: a pediatric brain tumor consortium study. Journal of neuro-oncology. 2013;114(2):173–9. doi: 10.1007/s11060-013-1166-7. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Combs SE, Heeger S, Haselmann R, et al. Treatment of primary glioblastoma multiforme with cetuximab, radiotherapy and temozolomide (GERT)--phase I/II trial: study protocol. BMC cancer. 2006;6:133. doi: 10.1186/1471-2407-6-133. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee JC, Vivanco I, Beroukhim R, et al. Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain. PLoS medicine. 2006;3(12):e485. doi: 10.1371/journal.pmed.0030485. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wen PY, Chang SM, Lamborn KR, et al. Phase I/II study of erlotinib and temsirolimus for patients with recurrent malignant gliomas: North American Brain Tumor Consortium trial 04-02. Neuro-oncology. 2014;16(4):567–78. doi: 10.1093/neuonc/not247. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reardon DA, Groves MD, Wen PY, et al. A phase I/II trial of pazopanib in combination with lapatinib in adult patients with relapsed malignant glioma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(4):900–8. doi: 10.1158/1078-0432.CCR-12-1707. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 43.Karavasilis V, Kotoula V, Pentheroudakis G, et al. A phase I study of temozolomide and lapatinib combination in patients with recurrent high-grade gliomas. Journal of neurology. 2013;260(6):1469–80. doi: 10.1007/s00415-012-6812-z. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 44.Greenall SA, Donoghue JF, Gottardo NG, et al. Glioma-specific Domain IV EGFR cysteine mutations promote ligand-induced covalent receptor dimerization and display enhanced sensitivity to dacomitinib in vivo. Oncogene. 2015;34(13):1658–66. doi: 10.1038/onc.2014.106. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 45.Reardon DA, Nabors LB, Mason WP, et al. Phase I/randomized phase II study of afatinib, an irreversible ErbB family blocker, with or without protracted temozolomide in adults with recurrent glioblastoma. Neuro-oncology. 2015;17(3):430–9. doi: 10.1093/neuonc/nou160. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gan HKPK, Fichtel L, Lassman AB, Merrell R, Van Den Bent MJ, Kumthekar P, Scott AM, Pedersen M, Gomez EJ, Fischer JS, Ames W, Xiong H, Lee HJ, Munasinghe W, Roberts-Rapp L, Ansell P, Holen KD, Lai R, Reardon DA. Phase I study of ABT-414 mono- or combination therapy with temozolomide (TMZ) in recurrent glioblastoma (GBM) Journal of Clinical Oncology, 2015 ASCO Annual Meeting. 2015;33(15_suppl) (May 20 Supplement), 2015: 2016. [Google Scholar]
- 47.Brennan CW, Verhaak RG, McKenna A, et al. The somatic genomic landscape of glioblastoma. Cell. 2013;155(2):462–77. doi: 10.1016/j.cell.2013.09.034. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reardon DA, Dresemann G, Taillibert S, et al. Multicentre phase II studies evaluating imatinib plus hydroxyurea in patients with progressive glioblastoma. British journal of cancer. 2009;101(12):1995–2004. doi: 10.1038/sj.bjc.6605411. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wen PY, Yung WK, Lamborn KR, et al. Phase I/II study of imatinib mesylate for recurrent malignant gliomas: North American Brain Tumor Consortium Study 99-08. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12(16):4899–907. doi: 10.1158/1078-0432.CCR-06-0773. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 50.Razis E, Selviaridis P, Labropoulos S, et al. Phase II study of neoadjuvant imatinib in glioblastoma: evaluation of clinical and molecular effects of the treatment. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15(19):6258–66. doi: 10.1158/1078-0432.CCR-08-1867. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 51.Iwamoto FM, Lamborn KR, Robins HI, et al. Phase II trial of pazopanib (GW786034), an oral multi-targeted angiogenesis inhibitor, for adults with recurrent glioblastoma (North American Brain Tumor Consortium Study 06-02) Neuro-oncology. 2010;12(8):855–61. doi: 10.1093/neuonc/noq025. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lassman AB, Pugh SL, Gilbert MR, et al. Phase 2 trial of dasatinib in target-selected patients with recurrent glioblastoma (RTOG 0627) Neuro-oncology. 2015;17(7):992–8. doi: 10.1093/neuonc/nov011. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Franceschi E, Stupp R, van den Bent MJ, et al. EORTC 26083 phase I/II trial of dasatinib in combination with CCNU in patients with recurrent glioblastoma. Neuro-oncology. 2012;14(12):1503–10. doi: 10.1093/neuonc/nos256. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu-Emerson C, Norden AD, Drappatz J, et al. Retrospective study of dasatinib for recurrent glioblastoma after bevacizumab failure. Journal of neuro-oncology. 2011;104(1):287–91. doi: 10.1007/s11060-010-0489-x. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 55.Nehoff H, Parayath NN, McConnell MJ, et al. A combination of tyrosine kinase inhibitors, crizotinib and dasatinib for the treatment of glioblastoma multiforme. Oncotarget. 2015;6(35):37948–64. doi: 10.18632/oncotarget.5698. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schiff D, Sarkaria J. Dasatinib in recurrent glioblastoma: failure as a teacher. Neuro-oncology. 2015;17(7):910–1. doi: 10.1093/neuonc/nov086. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Laack NNGE, Anderson SK, Leinweber C, Buckner JC, Giannini C, Geoffroy FJ, Johnson DR, Lesser GJ, Jaeckle KA, Sarkaria J. Randomized, placebo-controlled, phase II study of dasatinib with standard chemo-radiotherapy for newly diagnosed glioblastoma (GBM), NCCTG N0877 (Alliance) ACSO. 2015 [Google Scholar]
- 58.Chen Y, Agarwal S, Shaik NM, et al. P-glycoprotein and breast cancer resistance protein influence brain distribution of dasatinib. The Journal of pharmacology and experimental therapeutics. 2009;330(3):956–63. doi: 10.1124/jpet.109.154781. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 59.Agarwal S, Mittapalli RK, Zellmer DM, et al. Active efflux of Dasatinib from the brain limits efficacy against murine glioblastoma: broad implications for the clinical use of molecularly targeted agents. Molecular cancer therapeutics. 2012;11(10):2183–92. doi: 10.1158/1535-7163.MCT-12-0552. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and functions in development, organ regeneration and cancer. Nature reviews. Molecular cell biology. 2010;11(12):834–48. doi: 10.1038/nrm3012. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 61.Abounader R, Laterra J. Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neuro-oncology. 2005;7(4):436–51. doi: 10.1215/S1152851705000050. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Murray DW, Didier S, Chan A, et al. Guanine nucleotide exchange factor Dock7 mediates HGF-induced glioblastoma cell invasion via Rac activation. British journal of cancer. 2014;110(5):1307–15. doi: 10.1038/bjc.2014.39. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim KJ, Wang L, Su YC, et al. Systemic anti-hepatocyte growth factor monoclonal antibody therapy induces the regression of intracranial glioma xenografts. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12(4):1292–8. doi: 10.1158/1078-0432.CCR-05-1793. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 64.Wen PY. American Society of Clinical Oncology 2010: report of selected studies from the CNS tumors section. Expert review of anticancer therapy. 2010;10(9):1367–9. doi: 10.1586/era.10.117. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 65.Wen PY, Schiff D, Cloughesy TF, et al. A phase II study evaluating the efficacy and safety of AMG 102 (rilotumumab) in patients with recurrent glioblastoma. Neuro-oncology. 2011;13(4):437–46. doi: 10.1093/neuonc/noq198. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chamberlain MC. Bevacizumab for the treatment of recurrent glioblastoma. Clinical Medicine Insights. Oncology. 2011;5:117–29. doi: 10.4137/CMO.S7232. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wick WBA, Gorlia T, Bendszus M, Sahm F, Taal W, Taphoorn M, Domont J, Idbaih A, Campone M, Clement PM, Stupp R, Fabbro R, Dubois F, Bais C, Musmeci D, Platten M, Weller M, Golfinopoulos V, van den Bent M. Phase III Trial Exploring The Combination of Bevacizumab and Lomustine in Patients With First Recurrence of a Glioblastoma: THE EORTC 26101 TRIAL. Neuro-Oncology Volume 17. 2015;17(suppl 5) [Google Scholar]
- 68.Reardon DA, Vredenburgh JJ, Desjardins A, et al. Effect of CYP3A-inducing anti-epileptics on sorafenib exposure: results of a phase II study of sorafenib plus daily temozolomide in adults with recurrent glioblastoma. Journal of neuro-oncology. 2011;101(1):57–66. doi: 10.1007/s11060-010-0217-6. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zustovich F, Landi L, Lombardi G, et al. Sorafenib plus daily low-dose temozolomide for relapsed glioblastoma: a phase II study. Anticancer research. 2013;33(8):3487–94. [PubMed] [Google Scholar]
- 70.Lee EQ, Kuhn J, Lamborn KR, et al. Phase I/II study of sorafenib in combination with temsirolimus for recurrent glioblastoma or gliosarcoma: North American Brain Tumor Consortium study 05-02. Neuro-oncology. 2012;14(12):1511–8. doi: 10.1093/neuonc/nos264. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Peereboom DM, Ahluwalia MS, Ye X, et al. NABTT 0502: a phase II and pharmacokinetic study of erlotinib and sorafenib for patients with progressive or recurrent glioblastoma multiforme. Neuro-oncology. 2013;15(4):490–6. doi: 10.1093/neuonc/nos322. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Galanis E, Anderson SK, Lafky JM, et al. Phase II study of bevacizumab in combination with sorafenib in recurrent glioblastoma (N0776): a north central cancer treatment group trial. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(17):4816–23. doi: 10.1158/1078-0432.CCR-13-0708. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pan E, Yu D, Yue B, et al. A prospective phase II single-institution trial of sunitinib for recurrent malignant glioma. Journal of neuro-oncology. 2012;110(1):111–8. doi: 10.1007/s11060-012-0943-z. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Balana C, Gil MJ, Perez P, et al. Sunitinib administered prior to radiotherapy in patients with non-resectable glioblastoma: results of a phase II study. Targeted oncology. 2014;9(4):321–9. doi: 10.1007/s11523-014-0305-1. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 75.Hottinger AF, Aissa AB, Espeli V, et al. Phase I study of sorafenib combined with radiation therapy and temozolomide as first-line treatment of high-grade glioma. British journal of cancer. 2014;110(11):2655–61. doi: 10.1038/bjc.2014.209. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Batchelor TT, Duda DG, di Tomaso E, et al. Phase II study of cediranib, an oral pan-vascular endothelial growth factor receptor tyrosine kinase inhibitor, in patients with recurrent glioblastoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2010;28(17):2817–23. doi: 10.1200/JCO.2009.26.3988. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Batchelor TT, Mulholland P, Neyns B, et al. Phase III randomized trial comparing the efficacy of cediranib as monotherapy, and in combination with lomustine, versus lomustine alone in patients with recurrent glioblastoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2013;31(26):3212–8. doi: 10.1200/JCO.2012.47.2464. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Reardon DAPE, Fan J, Mink J, Barboriak JP, Vredenburgh JJ, Desjardins A, Peters K, O'Brien J, Wen PY. A phase 2 trial of the multitargeted kinase inhibitor lenvatinib (E7080) in patients (pts) with recurrent glioblastoma (GBM) and disease progression. ESMO Congress. 2012 [Google Scholar]
- 79.Neyns B, Duerinck J, Du Four S, et al. Randomized phase II study of axitinib versus standard of care in patients with recurrent glioblastoma. Journal of Clinical Oncology, ASCO Annual Meeting Abstracts. 2014;32(15_suppl) (May 20 Supplement) [Google Scholar]
- 80.Furnari FB, Cloughesy TF, Cavenee WK, et al. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nature reviews. Cancer. 2015;15(5):302–10. doi: 10.1038/nrc3918. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Szerlip NJ, Pedraza A, Chakravarty D, et al. Intratumoral heterogeneity of receptor tyrosine kinases EGFR and PDGFRA amplification in glioblastoma defines subpopulations with distinct growth factor response. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(8):3041–6. doi: 10.1073/pnas.1114033109. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cloughesy TF, Yoshimoto K, Nghiemphu P, et al. Antitumor activity of rapamycin in a Phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS medicine. 2008;5(1):e8. doi: 10.1371/journal.pmed.0050008. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chinnaiyan P, Won M, Wen PY, et al. RTOG 0913: a phase 1 study of daily everolimus (RAD001) in combination with radiation therapy and temozolomide in patients with newly diagnosed glioblastoma. International journal of radiation oncology, biology, physics. 2013;86(5):880–4. doi: 10.1016/j.ijrobp.2013.04.036. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sarkaria JN, Galanis E, Wu W, et al. North Central Cancer Treatment Group Phase I trial N057K of everolimus (RAD001) and temozolomide in combination with radiation therapy in patients with newly diagnosed glioblastoma multiforme. International journal of radiation oncology, biology, physics. 2011;81(2):468–75. doi: 10.1016/j.ijrobp.2010.05.064. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hainsworth JD, Shih KC, Shepard GC, et al. Phase II study of concurrent radiation therapy, temozolomide, and bevacizumab followed by bevacizumab/everolimus as first-line treatment for patients with glioblastoma. Clinical advances in hematology & oncology : H&O. 2012;10(4):240–6. [PubMed] [Google Scholar]
- 86.Galanis E, Buckner JC, Maurer MJ, et al. Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a North Central Cancer Treatment Group Study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2005;23(23):5294–304. doi: 10.1200/JCO.2005.23.622. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 87.Sarkaria JN, Galanis E, Wu W, et al. Combination of temsirolimus (CCI-779) with chemoradiation in newly diagnosed glioblastoma multiforme (GBM) (NCCTG trial N027D) is associated with increased infectious risks. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16(22):5573–80. doi: 10.1158/1078-0432.CCR-10-1453. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ma DJ, Galanis E, Anderson SK, et al. A phase II trial of everolimus, temozolomide, and radiotherapy in patients with newly diagnosed glioblastoma: NCCTG N057K. Neuro-oncology. 2015;17(9):1261–9. doi: 10.1093/neuonc/nou328. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lassen U, Sorensen M, Gaziel TB, et al. Phase II study of bevacizumab and temsirolimus combination therapy for recurrent glioblastoma multiforme. Anticancer research. 2013;33(4):1657–60. [PubMed] [Google Scholar]
- 90.Koul D, Fu J, Shen R, et al. Antitumor activity of NVP-BKM120--a selective pan class I PI3 kinase inhibitor showed differential forms of cell death based on p53 status of glioma cells. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18(1):184–95. doi: 10.1158/1078-0432.CCR-11-1558. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Koul D, Shen R, Kim YW, et al. Cellular and in vivo activity of a novel PI3K inhibitor, PX-866, against human glioblastoma. Neuro-oncology. 2010;12(6):559–69. doi: 10.1093/neuonc/nop058. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nicolaides TP, Li H, Solomon DA, et al. Targeted therapy for BRAFV600E malignant astrocytoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17(24):7595–604. doi: 10.1158/1078-0432.CCR-11-1456. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Penman CL, Faulkner C, Lowis SP, et al. Current Understanding of BRAF Alterations in Diagnosis, Prognosis, and Therapeutic Targeting in Pediatric Low-Grade Gliomas. Frontiers in oncology. 2015;5:54. doi: 10.3389/fonc.2015.00054. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chapman PB, Hauschild A, Robert C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. The New England journal of medicine. 2011;364(26):2507–16. doi: 10.1056/NEJMoa1103782. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rochet NM, Dronca RS, Kottschade LA, et al. Melanoma brain metastases and vemurafenib: need for further investigation. Mayo Clinic proceedings. 2012;87(10):976–81. doi: 10.1016/j.mayocp.2012.07.006. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96•.Robinson GW, Orr BA, Gajjar A. Complete clinical regression of a BRAF V600E-mutant pediatric glioblastoma multiforme after BRAF inhibitor therapy. BMC cancer. 2014;14:258. doi: 10.1186/1471-2407-14-258. [published Online First: Epub Date]|. first to show complete regression of a BRAFV600E positive GBM in response to a BRAF inhibitor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97•.Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. The New England journal of medicine. 2012;367(18):1694–703. doi: 10.1056/NEJMoa1210093. [published Online First: Epub Date]|. first to show that the combined use of BRAF kinase inhibitors maybe beneficial and perhaps reduce the rate of skin toxicities. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nazarian R, Shi H, Wang Q, et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468(7326):973–7. doi: 10.1038/nature09626. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhao Y, Adjei AA. The clinical development of MEK inhibitors. Nature reviews. Clinical oncology. 2014;11(7):385–400. doi: 10.1038/nrclinonc.2014.83. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 100.Widemann BCML, Fisher MJ, Weiss BA, Kim AR, Dombi E, Baldwin A, Whitcomb P, Martin S, Gillespie A. Doyle A Phase I study of the MEK1/2 inhibitor selumetinib (AZD6244) hydrogen sulfate in children and young adults with neurofibromatosis type 1 (NF1) and inoperable plexiform neurofibromas (PNs) J Clin Oncol. 2014;32(suppl):5s. (abstr 10018) 2014. [Google Scholar]
- 101••.Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 mutations in gliomas. The New England journal of medicine. 2009;360(8):765–73. doi: 10.1056/NEJMoa0808710. [published Online First: Epub Date]|. first to show that IDH1 mutations are predominantly found in lower grade gliomas. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hartmann C, Meyer J, Balss J, et al. Type and frequency of IDH1 and IDH2 mutations are related to astrocytic and oligodendroglial differentiation and age: a study of 1,010 diffuse gliomas. Acta neuropathologica. 2009;118(4):469–74. doi: 10.1007/s00401-009-0561-9. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 103••.Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–44. doi: 10.1038/nature08617. [published Online First: Epub Date]|. first to reveal a part of the IDH1 mechanism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fathi AT, Abdel-Wahab O. Mutations in epigenetic modifiers in myeloid malignancies and the prospect of novel epigenetic-targeted therapy. Advances in hematology. 2012;2012:469592. doi: 10.1155/2012/469592. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Borodovsky A, Salmasi V, Turcan S, et al. 5-azacytidine reduces methylation, promotes differentiation and induces tumor regression in a patient-derived IDH1 mutant glioma xenograft. Oncotarget. 2013;4(10):1737–47. doi: 10.18632/oncotarget.1408. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Turcan S, Fabius AW, Borodovsky A, et al. Efficient induction of differentiation and growth inhibition in IDH1 mutant glioma cells by the DNMT Inhibitor Decitabine. Oncotarget. 2013;4(10):1729–36. doi: 10.18632/oncotarget.1412. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107••.Rohle D, Popovici-Muller J, Palaskas N, et al. An inhibitor of mutant IDH1 delays growth and promotes differentiation of glioma cells. Science. 2013;340(6132):626–30. doi: 10.1126/science.1236062. [published Online First: Epub Date]|. first to show that IDH1 directed targeting indeed results in growth delay and cell differentiation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Davis M, Pragani R, Popovici-Muller J, et al. Probe Reports from the NIH Molecular Libraries Program. Bethesda (MD): 2010. ML309: A potent inhibitor of R132H mutant IDH1 capable of reducing 2-hydroxyglutarate production in U87 MG glioblastoma cells. [PubMed] [Google Scholar]
- 109.AACR-NCI-EORTC Investor Lunch. Hynes Convention Center; 2015. The First Reported Results of AG-120, a First-in-class, Potent Inhibitor of the IDH1 Mutant Protein, in a Phase 1 Study of Patients with Advanced IDH1-Mutant Solid Tumors, Including Gliomas. [Google Scholar]
- 110.Ferriero R, Iannuzzi C, Manco G, et al. Differential inhibition of PDKs by phenylbutyrate and enhancement of pyruvate dehydrogenase complex activity by combination with dichloroacetate. Journal of inherited metabolic disease. 2015;38(5):895–904. doi: 10.1007/s10545-014-9808-2. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Michelakis ED, Sutendra G, Dromparis P, et al. Metabolic modulation of glioblastoma with dichloroacetate. Science translational medicine. 2010;2(31):31ra34. doi: 10.1126/scitranslmed.3000677. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 112.Tonjes M, Barbus S, Park YJ, et al. BCAT1 promotes cell proliferation through amino acid catabolism in gliomas carrying wild-type IDH1. Nature medicine. 2013;19(7):901–8. doi: 10.1038/nm.3217. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Sokolovic M, Wehkamp D, Sokolovic A, et al. Fasting induces a biphasic adaptive metabolic response in murine small intestine. BMC genomics. 2007;8:361. doi: 10.1186/1471-2164-8-361. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Champ CE, Palmer JD, Volek JS, et al. Targeting metabolism with a ketogenic diet during the treatment of glioblastoma multiforme. Journal of neuro-oncology. 2014;117(1):125–31. doi: 10.1007/s11060-014-1362-0. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 115.Schwartz K, Chang HT, Nikolai M, et al. Treatment of glioma patients with ketogenic diets: report of two cases treated with an IRB-approved energy-restricted ketogenic diet protocol and review of the literature. Cancer & metabolism. 2015;3:3. doi: 10.1186/s40170-015-0129-1. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rieger J, Bahr O, Maurer GD, et al. ERGO: a pilot study of ketogenic diet in recurrent glioblastoma. International journal of oncology. 2014;44(6):1843–52. doi: 10.3892/ijo.2014.2382. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117••.Mashimo T, Pichumani K, Vemireddy V, et al. Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell. 2014;159(7):1603–14. doi: 10.1016/j.cell.2014.11.025. [published Online First: Epub Date]|. discovered acetate as an important alternative energy source used by GBMs and other brain metastasis, with potential for metabolic targeting in the future. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Comerford SA, Huang Z, Du X, et al. Acetate dependence of tumors. Cell. 2014;159(7):1591–602. doi: 10.1016/j.cell.2014.11.020. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wade PA. Transcriptional control at regulatory checkpoints by histone deacetylases: molecular connections between cancer and chromatin. Human molecular genetics. 2001;10(7):693–8. doi: 10.1093/hmg/10.7.693. [DOI] [PubMed] [Google Scholar]
- 120.Lin HY, Chen CS, Lin SP, et al. Targeting histone deacetylase in cancer therapy. Medicinal research reviews. 2006;26(4):397–413. doi: 10.1002/med.20056. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 121.Eyupoglu IY, Hahnen E, Buslei R, et al. Suberoylanilide hydroxamic acid (SAHA) has potent anti-glioma properties in vitro, ex vivo and in vivo. Journal of neurochemistry. 2005;93(4):992–9. doi: 10.1111/j.1471-4159.2005.03098.x. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 122.Yin D, Ong JM, Hu J, et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor: effects on gene expression and growth of glioma cells in vitro and in vivo. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13(3):1045–52. doi: 10.1158/1078-0432.CCR-06-1261. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 123.Lucio-Eterovic AK, Cortez MA, Valera ET, et al. Differential expression of 12 histone deacetylase (HDAC) genes in astrocytomas and normal brain tissue: class II and IV are hypoexpressed in glioblastomas. BMC cancer. 2008;8:243. doi: 10.1186/1471-2407-8-243. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Drappatz J, Lee EQ, Hammond S, et al. Phase I study of panobinostat in combination with bevacizumab for recurrent high-grade glioma. Journal of neuro-oncology. 2012;107(1):133–8. doi: 10.1007/s11060-011-0717-z. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 125.Friday BB, Anderson SK, Buckner J, et al. Phase II trial of vorinostat in combination with bortezomib in recurrent glioblastoma: a north central cancer treatment group study. Neuro-oncology. 2012;14(2):215–21. doi: 10.1093/neuonc/nor198. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Weller M, Gorlia T, Cairncross JG, et al. Prolonged survival with valproic acid use in the EORTC/NCIC temozolomide trial for glioblastoma. Neurology. 2011;77(12):1156–64. doi: 10.1212/WNL.0b013e31822f02e1. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wu G, Broniscer A, McEachron TA, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nature genetics. 2012;44(3):251–3. doi: 10.1038/ng.1102. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128••.Schwartzentruber J, Korshunov A, Liu XY, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature. 2012;482(7384):226–31. doi: 10.1038/nature10833. [published Online First: Epub Date]|. established the role of H3 histone alterations in pediatric HGGs. [DOI] [PubMed] [Google Scholar]
- 129.Wu G, Diaz AK, Paugh BS, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nature genetics. 2014;46(5):444–50. doi: 10.1038/ng.2938. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lewis PW, Allis CD. Poisoning the "histone code" in pediatric gliomagenesis. Cell cycle. 2013;12(20):3241–2. doi: 10.4161/cc.26356. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131••.Lewis PW, Muller MM, Koletsky MS, et al. Inhibition of PRC2 activity by a gain-of-function H3 mutation found in pediatric glioblastoma. Science. 2013;340(6134):857–61. doi: 10.1126/science.1232245. [published Online First: Epub Date]|. revealed the mechanism behind the H3 histone alterations in pediatric HGGs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132•.Grasso CS, Tang Y, Truffaux N, et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nature medicine. 2015;21(7):827. doi: 10.1038/nm0715-827a. [published Online First: Epub Date]|. first to show that panobinostat may be beneficial in the therapy of DIPG. [DOI] [PubMed] [Google Scholar]
- 133.Hashizume R, Andor N, Ihara Y, et al. Pharmacologic inhibition of histone demethylation as a therapy for pediatric brainstem glioma. Nature medicine. 2014;20(12):1394–6. doi: 10.1038/nm.3716. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bagcchi S. Panobinostat active against diffuse intrinsic pontine glioma. The Lancet. Oncology. 2015;16(6):e267. doi: 10.1016/S1470-2045(15)70230-5. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 135.Heimberger A, Hussain S, Aldape K, et al. Tumor-specific peptide vaccination in newly-diagnosed patients with GBM. J Clin Oncol. 2006;24:107S–07S. (2006) [Google Scholar]
- 136.Sampson JH, Aldape KD, Archer GE, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro-oncology. 2011;13(3):324–33. doi: 10.1093/neuonc/noq157. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Schuster J, Lai RK, Recht LD, et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro-oncology. 2015;17(6):854–61. doi: 10.1093/neuonc/nou348. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lai RKRL, Reardon DA, Paleologos N, Groves M, Rosenfeld MR, et al. Long-term follow-up of act III: a phase II trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma. Neuro Oncol. 2011;13:34–34. doi: 10.1093/neuonc/nou348. (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139••.Celldex. Data Safety and Monitoring Board Recommends Celldex's Phase 3 Study of RINTEGA® (rindopepimut) in Newly Diagnosed Glioblastoma be Discontinued as it is Unlikely to Meet Primary Overall Survival Endpoint in Patients with Minimal Residual Disease. Secondary Data Safety and Monitoring Board Recommends Celldex's Phase 3 Study of RINTEGA® (rindopepimut) in Newly Diagnosed Glioblastoma be Discontinued as it is Unlikely to Meet Primary Overall Survival Endpoint in Patients with Minimal Residual Disease. 2016 http://ir.celldex.com/releasedetail.cfm?ReleaseID=959021. latest update on the Phase III trial revealing that endpoints are unlikely to be met.
- 140.Shrimali RK, Yu Z, Theoret MR, et al. Antiangiogenic agents can increase lymphocyte infiltration into tumor and enhance the effectiveness of adoptive immunotherapy of cancer. Cancer research. 2010;70(15):6171–80. doi: 10.1158/0008-5472.CAN-10-0153. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Reardon D, Schuster J, Tran DD, Fink KL, Nabors LB, Li G, Bota DA, Lukas RV, Desjardins A, Ashby LS, Duic JP, Mrugala MM, Werner A, Hawthorne T, He Y, Green JA, Yellin MJ, Turner CD, Davis TA, Sampson JH. ReACT: Overall survival from a randomized phase II study of rindopepimut (CDX-110) plus bevacizumab in relapsed glioblastoma. J Clin Oncol. 2015;33(suppl) (abstr 2009) 2015. [Google Scholar]
- 142.Li G, Mitra SS, Monje M, et al. Expression of epidermal growth factor variant III (EGFRvIII) in pediatric diffuse intrinsic pontine gliomas. Journal of neuro-oncology. 2012;108(3):395–402. doi: 10.1007/s11060-012-0842-3. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143••.Schumacher T, Bunse L, Pusch S, et al. A vaccine targeting mutant IDH1 induces antitumour immunity. Nature. 2014;512(7514):324–7. doi: 10.1038/nature13387. [published Online First: Epub Date]|. first to show that vaccines targeting IDH1mutant gliomas are a treatment startegy. [DOI] [PubMed] [Google Scholar]
- 144.Ampie L, Choy W, Lamano JB, et al. Heat shock protein vaccines against glioblastoma: from bench to bedside. Journal of neuro-oncology. 2015;123(3):441–8. doi: 10.1007/s11060-015-1837-7. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bloch O, Crane CA, Fuks Y, et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: a phase II, single-arm trial. Neuro-oncology. 2014;16(2):274–9. doi: 10.1093/neuonc/not203. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Phuphanich SWC, Rudnick J. Long-term remission over 5 years in patients with newly diagnosed glioblastoma (GBM) treated with ICT-107 vaccine: A follow-up study. Neuro Oncol. 2013;15(suppl 3):iii68–iii74. Abstract#: IT-015 2013. [Google Scholar]
- 147.Therapeutics I. ImmunoCellular Therapeutics Presents Updated ICT-107 Phase 2 Survival and Immune Response Data at the Society for Neuro-Oncology Annual Meeting 2015. Secondary ImmunoCellular Therapeutics Presents Updated ICT-107 Phase 2 Survival and Immune Response Data at the Society for Neuro-Oncology Annual Meeting 2015. 2015 [Google Scholar]
- 148.Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nature reviews. Cancer. 2012;12(4):265–77. doi: 10.1038/nrc3258. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Liau LM, Prins RM, Kiertscher SM, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11(15):5515–25. doi: 10.1158/1078-0432.CCR-05-0464. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 150.Yu JS, Liu G, Ying H, et al. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer research. 2004;64(14):4973–9. doi: 10.1158/0008-5472.CAN-03-3505. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 151••.Mitchell DA, Batich KA, Gunn MD, et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature. 2015;519(7543):366–9. doi: 10.1038/nature14320. [published Online First: Epub Date]|. first to suggest and show that preconditioning with a strong recall antigen may enhance the effects of immunotherapies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152•.Louveau A, Smirnov I, Keyes TJ, et al. Structural and functional features of central nervous system lymphatic vessels. Nature. 2015;523(7560):337–41. doi: 10.1038/nature14432. [published Online First: Epub Date]|. discovered a lymphatic system in the CNS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Preusser M, Lim M, Hafler DA, et al. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nature reviews. Neurology. 2015;11(9):504–14. doi: 10.1038/nrneurol.2015.139. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Taube JM, Klein A, Brahmer JR, et al. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20(19):5064–74. doi: 10.1158/1078-0432.CCR-13-3271. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Herbst RS, Soria JC, Kowanetz M, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–7. doi: 10.1038/nature14011. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Fecci PE, Ochiai H, Mitchell DA, et al. Systemic CTLA-4 blockade ameliorates glioma-induced changes to the CD4+ T cell compartment without affecting regulatory T-cell function. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13(7):2158–67. doi: 10.1158/1078-0432.CCR-06-2070. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 157.Kjaergaard J, Tanaka J, Kim JA, et al. Therapeutic efficacy of OX-40 receptor antibody depends on tumor immunogenicity and anatomic site of tumor growth. Cancer research. 2000;60(19):5514–21. [PubMed] [Google Scholar]
- 158.Zeng J, See AP, Phallen J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. International journal of radiation oncology, biology, physics. 2013;86(2):343–9. doi: 10.1016/j.ijrobp.2012.12.025. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Weber JS, Yang JC, Atkins MB, et al. Toxicities of Immunotherapy for the Practitioner. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015;33(18):2092–9. doi: 10.1200/JCO.2014.60.0379. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160••.Okada H, Weller M, Huang R, et al. Immunotherapy response assessment in neuro-oncology: a report of the RANO working group. The Lancet. Oncology. 2015;16(15):e534–42. doi: 10.1016/S1470-2045(15)00088-1. [published Online First: Epub Date]|. highlights the challenges in radiographic assessment of patients undergoing immunotherapy and re-defines imaging parameters of tumor progression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161••.Le DT, Uram JN, Wang H, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. The New England journal of medicine. 2015;372(26):2509–20. doi: 10.1056/NEJMoa1500596. [published Online First: Epub Date]|. first to show that mismatch repair deficiency and high mutational load correlate with response to immun checkpoint inhibitors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162••.Bouffet E, Larouche V, Campbell BB, et al. Immune Checkpoint Inhibition for Hypermutant Glioblastoma Multiforme Resulting From Germline Biallelic Mismatch Repair Deficiency. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2016 doi: 10.1200/JCO.2016.66.6552. [published Online First: Epub Date]|. one of the first reports to show durable responses to immune checkpoint inhibition in hypermutant recurrent GBMs. [DOI] [PubMed] [Google Scholar]
- 163.Wollmann G, Ozduman K, van den Pol AN. Oncolytic virus therapy for glioblastoma multiforme: concepts and candidates. Cancer journal. 2012;18(1):69–81. doi: 10.1097/PPO.0b013e31824671c9. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nature biotechnology. 2012;30(7):658–70. doi: 10.1038/nbt.2287. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Bischoff JR, Kirn DH, Williams A, et al. An adenovirus mutant that replicates selectively in p53-deficient human tumor cells. Science. 1996;274(5286):373–6. doi: 10.1126/science.274.5286.373. [DOI] [PubMed] [Google Scholar]
- 166.Harada N, Maniwa Y, Yoshimura M, et al. E1B-deleted adenovirus replicates in p53-deficient lung cancer cells due to the absence of apoptosis. Oncology reports. 2005;14(5):1155–63. [PubMed] [Google Scholar]
- 167.Fueyo J, Gomez-Manzano C, Alemany R, et al. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. 2000;19(1):2–12. doi: 10.1038/sj.onc.1203251. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 168.Markert JM, Medlock MD, Rabkin SD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene therapy. 2000;7(10):867–74. doi: 10.1038/sj.gt.3301205. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 169.Markert JM, Liechty PG, Wang W, et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Molecular therapy : the journal of the American Society of Gene Therapy. 2009;17(1):199–207. doi: 10.1038/mt.2008.228. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Markert JM, Razdan SN, Kuo HC, et al. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Molecular therapy : the journal of the American Society of Gene Therapy. 2014;22(5):1048–55. doi: 10.1038/mt.2014.22. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lang F, Conrad C, Gomez-Manzano C, et al. Phase I clinical trial of oncolytic virus Delta-24-RGD (DNX-2401) with biological endpoints: Implications for viro-immunotherapy. SNO Meeting Abstracts 2014. 2014 [Google Scholar]
- 172.Dobrikova EY, Broadt T, Poiley-Nelson J, et al. Recombinant oncolytic poliovirus eliminates glioma in vivo without genetic adaptation to a pathogenic phenotype. Molecular therapy : the journal of the American Society of Gene Therapy. 2008;16(11):1865–72. doi: 10.1038/mt.2008.184. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Desjardins A. Results of a phase 1 trial of an oncolytic polio/rhinovirus recombinant (PVS-RIPO) against recurrent glioblastoma. Secondary Results of a phase 1 trial of an oncolytic polio/rhinovirus recombinant (PVS-RIPO) against recurrent glioblastoma. 2014 [Google Scholar]
- 174.Zamarin D, Holmgaard RB, Subudhi SK, et al. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Science translational medicine. 2014;6(226):226ra32. doi: 10.1126/scitranslmed.3008095. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175•.Engeland CE, Grossardt C, Veinalde R, et al. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Molecular therapy : the journal of the American Society of Gene Therapy. 2014;22(11):1949–59s. doi: 10.1038/mt.2014.160. [published Online First: Epub Date]|. demonstrate that the use of immune checkpoint inhibitors may enhance the oncolytic virus response. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Reardon DA, Desjardins A, Vredenburgh JJ, et al. Safety and pharmacokinetics of dose-intensive imatinib mesylate plus temozolomide: phase 1 trial in adults with malignant glioma. Neuro-oncology. 2008;10(3):330–40. doi: 10.1215/15228517-2008-003. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Gerstner ER, Eichler AF, Plotkin SR, et al. Phase I trial with biomarker studies of vatalanib (PTK787) in patients with newly diagnosed glioblastoma treated with enzyme inducing anti-epileptic drugs and standard radiation and temozolomide. Journal of neuro-oncology. 2011;103(2):325–32. doi: 10.1007/s11060-010-0390-7. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Schiff D, Desjardins A, Cloughesy T, et al. Phase 1 dose escalation trial of the safety and pharmacokinetics of cabozantinib concurrent with temozolomide and radiotherapy or temozolomide after radiotherapy in newly diagnosed patients with high-grade gliomas. Cancer. 2015 doi: 10.1002/cncr.29798. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 179.Fields EC, Damek D, Gaspar LE, et al. Phase I dose escalation trial of vandetanib with fractionated radiosurgery in patients with recurrent malignant gliomas. International journal of radiation oncology, biology, physics. 2012;82(1):51–7. doi: 10.1016/j.ijrobp.2010.09.008. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 180.Broniscer A, Baker SD, Wetmore C, et al. Phase I trial, pharmacokinetics, and pharmacodynamics of vandetanib and dasatinib in children with newly diagnosed diffuse intrinsic pontine glioma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2013;19(11):3050–8. doi: 10.1158/1078-0432.CCR-13-0306. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Drappatz J, Norden AD, Wong ET, et al. Phase I study of vandetanib with radiotherapy and temozolomide for newly diagnosed glioblastoma. International journal of radiation oncology, biology, physics. 2010;78(1):85–90. doi: 10.1016/j.ijrobp.2009.07.1741. [published Online First: Epub Date]|. [DOI] [PubMed] [Google Scholar]
- 182.Lee EQ, Puduvalli VK, Reid JM, et al. Phase I study of vorinostat in combination with temozolomide in patients with high-grade gliomas: North American Brain Tumor Consortium Study 04-03. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012;18(21):6032–9. doi: 10.1158/1078-0432.CCR-12-1841. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Xu LW, Chow KK, Lim M, et al. Current vaccine trials in glioblastoma: a review. Journal of immunology research. 2014;2014:796856. doi: 10.1155/2014/796856. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Kicielinski KP, Chiocca EA, Yu JS, et al. Phase 1 clinical trial of intratumoral reovirus infusion for the treatment of recurrent malignant gliomas in adults. Molecular therapy : the journal of the American Society of Gene Therapy. 2014;22(5):1056–62. doi: 10.1038/mt.2014.21. [published Online First: Epub Date]|. [DOI] [PMC free article] [PubMed] [Google Scholar]