

Abstract
Background
The aim of this study was to explore the impact of LBH589 alone or in combination with proteasome inhibitor bortezomib on multiple myeloma (MM) cell proliferation and its mechanism.
Material/Methods
MM cell line U266 and RRMM-BMMNC were treated with different concentrations of LBH589 alone or in combination with bortezomib. Cell proliferation was detected by MTT assay. Cell cycle and apoptosis was analyzed by flow cytometry. The protein and mRNA level of related genes was determined by Western blotting and qRT-PCR respectively.
Results
U266 cell and RRMM-BMMNC proliferation were inhibited by different concentrations of LBH589 (0, 10, 20, and 50 nmol/L) alone or 50 nmol/L of LBH589 in combination with bortezomib (10 and 20 nmol/L) in a dose- and time-dependent manner. LBH589 significantly induced G0/G1phase arrest and apoptosis in RRMM-BMMNC in a dose-dependent manner. The effects were significantly higher in all combined groups than in single-agent groups (all P<0.05). The mRNA level of Caspase3 and APAF1 were up-regulated gradually, while TOSO gene expression in RRMM-BMMNC was down-regulated gradually in a dose- and time-dependent manner. Moreover, LBH589 significantly induced hyperacetylation of histone H4, the protein level of PARP notably increased, and the level of Bcl-X decreased.
Conclusions
LBH589 can inhibit MM cell growth, block the cell cycle, and induce cell apoptosis, which has an anti-resistant effect on multidrug-resistant cells. LBH589 in combination with bortezomib has a synergistic effect on myeloma cells; its mechanism and reversal of drug resistance mechanism is involved in multiple changes in gene expression.
MeSH Keywords: Apoptosis, Cell Cycle, Cell Proliferation, Multiple Myeloma
Background
Multiple myeloma (MM) is a plasma cell malignant proliferative disease that seriously affect patient health. The current MM treatment of conventional chemotherapy drugs have basically increased the risk of a second cancer, and multidrug resistance (MDR) phenomenon have appeared in MM cells [1]. Therefore, searching for an effective diagnostic strategy and new anti-tumor drugs for MM is becoming more necessary and urgent [2]. New anticancer treatment strategies focus on targeting intrinsic molecular mechanisms of tumor cells, which could offer higher efficacy with less adverse effects [3,4]. Among these novel anticancer family drugs, which provided promising results, are histone deacetylase (HDAC) inhibitors [5–9].
HDACi, a class of enzymes that participate in chromatin remodeling, play important roles in the regulation of numerous biological processes, mainly via transcription inhibition [10–12]. In recent years, a growing number of studies have confirmed HDAC inhibitors can be used as novel anti-tumor agents. As a new generation of HDAC inhibitor, LBH589 has been verified to have anti-tumor activity. LBH589 exerts its anti-tumor roles mainly via inducing tumor cell apoptosis and HDACs inhibition [13–15]. LBH589 has been found to prevent human renal cell carcinoma development by inducing cell cycle arrest and cell apoptosis [16]. Fortunati et al. reported that LBH589 has anti-proliferative and anti-invasive capabilities in triple-negative breast cancer (TNBC) [17]. Jeon et al. suggested that LBH589 could induce oral squamous cell carcinoma cell apoptosis via regulating of Sp1 gene expression [18]. LBH589 demonstrated anti-myeloma activity across a range of human myeloma cell lines and significantly repressed the growth of multiple myeloma cells and fresh cells from multiple myeloma patients (IC50 <40 nmol/L), including cells resistant to standard chemotherapeutic agents [19,20]. HDAC inhibitors combined with dexamethasone (Dex), bortezomib, and insulin-like growth factor-1 (IGF-1) inhibitors have synergistic cytotoxicity against MM, as have tyrosine kinase inhibitors [21–25]. Phase 1 and 2 clinical trials of hydroxamic acid-derived HDAC inhibitors LBH589 and suberoylanilide hydroxamic acid (SAHA) for the treatment of MM are now under way in the United States [26,27].
The results of the present study show that LBH589 notably suppressed growth of multiple myeloma cells and bone marrow mononuclear cells of relapsed or refractory multiple myeloma patients (RRMM-BMMNC) and enhanced the cytotoxicity triggered by bortezomib. LBH589 induced cell cycle arrest and apoptosis of multiple myeloma cells through caspase-dependent pathways. Furthermore, gene expression results uncovered a group of proteins, such as Caspase3, APAF1, and TOSO, which may play a relevant role in multiple myeloma and RRMM-BMMNC apoptosis.
Material and Methods
Cell culture
The human MM cell line U266 and was preserved by long-term liquid nitrogen storage in our department. Recurrence of refractory multiple myeloma patients with bone marrow mononuclear cells were obtained from the Department of Hematology, Second Hospital of Shanxi Medical University. U266 cells were cultured in RPMI 1640 (GIBCO, Grand Island, NY) containing 10% heat-inactivated fetal bovine serum (Sijiqing, Hangzhou, China) and incubated at 37°C with 5% CO2.
Bone marrow from patients with recurrent refractory multiple myeloma was collected, then bone marrow mononuclear cells were isolated. RRMM-BMMNC was cultured in RPMI-1640 medium containing 10% fetal bovine serum, 100 U/ml penicillin, and 100 U/ml streptomycin.
LBH589 (Biovision) was dissolved in DMSO and Bortezomib dissolved in saline, then diluted in media for cell culture experiments.
Cell proliferation assay
The proliferation of multiple myeloma U266 cells and RRMM-BMMNC was examined using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) colorimetric assay. U266 and RRMM-BMMNC cells (1×105) were plated in a 96-well plate and cultured overnight at 37°C. The cells were then incubated with 0, 10, 20, and 50 nM LBH589 or 10 and 20 nM Bortezomib combined with 50 nM LBH589 for 24 or 48 h, respectively. The control group was treated with same volume of RPMI 1640 medium. Every drug concentration was carried out by at least 4 replicate wells. Then, each well received 204 μl MTT solution, incubated for 4 h at 37°C, centrifuged (4°C, 1000 r/min, 10 min), and the supernatant was discarded. We added 200 μl DMSO per well, then light oscillation for 10 min, and the optical density (OD) values at 492 nm were detected with a microplate reader. Finally, the relative survival rate of the cells was calculated.
Cell cycle and cell apoptosis assay
The RRMM-BMMNC (1×106/ml) were seeded in 24-well plates and treated with different concentrations of drugs. After 48 h, the cells were collected and centrifuged to remove the supernatant, then washed with cold PBS (1000 r/min centrifuge for 5 min). The cells were re-suspended in 1× staining solution, 5 μl of propidium iodide, and 25 μg/ml RNaseA were added, and the cells were incubated at room temperature for 15 min. A BD Flow Cytometer was used to analyze the cell samples (1×104 cells).
Quantitative PCR
To detect the expression of APAF1, TOSO, and Caspase3, real-time quantitative RT-PCR was performed using the Lightcycler-Faststart DNA master SYBR green I PCR kit (Roche Applied Science, Indianapolis, IN) in a Roche Lightcycler 1.2 Real-Time PCR System (Roche) according to the manufacturer’s instructions. We used the following primers (forward/reverse, 5′-3′):
APAF1: TTCTGATGCTTCGCAAACAC/CTGGCAAATCTGCCTTCTTC,
TOSO: CACTTGCTTCCTTTCCAAGC/GAAGGATGCCCGAGACAATA,
Caspase3: TTTTTCAGAGGGGATCGTTG/CGGCCTCCACTGGTATTTTA,
β-actin: AGAGCTACGAGCTGCCTGAC/AGCACTGTGTTGGCGTACAG;
β-actin was used as an internal control. The comparative threshold cycle method was used for calculation of amplification fold. The expression level of each gene was normalized by dividing by the expression level of β-actin.
Western blotting
RRMM-BMMNC were treated with different concentrations of LBH589, and 24 h later, total cellular proteins were extracted by using RIPA lysis buffer (Beyotime Biotechnology, Shanghai, China). The protein samples were separated by using 10% SDS-PAGE gels, and then transferred to a PVDF membrane (Millipore, Bedford, MA). After blocking with 5% non-fat milk, the membranes were then probed with antibodies against Acetyl-H4 (1: 1000, Cell Signaling Technology), PARP (1: 1000, Cell Signaling Technology), and Bcl-x (1: 1000, Cell Signaling Technology). Membranes were washed 3 times and then incubated with horseradish peroxidase (HRP)- conjugated secondary antibodies. The enhanced chemiluminescence detection system was used to visualize the immunoreactive bands.
Statistical analysis
SPSS13.0 software was used to analyze the data. Values are expressed as mean±SD of experiments performed in triplicate. Data were analyzed by one-way ANOVA and Student’s t-test. Statistical significance was defined as p<0.05.
Results
Effects of LBH589 therapy or combination with bortezomib therapy on the proliferation of MM cells
MTT assay showed that the proliferation of U266 and RRMM-BMMNC was inhibited by the LBH589 or combination with bortezomib. At the same time point, different concentrations of LBH589 on the inhibition of cell proliferation was significantly different (P<0.01) (Table 1), the inhibition rate showed a dose-dependent effect. At the same concentration, the inhibition rate at 48 h was significantly higher than that at 24 h (P<0.01) (Table 1). The inhibitory effect of the combination of the 2 drugs was stronger than that in the single-drug group (P<0.01).
Table 1.
Inhibition effects of LBH589 monotherapy or in combination with bortezomib (Bor) on U266 and RRMM-BMMNC proliferation (%, χ̄± s).
| Groups | n | Inhibition rate of U266 | Inhibition rate of RRMM-BMMNC | ||
|---|---|---|---|---|---|
| 24 h | 48 h | 24 h | 48 h | ||
| Control group | 8 | 0 | 0 | 0 | 0 |
| 10 nmol/L LBH589 | 8 | 11.08±0.36a | 29.12±1.057a | 8.65±0.74a,b | 12.05±0.80a,b |
| 20 nmol/L LBH589 | 8 | 21.23±0.53a | 42.08±0.35a | 16.40±0.59a,b | 37.65±0.57a,b |
| 50 nmol/L LBH589 | 8 | 33.58±1.09a | 64.12±1.22a | 23.68±1.65a,b | 59.62±1.02a,b |
| 50 nmol/L LBH589+10 nmol/l Bor | 8 | 59.08±1.22a | 85.68±1.10a | 37.27±1.40a,b | 67.88±1.79a,b |
| 50 nmol/L LBH589+20 nmol/l Bor | 8 | 75.48±0.76a | 90.07±0.67a | 47.52±0.67a,b | 75.42±0.78a,b |
Compared with the control group,
P<0.01; compared with the U266 group,
P<0.05.
Effect of LBH589 monotherapy or combination with bortezomib on cell cycle and apoptosis
Cell cycle results showed that 48 h after LBH589 treatment, the cells in G0/G1 phase increased and G2/M phase and S phase decreased compared with control group (Figure 1, Table 2), indicating that the cells were arrested in G0/G1 phase and the blocking effect were enhanced with increasing LBH589 concentration. After 48-h treatment, the cells in the control group had no obvious apoptosis, and the apoptosis rate of the LBH589-treated group increased gradually in a concentration-dependent manner. Compared with LBH589 monotherapy group, using LBH589 combined with bortezomib significantly increased the apoptosis rate, and more cells were arrested in G0/G1 phase.
Figure 1.

Cell apoptosis and cell cycle changes in different groups. Flow cytometry was used for cell apoptosis and cell cycle determination. (A) Cell apoptosis in different groups; (B) Cell cycle distribution in different groups.
Table 2.
Effect of LBH589 monotherapy or combined with bortezomib on cell cycle and apoptosis of RRMM-BMMNC cells (%, χ̄± s).
| Groups | n | Cell cycle distribution | Apoptosis rate | ||
|---|---|---|---|---|---|
| G0/G1 phase | S phase | G2 phase | |||
| Control group | 8 | 33.60±1.82 | 7.63±1.12 | 58.76±2.10 | 6.57±1.41 |
| 10 nmol/L LBH589 | 8 | 45.70±1.70a | 12.60±1.00a | 41.70±2.00a | 33.41±1.24a |
| 20 nmol/L LBH589 | 8 | 55.40±0.90a | 8.60±0.54a | 35.90±1.80a | 39.67±1.06a |
| 50 nmol/L LBH589 | 8 | 58.10±1.90a | 7.10±0.75a | 34.70±1.60a | 46.47±0.90a |
| 50 nmol/L LBH589+10 nmol/l Bor | 8 | 74.48±1.00a | 5.5±0.42a | 20.01±0.45a | 75.20±0.78a |
| 50 nmol/L LBH589+20 nmol/l Bor | 8 | 80.73±1.20a | 3.23±0.48a | 15.92±0.48a | 85.37±1.33a |
Ccompared with control group,
P<0.05.
Effects of LBH589 treatment on Caspase3, APAF1, and TOSO expression in RRMM-BMMNC
To explore the mechanism by which LBH589 causes apoptosis in RRMM-BMMNC, we assessed the expression of Caspase3, APAF1, and TOSO, which are recognized markers of apoptosis by Q-PCR. The result showed that the expression of pro-apoptotic gene Caspase3 and APAF-1were increased after 24 h or 48 h of treatment with 20 nM and 50 nM LBH589 (Table 3), and the expression level of TOSO, a drug resistance gene, was decreased.
Table 3.
LBH589 regulate the expression of caspase-3, APAF-1 and TOSO gene in RRMM-BMMNC (%, χ̄± s).
| Groups | n | Caspase-3 | APAF-1 | TOSO | |||
|---|---|---|---|---|---|---|---|
| 24 h | 48 h | 24 h | 48 h | 24 h | 48 h | ||
| Control group | 8 | 1.00±0.00 | 1.00±0.00 | 1.00±0.00 | 1.00±0.00 | 1.00±0.00 | 1.00±0.00 |
| 20 nmol/L LBH589 | 8 | 1.36±0.24a | 2.11±0.02a | 1.16±0.05a | 1.68±0.21a | 0.57±0.01a | 0.42±0.04a |
| 50 nmol/L LBH589 | 8 | 1.68±0.08a | 2.81±0.08a | 1.58±0.16a | 3.74±0.24a | 0.43±0.01a | 0.12±0.04a |
Compared with control group,
P<0.05.
Effects of LBH589 treatment on Histone H4 acetylation, and PARP, Bcl-X protein expression in RRMM-BMMNC
The Western blotting results of the present study suggest that LBH589 markedly induced the hyperacetylation of histone H4, notably up-regulated the protein expression level of PARP, and down-regulated the protein expression level of Bcl-X (Figure 2, Table 4).
Figure 2.
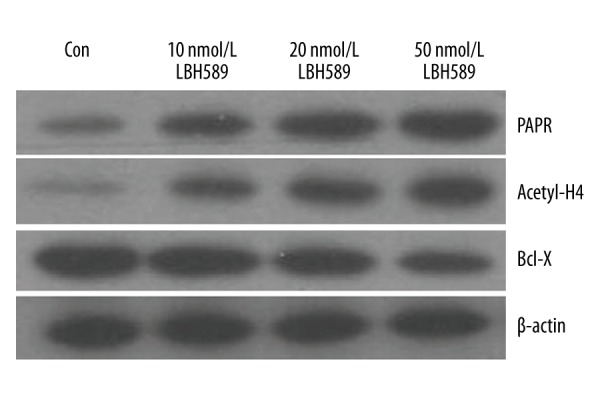
Western blot analysis was used to detect the expression of acetylated (Acetyl-H4), PARP, and Bcl-X protein of RRMM-BMMNC after 24 h of LBH589 treatment.
Table 4.
Effect of LBH589 on histone H4 acetylation and PARP and Bcl-X protein expression (%, χ̄± s).
| Groups | N | Histone H4 | PARP | Bcl-X |
|---|---|---|---|---|
| Control group | 8 | 0.252±0.038 | 0.225±0.055 | 1.120±0.060 |
| 10 nmol/L LBH589 | 8 | 0.375±0.003a | 0.498±0.030a | 0.780±0.067a |
| 20 nmol/L LBH589 | 8 | 0.574±0.085a | 0.657±0.048a | 0.720±0.007a |
| 50 nmol/L LBH589 | 8 | 0.896±0.032a | 0.876±0.044a | 0.477±0.039a |
Compared with control group,
P<0.05.
Discussion
Histone acetyltransferase (HAT) and histone deacetylase (HADC) regulate the acetylation and gene transcription of nuclear proteins. Histone lysine residues acetylation neutralize the positive charge of histone, make the affinity of histone and negatively charged DNA decreased, changes the nuclear body configuration, finally makes the transcription factor easy to bind DNA and promote gene transcription, while histone deacetylation inhibits transcription. The imbalance of histone acetylation regulated by these 2 enzymes breaks the original balance of gene expression, resulting in the expression imbalance of a number of cell proliferation and cell cycle related molecule, leading to cell malignancy [28,29]. Studies have shown that HDACi, as a class of anti-tumor drugs, can inhibit the activity of HDAC, induce highly acetylated histones in target cells, and regulate some specific gene expression, thus block the growth of tumor cells [30].
The hydroxamic acid LBH589 is a new type of HDACi, which has a strong effect on MM cells, with high efficiency, low toxicity, anti-drug resistance, and synergistic effect combined with other drugs [31–33].
Our results show that LBH589 monotherapy or combination with bortezomib can inhibit the growth of myeloma cells (U266 and RRMM-BMMNC) in a time- and dose-dependent manner. We observed the drug sensitivity of U266 cells were higher than RRMM-BMMNC, fully demonstrated RRMM-BMMNC have drug resistance, and proved that LBH589 is resistant to drug-resistant cells. LBH589 combined with 10 and 20 nmol/L bortezomib showed stronger inhibitory effect than LBH589 alone. It is clear that LBH589 has a synergistic effect with bortezomib and can exert a more potent effect. Using low concentrations of LBH589 can have significant inhibition effect on U266 and RRMM-BMMNC, indicating that LBH589 is a highly effective anti-MM drug able to overcome drug resistance, and it is superior to other HDACi, such as SAHA and NVP-LAQ824 [26,27,34]. In summary, LBH589 is a new type of chemotherapeutic agent with high efficiency, low dosage, and resistance to drug resistance.
In this study, we found that LBH589 as a single agent or in combination with bortezomib can cause RRMM-BMMNC cell cycle arrest in G0/G1 phase, and with increased drug dose, more cells were arrested in this phase. G2/M and S phase cells were gradually decreased, suggesting that LBH589 has the function of regulating G0/G1 phase detection point, indicating that the most sensitive point of drug action may be in the G1 phase. It is presumed that apoptosis may occur in G1 phase, apoptosis occurs after 48 h of drug action, and the apoptosis rate was also dose-dependent. This indicates that LBH589 has the effect of arresting cell cycle and inducing apoptosis.
Caspase-3 is a member of the caspase family and is the most important terminal cleavage enzyme in the process of apoptosis. It is also an important component of the cytotoxic T cell (CTL cell) killing mechanism. Caspase-3 is the core player in apoptosis and has an essential role in apoptosis. PARP is a DNA repair enzyme, a cutting substrate of Caspase-3, and the cut PARP can be used as a molecular marker of apoptosis, thus playing an important role in DNA damage repair and apoptosis [35,36]. The results showed that the expression of RRMM-BMMNC Caspase-3 and PARP was up-regulated after LBH589 treatment. The up-regulation effects were time- and dose-dependently, indicating that LBH589 induced myeloma cell apoptosis through the caspase pathway, which is one of the mechanisms of apoptosis induced by LBH589 in MM cells. In addition, APAF-1, an apoptotic protease activator, has been shown to be up-regulated in MM cells when treated with HDACi SAHA. Recent studies have also shown that down-regulation of APAF-1 can prevent SAHA-induced apoptosis [37]. Our results show that LBH589 treatment could up-regulate the expression of APAF-1 in RRMM-BMMNC, enhance the drug sensitivity of drug-resistant cells, and induce cell apoptosis, which may be an important factor for the induction of apoptosis of MM cells induced by LBH589.
Studies have shown that tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is a member of the tumor necrosis factor (TNF) family, which induces cell apoptosis by interacting with 2 apoptosis receptors: TRAIL-R1 and TRAIL-R2. Tumor cells have been shown to be tolerant to TRAIL-induced apoptosis and to inhibit apoptosis. Addition of HDACi trichostatin (TSA) up-regulates TRAIL receptors, enhancing the sensitivity of cells to TRAIL-mediated apoptosis [38]. Insinga et al. [39] found that HDACi redistributes death receptors, restoring the response of MM cells to the apoptotic signal, or by activating specific gene expression, and down-regulating the negative regulation of Fas ligand (FasL) or TRAIL.
The TOSO gene is a negative regulator of apoptotic signals in hematopoietic cells and is a member of the immunoglobulin super-family. It has been reported that the TOSO gene negatively regulates TRAIL-induced apoptosis in other cell types, and it inhibits tumor cell apoptosis [40–42]. Thus, the TOSO gene may be a drug-resistant gene, and LBH589 can down-regulate the expression of TOSO gene, which in turn can increase TRAIL-R1 and TRAIL-R2, recover the sensitivity of drug-resistant cells to TRAIL-mediated apoptosis, and re-promote cell apoptosis. In addition, Bcl-X gene, a member of the Bcl-2 gene family, is an anti-apoptotic gene. Studies have confirmed that Bcl-X gene has an important role in the regulation of apoptosis and cell resistance. For example, Bcl-X gene was observed to be highly expressed in the tissue sections in dexamethasone and melphalan treatment failure MM patients, resulting in apoptosis inhibition and drug resistance [43]. In the present study, we found that LBH589 down-regulates the expression of Bcl-X gene, and also promotes cell apoptosis. The acetylation of histone H4 was more and more obvious with increasing drug dose, which indicated that LBH589 could highly activate acetylate histone in MM cells, and the expression of TOSO and Bcl-X gene were down-regulated. Therefore, LBH589 can induce the down-regulation of TOSO and Bcl-X gene expression by inducing a high degree of acetylation of histone in MM cells, so as to restore the sensitivity of drug-resistant MM cells, re-promote the apoptosis of MM cells, and finally achieve the goal of anti-drug resistance.
In summary, LBH589 is a new effective MM targeted therapy drug, but reversing the drug resistance through changing the relevant gene expression via inducing the histone hyperacetylation in MM cells is only one aspect. Currently, research on LBH589 in the reversal of drug resistance in drug-resistant MM cells is still in the initial stages, and further experiments and explorations are needed.
Conclusions
Our data suggest that LBH589 can inhibit the growth of MM cells, block the cell cycle, and induce cell apoptosis, which has an anti-resistant effect on multidrug-resistant cells. LBH589 in combination with bortezomib has a synergistic effect on myeloma cells, and its mechanism and reversal of drug resistance involves in multiple changes in gene expression. LBH589 is a new effective MM targeted therapy drug for multiple myeloma treatment; however, due to some limits in the present study, further experiments and explorations are still needed.
Footnotes
Conlfict of interest
None.
Source of support: This study was supported by the International Science and Technology Cooperation Program of Shanxi province (No. 2010081064)
References
- 1.Pérez-Tomás R. Retrospect and prospects in anti-cancer drug treatment. Curr Med Chem. 2006;13:1859–76. doi: 10.2174/092986706777585077. [DOI] [PubMed] [Google Scholar]
- 2.Ayaz S, Ayaz ÜY. Fluorodeoxyglucose positron emission tomography-computed tomography (FDG PET/CT) findings in an unusual case of multiple myeloma presenting with a large extra-axial intracranial mass. Pol J Radiol. 2016;81:602–5. doi: 10.12659/PJR.899444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yin Q, Chen L, Mi R, et al. Efficacy and safety of danshen compound tablets in preventing thalidomide-associated thromboembolism in patients with multiple myeloma: A multicenter retrospective study. Med Sci Monit. 2016;22:3835–42. doi: 10.12659/MSM.900575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mendonca NP, Deepak K. Pulmonary nocardiosis in a multiple myeloma patient treated with proteasome inhibitors. Am J Case Rep. 2016;17:76–78. doi: 10.12659/AJCR.896280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller TA, Witter DJ, Belvedere S. Histone deacetylase inhibitors. J Med Chem. 2003;46:5097–116. doi: 10.1021/jm0303094. [DOI] [PubMed] [Google Scholar]
- 6.Drummond DC, Noble CO, Kirpotin DB, et al. Clinical development of histone deacetylase inhibitors as anticancer agents. Annu Rev Pharmacol Toxicol. 2005;45:495–528. doi: 10.1146/annurev.pharmtox.45.120403.095825. [DOI] [PubMed] [Google Scholar]
- 7.Marks P, Rifkind RA, Richon VM, et al. Histone deacetylases and cancer: Causes and therapies. Nat Rev Cancer. 2001;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 8.Shieh JM, Tang YA, Hu FH, et al. A histone deacetylase inhibitor enhances expression of genes inhibiting Wnt pathway and augments activity of DNA demethylation reagent against non-small cell lung cancer. Int J Cancer. 2017;140(10):2375–86. doi: 10.1002/ijc.30664. [DOI] [PubMed] [Google Scholar]
- 9.Lee HS, Park SB, Kim SA, et al. A novel HDAC inhibitor, CG200745, inhibits pancreatic cancer cell growth and overcomes gemcitabine resistance. Sci Rep. 2017;7:41615. doi: 10.1038/srep41615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bahari-Javan S, Maddalena A, Kerimoglu C, et al. HDAC1 regulates fear extinction in mice. J Neurosci. 2012;32:5062–73. doi: 10.1523/JNEUROSCI.0079-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bellucci L, Dalvai M, Kocanova S, et al. Activation of p21 by HDAC inhibitors requires acetylation of H2A.Z. PLoS One. 2013;8:e54102. doi: 10.1371/journal.pone.0054102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brilli LL, Swanhart LM, de Caestecker MP, Hukriede NA. HDAC inhibitors in kidney development and disease. Pediatr Nephrol. 2013;28:1909–21. doi: 10.1007/s00467-012-2320-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan WW, Allred JB, Moreno-Aspitia A, et al. Phase I study of panobinostat (LBH589) and letrozole in postmenopausal metastatic breast cancer patients. Clin Breast Cancer. 2016;16:82–86. doi: 10.1016/j.clbc.2015.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wasim L, Chopra M. Panobinostat induces apoptosis via production of reactive oxygen species and synergizes with topoisomerase inhibitors in cervical cancer cells. Biomed Pharmacother. 2016;84:1393–405. doi: 10.1016/j.biopha.2016.10.057. [DOI] [PubMed] [Google Scholar]
- 15.Greve G, Schiffmann I, Pfeifer D, et al. The pan-HDAC inhibitor panobinostat acts as a sensitizer for erlotinib activity in EGFR-mutated and -wildtype non-small cell lung cancer cells. BMC Cancer. 2015;15:947. doi: 10.1186/s12885-015-1967-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu M, Hong M, Xie H. Histone deacetylase inhibitors induce human renal cell carcinoma cell apoptosis through p-JNK activation. Journal of Southern Medical University. 2013;33:1409–15. [PubMed] [Google Scholar]
- 17.Fortunati N, Marano F, Bandino A, et al. The pan-histone deacetylase inhibitor LBH589 (panobinostat) alters the invasive breast cancer cell phenotype. Int J Oncol. 2014;44:700–8. doi: 10.3892/ijo.2013.2218. [DOI] [PubMed] [Google Scholar]
- 18.Jeon YJ, Ko SM, Cho JH, et al. The HDAC inhibitor, panobinostat, induces apoptosis by suppressing the expresssion of specificity protein 1 in oral squamous cell carcinoma. Int J Mol Med. 2013;32:860–66. doi: 10.3892/ijmm.2013.1451. [DOI] [PubMed] [Google Scholar]
- 19.Maiso P, Carvajal-Vergara X, Ocio EM, et al. The histone deacetylase inhibitor LBH589 is a potent antimyeloma agent that overcomes drug resistance. Cancer Res. 2006;66:5781–89. doi: 10.1158/0008-5472.CAN-05-4186. [DOI] [PubMed] [Google Scholar]
- 20.Catley L, Weisberg E, Kiziltepe T, et al. Aggresome induction by proteasome inhibitor bortezomib and alpha-tubulin hyperacetylation by tubulin deacetylase (TDAC) inhibitor LBH589 are synergistic in myeloma cells. Blood. 2006;108:3441–49. doi: 10.1182/blood-2006-04-016055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitsiades N, Mitsiades CS, Richardson PG, et al. Molecular sequelae of histone deacetylase inhibition in human malignant B cells. Blood. 2003;101:4055–62. doi: 10.1182/blood-2002-11-3514. [DOI] [PubMed] [Google Scholar]
- 22.Mitsiades CS, Mitsiades NS, McMullan CJ, et al. Transcriptional signature of histone deacetylase inhibition in multiple myeloma: Biological and clinical implications. Proc Natl Acad Sci USA. 2004;101:540–45. doi: 10.1073/pnas.2536759100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pei XY, Dai Y, Grant S. Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin Cancer Res. 2004;10:3839–52. doi: 10.1158/1078-0432.CCR-03-0561. [DOI] [PubMed] [Google Scholar]
- 24.Vogl DT, Raje NS, Jagannath S, et al. Ricolinostat, the first selective histone deacetylase 6 inhibitor, in combination with bortezomib and dexamethasone for relapsed or refractory multiple myeloma. Clin Cancer Res. 2017 doi: 10.1158/1078-0432.CCR-16-2526. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang H, Liu N, Yang C, et al. HDAC inhibitor L-carnitine and proteasome inhibitor bortezomib synergistically exert anti-tumor activity in vitro and in vivo. PLoS One. 2012;7:e52576. doi: 10.1371/journal.pone.0052576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Qian DZ, Wang X, Kachhap SK, et al. The histone deacetylase inhibitor NVP-LAQ824 inhibits angiogenesis and has a greater antitumor effect in combination with the vascular endothelial growth factor receptor tyrosine kinase inhibitor PTK787/ZK222584. Cancer Res. 2004;64:6626–34. doi: 10.1158/0008-5472.CAN-04-0540. [DOI] [PubMed] [Google Scholar]
- 27.Fuino L, Bali P, Wittmann S, et al. Histone deacetylase inhibitor LAQ824 down-regulates Her-2 and sensitizes human breast cancer cells to trastuzumab, taxotere, gemcitabine, and epothilone B. Mol Cancer Ther. 2003;2:971–84. [PubMed] [Google Scholar]
- 28.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–84. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 29.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 30.Martinez-Lglesias O, Ruiz-Llorente L, Sanchez-Martinez R, et al. Histone deacetylase inhibitiors: Mechanism of action and therapeutic use in cancer. Clin Transl Oncol. 2008;10:395–98. doi: 10.1007/s12094-008-0221-x. [DOI] [PubMed] [Google Scholar]
- 31.George P, Bali P, Annavarapu S, et al. Combination of the histone deacetylase inhibitor LBH589 and the hsp90 inhibitor 17AAG is highly active against human CML BC cells and AML cells with activating mutation of FLT-3. Blood. 2005;105:1768–76. doi: 10.1182/blood-2004-09-3413. [DOI] [PubMed] [Google Scholar]
- 32.Sivaraj D, Green MM, Gasparetto C. Panobinostat for the management of multiple myeloma. Future Oncol. 2017;13:477–88. doi: 10.2217/fon-2016-0329. [DOI] [PubMed] [Google Scholar]
- 33.Greig SL. Panobinostat: A review in relapsed or refractory multiple myeloma. Target Oncol. 2016;11:107–14. doi: 10.1007/s11523-015-0413-6. [DOI] [PubMed] [Google Scholar]
- 34.Nimmanapalli R, Fuino L, Bali P, et al. Histone deacetylase inhibitor LAQ824 both lowers expression and promotes proteasomal degradation of Bcr-Abl and induces apoptosis of imatinib mesylate-sensitive or refractory chronic myelogenous leukemia-blast crisis cells. Cancer Res. 2003;63:5126–35. [PubMed] [Google Scholar]
- 35.Danial NN, Korsmeyer SJ. Cell death: Critical control points. Cell. 2004;116:205–19. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 36.Chaitanya GV, Alexander JS, Babu PP. PARP-1 cleavage fragments: Signatures of cell-death proteases in neurodegeneration. Cell Commun Signal. 2010;8:31. doi: 10.1186/1478-811X-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kroemer G, Martin SJ. Caspase-independent cell death. Nat Med. 2005;11:725–30. doi: 10.1038/nm1263. [DOI] [PubMed] [Google Scholar]
- 38.Mahalingam D, Medina E. Effect of sunitinib on TRAIL-induced apoptosis in preclinical colon cancer models. J Clin Oncol. 2009;27:e14633. [Google Scholar]
- 39.Insinga A, Monestimli S, Ronzuni S, et al. Inhibitors of histone deacetylases induce tumor-selective apoptosis through activation of the death receptor pathway. Nat Med. 2005;11:71–76. doi: 10.1038/nm1160. [DOI] [PubMed] [Google Scholar]
- 40.Hitoshi Y, Lorens J, Kitada SI, et al. Toso, a cell surface, specific regulator of Fas-induced apoptosis in T cells. Immunity. 1998;8:461–71. doi: 10.1016/s1074-7613(00)80551-8. [DOI] [PubMed] [Google Scholar]
- 41.Song Y, Jacob CO. The mouse cell surface pmtein TOSO regulates Fas/Fas ligand-induced apoptosis through its binding to Fas-associated death domain. J Biol Chem. 2005;280:9618–26. doi: 10.1074/jbc.M413609200. [DOI] [PubMed] [Google Scholar]
- 42.Mitsiades N, Mitsiades CS, Poulaki V, et al. Molecular sequelae of proteasome inhibition in human multiple myeloma cells. Proc Natl Acad Sci USA. 2002;99:14374–79. doi: 10.1073/pnas.202445099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Khan SB, Maududi T, Barton K, et al. Analysis of histone deacetylase inhibitor, depsipeptide(FR901228), effect on multiple myeloma. Br J Haematol. 2004;125:156–61. doi: 10.1111/j.1365-2141.2004.04882.x. [DOI] [PubMed] [Google Scholar]