

ABSTRACT
Programmed death ligand 1 (PD-L1) functions as a key immune inhibitory factor by binding with its receptor, programmed death 1 (PD-1), to induce immune cell dysfunction and escape of the immune system. However, the mechanisms of PD-L1 expression under growth factor stimulation are not well characterized. Here, we demonstrate a novel role for glial cell line-derived neurotrophic factor (GDNF) in upregulating PD-L1 expression in head and neck squamous cell carcinoma (HNSCC). The expression and correlation of PD-L1, GDNF and perineural invasion (PNI) status were evaluated by bioinformatics analysis of TCGA database and IHC assays from 145 HNSCC patients. PD-L1 expression was investigated by flow cytometry, Western blot and real-time PCR analyses in HNSCC cells after GNDF incubation. The cell signaling pathways activated by GDNF were analyzed with an antibody array and blocked by specific signaling inhibitors in cancer cell lines. PD-L1 expression was significantly higher in cancer cells that exhibited PNI in the HNSCC specimens, and elevated PD-L1 expression was significantly correlated with GDNF levels. GDNF not only enhanced cancer cell PNI in a co-culture of dorsal root ganglions and cancer cells but also had a potent role in inducing PD-L1 expression through the JAK2-STAT1 signaling pathway. Moreover, a JAK2 inhibitor attenuated GDNF-induced PD-L1 and enhanced tumor cell susceptibility to NK cell killing. Our findings provide clinically novel evidence that nerve-derived GDNF can increase PD-L1 levels in cancer cells around the perineural niche and that regulatory signaling is critical for cancer cell escape from immune surveillance in the nerve-cancer microenvironment.
KEYWORDS: GDNF, PD-L1, JAK2, PNI, HNSCC
Introduction
The PD-1/PD-L1 immune checkpoint has been demonstrated as a key immune escape mechanism that is used by tumors.1 Programmed death-ligand 1 (PD-L1), an inhibitory member of the B7 family, is upregulated in various types of solid tumors, such as melanoma, non-small cell lung cancer, and head and neck squamous cell carcinoma (HNSCC).2 PD-L1 binds to its receptors, programmed death-1 (PD-1) or B7.1 (CD80), on activated immune cells to inhibit T-cell activation or proliferation and mediate the suppression of local immune responses, thus leading to the immune evasion of tumor cells.3-5 Indeed, recent clinical trials targeting the PD-1/PD-L1 axis with blocking antibodies have shown encouraging results in patients with several types of cancer, including HNSCC, despite the low response rate.6,7 Therefore, understanding the stimuli and signaling pathways that regulate tumor PD-L1 expression may help to predict the response to PD-1/PD-L1 blockade therapy, permitting more effective therapeutic approaches.
Aberrant PD-L1 expression in tumor cells may be regulated by 2 general mechanisms. The first is the “innate immune resistance” mechanism, in which intrinsic cellular changes associated with carcinogenesis can promote tumor cell PD-L1 expression. Upregulated PD-L1 expression can be driven by activation of constitutive oncogenic signaling pathways, such as MAPK and PI3K-AKT, as well as transcriptional factors, such as HIF-1, STAT3 and NF-κb.8-10 The second mechanism is an “adaptive immune resistance” mechanism where tumors upregulate PD-L1 as an adaptive response to endogenous antitumor immunity. Emerging studies have revealed that PD-L1 is characteristically associated with intratumoral immune infiltrates. Inflammatory mediators, especially IFN-γ secreted by tissue-recruiting immune cells, including NK cells and CD8+ TILs, can induce PD-L1 expression in tumor cells.2,11 In HNSCC, limited studies revealed that positive PD-L1 expression in tumor cells ranged variously from 18% to 87% and that PD-L1 expression contributed to immune resistance in HPV+ HNSCC.12-14 PD-L1 upregulation was demonstrated in response to EGFR activation or the pro-inflammatory cytokines, INF-γ, TNF-α, and the JAK2-STAT1 pathway was a significant signaling node.15 However, the mechanisms mediating PD-L1 expression remain largely unknown in consideration of intratumoral heterogeneity.
Perineural invasion (PNI) is the presence of tumor cell invasion in, around, or along nerve bundles.16 The prevalence of PNI varies considerably among cancer types and reaches 30–82% in HNSCC.17,18 Recent studies demonstrated that in the nerve-cancer microenvironment, tumor cells may potentially be attracted by neurotrophic factors and chemokines secreted by nerves to facilitate its local spread into adjacent nerves.19 Glial cell line-derived neurotrophic factor (GDNF) has been demonstrated to be a potent chemoattractant for cancer cells in the PNI process. Nerve-secreted GDNF can activate its tyrosine kinase receptor, RET, promoting neural invasion by pancreatic cancer cells.20,21 Our previous study demonstrated that elevated RET levels were correlated with poor clinical outcomes in HNSCC, although no direct evidence links RET and PNI.22 Meanwhile, recent studies revealed that the perineural niche is rich in immunocytes and has a unique inflammatory profile, which implicate GDNF and RET. Perineural macrophages can upregulate and activate RET, inducing PNI of pancreatic ductal adenocarcinoma.23,24 In breast cancer, RET stimulated by GDNF increases pro-inflammatory cytokine expression levels.25 Hence, it is of interest to investigate whether there is a role of the nerve-cancer microenvironment in contributing to tumor immune suppression.
In the present study, we demonstrated that PD-L1 expression is significantly higher in cancer cells that exhibited PNI in HNSCC tumor specimens. GDNF not only enhances cancer cell PNI but also has a potent role in mediating PD-L1 upregulation mainly via the JAK2-STAT1 signaling pathway. Moreover, specific JAK2 inhibition prevented GDNF-mediated PD-L1 upregulation and enhanced tumor cell susceptibility to NK cell killing.
Results
Elevated PD-L1 mRNA expression correlates with GDNF in HNSCC
To investigate the PD-L1 and GDNF expression in HNSCC, we first accessed information regarding gene mutations, deletions, amplification and mRNA expression levels of PD-L1 and GDNF in a large cohort of HNSCC specimens provided by The Cancer Genome Atlas (TCGA) (Fig. 1A). PD-L1 and GDNF gene mutations, deletions or amplifications were observed in a small proportion of patients (Fig. S1A). The PD-L1 and GDNF genes were expressed significantly higher in the HNSCC samples compared with normal tissues. Furthermore, when segregated by tumor perineural invasion (PNI) status, we found that GDNF expression was elevated in PNI+ specimens, and there was no difference of PD-L1 expression between PNI+ and PNI- specimens (Fig. S1B). Interestingly, when we investigated PD-L1 expression levels in GDNF-positive versus GDNF-negative specimens according to TCGA, we noted that PD-L1 expression levels were higher in the GDNF-positive specimens (Fig. 1B).
Figure 1.

PD-L1 protein expression is elevated in PNI+ GDNF-positive tumor specimens (A) PD-L1 and GDNF mRNA levels in normal tissues or HNSCC samples according to TCGA database. (B) PD-L1 mRNA expression in GDNF-positive or GDNF-negative tumor specimens according to TCGA database. (C) PD-L1 protein expression is significantly correlated with GDNF in HNSCC specimens (Pearson r = 0.38, p< 0.001). (D) IHC assays were performed to detect PD-L1, GDNF and PGP9.5 (nerve marker) in HNSCC PNI samples. IHC staining and H&E staining indicated that tumor cells were morphologically present within and around the peripheral nerve (labeled with “N”). PD-L1 was expressed in tumor cells around the nerve, while GDNF was detected in the nerve. (E) The PD-L1 staining scores in tumor cells around peripheral nerve (PNI+) compared with tumor cells without PNI in matched specimen (p < 0.001). (F) Kaplan–Meier analyses of the overall survival Indicated that patients with a high level of GDNF were associated with a significantly lower overall survival rate than patients with a low level of GDNF expression (p < 0.001).
PD-L1 protein expression is higher in PNI+ GDNF-positive tumor specimens
We then analyzed PD-L1 and GDNF protein expression and PNI status by conducting immunohistochemistry (IHC) assays of paraffin sections from 145 HNSCC patients. Representative images of negative and positive PD-L1 and GDNF staining are presented in Fig. S1C. The PD-L1 and GDNF expression level in the HNSCC samples were also significantly correlated (r = 0.38, p < 0.001, Fig. 1C). Furthermore, as shown in Fig. 1D, IHC staining with an anti-PGP9.5 antibody (nerve marker) and H&E staining showed that tumor cells were morphologically present within and around the peripheral nerve in the HNSCC PNI+ tissue. When considering the PNI status, GDNF expression was elevated in the PNI+ specimens (p = 0.0193, Fig. S1D), whereas there was no difference in PD-L1 expression between the PNI+ and PNI- specimens (p = 0.644, Fig. S1E), which was similar to TCGA data. Given that PNI is a local extension of cancer cell dissemination along nerves, we further investigated the PD-L1 and GDNF expression levels in the PNI+ vs. PNI- areas in the same tumor specimens. Interestingly, although PD-L1 and GDNF exhibited diffuse staining in the HNSCC specimens, the tumor cells around the nerve demonstrated stronger PD-L1 staining and GDNF was strongly stained in the nerves (Fig. 1D). Additionally, the relative PD-L1 expression was significantly elevated in the PNI+ areas compared with PNI- areas in matched tissues (p<0.001, Fig. 1E).
We also investigated the correlations between PD-L1 expression, GDNF expression and PNI status with clinical and histopathological parameters. As shown in Table S1, positive GDNF expression and the PNI status were significantly correlated with lymphatic metastasis (p = 0.012 and p = 0.007, respectively) and advanced tumor stage (p = 0.007 and p = 0.032, respectively), whereas GDNF expression and the PNI status were not significantly associated with the other parameters evaluated, including age, gender, histology and tumor size. Kaplan-Meier and Cox regression analyses further revealed that GDNF and the PNI status were significantly correlated with decreased overall survival in HNSCC patients (Fig. 1F, Fig. S1E) and that GDNF was an independent predictor of overall survival in the HNSCC patients (Table S2). In contrast, PD-L1 expression was found to not be relevantly correlated with the clinical and histopathological parameters and overall survival in the HNSCC patients (Fig. S1G).
These data suggest that PD-L1 protein expression is higher in cancer cells that exhibit perineural invasion in HNSCC tumor specimens and that GDNF not only participates in cancer cell PNI but also correlates with PD-L1 upregulation.
GDNF incubation stimulates PD-L1 mRNA and protein expression
To further investigate the role of GDNF in mediating PD-L1 upregulation, we treated HNSCC cell lines with a panel of neural growth factors, including GDNF, brain derived neurotrophic factor (BDNF), neurturin (NRTN), artemin (ARTN), neurotrophin 3 (NT-3) and nerve growth factor (NGF), which all play significant roles in promoting PNI of cancer cells. GDNF treatment obviously stimulated a 5–8-fold upregulation of PD-L1 mRNA in the HNSCC cell lines (Fig. 2A). Membranous PD-L1 expression was further examined in the HNSCC cell lines treated with the different neural growth factors, and only GDNF significantly induced upregulation of membranous PD-L1 (Fig. 2B–C). This effect of GDNF was also validated by Western blot analysis of total PD-L1 expression in the HN4, HN6 and HN30 cell lines (Fig. 2D).
Figure 2.

GDNF incubation stimulates PD-L1 mRNA and protein expression. (A) Real-time PCR assays to demonstrate PD-L1 mRNA upregulation in HNSCC cells. Cell lines were treated with vehicle control, NT-3 (30 ng/mL), NGF (30 ng/mL), GDNF (30 ng/mL), ARTN (30 ng/mL), BDNF (30 ng/mL) or NRTN (30 ng/mL) for 24 hours. (B) The flow cytometry examination indicated that GDNF increased PD-L1 protein expression. Cell lines were treated with vehicle control, ARTN (30 ng/mL), GDNF (30 ng/mL) or NGF (30 ng/mL) for 48 hours. (C) Representative flow cytometry analysis of PD-L1 on HN6, HN4 and HN30 cells in (B). (D) Western blot detection showed that PD-L1 protein expression was increased under stimulation. Cell lines were treated with a vehicle control, GDNF (30 ng/mL) or IFN-γ (20 ng/mL) for 48 hours. (E) A real-time PCR assay indicated that RET inhibitors abrogated GDNF-induced PD-L1 upregulation. Cell lines were treated with vehicle control, RETi (5 μmol/L), GDNF (30 ng/mL), or their combination for 24 hours. ***, p < 0.001 compared with the vehicle group. (F) PD-L1 protein expression was determined by flow cytometry after cells were treated with vehicle control, RETi (5 μmol/L), GDNF (30 ng/mL), or their combination for 48 hours. ***, p < 0.001 compared with the control group.
A previous study revealed that rearranged during transfection (RET) was a transmembrane receptor tyrosine kinase (RTK) activated by GDNF family ligands. The multi-kinase inhibitor, regorafenib, exhibits great selectivity for RET and can effectively prevent RET phosphorylation.22 Therefore, we used regorafenib to further demonstrate the role of GDNF in mediating PD-L1 upregulation. Indeed, regorafenib-mediated RET inhibition (RETi) indicated a significant downregulation of GDNF-mediated PD-L1 mRNA and protein expression (Fig. 2E-F). Taken together, these data indicate that GDNF plays a significant role in mediating PD-L1 expression.
Dorsal root ganglion (DRG)-released GDNF mediates HNSCC cell PNI and PD-L1 expression
A previous co-culture assay of cancer cells with dorsal root ganglion (DRG) revealed that neural-released GDNF was a potent chemoattractant that could facilitate neural tracking of pancreatic cancer cells.20 To assess the role of GDNF during neural invasion in HNSCC, a co-culture assay of cancer cells with DRG was optimized to investigate cancer-nerve interactions. Excised DRG were grown in matrigel, and HN4 cancer cells were added to the media. HN4 cells invaded the matrigel and migrated along the neurites in the control group at day 4, while the cells treated with an RET inhibitor, regorafenib, obviously reduced the nerve invasion area by the cancer cells (Fig. 3A), indicating that neural-released GDNF also plays a role in neural invasion of HNSCC cells. To further determine whether DRG-released GDNF promotes PD-L1 protein expression, conditioned media from DRG cultures that were collected 4 d after excised murine DRG that were placed in matrigel were added to HNSCC cell lines. As shown in Fig. 3B-D, the DRG-driven conditioned media significantly enhanced PD-L1 mRNA and protein expression, while RETi showed a significant abrogation of PD-L1 upregulation both at the mRNA and protein levels. Taken together, these results suggest that GDNF secreted by nerves may not only promote cancer cell neural invasion but also enhance PD-L1 expression.
Figure 3.
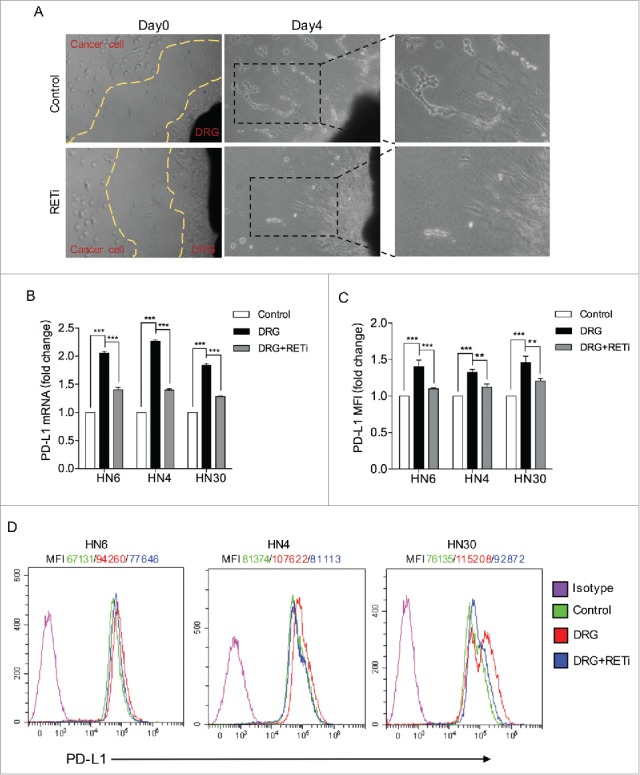
GDNF signaling inhibition in dorsal root ganglion (DRG) promoted PD-L1 protein expression. (A) In a nerve-cancer co-culture in vitro assay, co-culture of rat DRG with HN4 cells in matrigel permitted the assessment of the degree of PNI. By day 4, the HN4 cells exhibited invasion that extended along the neuritis in the control group, while the RET inhibitor treatment blocked the invasion. (B) Real-time PCR detection indicated that RETi abrogated DRG-mediated PD-L1 mRNA upregulation. Cell lines were treated with vehicle control, conditioned media from dissociated DRG nerve cell cultures or the conditioned media combined with RETi (5 μmol/L) for 24 hours. *** means p < 0.001 compared with the control group. (C) The flow cytometry examination indicated the PD-L1 protein levels. Cell lines were treated with vehicle control, conditioned media from dissociated DRG nerve cell cultures or the conditioned media combined with RETi (5 μmol/L) for 48 hours. ** means p < 0.01; *** means p < 0.001 compared with the control group. (D) Representative flow cytometry analysis of PD-L1.
GDNF-induced PD-L1 expression is dependent on the JAK2-STAT1 signaling pathway
Because GDNF activates several signaling pathways, it is of great interest to pinpoint which signaling cascades are involved in GDNF-mediated PD-L1 expression. To this end, we first treated HNSCC cell lines with GDNF (30 ng/mL) or vehicle control for 15 min and cell signaling pathways were analyzed. The GDNF treatment obviously activated signaling mediators, such as Erk1/2 (Thr202/Tyr204), Akt (Ser473), Stat1 (Tyr701) and Stat3 (Ser727) phosphorylation (Fig. 4A-B). Given that PD-L1 upregulation was linked with multiple mechanisms, such as AKT-mTOR and JAK-STAT signaling activation, we then applied several inhibitors to further investigate the signaling pathways by which GDNF upregulates PD-L1 expression. We found that a specific STAT1 inhibitor, fludarabine (STAT1i), had the highest abrogation effect on GDNF-induced PD-L1 mRNA expression, and cryptotanshinone (STAT3i) also exhibited a lesser extent of inhibition effect. However, other kinase inhibitors, including SCH772984 (ERKi), AZD5363 (AKTi), Roxadustat (p38MAPKi), SP600125 (JNKi) and Pictilisib (PI3Ki) had no obvious inhibitory effect on GDNF-induced PD-L1 mRNA expression, under conditions in which these inhibitors effectively prevented kinase phosphorylation (Fig. 4B, Figs. S2A-D). Consistently, the PD-L1 protein expression level was nearly suppressed by STAT1i and to a lesser extent by STAT3i. To further determine whether GDNF-mediated PD-L1 upregulation was solely STAT1 dependent, we silenced STAT1 and STAT3 expression using shRNA and siRNA technology, respectively (Fig. 4E-F). Interestingly, the STAT1 knockdown potently abolished the GDNF-induced upregulation of PD-L1 protein in both cell lines (Fig. 4G), while STAT3 knockdown did not show an obvious downregulation of GDNF-mediated PD-L1 expression in HN6 cells (Fig. 4H). Moreover, the GDNF incubation also stimulated STAT1 to bind with the promoter region of PD-L1 in the ChIP assays (Fig. S2E). These data indicate that the GDNF-regulated PD-L1 expression was mostly dependent on STAT1 activation in HNSCC cells.
Figure 4.

STAT1 activation regulates GDNF-mediated PD-L1 upregulation. (A) An antibody CHIP assay was used to explore the influence of GDNF on signaling pathways in HNSCC cells. Cell lines were starved for 24 hours and then treated with a negative control or GDNF (30 ng/mL) for 15 min and then harvested for cell signaling analysis. (B) The quantification data are shown for Erk1/2 (Thr202/Tyr204), Akt (Ser473), Stat1 (Tyr701) and Stat3 (Ser727) phosphorylation levels in GDNF-treated cell lines. (C) PD-L1 mRNA determined by real-time PCR analysis in HN6 and HN4 cells pretreated with vehicle control, AKTi (5 μmol/L), ERKi (5 μmol/L), STAT1i (5 μmol/L), STAT3i (5 μmol/L), p38MAPKi (5 μmol/L), JNKi (5 μmol/L) or PI3Ki (5 μmol/L) for 1 hour, flowed by treatment with vehicle control or GDNF (30 ng/mL) for 24 hours. *** means p < 0.001 compared with the control group. (D) PD-L1 protein expression was determined by flow cytometry analysis in HN6 and HN4 cells pretreated with vehicle control, AKTi (5 μmol/L), ERKi (5 μmol/L), STAT1i (5 μmol/L), STAT3i (5 μmol/L), p38MAPKi (5 μmol/L), JNKi (5 μmol/L) or PI3Ki (5 μmol/L) for 1 hour, flowed by treatment with vehicle control or GDNF (30 ng/mL) for 48 hours. *** means p < 0.001 compared with the control group. (E) STAT1 protein expression was evaluated by WB after shRNA transfection. β-actin was used as a loading control. (F) STAT3 protein expression was evaluated by WB after siRNA transfection. β-actin was used as a loading control. (G) A flow cytometry assay indicated that GDNF-mediated PD-L1 protein upregulation despite STAT3 knockdown. Cells lines were treated with SiNC or SiSTAT3 for 48 hours and then treated with vehicle control or GDNF (30 ng/mL) for 48 hours. (H) A flow cytometry assay indicated that GDNF-mediated PD-L1 protein upregulation was diminished when STAT1 was knocked down. ShNC and shSTAT1 cell lines were treated with vehicle control or GDNF (30 ng/mL) for 48 hours.
Given that a significant role of STAT1 in GDNF-mediated PD-L1 expression has been previously demonstrated, we then tested whether GDNF-mediated PD-L1 upregulation was JAK2 dependent. We used a selective JAK2 inhibitor, fedratinib (JAK2i), and a JAK1/3 inhibitor, ZM 39923 HCl (JAK1/3i). Indeed, the JAK2 inhibition demonstrated a strong abrogation of basal and GDNF-induced PD-L1 expression in the HNSCC cell lines examined, both at the mRNA and protein levels, while specific JAK1/3 inhibition did not show an obvious downregulation of GDNF-mediated PD-L1 expression (Fig. 5A-C). Considering this result and that a correlation between JAK2 and STAT1 has been fully demonstrated, we hypothesized that GDNF-activated STAT1 phosphorylation may be mediated by JAK2. Indeed, administration of GDNF to the HNSCC cell lines induced activation of downstream signaling, such as AKT (Ser473), ERK1/2 (Thr202/Tyr204), STAT1 (Tyr701), and STAT3 (Ser727), JAK2 inhibition effectively prevented STAT1 (Tyr701) and STAT3 (Ser727) activation in response to GDNF, and it had no effect on AKT (Ser473) and ERK1/2 (Thr202/Tyr204) phosphorylation (Fig. 5D). Meanwhile, the JAK1/3 inhibitor did not prevent the GDNF-stimulated STAT1 phosphorylation, despite obvious inhibitory effect on JAK1 and JAK3 phosphorylation (Fig. 5E). Taken together, these results indicate that despite the activation effect of GDNF-JAK2-STAT1 or STAT3, GDNF-induced PD-L1 upregulation may mainly depend on JAK2-STAT1 pathway activation.
Figure 5.

GDNF induces PD-L1 expression via JAK2-STAT1 activation. (A) PD-L1 mRNA determined by real-time PCR analysis in HN6 and HN4 cells pretreated with vehicle control, JAK2i (5 μmol/L) or JAK1/3i (5 μmol/L) for 1 hour, followed by treatment with vehicle control or GDNF (30 ng/mL) for 24 hours. *** means p < 0.001 compared with the control group. (B) PD-L1 protein expression was determined by flow cytometry analysis in HN6 and HN4 cells pretreated with vehicle control, JAK2i (5 μmol/L) or JAK1/3i (5 μmol/L) for 1 hour, followed by treatment with vehicle control or GDNF (30 ng/mL) for 48 hours. * means p < 0.05; ***means p < 0.001; “ns” means not significant compared with the control group. (C) Western blot analysis of cell signaling response to GDNF (30 ng/mL) in HN6 and HN4 cells pretreated with JAK2i (5 μmol/L) or a vehicle control for 1 hour. (D) Representative flow cytometry analysis of PD-L1 in HN6 and HN4 cells. (E) Western blot analysis of cell signaling response to GDNF (30 ng/mL) in HN6 and HN4 cells pretreated with JAK1/3i (5 μmol/L) or a vehicle control for 1 hour.
Inhibition of GDNF-JAK2 signaling enhances tumor cell susceptibility to NK cell killing
The expression of PD-1 on T cells and NK cells has been well characterized, and PD-L1 is known to inhibit T-cell immunity and NK cell killing. Given that GDNF represented a significant role in PD-L1 upregulation in HNSCC, we sought to determine the involvement of the GDNF-JAK2 signaling pathway in modulating susceptibility to NK cell activity. Tumor cells were pretreated with RETi (5 μmol/L), GDNF (30 ng/mL), or their combination for 48 hours, and then purified NK cells from different healthy donors were added in at 5:1 ratio for co-culture for 3 hours and NK cell lysis efficiency was detected with an LDH assay. As presented in Fig. 6A, NK cells lysis of HNSCC cell lines was significantly decreased when cells were pretreated with GDNF, although NK cells showed obviously higher specific lysis of HNSCC cells that were pretreated with the combination of RETi and GDNF than those pretreated with GDNF alone. However, the level of tumor cell killing was still lower than the vehicle control. Furthermore, we also investigated whether JAK2 inhibition would prevent the resistance to NK cell killing of HNSCC cells induced by GDNF. As shown in Fig. 6B, the JAK2 inhibitor pretreatment resulted in significantly increased lysis by NK cells compared with the vehicle control, and the resistance to NK cell killing induced by GDNF pretreatment was generally reversed by JAK2 inhibition, in the setting of reduced PD-L1 expression.
Figure 6.

Inhibition of GDNF-RET-JAK2 signaling enhances NK cytotoxicity of HNSCC cells. (A) LDH assay data demonstrated NK cytotoxicity. Tumor cells were pretreated with RETi (5 μmol/L), GDNF (30 ng/ml), or their combination for 48 hours, then purified NK cells were added at a 5:1 ratio for co-culturing for 3 hours. * means p < 0.05; ***means p < 0.001; “ns” means not significant compared with the control group. (B) LDH assay data demonstrated higher NK cell lysis of JAK2i-pretreated cells. Tumor cells were pretreated with JAK2i (5 μmol/L), GDNF (30 ng/ml), or their combination for 48 hours, then purified NK cells were added at a 5:1 ratio for co-culturing for 3 hours. * means p < 0.05; ***means p < 0.001 compared with the control group. (C) Proposed model of nerve-secreted GDNF-mediated PD-L1 upregulation by JAK2-STAT1 signaling contributing to immune surveillance escape.
Collectively, these results suggest that GDNF is mainly secreted by nerves and can activate JAK2-STAT1 signaling phosphorylation to upregulate PD-L1 expression in HNSCC cells, thereby promoting cancer cell escape from NK cell cytotoxicity (Fig. 6C).
Discussion
Malignant tumors can escape immunological surveillance by a process termed “immunoediting,” which turns the immune microenvironment into an immunosuppressive state.26,27 The PD-1/PD-L1 immune checkpoint has been demonstrated as a potent factor that promotes cancer cells to escape the immune system. PD-L1 is expressed in multiple malignancies, such as melanoma, NSCLC, HNSCC, glioblastoma, and ovarian carcinoma. The interaction between PD-L1 and its receptor, PD-1, inhibits T cell-mediated cytolysis and generates an immunosuppressive tumor microenvironment.2,3 Indeed, in a randomized trial of R/M HNSCC, the PD-1 monoclonal antibody, nivolumab, demonstrated a better quality of life and an increased overall survival in a 7.5-month treatment group vs patients treated for 5.1 months with standard therapy.6 However, information regarding the expression levels and regulation mechanisms of PD-L1 in HNSCC is confounded.
Our data indicate that PD-L1 expression is heterogeneous in primary HNSCC tissues, and it is higher in cancer cells with perineural invasion (PNI) in HNSCC tumor specimens. PNI is a mode of cancer progression whereby cancer cells invade in, around, or along the nerves, and it is associated with poor prognosis in patients having pancreatic, prostate, gastric, head and neck, and stomach cancers.28-31 Emerging studies have demonstrated that the nerve microenvironment secretes neurotrophic factors and chemokines that attract cancer cells. GDNF has been demonstrated as an important chemo-attractant for cancer cell neural invasion, including pancreatic cancer and bile duct carcinoma.20,21 We revealed that positive GDNF expression was significantly associated with PNI, nodal metastasis, advanced tumor stage and reduced survival of HNSCC. These suggested that GDNF functions as a critical tumor progression factor. However, the role of GDNF in promoting immunoresistance is rarely touched on. Recently, studies have revealed that the perineural niche is rich in immunocytes that are involved in mediating the PNI by activating RET in cancer cells.23,24 In the current study, we provided clinically novel evidence that GDNF secreted by nerves mediated PD-L1 upregulation in cancer cells in the perineural niche.
We also investigated the mechanism of GDNF-stimulated PD-L1 expression. GDNF has been reported to activate the AKT and ERK1/2 signal pathways.22,32 Although the AKT-mTOR and EGFR/RAS/MAPK pathways have been reported to mediate PD-L1 expression,9,33,34 we found that JAK2/STAT1 signaling was the main pathway for GDNF-induced PD-L1 expression. JAK2/STAT1 signaling is a major common regulator for PD-L1 transcription induced by the INF-γ and EGFR pathways in HNSCC.15 Here, our studies revealed a significant association between PD-L1 expression and activation of the JAK2-STAT1 pathway by GDNF in HNSCC, even though GDNF activated several signaling pathways, including the AKT, ERK and JAK pathways, with a signaling pathway screening assay. Indeed, JAK2 inhibition could obviously abolish GDNF-mediated PD-L1 upregulation, therefore reversing PD-L1 mediated immunoescape of HNSCC cells to NK cell killing. As JAK2 inhibitors are evaluated in clinical trials to treat haematopoietic diseases,35,36 our data will provide a novel approach to improve NK cell cytotoxicity and extend new fields for these kinds of chemicals.
To date, limited studies have evaluated PD-L1 expression in HNSCC, as they have positive rates ranging from 18% to 87%.12-14 In the present study, we observed PD-L1 expression in 46.2% of 145 primary HNSCC specimens. This PD-L1 expression incidence was similar to that observed by Straub et al37 and is more than that observed by Laveniya et al.12 These variable positive rates may be due to several factors, such as the use of different cutoff values for the positivity definition, the utilization of different immunohistochemical antibodies and staining protocols, and the inclusion of a high percentage of HPV+ cases. There is also variability in the prognostic and clinicopathological features of PD-L1 expression in HNSCC. Straub et al and Joao et al demonstrated that PD-L1 positive HNSCC patients had a significantly higher risk for lymph node metastasis as well as disease-and overall tumor-related deaths.37,38 In comparison, similar results were not observed by Laveniya et al,12 who observed a higher frequency of PD-L1 positivity in HNSCC female patients. In the present study, despite the poor overall survival in the HNSCC patients with positive PD-L1 expression, there was no significant correlation between PD-L1 expression and prognostic or clinicopathological features.
In summary, we demonstrated that PD-L1 protein expression is higher in HNSCC tumor cells in the perineural niche. GDNF secreted by nerves can upregulate PD-L1 expression by activating JAK2-STAT1 signaling phosphorylation in HNSCC cells, thereby promoting cancer cell escape from NK cell cytotoxicity. Thus, targeting cancer cell PD-L1 expression through GDNF-JAK2 inhibition represents a potential strategy to treat cancers that are associated with PNI.
Materials and methods
HNSCC patients and tissue samples
A total of 145 HNSCC paraffin-embedded tissue samples were randomly selected from the Department of Oral and Maxillofacial Surgery-Head and Neck Oncology, Shanghai Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine between September 2007 and June 2009. The pathological tumor stages were classified according to the Union for International Cancer Control (UICC) system. The tumor histological grades were determined according to the criteria recommended by the World Health Organization. All patients were treated with surgery with a curative intent and exhibited negative resection margins. Patients with T3 or T4 stages of disease and lymph node metastasis were further treated with radiation with or without chemotherapy after surgery. Written informed consent from all patients and approval of the Hospital Ethic Review Committees was obtained.
Cell culture
HNSCC cell lines (HN4, HN6, HN30) were purchased from NIH. Cells were maintained in Dulbecco's Minimum Essential Medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin and 100 μg/ml streptomycin, and incubated in a humidified atmosphere with 5% CO2 at 37°C.
Chemical compound and cytokine preparation
The kinase inhibitors, including the RET receptor inhibitor, regorafenib (S1178), the STAT1 inhibitor, fludarabine (S1491), the STAT3 inhibitor, cryptotanshinone (S2285), the ERK inhibitor, SCH772984 (S7101), the AKT inhibitor, AZD5363 (S8019), the p38MAPK inhibitor, SB202190 (S1077), the JNK inhibitor, SP600125 (S1460), the PI3K inhibitor, pictilisib (S1065), the JAK2 inhibitor, fedratinib (S2736) and the JAK1/3 inhibitor, ZM39923 HCl (S8004) were purchased from Selleck, China. The compounds were dissolved in DMSO at a concentration of 5 mM and maintained at 4°C. Recombinant human cytokines, including GDNF (#450–10), BDNF (#450–02), NGF (#450–01), ARTN (#450–17), NRTN (#450–11), NT-3 (#450–03) and IFN-γ (#300–02) were purchased from Peprotech, USA and were diluted in sterile PBS.
Immunohistochemistry
Immunohistochemistry was performed as described previously.22 Slides were deparaffinized, rehydrated and heated with citric acid buffer at 95°C for 20 min for antigen retrieval. Sections were cooled and immersed in 0.3% hydrogen peroxide for 20 min to block endogenous peroxidase activity, rinsed in phosphate-buffered saline (PBS) for 5 min and blocked with 3% bovine serum albumin (BSA) at room temperature for 20 min. Tissues were incubated with the indicated primary antibodies in a humidified chamber overnight at 4°C. After several washes with PBS, the sections were incubated with horseradish peroxidase (HRP)-labeled goat anti-mouse or goat anti-rabbit secondary antibody (Gene Tech, China) for 45 min at 37°C. Diaminobenzene was used as the chromogen, and hematoxylin was used to counterstain nuclei. The sections were dehydrated, cleared and mounted. Tumor cells exhibiting brown staining in the cytoplasm, nucleus or membrane were considered positive and were classified into 1 of 4 categories: none (0), weak brown (1+), moderate brown (2+), and strong brown (3+). The proportion of cells with positive staining was divided into 5 categories: 0 (0–5%), 1 (6–25%), 2 (26–50%), 3 (51–75%), and 4 (76–100%).
siRNA and shRNA transfection
For transient RNA silencing, HN4 and HN6 cells were transfected with STAT3 (STAT3-siRNA1: 5′-CGU CAU UAG CAG AAU CUC AdTdT- 3′) or control siRNA (NC: 5′-UUC UCC GAA CGU GUC ACG UdTdT-3′) obtained from Biotend, China, using Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer's instruction. Assays were performed 48 hours after transfection. For stable RNA silencing, we introduced a lentiviral vector with STAT1-shRNA (5′-TTC TCG TCC TGA TAC TTT GGG AdTdT-3′) or STAT1-shNC (5′-UUC UCC GAA CGU GUC ACG UdTdT-3′) and obtained cells using puromycin selection.
Flow cytometry analysis
Membranous PD-L1 was analyzed by flow cytometry analysis. A total of 1 × 106 HNSCC cells were harvested and incubated with PE-anti-human CD274 antibody (BioLegend, USA) for 30 minutes at 4°C and then washed twice. Samples were determined using a flow cytometer (BD Biosciences, USA). All experiments were performed in triplicate. Mean fluorescence intensity (MFI) fold change was calculated by normalizing after subtracting the isotype control MFI.
Western blot analysis
Western blotting was performed as described previously.22 Antibodies against the following proteins were used: PD-L1 (1:1000, #A11893, ABclonal, China), β-actin (1:5000, #AC004, ABclonal), STAT1 (1:1000, #14994, Cell Signaling Technology, USA), p-STAT1 (Tyr701) (1:1000, #7649, Cell Signaling Technology), STAT3 (1:1000, #4904, Cell Signaling Technology), p-STAT3 (Ser727) (1:1000, #94994, Cell Signaling Technology), JAK2 (1:1000, #3230, Cell Signaling Technology), p-JAK2 (Tyr1007) (1:1000, #AP0594, ABclonal) ERK1/2 (1:1500, #4695, Cell Signaling Technology), p-ERK1/2 (Thr202/Tyr204) (1:1500, #4370, Cell Signaling Technology), AKT (1:1000, #4691, Cell Signaling Technology), and p-AKT (ser473) (1:1000, #4060, Cell Signaling Technology). The immunoreactive bands were visualized using an Odyssey® Infrared Imaging System (Bioscience, USA). β-actin was used as a loading control.
Real-time PCR
Total HNSCC cell RNA was extracted with Trizol Reagent (Invitrogen) following the manufacturer's instructions. cDNA was synthesized with a PrimeScript™ RT Reagent Kit (TaKaRa Bio Company, Japan) and stored at −80°C for further used. The cDNA was amplified with a SYBR Premix Ex Taq™ real-time PCR kit (TaKaRa Bio Company, Japan) according to the standard protocol. Specific primers for PCR were the following: PD-L1: Forward: 5′-TGGCATTTGCTGAACGCATTT-3′; Reverse: 5′-TGCAGCCAGGTCTAATTGTTTT-3′, β-actin: Forward: 5′-CATGTACGTTGCTATCCAGGC-3′; Reverse: 5′-CTCCTTAATGTCACGCACGAT-3′.
Chromatin immunoprecipitation (ChIP) assay
Chromatin immunoprecipitation assays were performed as described previously.39 HN4 Cells were transfected with pcDNA3.1-Flag-STAT1 plasmid for 48 hours and serum starved for 12 hours perior to incubation with GDNF (30 ng/ml) for 10 hours. Then formaldehyde-cross-linked chromatin was prepared from 2 × 107 cells, and ChIP assay was performed using the ChIP Assay Kit (P2078, Beyotime, China) according to the standard protocol. Purified DNA was used in the PCR examination with the following primers for the PD-L1 promoter: Forward: 5′-TGGACTGACATGTTTCACTTTCT-3′; Reverse: 5′-CAAGGCAGCAAATCCAGTTT-3′.
Separation of dorsal root ganglions (DRGs) and co-culture
The in vitro co-culture model was done essentially as described previously.40 Briefly, mice (BALB/c, 4 to 6 weeks old) were killed by cervical dislocation. DRGs were harvested rapidly and stored on ice in DMEM, and then implanted in the center of a 20 μL drop of matrigel (BD, USA) in a 6-well plate. At day 2 after DRG implantation, 3 × 104 HNSCC cancer cells were added to the media around the DRG. The RET inhibitor, regorafenib (5 μmol/L), was also added to media daily thereafter. The co-cultures were grown in DMEM without FCS in 37°C and 5% CO2 incubation conditions. Plates were examined every day after the cancer cells were added. Animal welfare and experimental procedures followed the Guide for Care and Use of Laboratory Animals (The Ministry of Science and Technology of China, 2006) and the appropriate ethical regulations of the hospital.
Cell signaling array
The cell signaling pathways activated by GDNF were analyzed with an immune cell signaling antibody array kit (#13792, Cell Signaling Technology) according to the manufacture's introductions. The array kit allows for the simultaneous detection of 19 signaling molecules that are involved in the regulation of the immune and inflammatory responses. Cell lines were starved for 24 hours and then treated with a negative control or GDNF (30 ng/ml) for 15 min, then harvested for signaling assay.
Cellular cytotoxicity assays
NK cell cytotoxicity was determined by cell lysis quantified with an LDH Cytotoxicity Assay Kit (C0017, Beyotime, China) according to the manufacture's introductions. Briefly, HNSCC cells were seeded in 96-well plates at a density of 1 × 103 cells/well. Cells were pretreated with RETi (5 μmol/L), JAK2i (5 μmol/L), GDNF (30 ng/ml), or their combination for 48 hours. Then, purified NK cells at 5:1 ratio were added to the co-culture for 3 hours and cell lysis was analyzed. Specific lysis = (experimental lysis - spontaneous lysis)/(maximal lysis - experimental lysis) × 100. All experiments were performed in triplicate.
Statistical analysis
SPSS version 21 (SPSS Inc., Chicago, IL, USA) was used for the statistical analysis. The associations between GDNF expression, PD-L1 expression, and PNI status and clinicopathologic parameters were analyzed using the Chi-square or Fisher's exact tests when appropriate. The association between the GDNF and PD-L1 was assessed with the Spearman's rank correlation test. The Kaplan-Meier method was used to calculate survival and differences were analyzed with the log-rank test. The Cox proportional hazards model was used to estimate variables related to overall survival. Differences in means were evaluated with the student's t-test and ANOVA. A p value (2-sided) <0.05 was considered significant.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported by National Natural Science Foundation of China (81572646, 81672745); Natural Science Foundation of Shanghai Municipality (15ZR1424600); Project of the Shanghai Science and Technology Committee (14431905800); Cross Research Foundation of Medicine and Science of Shanghai Jiao Tong University (YG2012MS58).
References
- 1.Danilova L, Wang H, Sunshine J, Kaunitz GJ, Cottrell TR, Xu H, Esandrio J, Anders RA, Cope L, Pardoll DM, et al.. Association of PD-1/PD-L axis expression with cytolytic activity, mutational load, and prognosis in melanoma and other solid tumors. Proc Natl Acad Sci U S A 2016; 113:E7769-E77; PMID:27837027; https://doi.org/ 10.1073/pnas.1607836113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer 2016; 16:275-87; PMID:27079802; https://doi.org/ 10.1038/nrc.2016.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boussiotis VA. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N Engl J Med 2016; 375:1767-78; PMID:27806234; https://doi.org/ 10.1056/NEJMra1514296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bellucci R, Martin A, Bommarito D, Wang K, Hansen SH, Freeman GJ, Ritz J. Interferon-gamma-induced activation of JAK1 and JAK2 suppresses tumor cell susceptibility to NK cells through upregulation of PD-L1 expression. Oncoimmunology 2015; 4:e1008824; PMID:26155422; https://doi.org/ 10.1080/2162402X.2015.1008824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blank C, Mackensen A. Contribution of the PD-L1/PD-1 pathway to T-cell exhaustion: An update on implications for chronic infections and tumor evasion. Cancer Immunol Immunother 2007; 56:739-45; PMID:17195077; https://doi.org/ 10.1007/s00262-006-0272-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferris RL, Blumenschein G Jr, Fayette J, Guigay J, Colevas AD, Licitra L, Harrington K, Kasper S, Vokes EE, Even C, et al.. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med 2016; 375:1856-67; PMID:27718784; https://doi.org/ 10.1056/NEJMoa1602252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chow LQ, Haddad R, Gupta S, Mahipal A, Mehra R, Tahara M, Berger R, Eder JP, Burtness B, Lee SH, et al.. Antitumor activity of pembrolizumab in biomarker-unselected patients with recurrent and/or metastatic head and neck squamous cell carcinoma: Results from the phase Ib KEYNOTE-012 expansion cohort. J Clin Oncol 2016; 34:3838-3845; PMID:27646946; https://doi.org/ 10.1200/JCO.2016.68.1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen J, Jiang CC, Jin L, Zhang XD. Regulation of PD-L1: A novel role of pro-survival signalling in cancer. Ann Oncol 2016; 27:409-16; PMID:26681673; https://doi.org/ 10.1093/annonc/mdv615 [DOI] [PubMed] [Google Scholar]
- 9.Lastwika KJ, Wilson W, Li QK 3rd, Norris J, Xu H, Ghazarian SR, Kitagawa H, Kawabata S, Taube JM, Yao S, et al.. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res 2016; 76:227-38; PMID:26637667; https://doi.org/ 10.1158/0008-5472.CAN-14-3362 [DOI] [PubMed] [Google Scholar]
- 10.Atefi M, Avramis E, Lassen A, Wong DJ, Robert L, Foulad D, Cerniglia M, Titz B, Chodon T, Graeber TG, et al.. Effects of MAPK and PI3K pathways on PD-L1 expression in melanoma. Clin Cancer Res 2014; 20:3446-57; PMID:24812408; https://doi.org/ 10.1158/1078-0432.CCR-13-2797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu Y, Chang SS, Lin WC, Hsu JM, Hsu YH, et al.. Deubiquitination and Stabilization of PD-L1 by CSN5. Cancer Cell 2016; 30:925-39; PMID:27866850; https://doi.org/ 10.1016/j.ccell.2016.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Satgunaseelan L, Gupta R, Madore J, Chia N, Lum T, Palme CE, Boyer M, Scolyer RA, Clark JR. Programmed cell death-ligand 1 expression in oral squamous cell carcinoma is associated with an inflammatory phenotype. Pathology 2016; 48:574-80; PMID:27590194; https://doi.org/ 10.1016/j.pathol.2016.07.003 [DOI] [PubMed] [Google Scholar]
- 13.Badoual C, Hans S, Merillon N, Van Ryswick C, Ravel P, Benhamouda N, Levionnois E, Nizard M, Si-Mohamed A, Besnier N, et al.. PD-1-expressing tumor-infiltrating T cells are a favorable prognostic biomarker in HPV-associated head and neck cancer. Cancer Res 2013; 73:128-38; PMID:23135914; https://doi.org/ 10.1158/0008-5472.CAN-12-2606 [DOI] [PubMed] [Google Scholar]
- 14.Cho YA, Yoon HJ, Lee JI, Hong SP, Hong SD. Relationship between the expressions of PD-L1 and tumor-infiltrating lymphocytes in oral squamous cell carcinoma. Oral Oncol 2011; 47:1148-53; PMID:21911310; https://doi.org/ 10.1016/j.oraloncology.2011.08.007 [DOI] [PubMed] [Google Scholar]
- 15.Concha-Benavente F, Srivastava RM, Trivedi S, Lei Y, Chandran U, Seethala RR, Freeman GJ, Ferris RL. Identification of the cell-intrinsic and -extrinsic pathways downstream of EGFR and IFNgamma that induce PD-L1 expression in head and neck cancer. Cancer Res 2016; 76:1031-43; PMID:26676749; https://doi.org/ 10.1158/0008-5472.CAN-15-2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Amit M, Na'ara S, Gil Z. Mechanisms of cancer dissemination along nerves. Nat Rev Cancer 2016; 16:399-408; PMID:27150016; https://doi.org/ 10.1038/nrc.2016.38 [DOI] [PubMed] [Google Scholar]
- 17.Kurtz KA, Hoffman HT, Zimmerman MB, Robinson RA. Perineural and vascular invasion in oral cavity squamous carcinoma: Increased incidence on re-review of slides and by using immunohistochemical enhancement. Arch Pathol Lab Med 2005; 129:354-9; PMID:15737030; https://doi.org/ [DOI] [PubMed] [Google Scholar]
- 18.Shen WR, Wang YP, Chang JY, Yu SY, Chen HM, Chiang CP. Perineural invasion and expression of nerve growth factor can predict the progression and prognosis of oral tongue squamous cell carcinoma. J Oral Pathol Med 2014; 43:258-64; PMID:24822265; https://doi.org/ 10.1111/jop.12133 [DOI] [PubMed] [Google Scholar]
- 19.Airaksinen MS, Saarma M. The GDNF family: Signalling, biological functions and therapeutic value. Nat Rev Neurosci 2002; 3:383-94; PMID:11988777; https://doi.org/ 10.1038/nrn812 [DOI] [PubMed] [Google Scholar]
- 20.Gil Z, Cavel O, Kelly K, Brader P, Rein A, Gao SP, Carlson DL, Shah JP, Fong Y, Wong RJ. Paracrine regulation of pancreatic cancer cell invasion by peripheral nerves. J Natl Cancer Inst 2010; 102:107-18; PMID:20068194; https://doi.org/ 10.1093/jnci/djp456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He S, Chen CH, Chernichenko N, He S, Bakst RL, Barajas F, Deborde S, Allen PJ, Vakiani E, Yu Z, et al.. GFRalpha1 released by nerves enhances cancer cell perineural invasion through GDNF-RET signaling. Proc Natl Acad Sci U S A 2014; 111:E2008-17; PMID:24778213; https://doi.org/ 10.1073/pnas.1402944111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lin C, Lu W, Ren Z, Tang Y, Zhang C, Yang R, Chen Y, Cao W, Wang L, Wang X, et al.. Elevated RET expression enhances EGFR activation and mediates EGFR inhibitor resistance in head and neck squamous cell carcinoma. Cancer Lett 2016; 377:1-10; PMID:27090738; https://doi.org/ 10.1016/j.canlet.2016.04.023 [DOI] [PubMed] [Google Scholar]
- 23.Amit M, Na'ara S, Leider-Trejo L, Binenbaum Y, Kulish N, Fridman E, Shabtai-Orbach A, Wong RJ, Gil Z. Upregulation of RET induces perineurial invasion of pancreatic adenocarcinoma. Oncogene 2017; 36(23):3232-9; PMID:28092668; https://doi.org/ 10.1038/onc.2016.483 [DOI] [PubMed] [Google Scholar]
- 24.Cavel O, Shomron O, Shabtay A, Vital J, Trejo-Leider L, Weizman N, Krelin Y, Fong Y, Wong RJ, Amit M, et al.. Endoneurial macrophages induce perineural invasion of pancreatic cancer cells by secretion of GDNF and activation of RET tyrosine kinase receptor. Cancer Res 2012; 72:5733-43; PMID:22971345; https://doi.org/ 10.1158/0008-5472.CAN-12-0764 [DOI] [PubMed] [Google Scholar]
- 25.Gattelli A, Nalvarte I, Boulay A, Roloff TC, Schreiber M, Carragher N, Macleod KK, Schlederer M, Lienhard S, Kenner L, et al.. Ret inhibition decreases growth and metastatic potential of estrogen receptor positive breast cancer cells. EMBO Mol Med 2013; 5:1335-50; PMID:23868506; https://doi.org/ 10.1002/emmm.201302625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jie HB, Gildener-Leapman N, Li J, Srivastava RM, Gibson SP, Whiteside TL, Ferris RL. Intratumoral regulatory T cells upregulate immunosuppressive molecules in head and neck cancer patients. Br J Cancer 2013; 109:2629-35; PMID:24169351; https://doi.org/ 10.1038/bjc.2013.645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: Integrating immunity's roles in cancer suppression and promotion. Science 2011; 331:1565-70; PMID:21436444; https://doi.org/ 10.1126/science.1203486 [DOI] [PubMed] [Google Scholar]
- 28.Chatterjee D, Katz MH, Rashid A, Wang H, Iuga AC, Varadhachary GR, Wolff RA, Lee JE, Pisters PW, Crane CH, et al.. Perineural and intraneural invasion in posttherapy pancreaticoduodenectomy specimens predicts poor prognosis in patients with pancreatic ductal adenocarcinoma. Am J Surg Pathol 2012; 36:409-17; PMID:22301497; https://doi.org/ 10.1097/PAS.0b013e31824104c5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zareba P, Flavin R, Isikbay M, Rider JR, Gerke TA, Finn S, Pettersson A, Giunchi F, Unger RH, Tinianow AM, et al.. Perineural invasion and risk of lethal prostate cancer. Cancer Epidemiol Biomarkers Prev 2017; 26:719-26; PMID:28062398; https://doi.org/ 10.1158/1055-9965.EPI-16-0237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xia Q, Bai QR, Dong M, Sun X, Zhang H, Cui J, Xi H, Hu XL, Shen Q, Chen L. Interaction between gastric carcinoma cells and neural cells promotes perineural invasion by a pathway involving VCAM1. Dig Dis Sci 2015; 60:3283-92; PMID:26108418; https://doi.org/ 10.1007/s10620-015-3758-x [DOI] [PubMed] [Google Scholar]
- 31.Binmadi NO, Basile JR. Perineural invasion in oral squamous cell carcinoma: A discussion of significance and review of the literature. Oral Oncol 2011; 47:1005-10; PMID:21865078; https://doi.org/ 10.1016/j.oraloncology.2011.08.002 [DOI] [PubMed] [Google Scholar]
- 32.Mulligan LM. RET revisited: Expanding the oncogenic portfolio. Nat Rev Cancer 2014; 14:173-86; PMID:24561444; https://doi.org/ 10.1038/nrc3680 [DOI] [PubMed] [Google Scholar]
- 33.Azuma K, Ota K, Kawahara A, Hattori S, Iwama E, Harada T, Matsumoto K, Takayama K, Takamori S, Kage M, et al.. Association of PD-L1 overexpression with activating EGFR mutations in surgically resected nonsmall-cell lung cancer. Ann Oncol 2014; 25:1935-40; PMID:25009014; https://doi.org/ 10.1093/annonc/mdu242 [DOI] [PubMed] [Google Scholar]
- 34.Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, et al.. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med 2007; 13:84-8; PMID:17159987; https://doi.org/ 10.1038/nm1517 [DOI] [PubMed] [Google Scholar]
- 35.Jamieson C, Hasserjian R, Gotlib J, Cortes J, Stone R, Talpaz M, Thiele J, Rodig S, Pozdnyakova O. Effect of treatment with a JAK2-selective inhibitor, fedratinib, on bone marrow fibrosis in patients with myelofibrosis. J Transl Med 2015; 13:294; PMID:26357842; https://doi.org/ 10.1186/s12967-015-0644-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pardanani A, Tefferi A, Jamieson C, Gabrail NY, Lebedinsky C, Gao G, Liu F, Xu C, Cao H, Talpaz M. A phase 2 randomized dose-ranging study of the JAK2-selective inhibitor fedratinib (SAR302503) in patients with myelofibrosis. Blood Cancer J 2015; 5:e335; PMID:26252788; https://doi.org/ 10.1038/bcj.2015.63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Straub M, Drecoll E, Pfarr N, Weichert W, Langer R, Hapfelmeier A, Götz C, Wolff KD, Kolk A, Specht K. CD274/PD-L1 gene amplification and PD-L1 protein expression are common events in squamous cell carcinoma of the oral cavity. Oncotarget 2016; 7:12024-34; PMID:26918453; https://doi.org/ 10.18632/oncotarget.7593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Oliveira-Costa JP, de Carvalho AF, da Silveira da GG, Amaya P, Wu Y, Park KJ, Gigliola MP, Lustberg M, Buim ME, Ferreira EN, et al.. Gene expression patterns through oral squamous cell carcinoma development: PD-L1 expression in primary tumor and circulating tumor cells. Oncotarget 2015; 6:20902-20; PMID:26041877; https://doi.org/ 10.18632/oncotarget.3939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang X, Cao W, Zhang J, Yan M, Xu Q, Wu X, Wan L, Zhang Z, Zhang C, Qin X, et al.. A covalently bound inhibitor triggers EZH2 degradation through CHIP-mediated ubiquitination. EMBO J 2017; 36:1243-60; PMID:28320739; https://doi.org/ 10.15252/embj.201694058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gil Z, Rein A, Brader P, Li S, Shah JP, Fong Y, Wong RJ. Nerve-sparing therapy with oncolytic herpes virus for cancers with neural invasion. Clin Cancer Res 2007; 13:6479-85; PMID:17975160; https://doi.org/ 10.1158/1078-0432.CCR-07-1639 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.