

ABSTRACT
Although recent therapeutic approaches have revitalized the enthusiasm of the immunological way to combat cancer, still the comprehension of immunity against tumors is largely incomplete. Due to their specific function, CD8+ T cells with cytolytic activity (CTL) have attracted the attention of most investigators because CTL are considered the main effectors against tumor cells. Nevertheless, CTL activity and persistence is largely dependent on the action of CD4+ T helper cells (TH). Thus establishment of tumor-specific TH cell response is key to the optimal response against cancer. Here we describe emerging new strategies to increase the TH cell recognition of tumor antigens. In particular, we review recent data indicating that tumor cells themselves can act as surrogate antigen presenting cells for triggering TH response and how these findings can help in constructing immunotherapeutic protocols for anti-cancer vaccine development.
KEYWORDS: MHC class II transactivator, APC, T helper cells, anti-tumor vaccines
Abbreviations
- APC
Antigen Presenting Cell
- CIITA
class II Transactivator
- CTL
Cytotoxic T Lymphocytes
- DC
dendritic cells
- HCC
Hepatocellular carcinoma
- MHC
Major Histocompatibility complex
- TH
T helper cells
- TAA
tumor associated antigens
Introduction
The idea that the immune system has a prominent role in combating cancer is, at present, a consolidated concept.1 On the other hand, it is also known that the immune system may also promote tumor growth.2 The dual host-protective and tumor-promoting role of the immune system became apparent after the demonstration that tumors may escape the recognition of the immune system by creating tumor antigen-loss variants and also by generating a tissue microenvironment that suppresses the action of anti-tumor effectors. These evidences have been elegantly incorporated in the three Es (Elimination, Equilibrium and Escape) immunoediting model of Shreiber and coll.1
Different mechanisms could lead to the tumor escape. At the tumor cell level, the loss of tumor associated antigens (TAAs) expression may be the result of a progressive adaptation/selection process leading to absence of Major Histocompatibility Complex (MHC)-tumor peptide complexes available for scrutiny by T cells, in particular MHC- class I (MHC-I) tumor peptides recognized by CTL.3 This can be due to downregulation of tumor antigen expression, downregulation of MHC-I expression, defects in antigen processing and loading of tumor peptides onto MHC-I molecules.4 Thus the final result of the immunoediting process could be the emergence of poorly immunogenic tumor cells that are almost invisible to the immune system and consequently being able to escape, progress and spread.5,6 In the tumor microenvironment, tumor cells may secrete immunosuppressive cytokines like Tumor Growth Factor β (TGFβ) and Vascular Endothelial Growth Factor (VEGF),7 recruit regulatory T cells with suppressive function (Treg) and myeloid-derived suppressor cells (MDSC) that block the host-protective antitumor immune response by inhibiting, in different ways, the anti-tumor T lymphocytes.8,9
Role of CD4+ TH cells in anti-tumor immune response
CD4+ TH lymphocytes are conventionally primed and activated against antigens, including TAAs, by professional antigen presenting cells (APCs). Priming is mainly induced by dendritic cells (DC) and less efficiently by macrophages.10 DC endocytose, process and present antigens to CD4+ T cells only in the context of MHC-II cell surface molecules. Depending on their cytokine secretion profile, the primed TH cells can differentiate into several subsets such as T helper 1 (TH1), TH2, and TH17.7,11,12 TH cells were largely ignored as a target in cancer immunotherapy for the old belief that they had a limited role in the direct anti-tumor immune response. But in fact, one of the possible reasons why cancer immunotherapies achieved clinically limited results was likely the lack of a successful triggering for the anti-tumor TH CD4+ T lymphocytes. At present, the crucial role of TH cells in anti-tumor immunity is very well established.13,14 It has become apparent that the preferential polarization of TH cells toward the TH1 phenotype plays a crucial role in the tumor microenvironment by secretion of selected cytokines that support the clonal expansion, proliferation and acquisition of cytolytic activity of CD8+ T cells. Lack of polarized TH cell help results, indeed, in abortion of CTL function.15,16,17,18
Importantly, one of these cytokines, INF-γ, secreted mainly by TH1 cells, exerts additionally strong anti-tumor effects through a variety of mechanisms that include: (a) the enhancement of MHC-I and MHC-II antigen presentation and cross-presentation pathways in many cells including the target cells; (b) the induction of MHC-II and co-stimulatory molecules expression especially on the surface of professional APCs; (c) the production of reactive oxygen species (ROS) and nitric oxide (NO); (d) the stimulation of potent chemoattractant production; (e) the recruitment of monocytes/macrophages and T cells; and (6) the augmentation of cancer cells' susceptibility to extrinsic and intrinsic pathways of apoptosis.19,20,21 TH cells can also mediate tumor cells destruction by releasing additional cytokines, such as IL-4 that, in concert with IFN-γ, recruits both macrophages and eosinophils into the tumor microenvironment. As a matter of fact, the cooperation between these two types of cells portrays an efficient tumoricidal action. The eosinophil peroxidase can synergize with macrophage reactive oxygen intermediates to kill tumor cells and peroxidases can catalyze the oxidation of nitrite to generate further cytotoxic radicals which initiate third party cell killing of tumor cells.22
Thus, even though the CD8+ T cells are the most effective cytotoxic arm of the adaptive immune response, the importance of CD4+ TH cells in anti-tumor immunity, independently from CD8+ T cells, confers an additional crucial level that could complement the action of direct CD8+ T cell mediated cytotoxicity, partially avoiding escape mechanism due to loss of MHC-I-mediated TAA presentation.14
The anti-tumor vaccination approach with CIITA-driven MHC-II-positive cancer cells
The previously described pleiotropic and central role of TH cells in the anti-tumor immune response stimulated our interest toward strategies that may implement the triggering and proliferation of tumor specific TH cells. We reasoned that the initial crucial step was to provide the tumor-specific TH cells with adequate antigen availability (AAA), through not only sufficient amount of TAAs presented by MHC-II molecules but also in a putatively more suitable setting of antigen presentation, that is by the tumor cell itself.23 To this end, we used the MHC-II Transactivator (CIITA) discovered in our laboratory24,25,26,27 to render the tumor cells, most of which are MHC-II-negative, MHC-II-positive in a constitutive manner. The idea underlying this approach was that CIITA-driven MHC-II-positive tumor cells may present their tumor antigens directly to TH cells. The use of CIITA to make tumor cells possible “surrogate providers” for tumor antigen presentation was additionally motivated by previous findings that CIITA controls also the expression of other crucial molecules needed for antigen processing and loading of peptides onto MHC-II molecules such as DM and the invariant chain (In),28 in absence of which an unstable association of the MHC-II molecule ensues with poor antigen presentation properties.29 Moreover, it was elegantly demonstrated that endogenous proteins, thus including tumor antigens, can access in certain circumstances the antigen presentation pathway of MHC-II.30,31,32 Indeed, effective rejection or significant growth retardation of the CIITA-driven MHC-II-positive tumor cells of distinct histological origins was accomplished. Moreover, successfully “vaccinated” mice were shown to be protected from challenge with the MHC-II negative parental tumor.32,33,34
These results verified the effectiveness of our approach not only in triggering an immune response capable of rejecting the tumor or significantly retarding its growth, but also in developing a specific, strong, and long lasting anti-tumor memory. The cellular elements implicated in this response were investigated through adoptive cell transfer and, importantly, it was demonstrated that, beside CD8+ T cells, TH cells alone could transfer full protection against parental tumor cells in naïve recipients.32,33,34 Tumors derived from parental cells showed little infiltrate, represented mainly by macrophages and neutrophils, very few TH cells and absence of DC and CD8+ T cells. In contrast the site of CIITA-transfected tumor cell injection was rapidly infiltrated by TH cells. This was followed by the appearance of DC and CD8+ T cells and by the generation of extensive areas of tumor cell necrosis. Thus TH cells colonized CIITA-tumor tissue before CD8+ T cells and DC, supporting the idea that CIITA-driven MHC-II-positive tumor cells were crucial elements in recruiting and triggering tumor specific TH cells.16 Interestingly, CIITA-tumor vaccinated mice displayed a polarized CD4+ TH1 cell phenotype in tumor-draining lymph nodes with a large percentage of cells secreting IFNγ, as compared to an IL-4-secreting TH2-like cell phenotype found in parental tumor-bearing mice. A relevant finding of our studies was the a strong reduction of CD4+CD25+ regulatory T cells (Tregs) in tumor-draining lymph nodes of CIITA-vaccinated mice.33 This suggests that the anti-tumor polarization generated by the treatment counteracted or prevented the increase and/or the recruitment of Tregs in tumor-draining tissues. Therefore modulating the number and/or the function of Tregs may act as an important “adjuvant” to synergize the protective effect of our vaccination strategy.
CIITA-transfected MHC-II-positive tumor cells prime naïve tumor-specific TH cells in vivo: reassessing an immunological dogma
The above described results, obtained in Balb/C mice displaying an H-2d genetic background, were strongly suggestive of the role of “surrogate APC” by CIITA-driven MHC-II expressing tumor cells. Nevertheless, a basic fundamental question remained: was the initial priming of naïve tumor-specific TH cells triggered only by MHC-II positive tumor cells? Indeed, it could still be possible that MHC-II molecules from tumor cells loaded with TAA peptides derived from necrotic cells or from extracellular vesicles secreted in the tumor microenvironment could be taken up by professional APCs, such as DC, and efficiently presented to naïve TH cells.35,36 To test the effectiveness of CIITA-transfected cells, we investigated the response of C57BL/6 mice, with H-2b genetic background, to syngeneic tumor cells. Interestingly, C57BL/6 mice are characterized by the expression of only one MHC-II molecular subset, the I-A molecule, due to a structural defect in the I-E alpha gene promoter.37 Moreover, in the C57BL/6 H-2b genetic background, a transgenic strain is available, the CD11c.DTR mouse, that expresses the diphtheria toxin receptor (DTR) downstream the CD11c promoter, which is highly expressed in DC.38 Upon treatment with diphtheria toxin (DT) DC are depleted, allowing the assessment of the direct priming of naïve CD4+ T cells by MHC-II-expressing tumor cells. The recent results have clearly demonstrated that C57BL/6 tumor cells expressing CIITA-driven MHC-II molecules are rejected or highly retarded in their growth in syngeneic mice despite the absence of I-E molecules. More importantly, DC depletion in CD11c.DTR mice, did not abrogate the capacity of these mice to reject or reduce the growth of MHC-II positive tumors.39
Taken together, these observations unequivocally demonstrated the validity of our approach as a general strategy to increase the immunogenicity of tumor cells, to stimulate a strong and long lasting adaptive anti-tumor immunity and, importantly, to make a tumor cell an antigen presenting cell of its own TAAs for priming naïve, tumor-specific TH cells (Fig. 1). The latter fact challenges the immunological dogma that dendritic cells are the exclusive cells capable of inducing T cell priming.
Figure 1.
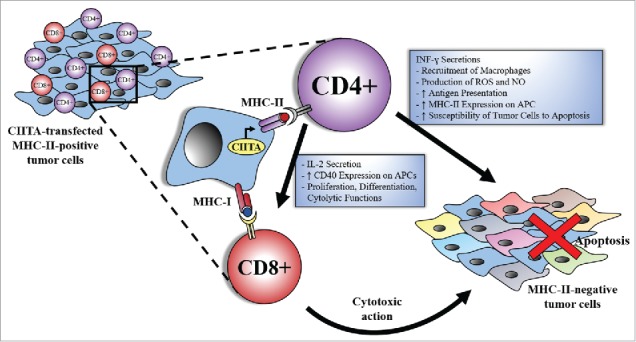
CIITA-transfected MHC-II-positive tumor cells trigger an optimal, CD4+ TH cell dependent, anti-tumor immune response. CIITA-transfected MHC-II-positive tumor cells act as surrogate APCs for TH cell priming against tumor antigens. TH cells exert their function through different mechanisms that could be CD8+ T cell-dependent or independent. Within the first category, TH cells secrete IL-2, that is important for proper activation and proliferation of CD8+T cells and enhances the cytolytic function of CD8+ T cells. On the other hand, TH cells boost the immune response through CD8-independent mechanisms, such as INF-γ secretion. The latter is responsible of the recruitment of macrophages, production of ROS and NO, enhancement of antigen presentation by increasing CIITA expression and consequently MHC-II expression on newly infiltrated APCs, and augmentation of the tumor cells' susceptibility to apoptosis. All of these mechanisms allow the immune system to reject not only the MHC-II-positive tumor cells but also the MHC-II negative tumor cells and possibly better counteracting tumor escape variants that may originate due to the process of immunoediting.
It was of interest that the tumor cells used in the above studies, both parental and CIITA-transfected, did not express co-stimulatory molecules such as the B7.1 (CD80) and B7.2 (CD86) usually required in APC to support optimal stimulation of naïve TH cells. Thus, either CIITA-tumors do not need accessory molecules to perform their APC function in vivo, or other accessory molecules are involved to provide the co-stimulatory signal. This important issue needs to be further investigated to better understand the intimate mechanisms of APC function of CIITA-dependent MHC class II expressing tumor cells.
An additional crucial issue raised by the above studies relates to tissue site where antigen priming takes place. The classical CD4+ TH cells priming occurs in the lymph nodes where DC migrate after having captured the antigen in the periphery.40 As mentioned above, previous studies have shown that CIITA-tumors rapidly become infiltrated first by CD4+ TH cells and subsequently by other cells such as DC, CD8+ T cells and macrophages.33 Thus it is possible that the non-classical priming of naïve tumor-specific CD4+ T cells by CIITA-transfected MHC-II-positive tumor cells may take place at the tumor site. Within this frame it is important to underline the existence of ectopic lymphoid aggregations outside the canonical lymphoid tissues. These structures are referred to as tertiary lymphoid organs or ectopic lymphoid-like structures and have been observed in inflamed and, more interestingly, in tumor tissues.41,42 These peculiar lymphoid formations show many characteristics of lymph nodes associated with the generation of an adaptive immune response.43 Thus, it is possible that tumor cells endowed with CIITA-driven MHC-II expression not only may exert full APC function for priming naive tumor-specific CD4+ T cells but may also perform this activity within the tumor tissue itself where a rudiment of organized lymphoid structure can be generated.
From bench to bedside
Can these experimental observations be integrated in a strategy applicable to human cancer? The straightforward conclusion from the above studies is the evidence of the strong immunogenicity of the MHC-II-tumor peptide complexes present on CIITA-positive tumor cells. Based on this evidence a European consortium (HepaVAC) of laboratories and clinical centers was organized to construct an innovative tumor vaccine against the human hepatocellular carcinoma (HCC) that could include both MHC-I-bound tumor peptides and MHC-II-bound tumor peptides (TUMAPs) isolated from CIITA-modified tumor cells following a methodological purification strategy already described.44
The reason to focus on HCC was motivated by the increasing incidence, severity and lack of resolutive therapeutic tools against this tumor. Indeed, HCC is the most common primary liver malignancy accounting for 6% of all new cancer cases diagnosed worldwide and the third and fifth leading cause of cancer death in men and women respectively.45
The innovation of the HepaVAC approach consists in being the first multi-epitope, multi-target and multi-MHC allele cancer vaccine aimed at stimulating both CD4+ and CD8+ T cells. The combination of both MHC-I and MHC-II-associated peptides (MHC ligandome), will hopefully optimize the efficiency of the vaccine by inducing synergistic effects of tumor-specific cytotoxic CD8+ T cells, CD4+ T helper cells and memory immune responses in the patients (Fig. 2). The concept of analyzing the MHC-II ligandome directly in HCC tumor cells rendered MHC-II-positive by genetic transfer of CIITA, stems directly from our studies and represents a key advance as compared to the classical approach of eluting MHC-II bound peptides from professional APCs (i.e. dendritic cells and macrophages). It is our belief that such a strategy will allow the display and identification of a much broader as well as more representative array of tumor peptides compared to those that professional APCs can display, due to their intrinsic limitation to process and present peptides derived only from exogenously engulfed, phagocytosed material. Tumor-specific peptides have been already identified, the peptide vaccine cocktail has been selected and phase I/II clinical trial is about to start. Results are expected by the end of 2018.
Figure 2.
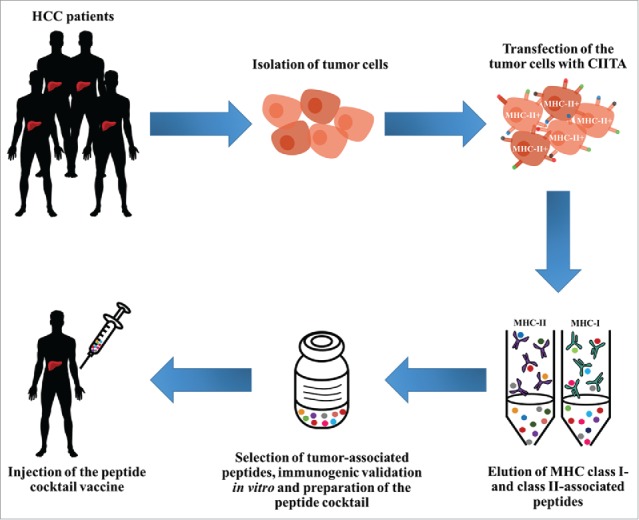
From bench to bedside: CIITA-modified tumor cells as source of MHC-II-bound tumor peptides for innovative vaccines. Human hepatocellular carcinoma (HCC) cells isolated from tumor-bearing patients are modified by genetic transfer of CIITA. Resulting MHC-II-positive cells are then submitted to purification of MHC-II-peptide complexes following the XPRESIDENT™ protocol,44 in association with purification of MHC-I-bound peptides. Peptides bound to both MHC-II and MHC-I are eluted, purified, sequenced and selected on the basis of their specific expression on tumor cells and not normal liver cells or tumors of different histotype. Immunogenic validation of selected tumor-specific peptides is performed in vitro by stimulation of lymphocytes from tumor-bearing patients sharing the MHC genotype from which peptides have been purified. Most immunogenic MHC-II- and MHC-I-bound tumor peptides will compose the “peptide vaccine cocktail” that will be injected into HCC patients. For additional detail on the vaccination strategy and clinical trial see also http://www.hepavac.eu/.
Thus, the study of anti-tumor immune response in animal models has paved the way not only to increase our knowledge on basic mechanism of antigen presentation and triggering of CD4+ TH cells but also to apply the acquired knowledge for testing innovative strategies of anti-tumor vaccination in clinical setting.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
The work was supported by the following grants: European Community FP7 Grant no. 602893 “Cancer Vaccine Development for Hepatocellular Carcinoma-HepaVAC” http://www.hepavac.eu (to RSA, GT and GF); University of Insubria “FAR 2015 and 2016” (to RSA, GF and GT).
References
- 1.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329-60. doi: 10.1146/annurev.immunol.22.012703.104803. PMID:15032581 [DOI] [PubMed] [Google Scholar]
- 2.de Visser KE, Eichten A, Coussens LM. Paradoxical roles of the immune system during cancer development. Nat Rev Cancer. 2006;6(1):24-37. doi: 10.1038/nrc1782. PMID:16397525 [DOI] [PubMed] [Google Scholar]
- 3.Khong HT, Restifo NP. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat Immunol. 2002;3(11):999-1005. doi: 10.1038/ni1102-999. PMID:12407407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garrido F, Algarra I, García-Lora AM. The escape of cancer from T lymphocytes: immunoselection of MHC class I loss variants harboring structural-irreversible “hard” lesions. Cancer Immunol Immunother CII. 2010;59(10):1601-6. doi: 10.1007/s00262-010-0893-2. PMID:20625726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3(11):991-8. doi: 10.1038/ni1102-991. PMID:12407406 [DOI] [PubMed] [Google Scholar]
- 6.Zitvogel L, Tesniere A, Kroemer G. Cancer despite immunosurveillance: immunoselection and immunosubversion. Nat Rev Immunol. 2006;6(10):715-27. doi: 10.1038/nri1936. PMID:16977338 [DOI] [PubMed] [Google Scholar]
- 7.Kim H-J, Cantor H. CD4 T-cell Subsets and Tumor Immunity: The Helpful and the Not-so-Helpful. Cancer Immunol Res. 2014;2(2):91-8. doi: 10.1158/2326-6066.CIR-13-0216. PMID:24778273 [DOI] [PubMed] [Google Scholar]
- 8.Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol. 2004;22:531-62. doi: 10.1146/annurev.immunol.21.120601.141122. PMID:15032588 [DOI] [PubMed] [Google Scholar]
- 9.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253-68. doi: 10.1038/nri3175. PMID:22437938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steinman RM. Lasker Basic Medical Research Award. Dendritic cells: versatile controllers of the immune system. Nat Med. 2007;13(10):1155-9. doi: 10.1038/nm1643. PMID:17917664 [DOI] [PubMed] [Google Scholar]
- 11.Vyas JM, Van der Veen AG, Ploegh HL. The known unknowns of antigen processing and presentation. Nat Rev Immunol. 2008;8(8):607-18. doi: 10.1038/nri2368. PMID:18641646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Golubovskaya V, Wu L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers. 2016;8(3):36. doi: 10.3390/cancers8030036. PMID:26999211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pardoll DM, Topalian SL. The role of CD4+ T cell responses in antitumor immunity. Curr Opin Immunol. 1998;10(5):588-94. doi: 10.1016/S0952-7915(98)80228-8. PMID:9794842. [DOI] [PubMed] [Google Scholar]
- 14.Parish CR. Cancer immunotherapy: The past, the present and the future*. Immunol Cell Biol. 2003;81(2):106-13. doi: 10.1046/j.0818-9641.2003.01151.x. PMID:12631233 [DOI] [PubMed] [Google Scholar]
- 15.Janssen EM, Droin NM, Lemmens EE, Pinkoski MJ, Bensinger SJ, Ehst BD, Griffith TS, Green DR, Schoenberger SP. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature. 2005;434(7029):88-93. doi: 10.1038/nature03337. PMID:15744305 [DOI] [PubMed] [Google Scholar]
- 16.Accolla RS, Lombardo L, Abdallah R, Raval G, Forlani G, Tosi G. Boosting the MHC class II-restricted tumor antigen presentation to CD4+ T helper cells: a critical issue for triggering protective immunity and re-orienting the tumor microenvironment toward an anti-tumor state. Frontiers Oncol. 2014;4:32. doi: 10.3389/fonc.2014.00032. PMID:24600588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsuzaki J, Tsuji T, Luescher IF, Shiku H, Mineno J, Okamoto S, Old LJ, Shrikant P, Gnjatic S, Odunsi K. Direct tumor recognition by a human CD4+ T-cell subset potently mediates tumor growth inhibition and orchestrates anti-tumor immune responses. Sci Rep. 2015;5:14896. doi: 10.1038/srep14896. PMID:26447332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan MP, Dolton GM, Gerry AB, Brewer JE, Bennett AD, Pumphrey NJ, Jakobsen BK, Sewell AK. Human leucocyte antigen class I-redirected anti-tumour CD4(+) T cells require a higher T cell receptor binding affinity for optimal activity than CD8(+) T cells. Clin Exp Immunol. 2017;187(1):124-37. doi: 10.1111/cei.12828. PMID:27324616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kursunel MA, Esendagli G. The untold story of IFN-γ in cancer biology. Cytokine Growth Factor Rev. 2016;31:73-81. doi: 10.1016/j.cytogfr.2016.07.005. PMID:27502919 [DOI] [PubMed] [Google Scholar]
- 20.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75(2):163-89. doi: 10.1189/jlb.0603252. PMID:14525967 [DOI] [PubMed] [Google Scholar]
- 21.Billiau A, Matthys P. Interferon-γ: A historical perspective. Cytokine Growth Factor Rev. 2009;20(2):97-113. doi: 10.1016/j.cytogfr.2009.02.004. PMID:19268625 [DOI] [PubMed] [Google Scholar]
- 22.Hung K, Hayashi R, Lafond-Walker A, Lowenstein C, Pardoll D, Levitsky H. The Central Role of CD4+ T Cells in the Antitumor Immune Response. J Exp Med. 1998;188(12):2357-68. doi: 10.1084/jem.188.12.2357. PMID:9858522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Accolla RS, Tosi G. Adequate antigen availability: a key issue for novel approaches to tumor vaccination and tumor immunotherapy. J Neuroimmune Pharmacol Off J Soc NeuroImmune Pharmacol. 2013;8(1):28-36. doi: 10.1007/s11481-012-9423-7. PMID:23224729 [DOI] [PubMed] [Google Scholar]
- 24.Accolla RS, Carra G, Guardiola J. Reactivation by a trans-acting factor of human major histocompatibility complex Ia gene expression in interspecies hybrids between an Ia-negative human B-cell variant and an Ia-positive mouse B-cell lymphoma. Proc Natl Acad Sci U S A. 1985;82(15):5145-9. PMID:3875096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Accolla RS, Scarpellino L, Carra G, Guardiola J. Trans-acting element(s) operating across species barriers positively regulate expression of major histocompatibility complex class II genes. J Exp Med. 1985;162(4):1117-33. doi: 10.1084/jem.162.4.1117. PMID:3862745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Accolla RS, Jotterand-Bellomo M, Scarpellino L, Maffei A, Carra G, Guardiola J. aIr-1, a newly found locus on mouse chromosome 16 encoding a trans-acting activator factor for MHC class II gene expression. J Exp Med. 1986;164(1):369-74. doi: 10.1084/jem.164.1.369. PMID:3088202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steimle V, Otten LA, Zufferey M, Mach B. Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome). Cell. 1993;75(1):135-46. doi: 10.1016/S0092-8674(05)80090-X. PMID:8402893 [DOI] [PubMed] [Google Scholar]
- 28.Accolla RS, Carra G, Buchegger F, Carrel S, Mach JP. The human Ia-associated invariant chain is synthesized in Ia-negative B cell variants and is not expressed on the cell surface of both Ia-negative and Ia-positive parental cells. J Immunol Baltim Md 1950. 1985;134(5):3265-71. PMID:3872331 [PubMed] [Google Scholar]
- 29.Bikoff EK, Huang LY, Episkopou V, van Meerwijk J, Germain RN, Robertson EJ. Defective major histocompatibility complex class II assembly, transport, peptide acquisition, and CD4+ T cell selection in mice lacking invariant chain expression. J Exp Med. 1993;177(6):1699-12. doi: 10.1084/jem.177.6.1699. PMID:8098731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rudensky AY, Preston-Hurlburt P, Hong SC, Barlow A, Janeway CA. Sequence analysis of peptides bound to MHC class II molecules. Nature. 1991;353(6345):622-27. doi: 10.1038/353622a0. PMID:1656276 [DOI] [PubMed] [Google Scholar]
- 31.Schmid D, Pypaert M, Münz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. 2007;26(1):79-92. doi: 10.1016/j.immuni.2006.10.018. PMID:17182262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meazza R, Comes A, Orengo AM, Ferrini S, Accolla RS. Tumor rejection by gene transfer of the MHC class II transactivator in murine mammary adenocarcinoma cells. Eur J Immunol. 2003;33(5):1183-92. doi: 10.1002/eji.200323712. PMID:12731043 [DOI] [PubMed] [Google Scholar]
- 33.Mortara L, Castellani P, Meazza R, Tosi G, De Lerma Barbaro A, Procopio FA, Comes A, Zardi L, Ferrini S, Accolla RS. CIITA-induced MHC class II expression in mammary adenocarcinoma leads to a Th1 polarization of the tumor microenvironment, tumor rejection, and specific antitumor memory. 2006;12(11 Pt 1):3435-43. doi: 10.1158/1078-0432.CCR-06-0165. PMID:16740768 [DOI] [PubMed] [Google Scholar]
- 34.Frangione V, Mortara L, Castellani P, De Lerma Barbaro A, Accolla RS. CIITA-driven MHC-II positive tumor cells: preventive vaccines and superior generators of antitumor CD4+ T lymphocytes for immunotherapy. Int J Cancer. 2010;127(7):1614-24. doi: 10.1002/ijc.25183. PMID:20091859 [DOI] [PubMed] [Google Scholar]
- 35.Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17(2):97-111. doi: 10.1038/nri.2016.107. PMID:27748397 [DOI] [PubMed] [Google Scholar]
- 36.Pitt JM, Kroemer G, Zitvogel L. Extracellular vesicles: masters of intercellular communication and potential clinical interventions. J Clin Invest. 2016;126(4):1139-43. doi: 10.1172/JCI87316. PMID:27035805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mathis DJ, Benoist C, Williams VE, Kanter M, McDevitt HO. Several mechanisms can account for defective E alpha gene expression in different mouse haplotypes. Proc Natl Acad Sci U S A. 1983;80(1):273-7. PMID:6296871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hochweller K, Striegler J, Hämmerling GJ, Garbi N. A novel CD11c.DTR transgenic mouse for depletion of dendritic cells reveals their requirement for homeostatic proliferation of natural killer cells. Eur J Immunol. 2008;38(10):2776-83. doi: 10.1002/eji.200838659. PMID:18825750 [DOI] [PubMed] [Google Scholar]
- 39.Bou Nasser Eddine F, Forlani G, Lombardo L, Tedeschi A, Tosi G, Accolla RS. CIITA-driven MHC class II expressing tumor cells can efficiently prime naive CD4(+) TH cells in vivo and vaccinate the host against parental MHC-II-negative tumor cells. Oncoimmunology. 2017;6(1):e1261777. doi: 10.1080/2162402X.2016.1261777. PMID:28197387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Segura E, Villadangos JA. Antigen presentation by dendritic cells in vivo. Curr Opin Immunol. 2009;21(1):105-10. doi: 10.1016/j.coi.2009.03.011. PMID:19342210 [DOI] [PubMed] [Google Scholar]
- 41.Dieu-Nosjean M-C, Goc J, Giraldo NA, Sautès-Fridman C, Fridman WH. Tertiary lymphoid structures in cancer and beyond. Trends Immunol. 2014;35(11):571-80. doi: 10.1016/j.it.2014.09.006. PMID:25443495 [DOI] [PubMed] [Google Scholar]
- 42.Aloisi F, Pujol-Borrell R. Lymphoid neogenesis in chronic inflammatory diseases. Nat Rev Immunol. 2006;6(3):205-17. doi: 10.1038/nri1786. PMID:16498451 [DOI] [PubMed] [Google Scholar]
- 43.Irvine DJ, Stachowiak AN, Hori Y. Lymphoid tissue engineering: Invoking lymphoid tissue neogenesis in immunotherapy and models of immunity. Semin Immunol. 2008;20(2):137-46. doi: 10.1016/j.smim.2007.10.010. PMID:18035552 [DOI] [PubMed] [Google Scholar]
- 44.Singh-Jasuja H, Emmerich NPN, Rammensee H-G. The Tübingen approach: identification, selection, and validation of tumor-associated HLA peptides for cancer therapy. Cancer Immunol Immunother CII. 2004;53(3):187-95. doi: 10.1007/s00262-003-0480-x. PMID:14758508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buonaguro L, HEPAVAC Consortium . Developments in cancer vaccines for hepatocellular carcinoma. Cancer Immunol Immunother CII. 2016;65(1):93-9. doi: 10.1007/s00262-015-1728-y. PMID:26093657 [DOI] [PMC free article] [PubMed] [Google Scholar]