

ABSTRACT
Signal transducer and activator of transcription 1 (STAT1) mediates interferon gamma signaling which activates the expression of various genes related to apoptosis, inflammation, cell cycle and angiogenesis. Several experimental and clinical studies have investigated the role of STAT1 in primary tumor growth in breast cancer; however, its role in tumor metastasis remains to be determined. To determine the role of STAT1 in breast cancer metastasis, we analyzed growth and metastasis in WT or STAT1−/− mice orthotopically implanted with metastatic 4T1.2 cells. Primary tumor development was faster in STAT1−/− mice and these mice developed significantly bigger primary tumors and displayed more lung metastasis compared with WT counterparts. STAT1−/− mice showed elevated Ly6G+CD11b+ granulocytic MDSC infiltration in their primary tumors and spleens with concomitant upregulation of Mmp9 and Cxcl1 expression in tumors compared with WT counterparts. Blockade of IL-17A in primary tumor-bearing STAT1−/− mice suppressed accumulation of Ly6G+CD11b+ cells and markedly reduced lung metastasis. These data show that STAT1 is an important suppressor of primary breast tumor growth and metastasis. Importantly, we found anti-IL-17 treatment can rescue STAT1 deficient animals from developing exacerbated metastasis to the lungs which could be important for immunotherapies for immunocompromised breast cancer patients.
KEYWORDS: neutrophil, metastasis, breast cancer, IL-17
Abbreviations
- Cxcl-1
Chemokine (C-X-C motif) ligand 1; Vegf
- Erb2
Receptor tyrosine-protein kinase erbB-2
- G-CSF
Granulocyte-Colony Stimulating Factor
- MDSCs
Myeloid Derived Suppressor Cells
- Mmp9
Matrix metallopeptidase 9
- PDL1
Programmed Death Ligand-1
- TRAIL
Tumor Necrosis factor- Related Apoptosis-Inducing Ligand
- Vegf
vesicular endothelial growth factor
Introduction
Signal transducers and activators of transcription (STATs) are a family of molecules that mediate intracellular signaling triggered mainly by cytokines.1 STAT1 is critical for the expression of several genes in response to interferons2 and has recently been involved in mediating signaling by regulatory cytokines such as IL-353 and IL-27.4 STAT1 is important for induction of the transcription factor T-bet and development of an efficient Th1 response.4 In turn, Th1 cells produce IFN-γ which activates macrophages to produce nitric oxide and mediates tumoricidal activity.5-7 In tumors, STAT1 activation by IFNs affects the expression of genes related to antiproliferative and apoptotic functions such as downregulation of c-myc,8 induction of caspases9 and surface death ligands such as TRAIL.10 Additionally, STAT1 has been shown to be important for immunosurvelliance by enhancing MHCI expression that is required for efficient antigen presentation to cytolytic lymphocytes.11 Interestingly, tumor intrinsic-STAT1 can repress angiogenesis by downregulating the expression of matrix metallo-proteinases (MMPs).12,13 Despite these findings, the role of STAT1 in cancer progression is still controversial. For example STAT1 expression was categorized as an oncogene in endometrial cancer by upregulating MYC and PDL1 expression.14 Conversely, absence of STAT1 abrogated the suppressor activity of tumor associated macrophages (TAMs) and myeloid derived suppressor cells (MDSCs) on T cells.15,16 Also of note, MDSCs are important suppressors of the anti-tumor immune response and deletion of this population has been shown to result in tumor reduction by enhancing the infiltration of cytotoxic T cells.17
Previous studies investigating the role of STAT1 in primary tumor development and growth of breast cancer have reported conflicting findings. While studies using STAT1−/− mice have found that STAT1 plays a critical role in preventing mammary tumor development and restricting tumor growth,11,18-21 Hix et al recently reported that upregulation of STAT1 expression in tumors correlated with disease progression to invasive carcinoma in both humans and mice.22 This study also found that overexpression of STAT1 in breast cancer cells promotes tumor growth in mice by mediating the accumulation of MDSCs into the tumor, indicating a role for tumor cell-intrinsic STAT1 in promoting homing of MDSCs into the tumor.22 These conflicting results reported in the above studies could be due to differences in the experimental models or to the fact that different roles of STAT1 may depend on its expression in particular cell types. Nevertheless, to our knowledge, the role of STAT1 in breast cancer metastasis has never been evaluated.
In this study we examined the role of STAT1 in metastasis of breast cancer using STAT1−/− mice and an orthotopic mouse model of breast cancer metastasis to the lungs.23 Our results demonstrate that STAT1 plays a critical role in controlling primary tumor growth and inhibiting metastasis in breast cancer. Additionally, our studies reveal that the effect of host STAT1 deficiency on tumor metastasis to the lung can be reversed upon IL-17-blockade treatment which correlates with decreased expression of pro-angiogenic factors in the primary tumor.
Results
STAT1 gene deficient mice develop significantly larger primary tumors compared with WT mice
Following implantation of a highly metastatic 4T1.2 cells into mammary fat pads, STAT1−/− Balb/c female mice developed faster and significantly larger primary tumors compared with WT mice. Large primary tumors were evident at earlier time points in STAT1−/− mice compared with primary tumor masses in WT mice beginning at day 14 and thereafter (Fig. 1a). Furthermore, weights of primary tumors harvested from the STAT1−/− group at the end of the experiment were significantly higher compared with primary tumors from WT mice (Fig. 1b). These data indicate that STAT1 plays a critical role in limiting primary tumor growth in breast cancer.
Figure 1.
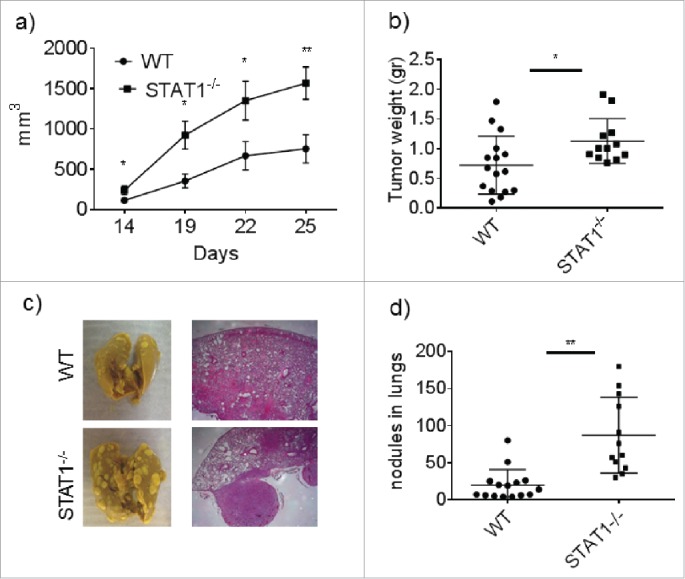
STAT1 deficient mice develop faster tumor growth and enhanced metastasis to the lungs. WT or STAT1−/− mice were injected with 4T1.2 cells in the mammary tissue. (a) Primary breast tumor measurements in WT and STAT1−/− mice during the course of the experiment. (b) Postmortem analysis of primary breast tumor weight in WT and STAT1−/− mice. (c) Representative images and H&E sections of lungs isolated from WT and STAT1−/− primary tumor bearing mice. (d) Counts of nodules per/lung in WT and STAT1−/− mice. Data from 2 independent experiments with an n-value of 5–6 mice per group. *p = 0.05, **p = 0.01.
Absence of STAT1 leads to enhanced tumor metastasis to the lungs
The main cause of death among breast cancer patients is the metastasis of the primary tumor to distant sites; most commonly the bone, lung and liver. Despite the accumulated experimental data regarding the role of STAT1 in breast cancer, to our knowledge no study has shown the impact of host STAT1 deficiency on breast cancer metastasis. Thus we further analyzed the specific role of STAT1 on spontaneous metastasis of breast cancer cells. To this end we analyzed the tumor metastatic nodules in the lung. At day 25, we found lungs from STAT1−/− mice contained significantly more metastatic nodules compared with WT mice (Fig. 1c and d), indicating that STAT1 also plays an important role in restricting metastasis of breast cancer cells.
STAT1−/− mice show increased granulocytic MDSCs accumulation into tumors and spleens during breast cancer
Myeloid derived suppressor cells (MDSCs) are known to play an important role in tumor growth by inhibiting the activation of tumoricidal cytotoxic and T helper cells.17 MDSCs were first characterized as cells expressing Gr-1 and CD11b on their membrane. However, anti Gr-1 antibody recognizes the epitopes of both Ly6C and Ly6G molecules. More recently, MDSCs have been divided into 2 different subsets, one monocytic subset which expresses Ly6Chi Ly6G−CD11b+ and another granulocytic subset which expresses Ly6G+ Ly6Cmed CD11b+ 16. Due to the importance of MDSCs in cancer progression, we evaluated the accumulation of MDSCs in the spleen and tumor by flow cytometry. The myeloid compartment in the spleens of WT and STAT1−/− mice was found to be expanded in breast cancer bearing mice compared with non-cancer bearing mice. However, the expansion of myeloid cells identified by CD11b expression was 3-fold higher in tumor-bearing STAT1−/− mice compared with WT mice (Fig. 2a and c). Next, we investigated surface markers associated with MDSCs. As shown in Fig. 2b, the CD11b+ gated cells were further subdivided by Ly6C and Ly6G expression. WT and STAT1−/− mice presented enhanced accumulation of Ly6ChiLy6G−CD11b+ and Ly6G+Ly6CmedCD11b+ cells in the spleens compared with naïve mice (Fig. 2b). However, the frequency of both myeloid subsets was higher in STAT1−/− mice compared with WT mice (Fig. 2d and e). Interestingly Ly6G+Ly6CmedCD11b+ cells were the predominant subset in WT and STAT1−/− mice (Fig. 2b). Thus, absence of STAT1 results in increased accumulation of myeloid cells that resemble the monocytic-like and granulocytic-like MDSCs. The presence of MDSCs usually correlates with diminished T cell response upon re-stimulation. We confirmed the identity of Ly6G+Ly6CmedCD11b+ cells as MDSCs by their ability to suppress T cell proliferation using an in vitro suppression assay. Ly6G+Ly6CmedCD11b+ cells from tumor bearing WT and STAT1−/− mice were able to suppress responder T cell proliferation compared with control CFSE labeled T cells (Fig. 2f). Interestingly, although STAT1−/− mice showed enhanced accumulation of MDSCs in the spleen, following in vitro stimulation with anti-CD3, splenocytes from tumor bearing STAT1−/− mice showed increased proliferation compared with similarly activated spleen cells from tumor bearing WT mice (Fig. 2g). We also observed increased frequency of CD11b+Ly6C−/Ly6G− cells in spleens of tumor bearing STAT1−/− mice compared with WT counterparts (Fig. 2b). Our analyses show that these cells are a diverse population, which are predominantly F4/80+, CD11c-, NK1.1 (data not shown). Similar to the spleens, tumors of STAT1−/− mice also showed enhanced accumulation of Ly6G+Ly6CmedCD11b+ cells compared with the tumors of WT mice (Fig. 2h and i). PDL1 expression on myeloid cells has been shown to be a mechanism of immunoregulation on breast cancer. Also, PDL1 expression is dependent on STAT1 expression in some circumstances. We found that Ly6ChiLy6G−CD11b+ and Ly6G+Ly6CmedCD11b+ MDSCs isolated from the spleens of WT and STAT1−/− mice expressed comparable levels of PDL1. On the other hand, Ly6ChiLy6G−CD11b+ and Ly6G+Ly6CmedCD11b+ cells in the primary tumors of STAT1−/− mice expressed significantly less PDL1 compared with their WT counterparts (Supplementary Fig. 1). Together, these data show enhanced MDSCs accumulation in absence of STAT1; however the proliferation response of splenic T cells was augmented in these mice. Interestingly, PDL1 expression was dependent on STAT1 only in MDSCs which accumulated in the tumor.
Figure 2.
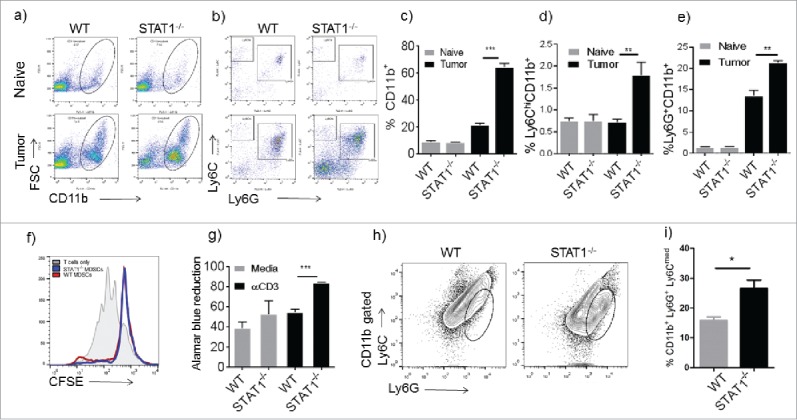
Enhanced recruitment of Ly6G+ cells in STAT1−/− tumor bearing mice. Flow cytometry analysis of splenocytes from WT or STAT1−/− mice. (a) Myeloid cells were identified by flow cytometry by labeling splenocytes with anti CD11b. (b) Myeloid cells gated in Fig. 2a were analyzed for Ly6G and Ly6C markers. (c) Frequencies of CD11b+ cell in the spleens of WT or STAT1−/− naïve or primary tumor bearing mice. (d) Frequencies of Ly6C+CD11b+ cells gated as in Fig. 2b. (e) Frequencies of Ly6G+CD11b+ cells in spleens of WT or STAT1−/− mice gated as in Fig. 2b. (f) Proliferation of CFSE labeled T cells incubated with either sorted WT or STAT1KO CD11b+ Ly6G+ cells. (g) Proliferation of splenocytes from WT or STAT1−/− tumor bearing mice re-stimulated with anti CD3 was evaluated by alamar blue reduction after 72h. (h) Tumors were harvested; myeloid cells were gated with CD11b and analyzed for Ly6G and Ly6C markers. (i) Frequencies of CD11b+Ly6G+Ly6Cmed cells in tumors of WT and STAT1−/− mice. *p = 0.05, **p = 0.01, ***p = 0.0001.
IL-17 neutralization reduces tumor metastasis to the lung in STAT1−/− mice
Ly6G+CD11b+ cell accumulation has been shown to be largely dependent on IL-17 production in several diseases.24,25 Recent reports have shown that IL-17 mediated accumulation of neutrophils has an important role in promoting breast cancer growth and metastasis in an experimental mode l26,27. To determine whether IL-17 was involved in the observed accumulation of Ly6G+Ly6CmedCD11b+ cells28 in the spleens and tumors, as well as in promoting primary tumor growth and/or metastasis in STAT1−/− mice, we first examined IL-17 levels in tumor bearing WT and STAT1−/− mice. Our results indicate that STAT1−/− mice expressed slightly higher Il-17 transcripts in their tumors compared with the tumors of WT mice, although not statistically significant (Fig. 3a). IL-17 cytokine production in stimulated splenocytes of tumor bearing STAT1−/− mice was also slightly higher than in WT mice, although this difference was not statistically significant (Fig. 3b). Interestingly, STAT1−/− mice intrinsically upregulate IL-17 in T cells stimulated with CD3 in vitro, which is enhanced in the presence of 4T1.2 breast cancer cell supernatants (Supplementary Fig. 2a, b). These studies corroborate previous findings that STAT1 deficiency promotes Th17 differentiation in vitro.29
Figure 3.
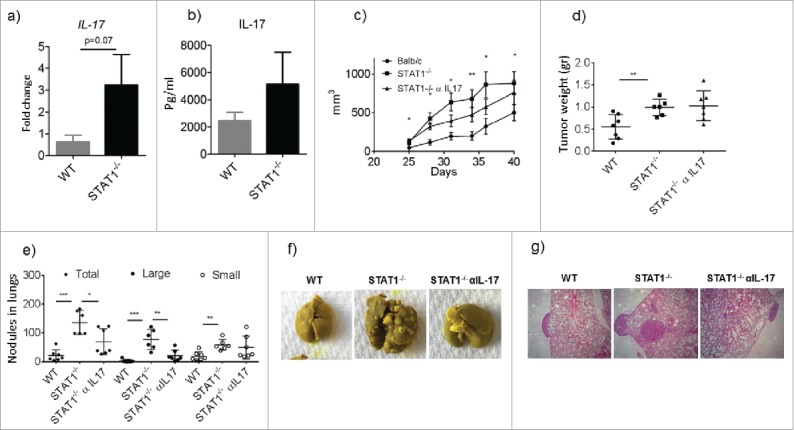
Enhanced metastasis in STAT1−/− mice is controlled by anti-IL-17 treatment. WT and STAT1−/− female mice were injected with 4T1.2 cells in the mammary tissue. After 25 d groups of STAT1−/− mice received either isotype control or anti-IL-17 neutralizing antibody every other day for the rest of the experiment. (a) Gene expression analysis of IL-17 in primary tumors from WT and STAT1−/− mice as determined by RT PCR. (b) IL-17 cytokine production from splenocytes of tumor bearing WT and STAT1−/− mice stimulated with CD3. (c) Progression of primary breast tumor volume in WT, STAT1−/− and STAT1−/− treated with anti-IL-17. * and ** represent significant differences between WT and STAT1−/− mice (d) Primary breast tumor weights at the end of the experiment. (e) Number of metastatic tumor nodules in the lungs of the different groups. (f) Representative pictures of lungs from different experimental groups. (g) H&E staining of lung sections. Representative data from one of 2 independent experiments with an n-value of 5–7 mice per group. *p = 0.05, **p = 0.01, ***p = 0.0001.
Our observed differences in IL-17 production in tumor bearing STAT1−/− and WT mice led us to investigate the impact of anti-IL-17 treatment on MDSC recruitment and subsequent tumor growth and metastasis. Female mice with similar primary tumor size were selected and randomized in groups that received anti-IL-17 treatment or IgG control antibody administration. Administration of IL-17 neutralizing antibody resulted in a slight reduction of primary tumor progression in STAT1−/− mice, however there were no statistical differences in any of the time points compared with STAT1−/− that received the isotype control antibody. Also, no differences were noted in the primary tumor sizes and weights between anti-IL-17 versus control antibody treated STAT1−/− mice at the termination of the experiment (Fig. 3c, d). Interestingly, STAT1−/− mice treated with anti-IL-17 contained significantly fewer metastatic nodules in their lungs compared with STAT1−/− mice that received isotype antibody (Fig. 3e, f and g).
Anti-IL-17 treatment affects immune populations in the spleen
We next evaluated the impact of IL-17 blockade on immune cell populations in the spleen. As expected we found decreased accumulation of Ly6G+CD11b+ cells in STAT1−/− mice treated with anti-IL-17 blocking antibody compared with STAT1−/− mice that received the isotype antibody (Fig. 4a, b). This is in line with previous reports showing reduced accumulation of neutrophils after IL-17 blockade in breast cancer bearing WT mice.26,27 Furthermore, we investigated whether blocking IL-17had an effect on T cell population and activation status by evaluating the expression of CD69. The proportion of both CD4+ and CD8+ cells was lower in tumor bearing STAT1−/− mice in comparison to tumor bearing WT mice (Fig. 4c and d). The frequency of CD4+ CD69+ activated T cells was similar between WT and STAT1−/− mice, but it was enhanced in STAT1−/− mice upon IL-17 neutralization (Fig. 4e). Unexpectedly, the frequency of CD8+CD69+ cells was higher in STAT1−/− mice and was unchanged upon anti-IL-17 treatment (Fig. 4f).
Figure 4.
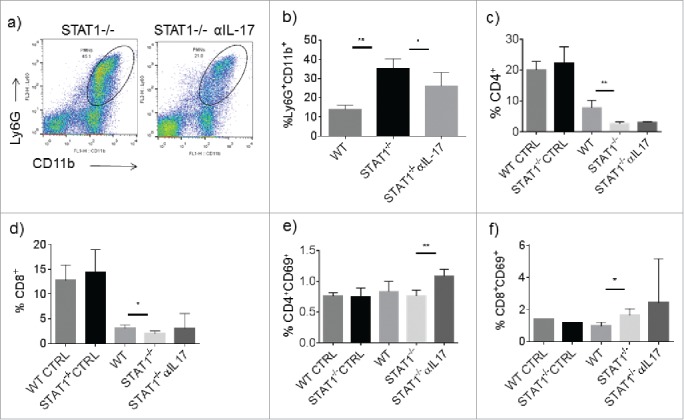
Treatment with anti-IL-17 affects the expansion of Ly6G+ cells in the spleens of STAT1−/− mice. Mice were treated as in Fig. 3. At the end of the experiment different populations of splenocytes were analyzed by flow cytometry. (a) Representative dot plot of Ly6G+CD11b+ cells from STAT1−/− and anti-IL-17 treated STAT1−/− primary tumor bearing mice. (b) Frequencies of Ly6G+CD11b+ cells in the spleens of primary tumor bearing WT, STAT1−/− or STAT1−/− treated with anti-IL-17 neutralizing antibody. Frequencies of total (c) CD4+ cells and (d) CD8+ T cells. Frequencies of activated (e) CD4+ and (f) CD8+ in the different experimental conditions (WT CNTRL: naive WT mice, STAT1−/− CNTRL: naïve STAT1−/− mice, WT: tumor bearing WT mice, STAT1−/−: tumor bearing STAT1−/− mice, STAT1−/−αIL-17: tumor bearing STAT1−/− mice treated with anti-IL-17. *p = 0.05, **p = 0.01.
STAT1−/− mice present enhanced tumor promoting microenvironment that is partially dependent on IL-17
STAT1 expression in tumor cells (fribrosarcoma) is necessary to regulate angiogenesis, tumorigenesis and metastasis,12 however the requirement of STAT1 expression in non-tumor cells for these functions has not been evaluated. Of note, vascularization of the tumor is largely associated with tumor metastasis.30 Because STAT1−/− mice showed enhanced lung metastasis, we investigated vascularization markers in the primary tumor. Interestingly, we found increased CD31 positive cells within the primary tumors of STAT1−/− mice compared with WT mice (Fig. 5a and b). STAT1−/− mice that received anti-IL-17 treatment showed slightly lower expression of CD31 compared with STAT1−/− mice that received the isotype antibody, however, the difference between the groups was statistically not significant (Fig. 5a and c). A recent report showed the critical importance of Ly6G+ cells for inducing tumor growth and metastasis during breast cancer.26,27 We therefore evaluated Ly6G+ cells in the primary tumor by immunofluorescence microscopy. As noted earlier, WT and STAT1−/− mice showed Ly6G+ cell accumulation in their primary tumors, which was higher in STAT1−/− mice compared with WT mice. Additionally, Ly6G+ cell accumulation was reduced by anti-IL-17 treatment, although this was not statistically significant (Fig. 5a, d and e). Since STAT1 is a critical mediator of IFN-γ signaling, we evaluated the expression of iNOS, a molecule known to be induced by IFN-γ/STAT1 pathway in macrophages and related to their tumoricidal functions.7 We found increased iNOS expression in primary breast tumors of WT and STAT1−/− compared with normal mammary gland tissue, however, the expression of iNOS in STAT1−/− mice was not affected upon IL-17 neutralization (Fig. 6a). We also evaluated the expression of genes related with tumor progression and metastasis. Interestingly, the expression of arginase, a molecule expressed in tumor promoting macrophages and canonical marker for M2 macrophages,5 was highly increased in STAT1−/− mice compared with WT mice. Of note, primary tumors from STAT1−/− mice treated with anti-IL-17 showed significantly reduced arginase expression (Fig. 6b). To further understand the mechanism of increased influx of Ly6G+ cells into primary tumors, we evaluated the expression of Cxcl1 a chemokine involved in neutrophil and granulocytic MDSC chemotaxis and poor breast cancer prognosis.31,32 In line with elevated numbers of granulocytic MDSCs in STAT1−/− mice, the expression of Cxcl1 was higher in STAT1−/− mice than WT mice; interestingly Cxcl1 expression in STAT1−/− mice was reduced upon treatment with anti-IL-17 (Fig. 6c). Since primary tumors of STAT1−/− showed enhanced vascularization compared with WT mice, we investigated the expression of genes related with angiogenesis such as Mmp9 and Vegf. Interestingly, Mmp9 expression was higher in STAT1−/− compared with WT tumor bearing mice (Fig. 6d). Anti-IL-17 treatment resulted in reduced the expression of Mmp9 in STAT1−/− tumor bearing mice. The expression of granzyme B and Vegf were similar in WT and STAT1−/− and unaffected upon anti IL17 treatment in primary tumors (Fig. 6e and f). It is also known that in addition to IL-17, G-CSF also plays an important role in neutrophil expansion.26 Hence, we measured the expression of G-CSF in the tumors. Our data indicates that tumors of STAT1−/− mice expressed significantly higher levels of G-CSF, compared with the tumors of WT mice (Fig. 6g). Anti-IL-17 treatment resulted in decreased expression of G-CSF in tumor bearing mice STAT1−/− mice. Next, we examined the effects of STAT1 deficiency on the expression of genes associated with granulocytic MDSC phenotype during breast cancer progression and metastasis. Gene expression of c-Kit, Nos2, Prok2, S100a8, S100a9, which have been shown to be associated with the granulocytic MDSC phenotype in some breast cancer models,26 were examined in sorted CD11b+ Ly6G+ cells in spleens of tumor bearing WT and STAT 1−/− mice. Interestingly, our data shows no significant differences in the expression of these genes between WT and STAT1−/− Ly6G+ CD11b+ cells (Supplementary Fig. 2c-g). It appears from our data that the role STAT1 plays in our breast cancer model is mostly associated with granulocytic MDSC accumulation, and does not affect the phenotype of these cells.
Figure 5.
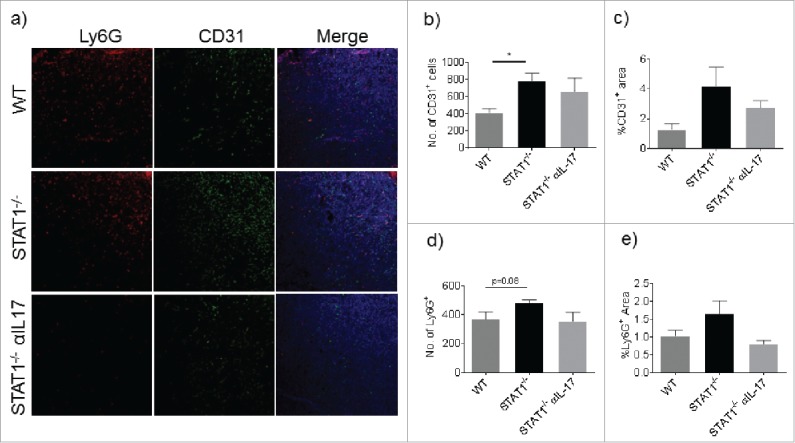
STAT1−/− mice present enhanced vasculature in the primary tumor. Cryosections of primary tumors were processed for immunostaining of CD31 and Ly6G. (a) Representative Z stack projections of sections immunostained with anti Ly6G (red) anti CD31 (Green) and stained with DAPI (Blue). (b) Number of CD31+ cells per section. (c) Percentage of area stained by anti CD31. (d) Number of Ly6G+ cells per section. (e) Percentage of positive area stained with anti Ly6G. Data represent analysis of sections from 3 mice per group. *p = 0.05.
Figure 6.
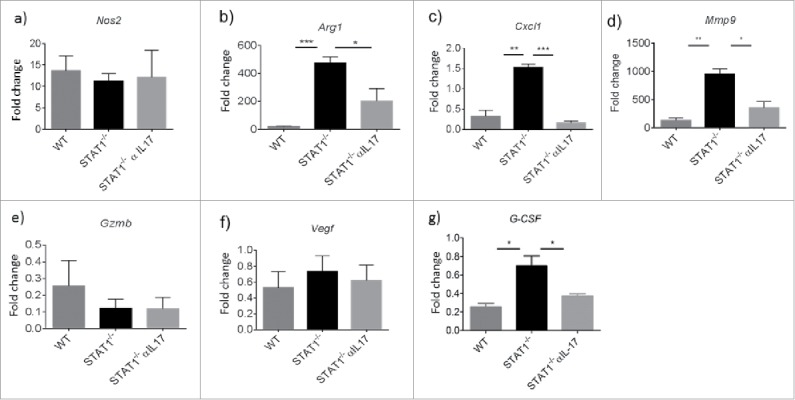
Dysregulated gene expression in the primary tumor of STAT1−/− is partially dependent on IL-17. Gene expression analysis of primary tumors from WT, STAT1−/− or STAT1−/− treated with anti-IL-17 neutralizing antibody by real time PCR. Gene expressions of (a) Nos2, (b) Arg1, (c) Cxcl1, (d) Mmp9, (e) Granzyme B, (f) Vegf (g) G-CSF respectively. Data obtained from 3 different mice per group. *p = 0.05, **p = 0.01, ***p = 0.0001.
Deficiency in CD8+ cell infiltration in primary tumor of STAT1−/− mice is restored upon anti-IL-17 treatment
A well-known mechanism for controlling tumor progression is the infiltration of T cells. We explored the infiltration of T cells into the tumors of WT and STAT1−/− mice by flow cytometry. STAT1−/− mice presented decreased accumulation of CD4+ T cells compared with WT mice (Fig. 7a). Additionally, CD4+CD69+ activated T cells were also less frequent in tumors from STAT1−/− mice and were unaffected by the treatment with anti-IL-17 (Fig. 7c). Similar to CD4+ T cells, the frequency of CD8+ T cells was lower in tumors of STAT1−/− mice compared with WT mice (Fig. 7b). The infiltrating CD8+ cells were found to be activated in WT and STAT1−/− mice as assessed by CD69 expression. Interestingly, the treatment with anti-IL-17 in STAT1−/− mice restored the frequencies of CD8+ cells similar to those found in tumors from WT mice (Fig. 7d). However, the expression of granzyme b, an important molecule that mediates tumor toxicity by CD8 and NK cells was similar between WT and STAT1−/− mice and unaffected upon anti-IL-17 treatment (Fig. 6e).
Figure 7.
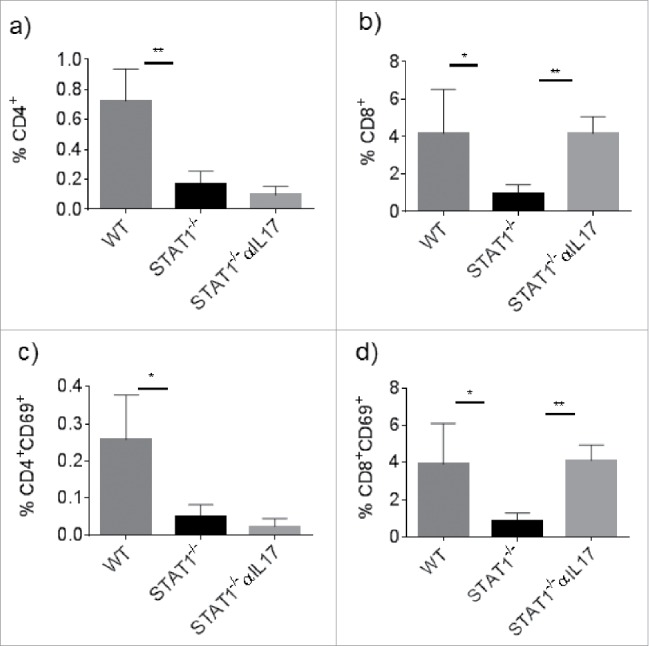
Impaired infiltration of CD4+ and CD8+ cells in the primary tumor of STAT1−/− mice. Primary tumors from WT, STAT1−/− or STAT1−/− anti IL-17 treated mice were processed to obtain single cell suspension and analyzed by flow cytometry. Frequency of total (a) CD4+ and (b) CD8+ T cells within the primary tumor. Percentage of (c) CD4+ or (d) CD8+ activated T cells. Data from 4 mice per group. *p = 0.05, **p = 0.01.
Discussion
STAT1 has important roles in the function and activation of immune cells. Here, we show that STAT1 is necessary to control tumor growth and metastasis in an orthotopic model of breast cancer. Mechanistically, host STAT1 deficiency resulted in increased accumulation of Ly6G+Ly6C−CD11b+ cells resembling granulocytic MDSCs and overexpression of molecules associated with tumor progression and metastasis such as Mmp9, Cxcl1, arginase and CD31. The accumulation of Ly6G+Ly6C−CD11b+ cells in the spleen of STAT1−/− mice was reduced upon IL-17 blockade along with Mmp9, Cxcl1 and arginase expression. These data identify that STAT1 deficiency in non-tumor cells generates a favorable microenvironment for tumor metastasis. Also of note, the uncontrolled metastasis found in STAT1−/− mice can be restored to basal levels upon IL-17 blockade, suggesting a negative regulation of the STAT1 on IL-17 induced genes involved in tumor metastasis.
Type I and II interferons are canonical inducers of STAT1 activation. The gene expression activated by IFNs in tumor cells has been shown to be mainly related to tumor apoptosis.33 Earlier studies have found beneficial effects of IFN therapy against tumors, however with high toxicity effects.34-36 Similarly, different studies using different models of breast cancer in mice have found an important role of STAT1 in suppressing tumor growth.18-20 Interestingly, STAT1−/− mice were reported to have an increased susceptibility of developing spontaneous breast tumors compared with WT mice.20,21 Also, targeted deletion of STAT1 in mammary epithelia cells resulted in accelerated tumor growth.18 Although these findings suggest a tumor intrinsic importance for STAT1 regulating tumor growth, our studies have found an important role for STAT1 in non-tumor cells for controlling tumor growth. Orthotopic injection of 4T1.2 cells resulted in increased tumor growth and metastasis in STAT1 deficient mice. In addition, we found a remarkable increase in lung metastasis in STAT1−/− mice. The relationship between STAT1 and metastasis suppression was shown to be important in a fibrosarcoma model, where loss of STAT1 in fibrosarcoma cells resulted in enhanced metastasis and angiogenesis.12 In fact, we found enhanced expression of angiogenic factors (MMP9) and enhanced micro vessel density in tumors of STAT1−/− mice which may favor the enhanced metastasis to the lungs. Increased Mmp9 expression in tumors of STAT1−/− mice is perhaps not surprising as IFN-β, a STAT1 activator, has been shown to downregulate MMP9 expression in neutrophils from tumor bearing mice to basal levels.37
An important cytokine for breast cancer growth and metastasis is IL-17 which drives the expansion of Ly6G+Ly6C−CD11b+ cells.26,27 In a model of melanoma, IFN-γ deficient mice showed enhanced tumor growth whereas IFN-γ−/−/IL-17−/− double KO mice showed smaller tumors38 indicating that IFN-γ could be inhibiting tumor growth by suppressing IL-17 production. In the present study, we found that IL-17 was increased in tumor bearing STAT1−/− mice, and host STAT1 deficiency also led to CD11b+Ly6G+ accumulation in the spleens which was at least in part mediated by IL-17, as STAT1−/− mice treated with anti-IL-17 antibody showed a marked reduction in CD11b+Ly6G+ accumulation. In addition, we found that neutralization of IL-17 diminished expression of proangiogenic factors such as Mmp9, which is overexpressed in tumor infiltrating CD11b+Ly6G+ cells in breast cancer.27 Importantly, it has been shown that MMP9+ CD11b+Ly6G+ cells induce tumor angiogenesis and vascularization.39 It is possible that the reduced expression of Mmp9 in tumors from anti-IL-17-treated STAT1−/− mice in the present study could be due to the IL-17 induced Mmp9 expression in CD11b+Ly6G+ cells. Previous studies have shown that CXCL1 enhances accumulation of CD11b+Ly6G+ cells into the tumors which promotes tumor cell survival,31 and that IL-17 is an important cytokine for CXCL1 induction.40,41 In the present study, we found that Cxcl1 expression was increased in the tumors of STAT1−/− mice in an IL-17 dependent manner. Collectively, these data suggest a pathologic loop of mediators of tumor growth and metastasis that are negatively regulated by STAT1.
The expansion of Ly6G+CD11b+ cells into the tumor and spleens has been reported to be IL-17 dependent by increasing the production of granulocytic colony stimulating-factor (G-CSF) that in turn expands and maintains an elevated number of Ly6G+CD11b+ cells.26 In line with these studies, we observed increased G-CSF expression in tumors of STAT1−/− mice compared with WT mice, which was decreased by anti-IL-17 treatment. Interestingly, the suppressor activity of monocytic MDSCs and TAMs has been reported to be dependent on STAT1 expression.15,16 In the present study, despite the higher accumulation of MDSCs in the spleen of tumor-bearing STAT1−/− mice, the proliferative capacity of splenic T cell in response to anti-CD3 stimulation was higher than that of WT mice, suggesting an intact capacity of T cells to proliferate even in presence of high numbers of MDSCs in STAT1−/− mice. This phenomenon could be explained by lower levels of PDL-1 expressing MDSCs observed in STAT1−/− tumor bearing mice compared with the WT tumor bearing mice (Supplementary Fig. 1c, d). These results are quite different from the spleen where similar levels of PDL1 are observed among monocytic and granulocytic MDSCs between tumor bearing WT and STAT−/− mice (Supplementary Fig. 1c, d). Indeed, granulocytic MDSCs from the spleen are able to suppress T cell proliferative responses equally between tumor bearing WT and STAT1−/− mice (Fig. 2f).
Additionally, neutralization of IL-17 in STAT1−/− mice resulted in increased frequencies of activated CD4+ T cells in the spleens along with increased infiltration of activated CD8+ T cells into the tumors. The increased infiltration of CD8+ T cells is in line with a recent report showing induction of CD8+ T cell infiltration after IL-17 neutralization in WT mice bearing breast cancer26 and correlates with the decreased accumulation of Ly6G+CD11b+ cells after IL-17 neutralization. Interestingly, despite increased infiltration of CD8+ T cells, granzyme b expression was not restored by anti-IL-17 treatment of STAT1−/− mice. Of course, granzyme b is also expressed by NK cells as well as other non-cytotoxic cells such as basophils and mast cells.42 Moreover, as a protease which is present in granules of activated CD8+ T cells, increased de novo gene expression of Gzmb may not be reflective of an increase in the number of activated CD8+ T cells. We observed interesting differences in activation marker expression between CD4+ and CD8+ T cells in the tumors and spleens of tumor bearing STAT1−/− mice treated with IL-17 blocking antibody. We previously showed a differential requirement for STAT1 in CD4+ vs. CD8+ T cell activation.43 In that study, STAT1−/− CD4+ and CD8+ T cells were activated via separate signaling pathways.43 It is possible that similar effects of IL-17 blockade in STAT1−/− CD4+ and CD8+ T cells are also occurring in our breast cancer model.
It has been largely thought that the control of tumor growth by STAT1 is mainly related to the ability to induce apoptosis and provide immunosurveillance of tumors.44 Our data provide evidence of a new mechanism of how STAT1 can repress tumor growth by limiting the accumulation of MDSCs and the expression of pro-angiogenic genes such as arginase1, Cxcl1 and Mmp9, which was reversed upon IL-17 blockade. This implies that anti-IL17 treatment may decrease the expression of pro-metastatic factors even under conditions where the host is immunologically compromised and is unable to control breast cancer metastasis. Finally, our study suggests investigating the role of inhibition of this inflammatory loop by using STAT1 agonists to control breast cancer growth and metastasis.
Methods
Mice and tumor injections
WT Balb/c mice were purchased from Envigo (previously Harlan laboratories, Indianapolis, IN). STAT1−/− mice were generated and maintained as described previously.45 All female mice were age matched and maintained in compliance with the guidelines of OSU ULAR. 4T1.2 tumor cells were obtained from 4T1.2, a subclone of murine cell line 4T1 cells23,46 and were kindly provided by Dr. Robin Anderson.47 The cells were cultured in RPMI1640 medium supplemented with 10% FBS and 1% penicillin and streptomycin (Life technologies, Carlsbad, CA) at 370C in 5%CO2. All experimental mice were injected with 105 4T1.2 cells in the mammary fat pad by following the OSU IACUC guidelines.
Tumor measurements and lung metastasis counts
Breast tumors were measured every 3 d by using electronic calipers and volume was calculated by using the following formula (volume = 0.52 X a2 X b), where “a” is the smaller superficial diameter and “b” is the larger superficial diameter (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3271140/). Experimental mice were killed at specified time intervals, and tumor weights and lung metastasis were calculated. Lungs were inflated and stored in Bouin's solution (Sigma Aldrich, St. Louis, MO). Lung metastasis was calculated by counting the total number of visible nodules present on the lungs.
Immunohistochemistry
Tumors from WT and the various treatment groups of STAT1−/− mice were cryopreserved in OCT Compound (Fisher Healthcare 23–730–571). Sections 7 micron thick were obtained (Leica Microsystems CM1850 Cryostat) and stained with anti CD31 (Santa Cruz biotechnologies) and anti Ly6G-PE conjugated antibody (Bio Legend) overnight at 4°C. Anti- goat secondary antibody was added during 2.5 hours to detect anti CD31. The sections were stained with DAPI (Bio Legend). Images were captured by using a confocal microscope Zeiss LSM700 (Zeiss, Dublin CA, USA). Z stack projections were obtained with the Zeiss black software and analysis of number of positive cells and percentage of positive area were analyzed by Image J software.48
Flow cytometry analysis
Mice were harvested at respective time points, single cell suspensions were prepared from spleens and tumors. Briefly, spleens were mashed and red blood cells were lysed by ACK lysis buffer. Tumors were pulverized and incubated in buffer containing 1mg/ml DNase and 1mg/ml collagenase at 450C for 15 minutes. Spleens and tumor suspensions were passed through 60micron filters and cells were stained with the respective stain mixes. CD3(clone:145–2C11), CD4(clone: GK1.5), CD8(clone:53–6.7), CD69(clone:H1.2F3), CD11b(clone:M1/70), Ly6g(clone:IA8), Ly6C(clone:HK1.4) and PDL1(clone:10F.9G2) antibodies were purchased from Bio Legend (San Diego, CA). Cells were acquired through Fluorescence Activated Cell Sorter (FACS, BD biosciences, San Jose, CA, USA). Analysis was performed with Flow Jo software (Tree Star Inc., Ashland, OR, USA).
ELISA and T cell proliferation assays
Single cell suspensions were prepared from the spleens at the concentration of 5 × 106/ml. Cells were incubated with 2µg/ml of LEAF purified anti-mouse CD3e (Purchased from Bio Legend, San Diego, CA) in RPMI 1640 supplemented with 10% FBS and 1% penicillin and streptomycin at 370C in 5%CO2 for 72hrs. Cell culture supernatants were collected, production of IL-17 cytokine was analyzed by ELISA. All the capture, detection antibodies and standards of IL-17 were purchased from Bio Legend (San Diego, CA, USA). Cell proliferation was measured by Alamar blue reduction method (Bio-Rad AbD Serotec Inc., Raleigh, NC) Briefly, 10% Alamar blue (Life technologies, Carlsbad, CA) was added at 60 hrs of incubation and reduction of Alamar blue was measured at 72 hrs by measuring the absorbance at 570nm and 670 nm by using spectramax microplate reader and Softmax pro software (Molecular Devices LLC, Sunnyvale, CA, USA).
RT-PCR analysis
Total RNA was extracted from both the spleens and tumors of the experimental mice by TRIzol extraction method (purchased from Life technologies (Carlsbad, CA)). cDNAs were prepared by iScript reverse transcriptase and RT PCR reactions were done by using I Q SYBR green super mix and CFX 96 RT-PCR cycler (purchased from Bio-Rad, Hercules, CA, USA). Primers were selected from the Primer bank website (http://pga.mgh.harvard.edu/primerbank). Data obtained was normalized by using housekeeping gene β-actin and presented as the fold induction over WT mice.
In vivo administrations of neutralizing antibodies
For the in vivo antibody administration, STAT1−/− mice with similar sized tumors were selected, divided into 2 groups. One group was treated with 200µg of InVivo MAb anti-mouse IL-17A (clone: 17F3), Bio X Cell, (Lebanon, NH, USA) and the second group was treated with a similar concentration of isotype control every alternate day for 2 weeks via intraperitoneal route.
Statistics
Statistical differences between groups were analyzed in Graph Pad Prism by using Student T test. Differences were considered statistically significant when the p value was below to 0.05.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to thank Department of Pathology, OSUMC for their kind support allowing us to use the confocal microscopes and other instruments. We thank Ms. Millie Sudhakar for her technical assistance.
Author's contributions
Conceived and design the study: CT and ARS. Performed the experiments: CT, SV, SO, ME and MN. Assisted with the experiments: GV, DA, JK, PA, RS, EM and MP. Acquisition of data: CT, SV, SO. Analysis and interpretation of data: CT, SV, SO. Wrote the manuscript: CT, ARS, SV, SO. Edited the manuscript: SV, SO and ME. Administrative, technical or material support: RKG and ARS Study supervision: CT and ARS.
Grant support
This work was supported by Department of Defense grant BC150072 to ARS and NIH R01 grants (CA109527 and CA153490) to RKG.
References
- 1.Levy DE, Darnell JE Jr.. Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651-62. doi: 10.1038/nrm909. PMID:12209125 [DOI] [PubMed] [Google Scholar]
- 2.Ramana CV, Gil MP, Schreiber RD, Stark GR. Stat1-dependent and -independent pathways in IFN-gamma-dependent signaling. Trends Immunol. 2002;23:96-101. doi: 10.1016/S1471-4906(01)02118-4. PMID:11929133 [DOI] [PubMed] [Google Scholar]
- 3.Collison LW, Delgoffe GM, Guy CS, Vignali KM, Chaturvedi V, Fairweather D, Satoskar AR, Garcia KC, Hunter CA, Drake CG, et al.. The composition and signaling of the IL-35 receptor are unconventional. Nat Immunol. 2012;13:290-9. doi: 10.1038/ni.2227. PMID:22306691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takeda A, Hamano S, Yamanaka A, Hanada T, Ishibashi T, Mak TW, Yoshimura A, Yoshida H. Cutting edge: role of IL-27/WSX-1 signaling for induction of T-bet through activation of STAT1 during initial Th1 commitment. J Immunol (Baltimore, Md: 1950). 2003;170:4886-90. doi: 10.4049/jimmunol.170.10.4886. PMID:12734330 [DOI] [PubMed] [Google Scholar]
- 5.Chang CI, Liao JC, Kuo L. Macrophage arginase promotes tumor cell growth and suppresses nitric oxide-mediated tumor cytotoxicity. Cancer Res. 2001;61:1100-6. PMID:11221839 [PubMed] [Google Scholar]
- 6.Vicetti Miguel RD, Cherpes TL, Watson LJ, McKenna KC. CTL induction of tumoricidal nitric oxide production by intratumoral macrophages is critical for tumor elimination. J Immunol (Baltimore, Md: 1950). 2010;185:6706-18. doi: 10.4049/jimmunol.0903411. PMID:21041723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dinapoli MR, Calderon CL, Lopez DM. The altered tumoricidal capacity of macrophages isolated from tumor-bearing mice is related to reduce expression of the inducible nitric oxide synthase gene. J Exp Med. 1996;183:1323-9. doi: 10.1084/jem.183.4.1323. PMID:8666890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dimberg A, Karlberg I, Nilsson K, Oberg F. Ser727/Tyr701-phosphorylated Stat1 is required for the regulation of c-Myc, cyclins, and p27Kip1 associated with ATRA-induced G0/G1 arrest of U-937 cells. Blood. 2003;102:254-61. doi: 10.1182/blood-2002-10-3149. PMID:12637327 [DOI] [PubMed] [Google Scholar]
- 9.Fulda S, Debatin KM. IFNgamma sensitizes for apoptosis by upregulating caspase-8 expression through the Stat1 pathway. Oncogene. 2002;21:2295-308. doi: 10.1038/sj.onc.1205255. PMID:11948413 [DOI] [PubMed] [Google Scholar]
- 10.Miura Y, Tsujioka T, Nishimura Y, Sakaguchi H, Maeda M, Hayashi H, Dong M, Hyodoh F, Yata K, Wada H, et al.. TRAIL expression up-regulated by interferon-gamma via phosphorylation of STAT1 induces myeloma cell death. Anticancer Res. 2006;26:4115-24. PMID:17201122 [PubMed] [Google Scholar]
- 11.Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ, Schreiber RD. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci U S A. 1998;95:7556-61. doi: 10.1073/pnas.95.13.7556. PMID:9636188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang S, Bucana CD, Van Arsdall M Fidler IJ. Stat1 negatively regulates angiogenesis, tumorigenicity and metastasis of tumor cells. Oncogene. 2002;21:2504-12. doi: 10.1038/sj.onc.1205341. PMID:11971185 [DOI] [PubMed] [Google Scholar]
- 13.Battle TE, Lynch RA, Frank DA. Signal transducer and activator of transcription 1 activation in endothelial cells is a negative regulator of angiogenesis. Cancer Res. 2006;66:3649-57. doi: 10.1158/0008-5472.CAN-05-3612. PMID:16585190 [DOI] [PubMed] [Google Scholar]
- 14.Kharma B, Baba T, Matsumura N, Kang HS, Hamanishi J, Murakami R, McConechy MM, Leung S, Yamaguchi K, Hosoe Y, et al.. STAT1 drives tumor progression in serous papillary endometrial cancer. Cancer Res. 2014;74:6519-30. doi: 10.1158/0008-5472.CAN-14-0847. PMID:25267067 [DOI] [PubMed] [Google Scholar]
- 15.Kusmartsev S, Gabrilovich DI. STAT1 signaling regulates tumor-associated macrophage-mediated T cell deletion. J Immunol (Baltimore, Md: 1950). 2005;174:4880-91. doi: 10.4049/jimmunol.174.8.4880. PMID:15814715 [DOI] [PubMed] [Google Scholar]
- 16.Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, De Baetselier P, Van Ginderachter JA. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111:4233-44. doi: 10.1182/blood-2007-07-099226. PMID:18272812 [DOI] [PubMed] [Google Scholar]
- 17.Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162-74. doi: 10.1038/nri2506. PMID:19197294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klover PJ, Muller WJ, Robinson GW, Pfeiffer RM, Yamaji D, Hennighausen L. Loss of STAT1 from mouse mammary epithelium results in an increased Neu-induced tumor burden. Neoplasia (New York, NY). 2010;12:899-905. doi: 10.1593/neo.10716. PMID:21076615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raven JF, Williams V, Wang S, Tremblay ML, Muller WJ, Durbin JE, Koromilas AE. Stat1 is a suppressor of ErbB2/Neu-mediated cellular transformation and mouse mammary gland tumor formation. Cell cycle (Georgetown, Tex). 2011;10:794-804. doi: 10.4161/cc.10.5.14956. PMID:21311224 [DOI] [PubMed] [Google Scholar]
- 20.Chan SR, Vermi W, Luo J, Lucini L, Rickert C, Fowler AM, et al.. STAT1-deficient mice spontaneously develop estrogen receptor alpha-positive luminal mammary carcinomas. Breast Cancer Res. 2012;14:R16. doi: 10.1186/bcr3100. PMID:22264274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schneckenleithner C, Bago-Horvath Z, Dolznig H, Neugebauer N, Kollmann K, Kolbe T, et al.. Putting the brakes on mammary tumorigenesis: loss of STAT1 predisposes to intraepithelial neoplasias. Oncotarget. 2011;2:1043-54. doi: 10.18632/oncotarget.371. PMID:22185785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hix LM, Karavitis J, Khan MW, Shi YH, Khazaie K, Zhang M. Tumor STAT1 transcription factor activity enhances breast tumor growth and immune suppression mediated by myeloid-derived suppressor cells. J Biol Chem. 2013;288:11676-88. doi: 10.1074/jbc.M112.441402. PMID:23486482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lelekakis M, Moseley JM, Martin TJ, Hards D, Williams E, Ho P, Lowen D, Javni J, Miller FR, Slavin J, et al.. A novel orthotopic model of breast cancer metastasis to bone. Clin Ex Metastasis. 1999;17:163-70. doi: 10.1023/A:1006689719505 [DOI] [PubMed] [Google Scholar]
- 24.Miyamoto M, Prause O, Sjostrand M, Laan M, Lotvall J, Linden A. Endogenous IL-17 as a mediator of neutrophil recruitment caused by endotoxin exposure in mouse airways. J Immunol (Baltimore, Md: 1950). 2003;170:4665-72. doi: 10.4049/jimmunol.170.9.4665. PMID:12707345 [DOI] [PubMed] [Google Scholar]
- 25.Ye P, Rodriguez FH, Kanaly S, Stocking KL, Schurr J, Schwarzenberger P, Oliver P, Huang W, Zhang P, Zhang J, et al.. Requirement of interleukin 17 receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense. J Exp Med. 2001;194:519-27. doi: 10.1084/jem.194.4.519. PMID:11514607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, Verstegen NJM, Ciampricotti M, Hawinkels LJAC, Jonkers J, et al.. IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522:345-8. doi: 10.1038/nature14282. PMID:25822788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benevides L, da Fonseca DM, Donate PB, Tiezzi DG, De Carvalho DD, de Andrade JM, de Andrade JM, Martins GA, Silva JS. IL17 promotes mammary tumor progression by changing the behavior of tumor cells and eliciting tumorigenic neutrophils recruitment. Cancer Res. 2015;75:3788-99. doi: 10.1158/0008-5472.CAN-15-0054. PMID:26208902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, Mandruzzato S, Murray PJ, Ochoa A, Ostrand-Rosenberg S, et al.. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150. doi: 10.1038/ncomms12150. PMID:27381735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peters A, Fowler KD, Chalmin F, Merkler D, Kuchroo VK, Pot C. IL-27 Induces Th17 differentiation in the absence of STAT1 signaling. J Immunol (Baltimore, Md: 1950). 2015;195:4144-53. doi: 10.4049/jimmunol.1302246. PMID:26408664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29:15-8. doi: 10.1053/sonc.2002.37263. PMID:12516034 [DOI] [PubMed] [Google Scholar]
- 31.Acharyya S, Oskarsson T, Vanharanta S, Malladi S, Kim J, Morris PG, Manova-Todorova K, Leversha M, Hogg N, Seshan VE, et al.. A CXCL1 paracrine network links cancer chemoresistance and metastasis. Cell. 2012;150:165-78. doi: 10.1016/j.cell.2012.04.042. PMID:22770218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zou A, Lambert D, Yeh H, Yasukawa K, Behbod F, Fan F, Cheng N. Elevated CXCL1 expression in breast cancer stroma predicts poor prognosis and is inversely associated with expression of TGF-beta signaling proteins. BMC Cancer. 2014;14:781. doi: 10.1186/1471-2407-14-781. PMID:25344051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meissl K, Macho-Maschler S, Muller M, Strobl B. The good and the bad faces of STAT1 in solid tumours. Cytokine. 2017;89:12-20. doi: 10.1016/j.cyto.2015.11.011. PMID:26631912 [DOI] [PubMed] [Google Scholar]
- 34.Gutterman JU, Blumenschein GR, Alexanian R, Yap HY, Buzdar AU, Cabanillas F, Hortobagyi GN, Hersh EM, Rasmussen SL, Harmon M, et al.. Leukocyte interferon-induced tumor regression in human metastatic breast cancer, multiple myeloma, and malignant lymphoma. Ann Intern Med. 1980;93:399-406. doi: 10.7326/0003-4819-93-3-399. PMID:6159812 [DOI] [PubMed] [Google Scholar]
- 35.Vial T, Descotes J. Clinical toxicity of the interferons. Drug safety. 1994;10:115-50. doi: 10.2165/00002018-199410020-00003. PMID:7516663 [DOI] [PubMed] [Google Scholar]
- 36.Parker BS, Rautela J, Hertzog PJ. Antitumour actions of interferons: implications for cancer therapy. Nat Rev Cancer. 2016;16:131-44. doi: 10.1038/nrc.2016.14. PMID:26911188 [DOI] [PubMed] [Google Scholar]
- 37.Jablonska J, Leschner S, Westphal K, Lienenklaus S, Weiss S. Neutrophils responsive to endogenous IFN-beta regulate tumor angiogenesis and growth in a mouse tumor model. J Clin Invest. 2010;120:1151-64. doi: 10.1172/JCI37223. PMID:20237412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med. 2009;206:1457-64. doi: 10.1084/jem.20090207. PMID:19564351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bekes EM, Schweighofer B, Kupriyanova TA, Zajac E, Ardi VC, Quigley JP, Deryugina EI. Tumor-recruited neutrophils and neutrophil TIMP-free MMP-9 regulate coordinately the levels of tumor angiogenesis and efficiency of malignant cell intravasation. Am J Pathol. 2011;179:1455-70. doi: 10.1016/j.ajpath.2011.05.031. PMID:21741942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Datta S, Novotny M, Pavicic PG Jr., Zhao C, Herjan T, Hartupee J, Hamilton T. IL-17 regulates CXCL1 mRNA stability via an AUUUA/tristetraprolin-independent sequence. J Immunol (Baltimore, Md: 1950). 2010;184:1484-91. doi: 10.4049/jimmunol.0902423. PMID:20042592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sun D, Novotny M, Bulek K, Liu C, Li X, Hamilton T. Treatment with IL-17 prolongs the half-life of chemokine CXCL1 mRNA via the adaptor TRAF5 and the splicing-regulatory factor SF2 (ASF). Nat Immunol. 2011;12:853-60. doi: 10.1038/ni.2081. PMID:21822258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Afonina IS, Cullen SP, Martin SJ. Cytotoxic and non-cytotoxic roles of the CTL/NK protease granzyme B. Immunol Rev. 2010;235:105-16. doi: 10.1111/j.0105-2896.2010.00908.x. PMID:20536558 [DOI] [PubMed] [Google Scholar]
- 43.Barbi J, Oghumu S, Lezama-Davila CM, Satoskar AR. IFN-gamma and STAT1 are required for efficient induction of CXC chemokine receptor 3 (CXCR3) on CD4+ but not CD8+ T cells. Blood. 2007;110:2215-6. doi: 10.1182/blood-2007-03-081307. PMID:17785588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dunn GP, Koebel CM, Schreiber RD. Interferons, immunity and cancer immunoediting. Nat Rev Immunol. 2006;6:836-48. doi: 10.1038/nri1961. PMID:17063185 [DOI] [PubMed] [Google Scholar]
- 45.Rosas LE, Keiser T, Pyles R, Durbin J, Satoskar AR. Development of protective immunity against cutaneous leishmaniasis is dependent on STAT1-mediated IFN signaling pathway. European J Immunol. 2003;33:1799-805. doi: 10.1002/eji.200323163. PMID:12811839 [DOI] [PubMed] [Google Scholar]
- 46.Jin L, Lim M, Zhao S, Sano Y, Simone BA, Savage JE, Wickstrom E, Camphausen K, Pestell RG, Simone NL. The metastatic potential of triple-negative breast cancer is decreased via caloric restriction-mediated reduction of the miR-17∼92 cluster. Breast Cancer Res and Treat. 2014;146:41-50. doi: 10.1007/s10549-014-2978-7. PMID:24863696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eckhardt BL, Parker BS, van Laar RK, Restall CM, Natoli AL, Tavaria MD, et al.. Genomic analysis of a spontaneous model of breast cancer metastasis to bone reveals a role for the extracellular matrix. Mol Cancer Res. 2005;3:1-13. PMID:15671244 [PubMed] [Google Scholar]
- 48.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671-5. doi: 10.1038/nmeth.2089. PMID:22930834 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.