

Abstract
Polarization transfer is demonstrated as a sensitive technique for the measurement of isotopic fractionation of protonated carbons at natural abundance. This method allows kinetic isotope effects (KIEs) to be determined with substantially less material or shorter acquisition time compared with traditional experiments. Computations quantitatively reproduce the KIEs in a Diels–Alder reaction and a catalytic glycosylation. The glycosylation is shown to occur by an effectively concerted mechanism.
Graphical Abstract
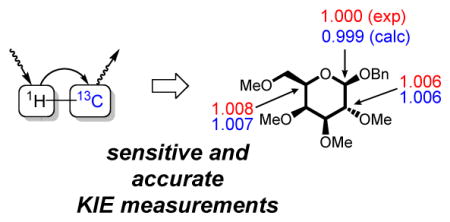
The accurate determination of kinetic isotope effects (KIEs) can provide uniquely powerful and sensitive information about reaction mechanisms, and has therefore been one of the primary tools of physical-organic chemistry for the past several decades.1 From a practical stand-point, application of KIE analyses is often limited by accessibility to the requisite isotopically labelled materials. In 1995, the Singleton group introduced a crucially important advance in this regard, through the development of methods for measuring KIEs at natural isotopic abundance.2 The Singleton method has been applied successfully to illuminate the mechanisms of many important reactions.3 However, to achieve sufficient fractionation for the accurate determination of KIEs by this method, reactions must be carried to either very low conversion (for product analysis) or high conversion (for starting material analysis). The requirement that small percentages of a reaction mixture be analyzed, combined with the intrinsically low sensitivity of the 13C nucleus in NMR spectroscopy, has hampered the application of KIE methodology to reactions in which the reagents or catalysts are precious.4 Here we show that 1H to 13C polarization transfer can reduce the time and material required for many KIE measurements substantially, and apply the improved protocol to analysis of the mechanism of a catalytic glycosylation reaction at the boundary between the SN1 and SN2 mechanisms.
Measurements of intermolecular 13C KIEs at natural abundance rely on kinetic resolution: at high conversions, the remaining starting material becomes enriched in the slower reacting isotope (usually 13C), while at low conversions, the product is enriched in the faster reacting isotope (usually 12C). Under pseudo-first-order conditions, the change in isotopic composition at a particular site is related to the fractional conversion (F) via:
where RSM is the ratio of heavy to light isotopes in recovered starting material, RPDT is the ratio in product, and R0 is the ratio in the unfractionated starting materials.5 In general, the degree of fractionation – and therefore the accuracy of KIE determinations – is higher for recovered starting material at high conversion than for product at low conversion. However, in cases where the starting material is unstable or where low reaction efficiency precludes attainment of high conversion, product analysis is the only practical option. Regardless of whether starting material or product is analyzed, the enrichments are small because 13C KIEs are themselves intrinsically small (0.98–1.10).5
The Singleton method employs quantitative single pulse 13C NMR spectroscopy to measure the isotopic ratios R and R0, each of which is determined using the signal of a carbon with negligible KIE as an internal standard. When the enrichment is calculated as the ratio of ratios R/R0, any discrepancy between the response factors, s and sref, cancels:
This cancellation of errors can be exploited to allow for the use of more sensitive NMR methods with non-uniform response factors. In particular, distortionless enhancement by polarization transfer (DEPT),6 which lever-ages the larger gyromagnetic ratio of 1H over 13C (43 and 11 MHz T−1, respectively), can theoretically yield a four-fold improvement in sensitivity or, equivalently, a sixteen-fold reduction in experimental time. In practice, quantitative implementations of DEPT sacrifice sensitivity to maximize response factor uniformity by using arrayed values of Δ (the magnetization transfer delay) and β (the read pulse angle).7 However, without the need for uniform response factors in KIE measurements, the maximum theoretical sensitivity improvement can be retained through a single choice of set values for the Δ and β parameters.
To explore this polarization transfer approach, we selected the Diels–Alder reaction between isoprene and maleic anhydride as a test case. Although Singleton and co-workers determined the 13C KIEs for this reaction by analyzing recovered isoprene at high conversion, we chose instead to analyze product at low conversion. For reactions that combine two molecules into one, product analysis is more efficient than starting material analysis because it allows the KIEs to be determined with respect to both reactants (maleic anhydride and isoprene) in one set of experiments. Additionally, because of reduced fractionation, product analysis constitutes a more challenging test of our methodology.4a,8
Given measurements of 1JCH obtained from coupled HSQC experiments, numerical optimization of Δ and β using product operator expressions (see SI)7b gave optimized values of Δ=3.319 ms (1JCH = 150.6 Hz) and β = 55.72°. These parameters correspond to positive enhancements of 3.3 for CH and CH3 groups and 3.7 for methylene groups, which translate into an approximately ten-fold reduction in instrument time. Although the optimal choices of Δ and β will vary slightly for other systems, the values above can be applied to most organic compounds due to the correlation between 1JCH values and hybridization. Both the traditional single-pulse and DEPT-55 methods gave statistically indistinguishable results with high precision for all intermolecular KIEs, and, in the case of isoprene, the measured KIEs match literature values (Table 1).9 On average, DEPT-55 produced a three-fold increase in signal to noise, which is consistent with the theoretical enhancements when relaxation is taken into account.
Table 1.
Diels–Alder Intermolecular KIEs
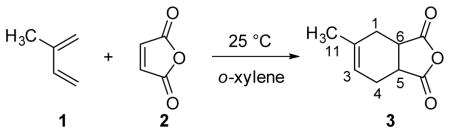
The choice to analyze product also allowed both the inter- and intra-molecular KIEs for maleic anhydride to be measured. Unlike the measurement of intermolecular KIEs, which benefits from the cancellation of response factors, the measurement of intramolecular KIEs requires direct knowledge of the relative fractionation.10 In this reaction, the average fractionation at C5 and C6 define the intermolecular KIE (Table 1). By contrast, their relative fractionation defines the intramolecular KIE because of the homotopic relationship between those carbons in maleic anhydride. By transferring polarization from proton to carbon, the excitation and detection spectral windows can be adjusted independently and therefore, the inter- and intra-molecular KIEs can be measured simultaneously. When routine steps are taken (see SI) to equalize the response factors at C5 and C6, the single-pulse and DEPT-55 methods give identical intramolecular KIEs of 1.004(1) and 1.005(1), respectively. This agreement demonstrates that DEPT-55 can be applied to the determination of inter- and intra-molecular KIEs simultaneously and with no loss of accuracy compared with less sensitive traditional integration methods.2,10
To determine the extent to which these KIEs define a unique transition structure,12 we located the endo transition state using 332 different combinations of electronic structure methods, basis sets, and solvation models. The standard functional B3LYP14 outperforms ab initio methods, with B3LYP/cc-pVQZ reproducing every KIE to within 0.002 (Table 2). Many other levels of theory also perform well: in approximately half of the calculations, every predicted KIE lies inside its corresponding experimental confidence interval (i.e., ±0.004, as shown in Figure 1a). Within this subset, the forming bond distances span a range of 0.10–0.15 Å (Figure 1b). This uncertainty is somewhat larger than that found by Singleton and coworkers for the epoxidation of alkenes (0.05–0.10 Å).13 Overall, these KIEs strongly support the canonical view of a concerted cycloaddition.
Table 2.
Diels–Alder KIE Predictions
| Methoda | C1 | C4 | C5 | C6 |
|---|---|---|---|---|
| HF/cc-pVQZ | 1.033 | 1.030 | 1.036 | 1.039 |
| MP2/cc-pVTZ | 1.015 | 1.016 | 1.019 | 1.019 |
| CCSD/cc-pVDZ | 1.024 | 1.022 | 1.028 | 1.029 |
| B3LYP/cc-pVQZ | 1.023 | 1.018 | 1.022 | 1.028 |
|
| ||||
| experimental | 1.022 | 1.019 | 1.024 | 1.028 |
Bigeleisen–Mayer predictions relative to C11 at 298 K with infinite parabola tunneling corrections and PCM solvation.
Figure 1. Consensus KIE Predictions and Geometries.
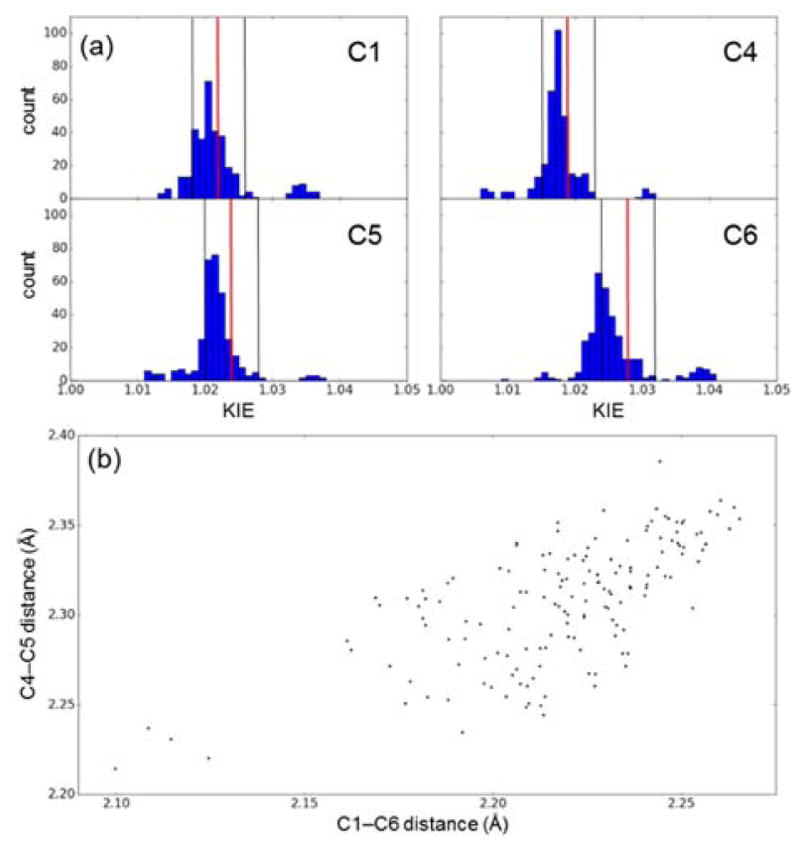
(a) Histograms of Diels–Alder KIE predictions at all levels of theory.
 and black vertical lines: experimental KIEs and standard errors, respectively. (b) Transition state forming bond distances for predictions within the ranges in panel a.
and black vertical lines: experimental KIEs and standard errors, respectively. (b) Transition state forming bond distances for predictions within the ranges in panel a.
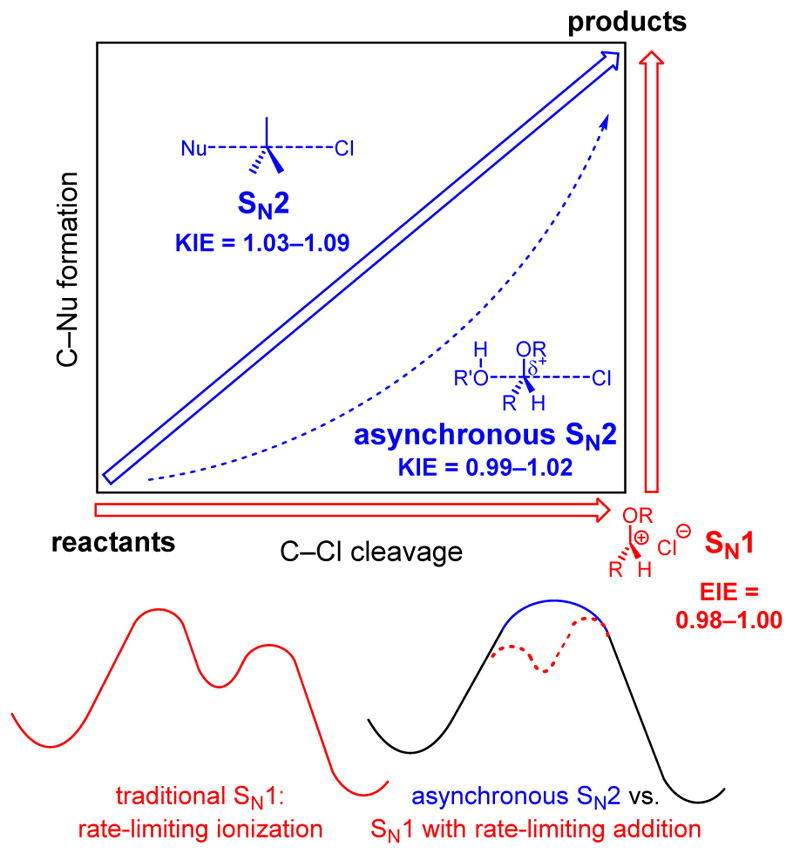
Computational models and KIEs are also useful for distinguishing between the SN1 and SN2 mechanisms (Scheme 1).5 In this context, α-12C/13C KIEs can be more diagnostic than α-1H/2H KIEs because the former are primary KIEs that probe bond-breaking and forming directly, while the latter are secondary KIEs that merely reflect the extent of hybridization changes.15 In both SN1 and loose SN2 reactions, where the central carbon is nearly sp2-hybridized in the transition state, large and normal secondary α-1H/2H KIEs are expected.
Scheme 1.
SN1 vs. SN2 Mechanisms
In general, SN1 mechanisms give rise to small normal or inverse 12C/13C KIEs, while SN2 mechanisms produce relatively large normal KIEs (Scheme 1). In a traditional SN1 mechanism with rate-limiting leaving group dissociation, the large degree of heterolysis in the transition state would ordinarily cause a large, normal isotope effect. However, the bonds in the cation are strengthened by hyperconjugation. These effects are nearly offsetting: for example, the equilibrium isotope effects (EIEs) for trityl chloride solvolysis and phenethyl bromide methanolysis are 0.98 and 1.01, respectively.16 By contrast, in an SN2 mechanism, the degrees of bond formation and bond cleavage are relatively equal, with a strong preference for a linear geometry. This can produce large KIEs; for example, the concerted addition of cyanide to methyl chloride gives a KIE of 1.07.14a
Glycosylation is a special case of nucleophilic substitution in which distinguishing between the SN1 and SN2 mechanisms is more challenging. This is because the presence of an oxygen adjacent to the site of displacement (i.e., C1) can stabilize incipient positive charge. Thus, any potential concerted pathway becomes asynchronous (dotted line in Scheme 1) and approaches the oxocarbenium ion intermediate of the SN1 mechanism (lower-right-hand corner). Accordingly, the SN2 KIEs are depressed relative to those of a synchronous mechanism. For example, the acidic hydrolysis of β-methylglucoside, which proceeds by an SN1 mechanism, displays a 13C KIE of 1.011, and the concerted enzymatic hydrolysis of the same substrate results in a KIE of 1.032.9
The thiourea-catalyzed glycosylation of galactose developed recently in our laboratories17 (Table 3) offers an opportunity to study this interesting borderline region between SN1 and SN2: the reaction occurs with stereochemical inversion, displays relatively large α-1H/2H KIEs, and is faster for sugars with cation-stabilizing axial substituents. In this reaction, the instability of the starting material requires product analysis. The DEPT-55 method described above is particularly well-suited for this case because its improved sensitivity offsets the difficulties of low fractionation associated with product analysis, all the KIEs of interest belong to protonated carbons, and the catalyst and products are relatively precious.
Table 3.
Measured vs. Predicted Glycosylation KIEs
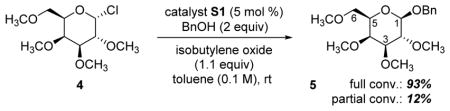
| ||||
|---|---|---|---|---|
|
| ||||
| C1 | C2 | C4 | C5 | |
|
| ||||
| experimentala | 1.000(4) | 1.006(5) | 1.000(4) | 1.008(4) |
| predictedb SN2 KIEs | ||||
| PBE0-D3(BJ) | 0.999 | 1.006 | 0.999 | 1.007 |
| B3LYP-D3(BJ) | 0.993 | 1.006 | 0.999 | 1.007 |
| M06-2X | 1.008 | 1.011 | 0.998 | 1.005 |
| SN1 (predictedb,c) | 0.981 | 1.007 | 1.000 | 1.011 |
DEPT-55 KIEs and standard errors over two trials. Other KIEs: 1.001(4) at C3, 1.000(3) at C6. KIEs are relative to the C2 methyl group.
6-31G*/PCM. All predicted KIEs at C3 and C6 are within 0.001 of unity.
PBE0–D3(BJ) EIEs for the ionization of galactosyl chloride 4.
Catalytic glycosylations were carried to 12% and 100% conversion and the products analyzed. As expected, small and normal KIEs were observed at C1, C2, and C5 (Table 3), indicating significant oxocarbenium character in the transition state. Calculations were performed in order to distinguish between the SN1 and SN2 mechanisms. The SN1 KIEs can be approximated by EIEs.18 The calculated value at C1 is 0.981. This value does not vary significantly with the choice of computational method and is lower than the KIE determined experimentally. By contrast, C1 KIEs for the SN2 mechanism are predicted to lie between 0.99–1.01. In particular, PBE0-D3(BJ)/6-31G*/PCM reproduces every experimental KIE to within 0.001. This agreement validates the picture of an asynchronous mechanism with a large degree of charge separation.
An alternative stepwise mechanism involving ionization followed by rate-limiting nucleophilic addition cannot be ruled out. When the commitment factor for the cationic intermediate is small, the apparent KIE is the product of the EIE for the ionization step (taking starting chloride as the reference) and the KIE of the nucleophilic addition step (taking cation as the reference). This is equivalent to calculating the KIE for the addition step with starting chloride as the reference (see SI). Because the latter procedure is also the one followed for an SN2 prediction, both predictions are necessarily identical.
Although SN1 and SN2 represent formally limiting mechanisms, the experimental characteristics of a stepwise process with rate-limiting addition and a loose-but-concerted displacement converge when the intermediate is high in energy (Scheme 1). Specifically, such reactions are positive order in both the nucleophile and electrophile and yield stereo-chemical inversion. While this KIE analysis cannot rule out the possibility of a shallow intermediate along the reaction coordinate, it confirms that the reaction occurs through a cooperative mechanism in which dual activation of the nucleophile and electrophile are required for catalysis.
In conclusion, polarization transfer offers a highly sensitive method for determining the KIEs of protonated carbons at natural abundance, reducing the amount of sample required by a factor of three or acquisition time by a factor of nine. In complex or catalytic systems, this method is particularly advantageous: the KIEs of interest usually belong to protonated carbons, time and material are at a premium, and high concentrations can be impractical due to material limitations. The DEPT-55 method was applied successfully to analyses of a Diels–Alder reaction and a catalytic glycosylation, with experimental natural abundance KIEs serving both to validate and differentiate DFT calculations.1a We hope that our method will enable the further application of KIE analyses as a general mechanistic tool, thereby offering new insights into the mechanisms of organic reactions.
Supplementary Material
Figure 2.
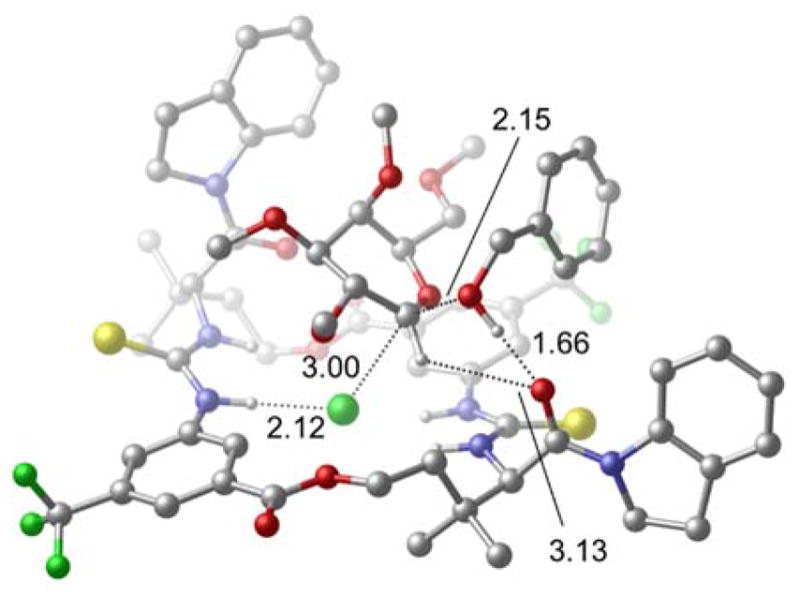
PBE0-D3(BJ)/6-31G*/PCM transition structure for the glycosylation of galactose with benzyl alcohol (distances in Å).19
Acknowledgments
Financial support from the NIH (UO1GM116249 and RO1GM43214), the NSF (predoctoral fellowship for Y.P.), and the ACS (SURF award for H.A.B.) are acknowledged. We thank Dr. Shaw Huang and Mr. William Collins for NMR assistance. We thank Professor William F. Reynolds for helpful discussions.
Footnotes
The authors declare no competing financial interests.
Experimental procedures, spectroscopic data, NMR data, and computational structures. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Bigeleisen J, Wolfsberg M. Adv Chem Phys. 1958;1:15–76. [Google Scholar]; (b) Westheimer FH. Chem Rev. 1961;61:265–273. [Google Scholar]; (c) Bell RP. Chem Soc Rev. 1974;3:513–544. [Google Scholar]; (d) Cleland WW. Arch Biochem Biophys. 2005;433:2–12. doi: 10.1016/j.abb.2004.08.027. [DOI] [PubMed] [Google Scholar]; (e) Westaway KC. Adv Phys Org Chem. 2006;41:217–273. [Google Scholar]; (e) DelMonte AJ, Haller J, Sharpless KB, Singleton DA, Strassner T, Thomas AA. J Am Chem Soc. 1997;119:9907–9908. [Google Scholar]; (f) Singleton DA, Wang Z. J Am Chem Soc. 2005;127:6679–6685. doi: 10.1021/ja0435788. [DOI] [PubMed] [Google Scholar]
- 2.Singleton DA, Thomas AA. J Am Chem Soc. 1995;117:9357–9358. [Google Scholar]
- 3.General review: Meyer MP. Adv Phys Org Chem. 2012;46:57–120.Singleton DA, Hang C, Szymanski MJ, Meyer MP, Leach Ag, Kuwata KT, Chen JS, Greer A, Foote CS, Houk KN. J Am Chem Soc. 2003;125:1319–1328. doi: 10.1021/ja027225p.Bandar JS, Sauer GS, Wulff WD, Lambert TH, Vetticatt MJ. J Am Chem Soc. 2014;136:10700–10707. doi: 10.1021/ja504532d.Crich D. Acc Chem Res. 2010;43:1144–1153. doi: 10.1021/ar100035r.Ryan SJ, Candish L, Lupton DW. J Am Chem Soc. 2011;133:4694–4697. doi: 10.1021/ja111067j.Dahlen A, Hilmersson G. J Am Chem Soc. 2005;127:8340–8347. doi: 10.1021/ja043323u.Yoshikai N, Nakamura E. J Am Chem Soc. 2004;126:12264–12265. doi: 10.1021/ja046616w.
- 4.Recent improvements: Xiang S, Meyer MP. J Am Chem Soc. 2014;136:5832–5835. doi: 10.1021/ja412827c.Manning KA, Sathyamoorthy B, Eletsky A, Szyperski T, Murkin AS. J Am Chem Soc. 2012;134:20589–20592. doi: 10.1021/ja310353c.Rabis A, Kaminski R, Ciepielowski G, Jankowski S, Paneth P. J Org Chem. 2011;76:8033–8035. doi: 10.1021/jo201226g.Chan J, Lewis AR, Gilbert M, Karwaski MF, Bennet AJ. Nat Chem Biol. 2010;6:405–407. doi: 10.1038/nchembio.352.
- 5.(a) Melander L, Saunders WH., Jr . Reactions Rates of Isotopic Molecules. Wiley; New York: 1980. [Google Scholar]; (b) Wolfsberg M, Van Hook WA, Paneth P, Rebelo LPN. Isotope Effects in the Chemical, Geological, and Bio Sciences. Springer; Dordrecht: 2010. [Google Scholar]
- 6.Doddrell DM, Pegg DT, Bendall MR. J Magn Reson. 1982;48:323–327. [Google Scholar]
- 7.(a) Henderson TJ. J Am Chem Soc. 2004;126:3682–3683. doi: 10.1021/ja039261+. [DOI] [PubMed] [Google Scholar]; (b) Jiang B, Xiao N, Liu H, Zhou Z, Mao X, Liu M. Anal Chem. 2008;80:8293–8298. doi: 10.1021/ac8015455. [DOI] [PubMed] [Google Scholar]; (c) Mäkelä AV, Kilpeläinen I, Heikkinen S. J Magn Reson. 2010;204:124–130. doi: 10.1016/j.jmr.2010.02.015. [DOI] [PubMed] [Google Scholar]
- 8.Vetticatt MJ, Singleton DA. Org Lett. 2012;14:2370–2703. doi: 10.1021/ol300789a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee JK, Bain AD, Berti PJ. J Am Chem Soc. 2004;126:3769–3776. doi: 10.1021/ja0394028. [DOI] [PubMed] [Google Scholar]
- 10.Singleton DA, Szymanski MJ. J Am Chem Soc. 1999;121:9455–9456. [Google Scholar]
- 11.Alternatively, DEPT-90 can moderately improve the sensitivity for methines, at the expense of the ability to measure any intermolecular KIEs.
- 12.Beno BR, Houk KN, Singleton DA. J Am Chem Soc. 1996;118:9984–9985. [Google Scholar]
- 13.Hirschi JS, Takeya T, Hang C, Singleton DA. J Am Chem Soc. 2009;131:2397–2403. doi: 10.1021/ja8088636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Becke AD. J Chem Phys. 1993;98:5648–5652. [Google Scholar]; (b) Lee CT, Yang WT, Parr RG. Phys Rev B. 1988;37:785. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]; (c) Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. J Phys Chem. 1994;98:11623–11627. [Google Scholar]
- 15.(a) Matsson O, Dybala-Defratyka A, Rostkowski M, Paneth P, Westaway KC. J Org Chem. 2005;70:4022–4027. doi: 10.1021/jo047987y. [DOI] [PubMed] [Google Scholar]; (b) Westaway KC, Pham TV, Fang Y. J Am Chem Soc. 1997;119:3670–3676. [Google Scholar]; (c) Poirier RA, Wang Y, Westa-way KC. J Am Chem Soc. 1994;116:2526–2533. [Google Scholar]; (d) Westaway KC, Pham TV, Fang Y. J Am Chem Soc. 1997;119:3670–3676. [Google Scholar]
- 16.Kresge AJ, Lichtin NN, Rao KN, Weston RE., Jr J Am Chem Soc. 1965;87:437–445. [Google Scholar]
- 17.Park Y, Harper KC, Kuhl N, Kwan EE, Liu RY, Jacobsen EN. Science. doi: 10.1126/science.aal1875. accepted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.There is no saddle point for bond heterolysis on the potential energy surface, so evaluation of the EIE represents the best approach to estimating the KIE for the SN1 pathway. See reference 9.
- 19.The experimental KIEs are closest to the values predicted by PBE0-D3BJ and B3LYP. Given that a dispersion-corrected model should be physically more reasonable, and that the un-corrected transition structure features an unusual non-planar chloride-binding geometry, the PBE0-D3(BJ structure is the model that best fits the observed data.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.