

Abstract
Agonists of immune cell receptors direct innate and adaptive immunity. These agonists range in size and complexity from small molecules to large macromolecules. Here, agonists of a class of immune cell receptors known as the Toll-Like Receptors (TLRs) are highlighted focusing on the distinctive molecular moieties that pertain to receptor binding and activation. How the structure and combined chemical signals translate into a variety of immune responses remain major questions in the field. In this structure-focused review, we outline potential areas where the tools of chemical biology could help decipher the emerging molecular codes that direct immune stimulation.
Introduction
Using the tools of chemical biology to understand the molecular cues of the innate immune system will provide insight into the design of future immunotherapies. Over the last decade, many of the biochemical and biophysical pathways through which our immune system senses foreign pathogens have been identified. This has resulted in both the commercial deployment of small-molecule immunostimulants and the discovery of macromolecules that activate immune cells. The “molecular fingerprints” used by the immune system to determine self from non-self are rapidly being catalogued. This review provides a guide to the growing body of literature on the molecular basis of immune cell stimulation with a focus on the structural and chemical aspects of these agonists as opposed to the receptors and cellular stimulation pathways commonly presented within immunology reviews.1
The innate immune system directs primary cellular immune responses that occur within the body. Innate immune cells are directed by pattern recognition receptors (PRRs) located on their cell surface, in the cytosol, and within endosomes. The molecular interactions between PRRs and the extracellular environment are the primary method of modulating the immune system (e.g. immunostimulants). Hyper-activation of these receptors is the main cause of many diseases including asthma, chronic obstructive pulmonary disease, arthritis, lupus, and is strongly implicated in atherosclerosis, diabetes, and inflammatory disorders.2 Conversely, PRRs and their agonists are responsible for the efficacy of almost every vaccine. The interaction of immune cell receptors with chemical structures is the “molecular language” through which the body determines self from non-self. Understanding and manipulating these structures holds the promise of creating better vaccines, modulating the inflammatory response to disease, and better understanding how molecular structure influences the immune response.3
As new entrants into the field, we sought a field-guide to immunostimulants from a chemical perspective. We wondered, “how does the molecular structure of immunostimulants influence and control the immune response”? This structure-guided review will benefit any like-minded researcher considering the molecular aspects of immune cell stimulation. Our intent for this guide is to spark interest in the creative use of structural interactions; a quality that is a hallmark of the chemical biology community. This review covers agonists for the most studied receptor class that interfaces between the innate immune system and the biochemical world, the Toll-Like Receptors (TLRs). We review agonists of TLRs 1–9 of the 13 total (13 murine, 10 human) TLRs known to date. We will present each set of molecules organized by their receptor, separating them into two general categories: small molecule binders and macromolecule binders. Where possible, we present primary structural information.
As the review is structure-based, we mention only briefly the immunological implications. There are already several excellent reviews to assist in understanding the immunological consequences of stimulation of these receptors.1,4,5 We introduce here only the concepts necessary to provide context. Finally, we cover the emerging field of receptor synergies - broadly defined as an increased immune response from stimulation by multiple agonists. These synergies are observed in a variety of experiments from increases in antibody titers, to distinctive increases in cytokine expression6 and cell signaling.7 Synergies represent a promising landscape for chemical biologists because structural evidence suggests that molecular level interactions drive synergistic immune responses.8 Some evidence suggests synergies may occur through the clustering of receptors.9 Synergies are a key element in effective vaccine formulations and therefore are an area of future study.10
Brief Primer of Immunological Response
Robert Koch described the first model of an immune response more than 100 years ago, and since then modulating the immune response, primarily through vaccines has led to the eradication of many of the world’s diseases.11 However, research into the molecular and structural understanding of the immune system is still in its infancy; the primary immune cell receptors have only been identified in the last 20 years. The Toll-Like Receptors discussed in this review occur in multiple cell types including dendritic cells (DCs - the primary antigen-presenting cell), macrophages, lung epithelial cells, and B-cells. Each cell type is a general cell class with many distinct sub-classes, and the location, identity, and cell-type all influence activity.12
As these receptors play different roles in different locations and cell-types within the body, we focus exclusively on Toll-Like Receptor (TLR) activation. TLR activation can change based on cellular compartmentalization and the signaling network within a cell. Although the cellular response varies depending on cell type, we review here the molecular interactions that comprise TLR activation by a variety of agonists.
Cellular Responses
There are two general pathways (innate and adaptive) through which the immune system responds to antigens.13,14 The adaptive immune response pathway involves signaling through antigen-presenting cells (APCs). The primary group of APCs is dendritic cells (DCs). DCs exist throughout the body including the lung, gut, blood and spleen.15 The three most referenced categories of DCs are myeloid (mDC) derived from bone marrow, plasmacytoid (pDC) free in blood, and Langerhans cells (LCs) that occupy the skin. Each DC subtype contains different combinations of TLRs; therefore each DC subset responds differently to TLR agonists. When a DC encounters a molecular entity, it must determine if the entity is foreign or self.16 The DC uses a series of pattern recognition receptors (PRRs), including the TLR family of receptors, to determine if the entity is foreign. The molecular basis for how this consensus is reached is an active area of research that centers on the activation of PRRs. This review focuses on the structural characteristics of TLR agonists that promote immune cell activation (Figure 1).
Figure 1.

Overall pathway of DC Activation. A series of molecular signals, each a specific agonist for different TLRs are presented alongside an antigen. Dimerization of TLRs activates downstream signaling “maturing” the dendritic cell. Mature cells present the antigen on the major histocompatibility complex along with co-stimulatory and recognition proteins (e.g. CD80/CD86) and signaling cytokines that further the immune response. A Th1 response involves secretion of many cytokines, but notable ones include IL-6 and IL-12. This then elicits a cytotoxic T cell response with antibodies directing cytotoxic cells towards a specific antigen. A Th2 response is the production of soluble antibodies via activated B cells. The marked cytokines include IL-5 and -7.
TLR – Toll-Like Receptors
Several subclasses of pattern recognition receptors have been identified including C-type lectin, RIG-I, DANGER, and dectins; however, Toll-Like Receptors are the best-characterized class of PRRs. TLRs are membrane-bound and occur on the cell surface or within endosomes. Upon binding an agonist, TLRs form higher order constructs including homodimers, heterodimers, and tetramers initiating a signaling cascade that results in maturation of the immune cell and expression of immunostimulatory cytokines (Figure 2). Crystal structures of TLR 8 reveal that receptors in the TLR family share many structural elements. Similarities include a “question mark” or horseshoe motif containing several leucine rich repeat units (LRRs) around which two TLRs form a constitutive dimer. The LRRs present hydrophobic surfaces that surround variable binding regions. LRRs combined with a TLR-specific variable region are responsible for the molecular level discrimination of TLR agonists.
Figure 2.
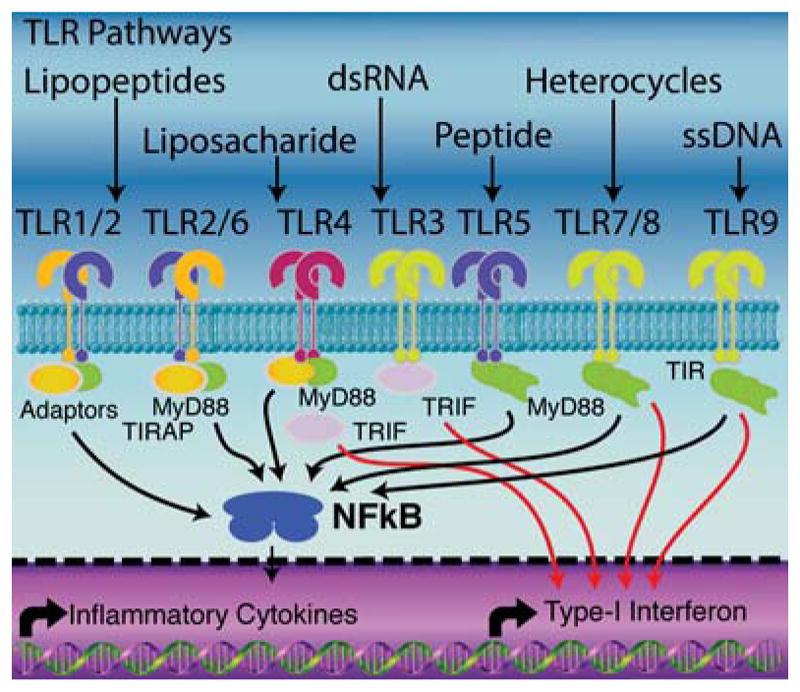
General signaling cascades responsible for downstream activation of TLRs. Activation of TLRs activates the NFκB pathway triggering expression and production of inflammatory cytokines and interferons. Activated cells also up-regulate the expression of MHCI/II, CD80/86 and CD40. MyD88 is the predominant adaptor protein between TLR activation and inflammatory cytokines. TRIF is the other major adaptor protein. Many synergies occur through the simultaneous activation of both pathways.
Generally, TLRs signal from two different regions of the cell, the cell surface and endosomal compartments. The first location, the cell surface, contains TLRs 1, 2, 4, 5 and 6. In each case, ligand binding causes association of at least two TLRs that initiate signaling (Figure 2). As examples, TLR2 can dimerize with itself, TLR1, or TLR6 whereas TLR5 forms a homodimer with itself. TLR4 can form an agonist-mediated heterotetramer with the stabilizing protein MD-2 to initiate activation. Each of these TLRs binds molecules present on the surfaces of pathogens ranging from zymosan to the flagellin of bacteria.17,18 Many agonists of surface TLRs also increase macro-pinocytosis.19 The remaining TLRs, TLR3, 7, 8 and 9 all signal from the endosome. Signaling of TLR9 is activated by a series of endosomal proteases,20 and a similar mode of action is hypothesized or proven for other endosomal TLRs.
Here we present an overview of TLRs 1–9 summarizing known crystal structures and related agonists. We present the receptors based, first, on the classification of their agonists, then, their signaling location and, finally, their arbitrary numbering scheme. To aid in the rational design of new immunostimulants, we include figures that compare structures based on the size and molecular interactions of co-crystallized agonists with TLRs.
Toll-Like Receptor 1
Major Agonists: Lipids, Lipidated Proteins, Hetero-aromatics
TLR1 is an extracellular receptor that binds lipidated proteins and oligosaccharides found on the exterior of bacterial cell walls. A variety of synthetic molecular mimics have also been developed to bind TLR1. TLR1 is hypothesized to be one of the first receptors activated when a DC encounters a foreign bacterial pathogen. Lipid chains on TLR1 agonists intercalate both TLR1 and TLR2 to form an “m” shaped TLR1/2 heterodimer that initiates up-regulation of NFκB in a MyD88 dependent manner.21
The main binding pocket for TLR1 involves hydrophobic interactions with lipid chains present on bacterial lipoproteins, and a range of tri-palmitylated lipopeptide sequences.22 Most synthetic TLR1 agonists to-date contain multiple lipid chains with maximum activation occurring for C-16 acyl groups; specifically, triacylated N-terminal cysteine comprises the component that is generally thought to interact with both TLR1 and TLR2 inducing hetero-dimerization and activation.23 Synthetic peptides, like PAM3CSK4, mimic the acylated peptides found in natural agonists.24
Activation through the TLR1/2 pathway specifically increases immune cell stimulation (abrogates the Treg response) relative to other TLR activation pathways.25 As such, the TLR1/2 pathway is used in several approaches to cancer immunotherapy.26 TLR1 detects the N-terminal acyl chains found in bacterial lipoproteins, but is also responsive to strains of LPS depending on the lipid A motif. Synthetic TLR1 agonists have, so far, all involved tri-acylated peptide sequences that mimic natural agonists.
Toll-Like Receptor 2
Major Agonists: Lipids, Lipidated Proteins, Polysaccharides, Hetero-aromatics
Toll-Like Receptor 2 is a promiscuous TLR implicated in activation/dimerization with both TLR1 and 6 initiating NFκB activation in a MyD88 dependent manner.27 Agonists for TLR2 therefore include those outlined for TLR1 and TLR6. The main binding mode for TLR2 involves hydrophobic interactions with molecules that contain 1–3 lipid chains such as lipoteichoic acids, lipoarabinomannans, macrophage activating lipopeptides (MALP) or mono, di, and tri-palmitylated peptide sequences.28 In the latter case, the structural subunit that interacts with TLR2 is the lipid chain connected to a peptide through a thioether bond required for activity. The remaining segment of the agonist determines the binding partner TLR; polar interactions recruit TLR6 whereas additional lipid chains induce dimerization with TLR1.29 Diacyl chains from C-6 to C-18 bind to the receptor, and C-16 lipids have the greatest activity.30 Interestingly, the chain must be hydrophobic, and replacement with other un-branched organic moieties such as polyethylene glycol greatly decreases activity.31 TLR2 can also bind agonists through non-lipid interactions. For example, zymosan (a mixture of β-1,3 glucans),32 and other polysaccharides33 activate TLR2 through polar interactions.34 TLR6 independent signaling can also occur for a range of traditional TLR2/6 immunostimulants, including PAM2CSK4, indicating that the peptide portion of the lipopeptide could contribute to TLR2 activation.35 Additionally, small molecule agonists including carbonothioylamino linked 3-carboxylbenzothiophenes and N1-(benzyl)-N2-(phenyl)-N2-(sulfonyl)glycinamides activate the TLR 1/2 pathway through binding motifs distinctly different from the PAMCSK series.36
Toll-Like Receptor 6
Major Agonists: Lipids, Lipidated Proteins, Polysaccharides
Toll-Like Receptor 6 serves as a compliment to the TLR1/2 heterodimer. While a TLR1/2 heterodimer is characterized primarily by lipid interactions, the TLR2/6 heterodimer involves hydrophobic (TLR2) and polar (TLR6) binding pockets.37 Natural TLR6 agonists are lipoteichoic acids. Lipoteichoic acids are a complex mixture of acylated glycopeptides featuring GlcNAc, hydroxyl, and γ-linked alanine residues along a polymeric phosphate backbone attached to a gentiobiose derived head group.38 In a synthetic tour de force, variants of lipoteichoic acids were synthesized providing evidence that the lipid, alanine, and gentiobiose groups all contribute to optimal activity.39 Smaller TLR2/6 agonists have been synthesized and they include mono or diacylated lipopeptides such as PAM2CSK4.40 In this case, TLR6 interacts primarily with the nitrogen of the N-terminal cysteine along with lesser contributions from the alcohol and amide bond of the neighboring serine; TLR2 interacts with the hydrophobic palmitic acid chains and may have additional activation via TLR6 independent pathways.35
Toll-Like Receptor 4
Major Agonists: Lipids, Proteins/Peptides, Hetero-aromatics
Toll-Like Receptor 4 is the most highly studied receptor, of the TLR family. Discovered as the target receptor of lipopolysaccharide (LPS), it can signal via both the MyD88 and TRIF pathways. TLR4 is also distinct in requiring a co-protein, MD2, for functional recognition of agonists and activation. The crystal structure of TLR4 with MD2 and LPS shows that the agonist binding pocket forms between MD2 and TLR4, with the hydrophobic tails of Lipid A inserted into the hydrophobic core of MD2.41 Two such dimers then complex with each other to form a tetramer consisting of two TLR4 and two MD2 subunits. The electrostatic interaction of the 4-phosphate with arginine and lysine residues of TLR4 appears critical for the activity of monophosphoryl lipid A (MPLA) and all other LPS derivatives.42 Substitution at the primary alcohol in MPLA does not seem to interrupt binding, as this is the position connected to the bulky saccharide chain of LPS. Several derivatives of Lipid A have been synthesized including Eritoran (E5564), which is an antagonist, 6202, E5531, and other aminoalkyl glucoaminide 4-phosphates (Figure 3).43,44 Reduced forms of MPLA have recently been synthesized, including Glucopyranosyl Lipid A (GLA).45 Also, E. coli have been engineered to provide multiple forms of Lipid A derivatives with distinct activities.46
Figure 3.

Small molecule binding TLRs. Structures of TLRs and structural information of their small molecule interactions. TLR2 agonists include palmitoylated peptides as well as polymeric saccharides. The structure of TLR2/6 bound with a PAMCys derivative is shown. TLR4, agonists include Lipid A derivatives, a pyrimido-[5,4-b]indole and the protein DerP2. The crystal structure of Lipid A bound within the TLR4 is shown. Hydrophobic interactions drive the association of MD2 and TLR4 mediated by the lipid tails of these derivatives. TLR8, several molecules containing an moiety bind TLR7 or TLR8. Variations in structure can increase specificity for either TLR7 or TLR8. In addition, in vivo, TLR7 and 8 bind ssRNA. The crystal structural of TLR8 is shown with the agonist R848, Resiqiumod, the amino acids responsible for binding are shown.
TLR4 selectivity in signaling between the TRIF/TRAM and MAL/MyD88 pathways is currently under investigation. Most exciting is that different TLR4 agonists change the signaling pathway from MAL/MyD88 to predominantly TRIF/TRAM, implying a structural basis for the activation of these signaling pathways.47,48 Intriguingly, the stereo-chemical configuration of a Lipid A mimic can direct the TLR4 downstream pathway implying that structure and stereochemistry will play a role in immunostimulant designs.49
Peptide-based agonists, with no lipid tail or phosphate moieties can signal through TLR4. One notable example is the activation of TLR4 by Der p 2, a dust-mite protein that causes dust allergies. The mode of action of Der p 2 has not been shown, however, its structural homology to MD2 has been suggested to aid in TLR4 binding.50,51 Recently, peptide agonists of TLR4 have been identified via phage display and antibody binding assays.52 It has yet to be determined how these peptides activate TLR4 and whether MD2 is necessary for binding. Endogenous molecules, such as heat shock proteins 6053 and 7054, fibrinogen55, fibronectin domain A56, extracellular DNA-binding proteins57, and other molecules activate TLR4, but the modes of binding are unclear. With the promiscuity of this TLR, many questions remain in both the design of selective TLR4 ligands, and the mode of action of known agonists.
Cottam and coworkers reported the first small molecule TLR4 agonist.58 Using a small molecule library, they identified pyrimido[5,4 b]-indoles as highly selective TLR4 agonists. Through in depth SAR studies, they found that substitutions of the pyrimidoindole core were as limiting as those derived from Lipid A. Using in silico docking studies, they predicted that the indole compounds bound a unique binding site yet still brought together TLR4 and MD2 (Figure 3).
Toll-Like Receptors 7 and 8
Major Agonists: Hetero-aromatics, Oligonucleotides
TLR7 and TLR 8 are endosomal TLRs that are receptors for ssRNA and the imidazoquinoline family of hetero-aromatics. These derivatives include the topical cancer therapeutics, Imiquimod (R837) and Resiquimod (R-848). The crystal structure of TLR8 was recently published showing TLR8 exists as a homodimer prior to ligand recognition and activation. This suggests a common mechanism in TLR pre-association and binding (Figure 3).59
Like other TLRs, TLR7 and TLR8 are composed of a region of leucine rich repeats(LRR) resembling a horseshoe shape.60 Prior to agonist binding, the C-terminal domains of the TLRs are 53 Å apart, preventing cytoplasmic domain association.61 When an agonist binds to the homodimer, the two TLR8 C-terminal subunits move together a distance of only 23 Å dimerizing the intracellular Toll-interleukin-1 receptor domain (TIR). Dimerization initiates the TIR signaling cascade resulting in immune cell stimulation. Alanine scanning studies have identified residues critical for activity. Aromatic rings in imidazoquinolines stack with Phe residues on both TLR8s. Alkyl and/or ether chains attached to the imidazoquinoline can protrude into a hydrophobic pocket. This pocket may be important for TLR8 specificity as Imiquimod, which has no chain, is TLR7 specific. Within the binding pocket of TLR8, Asp543, is essential for ligand recognition.62 The crystal structure of liganded TLR8 shows that hydrogen bonding with the nitrogen at the four position, of the agonist is critical in imidazoquinoline positioning and the subsequent conformational change of the TLR ligand.
Carson and coworkers modified 9-benzyl-8-hydroxy-2-(2-methoxyethoxy) adenine (TLR7 agonist) and conjugated phospholipid, PEG, or phospholipid-PEG on the benzoic acid functional group of the agonist. They reported that conjugates with PEG linkers of 18 units or longer, showed improved agonistic activity.63 When the agonist was conjugated with phospholipids, the activity was enhanced at least 100 fold while the phospholipid-PEG hybrid shows comparable potency to free agonists.64 Thiazolo[4,5-c]quinoline modifications presented by David and coworkers demonstrate that alkyl chains at C2 of up to 3 carbons have increasing TLR8-agonistic potencies whereas alkylation of any kind at C4 was not tolerated (removed TLR8 agonistic potency entirely).65 Furo[2,3-c]pyridines also showed TLR8 dependent NFκB signaling but failed to induce any proinflammatory cytokines. These compounds might be developed to lower the local or systemic reactogenicity (Figure 3).30 David also studied dimeric and dendritic agonist systems of TLR7 and 8. Imidazoquinoline dimers linked at C4, C8, and N1-aryl positions were agonists of TLR7 while the dimer with a 12-carbon linker attached at the N1-aryl position stimulated both TLR7 and 8.66 Dendrimers linked at N1-aryl positions had greater potency than the parent imidazoquinoline monomers likely due to spatial confinement.66
Toll-Like Receptor 3
Major Agonists: Oligonucleotides
Toll-Like Receptor 3 is unique among reported TLRs as it signals exclusively through the TRIF pathway.67 TLR3 is found in the endosomal/phagosomal compartments within DCs and macrophages.17 The native ligand for TLR3 is double stranded viral RNA. (dsRNA) The crystal structure of TLR3, published by Davies et. al in 2008, shows that the LRRs on the binding face, are selective for the negatively charged backbone of dsRNA.68 A common theme, it seems, in macromolecular TLR recognition is nucleic acid binding mediated by negative phosphates coupled with a modest degree of sequence specificity. (Figure 4)
Figure 4.

TLRs that bind macromolecules. Structures of agonists and structural information. TLR3, is shown binding dsRNA from top-down and front-forward structure. There are two sections of association. A) Binding interaction of TLR and dsRNA, relevant amino-acids and active phosphates are shown. TLR5 is shown associated with the FLiC, Flagellin peptide B-D) Different locations of interaction. The FliC elements are associated with three clusters of the TLR5 backbone. Each is important for recognition and dimerization.
The TLR3 binding domain has several unique features. First, the two units of the homodimer are separated by 120 Å. This separation accommodates a minimum of 40–50 base pairs of dsRNA required for binding. Originally, it was thought that prior to binding, the surfaces of the TLR3 homodimer subunits exhibited minimal contact,69 and polar interactions with the negatively charged dsRNA were the driving force of association. However, recent reports indicate that protease cleavage of TLR3 modulates signaling and that potentially fewer base pairs are required for activation. The binding mechanisms of the crystal structure may be just the beginning of the TLR3 story.70 Intriguingly, a recent report found that higher molecular weight Poly I:C stimulated macrophages to a greater extent than comparable lower molecular weight Poly I:Cs.71 This implies the synergistic activity of TLRs is dependent on agonist length. It also suggests that polymeric agonists may be a useful approach for TLR3.71 Additionally, selective delivery of TLR3 agonists to the endosome increases activity.72 To date, no synthetic, small-molecule (<1 kDa), agonists of TLR3 have been reported.
Toll-Like Receptor 5
Major Agonists: Proteins/Peptides
Toll-Like Receptor 5 binds flagellin and flagellin related peptides. Flagellin, a major component of the bacterial flagellar filament, confers motility to many bacterial species. TLR5 binds to the entire flagellin protein and reduced flagellin peptide mimics in a 2:2 stochiometry.73
The structural information about flagellin and TLR5 reveals a unique binding modality. Using a series of truncation experiments, a minimal binding element was discovered consisting of roughly 48 amino acids in an alpha helix within the flagellin peptide C-terminal domain of the FliC structure (Figure 4).74 While this minimal element binds, the signaling pathway may be different and truncated forms of flagellin have had mixed success as standalone adjuvants. In 2012, Wilson and coworkers published the structure of the FliC protein bound to a TLR5 of zebra-fish.75 (Figure 4) The LRRs align to form a binding pocket into which the 4 alpha-helical elements are inserted. The key residues involved in the activation of TLR5 are varied, but clearly critical for binding are D425, N445, and A417.76,77 The residues necessary for activation are not yet characterized in the dimer interface. Modifying these residues might inactivate the agonist or possibly convert it to an antagonist by preventing dimerization. As TLR5 senses peptides, many peptido-mimetic materials may also prove useful in forming longer lasting, more stable and safer agonists.
Toll-Like Receptor 9
Major Agonists: Oligonucleotides
TLR9 is an endosomal TLR, similar to TLR3, 7, and 8. TLR9 distinguishes between foreign and self DNA.78 This receptor is activated by bacterial oligonucleotides, specifically, unmethylated cytosine guanine (CpG) dinucleotide sequences. There are optimal CpG-ODN sequences, but the sequence differs depending on the species of TLR. CpG oligonucleotides (CpG-ODN) are categorized in classes A, B, and C. Depending on the sequence, different immune responses can result. Class A CpG elicits a large amount of IFN-α from plasmacytoid dendritic cells (pDCs). Class B CpG activates NK and B cells resulting in greater cytokine production. Lastly, the activity of class C CpG is a combination of classes A and B where both B cells are activated and IFN-α is produced.79–81 Downstream signaling of TLR9 proceeds through the MyD88 pathway and results in a polarized TH1 response.82
The operating paradigm is only vertebrate CpG-ODN sequences are methylated, so TLR9 distinguishes between bacterial and mammalian oligonucleotides by sensing methylation. The CpG dinucleotides must also be positioned next to two 5′-purines and two 3′-pyrimidines in the sequence, which is more common in bacteria than vertebrates.82–84 Synthetic DNA containing a phosphorothioated backbone is often employed for its resistance to nucleases. The synthetic modification increases cellular uptake and enhances immune system activation and binding affinity.85 However, if the phosphorothioated oligonucleotides do not contain the required CpG sequences, the resulting oligonucleotides act as antagonists implying that the thioate bond promotes tight binding regardless of sequence.
Interestingly, the CpG dinucleotide sequence is not the only factor that contributes to activation of TLR9. The 2′-hydroxy sugar backbone also plays a role in TLR9 stimulation.86 Though a crystal structure of TLR9 has not yet been published, TLR9 has been compared to TLR3 as both are endosomal TLRs that recognize nucleotides. Inferring from the crystal structure of TLR3, the 2′-hydroxy group may interact with an arginine residue, however, this has not been experimentally confirmed.87 The aggregation of CpG-ODN into multimeric complexes also promotes immune cell activation. It is proposed that the ODN-complexes may contribute to the clustering of TLR9, allowing for the initiation of TLR9 activation. Therefore, homodimers and higher order CpG-TLR9 complexes should be considered when designing synthetic ligands.
Mechanistically, CpG-ODNs are believed to directly interact with TLR9. Fluorescent microscopy experiments revealed one TLR9 activation mechanism where TLR9 is initially located in the endoplasmic reticulum (ER). In the ER, TLR9 is a full-length, non-functional protein. TLR9 then gets processed in the Golgi before being activated via protease cleavage in the endosome. The cleaved or shortened receptor is the active form and is activated by an oligonucleotide ligand to recruit MyD88.20 This may be a regulatory mechanism, so that TLR9 is selective for bacterial DNA, preventing “self-DNA” from stimulating TLR9.88,89
More recently, a hypothesis has been presented that TLR9 dimerization is independent of CpG sequence and that the phosphorothioate bond directs TLR9 dimerization.90 If true, synthetic polymers might mimic the TLR9 ligand and act as immune agonists. Obtaining a crystal structure of TLR9 will help determine the specific interactions necessary for the rational design of additional TLR9 agonists.
TLR Synergies – Compliments of Signaling and Structural Properties
So far, we have covered each TLR as an individual receptor. However, each receptor, and truly each PRR, works as part of a synergistic system that increases activity when multiple receptors are activated. A synergy is defined as increased immune activity (e.g. NFkB, cytokine secretion, or antibody titer) when the result of stimulation with two molecules together is stronger than the sum of their individual signals. Synergies are an exciting opportunity for chemical biologists. They are a key element to many of the most effective vaccines. Exploiting TLR synergies requires detailed structural and temporal information, the immunological implications of which are only beginning to be understood. These synergies are currently attributed to multiple elements including the activation of multiple signaling pathways within a dendritic cell,8,91,92 and the clustering of multiple receptors to form a hypothesized signalosome.93
Signaling Synergies
Signaling synergies involve the activation of multiple TLRs coordinating the activation of multiple signaling pathways.94 Beginning in 2005, many groups reported that combinations of soluble agonists had synergistic responses. Recent studies have explored the synergistic activation of signaling pathways with a focus on combining the predominant signaling pathways, MyD88 and TRIF (Figure 2). Agonists that stimulate TLR 3, 4 and 7, largely endosomal-elements, synergize with agonists that stimulate TLR 1/2/6. Intriguingly, several TLR combinations also act in an inhibitory manner including combinations of MPLA, Pam3CSK4 (TLR4, 2) and LPS or combinations of gardiquimod (TLR7) and CpGs (TLR9).95
Synergies appear to vary between humans and mice as well. A comprehensive study on synergies using PBMCs from human blood (PBMCs, distinct from plasmacytoid DC) was performed intentionally looking for variations between the PBMCs of multiple patients. In PBMCs, starkly different synergies are at work. Namely, in PBMCs, TLR5 is a dominant partner in synergistic activity with activity between TLR3 and TLR9; the so-called “TLR 3, 5, 9 highway”. Other PRR synergies are also present in a second, “TLR2/Dec1/NOD2/TLR3 highway” implying that synergies can be mapped across both membrane-bound TLRs and other PRRs including cytosolic (NOD2) receptors. These interactions demonstrate that pathways between DCs are highly specialized even between species. Therefore synergistic information generated from mouse models must be verified in human DCs. Are some synergies induced by chemical interaction or membrane modulation? Does timing play a role? These are all questions that chemical biologists are uniquely suited to answer as they begin designing immune responses.96
Signalosomes
Signalosomes are one potential explanation and a possible structural basis of synergies. The Wu group reported structural evidence for the observation that TLRs cluster when stimulated.97 Both MyD8898 and the downstream signal TRAF6 (Figure 2) form unique dimeric and trimeric complexes, implying the formation of a 2D lattice of TLRs arranged across the cell membrane in a hexagonal geometry (Figure 5).97 They posit that this mechanism lets TLR activation integrate signals into an on/off signaling mechanism. Could this mechanism also play a role in synergistic stimulation? Again, these are questions that chemical biologists might answer by developing molecular probes to interrogate these synergies using spatio-temporal control.
Figure 5.

Overview of PRR synergies. A) A proposed dynamic structure of the TLR signalasome. TLRs are shown in orange at the top. An example of TLR clusters are shown as well. B) Signaling Synergies. Interaction maps of different agonists portraying synergies as heat maps. Red indicates a greater interaction while blue represents inhibitory interactions. The thickness of the line indicates strength of synergistic activity. Tables of PRR synergies found in PBMCs. Color indicates type of interaction in at least 7 of 10 of the healthy volunteers. Red, synergistic effect; green, no effect/additive effect; blue, inhibitory effect; white, variable effect; black, experiment not performed.
Certain TLRs might be more prone to clustering and thereby increase the local concentration of signaling. Clustering in a signalosome would help explain the effectiveness of nano-particle systems in increasing immune responses.10 Additionally, for TLRs that detect long biomolecules (TLR3, TLR9), clusters may be used to augment their signal. The stronger signaling threshold induced by this clustering is one potential hypothesis for the synergistic activity reported between TLRs 3, 7, and 9. Remaining questions include: How do these structural elements translate to increased signaling, and what role do they play in determining the overall synergistic immune response? In the coming years, we believe that chemical probes will be a critical tool in understanding TLR synergies at a molecular and cellular level, and this understanding will be harnessed to map synergistic pathways.
Conclusions & Outlook
Stimulation of antigen presenting cells has seen immense progress in the past decade, and small molecules and biological polymers are poised to play a crucial role not only as vaccine candidates but also as biochemical tools to study the activation of the immune system. New studies are revealing the importance of LRR domains, TLR specificity, and TLR synergies. At the same time, the use of chemical tools to make structurally defined, polymeric or oligomeric agonist combinations is now possible. Using biochemical probes of TLR signaling could shed light on the spatio-temporal aspects of TLR synergies. The combination of all these techniques will lead to a deeper understanding of antigen presentation pathways, antigen processing, and cooperation between DCs and the adaptive immune system. Knowledge generated from these future studies promises to profoundly impact the design of the next generation of vaccines. Insight into these synergistic pathways might also reveal new treatments for diseases resulting from excessive inflammatory responses such as COPD, asthma, arthritis and generalized inflammation. Finally, optimization of the molecular structure of immune receptor agonists in terms of adjuvants or delivery systems will lead to improvements in efficacy of currently available vaccines and open perspectives for vaccines of diseases such as HIV, malaria, or cancer.11
Keywords
For historical and personal reasons, the field of immunology, like many others, contains unique terms that can intimidate new entrants. Acronyms, as always, serve two conflicting purposes, to quicken communication and to make it exclusive. We provide here a guide for terminology to be used as quick reference during this review.
- Antigen Presenting Cell (APC)
Cells activated by PAMPs interacting with PRRs that then present antigens to adaptive immune cells such as T cells. Dendritic Cells (DCs) are the most prominent and active form of APC with macrophages playing a similar role. There are many sub-classes of both. For an excellent review, see Kagan99 and Steinman15
- Pattern Recognition Receptor (PRR)
The general class of membrane and cytosolic protein receptors that recognize PAMPs . Each PRR described in this review will have a defined molecular agonist, but several PRRs, notably TLR4, are promiscuous binders, being activated by a large number of structurally dissimilar agonists. Sub-classes of PRRs include the Toll-Like Receptors (TLRs), the Nuclear Oligomerization Domain (NOD)-Like Receptors (NLRs), the Retenoic Acid Inducible Gene (RIG) Like Receptors (RLRs), and the C-type Lectin receptors (CLRs). Each PRR class is defined by their mode of action and molecular targets.
- Pathogen Associated Molecular Pattern (PAMP)
A molecule that activates the immune system. Originally postulated by Janeway,100 all PAMPs can be considered agonists of the innate immune system often operating through a PRR. However, their receptors or mechanism of action have not always been identified. PAMPs range from small molecules, to single and double-stranded oligos, up to full proteins. Not all PAMPS bind directly to a protein-based receptor, but we present only those that do.
- Cytokines
Cellular, proteinaceous signals secreted by cells of the immune system to communicate and direct one another. Within the context of APCs, cytokines can be considered as a measure of both overall immune stimulation and influencing the developmental pathway of T-cells.
- T Cells
A type of immune cell with subclasses including TH1, TH2, TH17, TH0, and Treg. Each T cell subclass interacts with a different set of immune cells and carries out a different set of instructions. T cells and their responses are described by general pathways using the THX designation.
- NFκB
A transcription factor that codes for immune cell stimulation. Generally, activation of a TLR increases NFκB resulting in immune cell stimulation. This can occur via a variety of pathways including TIR-domain-containing adapter-inducing interferon-β (TRIF) directly for TLR3 or indirectly for TLR4. Other TLRs including TLR4 activate NFκB via a pathway that includes myeloid differentiation primary response gene (88) (MyD88).
References
- 1.Kang JY, Lee JO. Annual Review of Biochemistry. 2011;80:917–941. doi: 10.1146/annurev-biochem-052909-141507. [DOI] [PubMed] [Google Scholar]
- 2.Marshak-Rothstein A. Nature Reviews Immunology. 2006;6:823–835. doi: 10.1038/nri1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmed R, Pulendran B. J Exp Med. 2011;208:2347–2349. doi: 10.1084/jem.20112321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumar H, Kawai T, Akira S. International reviews of immunology. 2011;30:16–34. doi: 10.3109/08830185.2010.529976. [DOI] [PubMed] [Google Scholar]
- 5.Mogensen TH. Clinical Microbiology Reviews. 2009;22:240–273. doi: 10.1128/CMR.00046-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tom JK, Mancini RJ, Esser-Kahn AP. Chem Commun. 2013 doi: 10.1039/c3cc45468a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kasturi SP, Skountzou I, Albrecht RA, Koutsonanos D, Hua T, Nakaya HI, Ravindran R, Stewart S, Alam M, Kwissa M, Villinger F, Murthy N, Steel J, Jacob J, Hogan RJ, García-Sastre A, Compans R, Pulendran B. Nature. 2011;470:543–547. doi: 10.1038/nature09737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu Q, Egelston C, Vivekanandhan A, Uematsu S, Akira S, Klinman DM, Belyakov IM, Berzofsky JA. PNAS. 2008;105:16260–16265. doi: 10.1073/pnas.0805325105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kiessling LL, Grim JC. Chem Soc Rev. 2013;42:4476–4491. doi: 10.1039/c3cs60097a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pulendran B, Ahmed R. Nature Immunology. 2011;131:509–517. doi: 10.1038/ni.2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nabel GJ. New England Journal of Medicine. 2013;368:551–560. doi: 10.1056/NEJMra1204186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Applequist SE, Wallin RPA, Ljunggren HG. Int Immunol. 2002;14:1065–1074. doi: 10.1093/intimm/dxf069. [DOI] [PubMed] [Google Scholar]
- 13.Berti F, Adamo R. ACS Chem Biol. 2013 doi: 10.1021/cb400423g. [DOI] [PubMed] [Google Scholar]
- 14.Huang Q, Liu D, Majewski P, Schulte LC, Korn JM, Young RA, Lander ES, Hacohen N. Science. 2001;294:870–875. doi: 10.1126/science.294.5543.870. [DOI] [PubMed] [Google Scholar]
- 15.Steinman RM. Annu Rev Immunol. 2012;30:1–22. doi: 10.1146/annurev-immunol-100311-102839. [DOI] [PubMed] [Google Scholar]
- 16.Rowley DA, Fitch FW. Cell Immunol. 2012;273:95–98. doi: 10.1016/j.cellimm.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 17.Barton GM, Kagan JC. Nat Rev Immunol. 2009;9:535–542. doi: 10.1038/nri2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takeuchi O, Akira S. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 19.West MA, Wallin RPA, Matthews SP, Svensson HG, Zaru R, Ljunggren HG, Prescott AR, Watts C. Science. 2004;305:1153–1157. doi: 10.1126/science.1099153. [DOI] [PubMed] [Google Scholar]
- 20.Ewald SE, Lee BL, Lau L, Wickliffe KE, Shi GP, Chapman HA, Barton GM. Nature. 2008;456:658–662. doi: 10.1038/nature07405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jin MS, Kim SE, Heo JY, Lee ME, Kim HM, Paik SG, Lee H, Lee JO. Cell. 2007;130:1071–1082. doi: 10.1016/j.cell.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 22.Manavalan B, Basith S, Choi S. Front Physiol. 2011;2 doi: 10.3389/fphys.2011.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berg M, Offermanns S, Seifert R, Schultz G. Am J Physiol. 1994;266:C1684–C1691. doi: 10.1152/ajpcell.1994.266.6.C1684. [DOI] [PubMed] [Google Scholar]
- 24.Wiesmuller K, Bessler W, Jung G. Hoppe-Seylers Zeitschrift Fur Physiologische Chemie. 1983;364:593–606. doi: 10.1515/bchm2.1983.364.1.593. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y, Luo F, Cai Y, Liu N, Wang L, Xu D, Chu Y. J Immunol. 2011;186:1963–1969. doi: 10.4049/jimmunol.1002320. [DOI] [PubMed] [Google Scholar]
- 26.Van Duin D, Medzhitov R, Shaw AC. Trends Immunol. 2006;27:49–55. doi: 10.1016/j.it.2005.11.005. [DOI] [PubMed] [Google Scholar]
- 27.Seong SY, Matzinger P. Nat Rev Immunol. 2004;4:469–478. doi: 10.1038/nri1372. [DOI] [PubMed] [Google Scholar]
- 28.Zaehringer U, Lindner B, Inamura S, Heine H, Alexander C. Immunobiology. 2008;213:205–224. doi: 10.1016/j.imbio.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 29.Ray A, Cot M, Puzo G, Gilleron M, Nigou J. Biochimie. 2013;95:33–42. doi: 10.1016/j.biochi.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 30.Salunke DB, Shukla NM, Yoo E, Crall BM, Balakrishna R, Malladi SS, David SA. J Med Chem. 2012;55:3353–3363. doi: 10.1021/jm3000533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agnihotri G, Crall BM, Lewis TC, Day TP, Balakrishna R, Warshakoon HJ, Malladi SS, David SA. J Med Chem. 2011;54:8148–8160. doi: 10.1021/jm201071e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sato M, Sano H, Iwaki D, Kudo K, Konishi M, Takahashi H, Takahashi T, Imaizumi H, Asai Y, Kuroki Y. J Immunol. 2003;171:417–425. doi: 10.4049/jimmunol.171.1.417. [DOI] [PubMed] [Google Scholar]
- 33.Tsai CC, Lin CR, Tsai HY, Chen CJ, Li WT, Yu HM, Ke YY, Hsieh WY, Chang CY, Wu CY, Chen ST, Wong CH. J Biol Chem. 2013;288:17689–17697. doi: 10.1074/jbc.M112.448381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sahoo BR, Basu M, Swain B, Dikhit MR, Jayasankar P, Samanta M. BioMed Research International. 2013;2013 doi: 10.1155/2013/185282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buwitt-Beckmann U, Heine H, Wiesmüller KH, Jung G, Brock R, Akira S, Ulmer A. European Journal of Immunology. 2005;35:282–289. doi: 10.1002/eji.200424955. [DOI] [PubMed] [Google Scholar]
- 36.Guan Y, Omueti-Ayoade K, Mutha SK, Hergenrother PJ, Tapping RI. J Biol Chem. 2010;285:23755–23762. doi: 10.1074/jbc.M110.116046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kang JY, Nan X, Jin MS, Youn SJ, Ryu YH, Mah S, Han SH, Lee H, Paik SG, Lee JO. Immunity. 2009;31:873–884. doi: 10.1016/j.immuni.2009.09.018. [DOI] [PubMed] [Google Scholar]
- 38.Ginsburg I. The Lancet Infectious Diseases. 2002;2:171–179. doi: 10.1016/s1473-3099(02)00226-8. [DOI] [PubMed] [Google Scholar]
- 39.Stadelmaier A, Morath S, Hartung T, Schmidt RR. Angewandte Chemie International Edition. 2003;42:916–920. doi: 10.1002/anie.200390243. [DOI] [PubMed] [Google Scholar]
- 40.Metzger JW, Beck-Sickinger AG, Loleit M, Eckert M, Bessler WG, Jung G. J Pept Sci. 1995;1:184–190. doi: 10.1002/psc.310010305. [DOI] [PubMed] [Google Scholar]
- 41.Ohto U, Fukase K, Miyake K, Shimizu T. PNAS. 2012;109:7421–7426. doi: 10.1073/pnas.1201193109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park BS, Song DH, Kim HM, Choi BS, Lee H, Lee JO. Nature. 2009;458:1191–1195. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- 43.Stover AG. Journal of Biological Chemistry. 2003;279:4440–4449. doi: 10.1074/jbc.M310760200. [DOI] [PubMed] [Google Scholar]
- 44.Coler RN, Baldwin SL, Shaverdian N, Bertholet S, Reed SJ, Raman VS, Lu X, DeVos J, Hancock K, Katz JM, Vedvick TS, Duthie MS, Clegg CH, Van Hoeven N, Reed SG. PLoS ONE. 2010;5:e13677. doi: 10.1371/journal.pone.0013677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coler RN, Bertholet S, Moutaftsi M, Guderian JA, Windish HP, Baldwin SL, Laughlin EM, Duthie MS, Fox CB, Carter D, Friede M, Vedvick TS, Reed SG. PLoS ONE. 2011;6:e16333. doi: 10.1371/journal.pone.0016333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Needham BD, Carroll SM, Giles DK, Georgiou G, Whiteley M, Trent MS. Proc Natl Acad Sci USA. 2013;110:1464–1469. doi: 10.1073/pnas.1218080110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, Mitchell TC. Science. 2007;316:1628–1632. doi: 10.1126/science.1138963. [DOI] [PubMed] [Google Scholar]
- 48.Gangloff M. Trends in Biochemical Sciences. 2012;37:92–98. doi: 10.1016/j.tibs.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bowen WS, Minns LA, Johnson DA, Mitchell TC, Hutton MM, Evans JT. Science Signaling. 2012;5:ra13–ra13. doi: 10.1126/scisignal.2001963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Trompette A, Divanovic S, Visintin A, Blanchard C, Hegde RS, Madan R, Thorne PS, Wills-Karp M, Gioannini TL, Weiss JP, Karp CL. Nature. 2008;457:585–588. doi: 10.1038/nature07548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bryant CE, Spring DR, Gangloff M, Gay NJ. Nature Reviews Microbiology. 2009 doi: 10.1038/nrmicro2266. [DOI] [PubMed] [Google Scholar]
- 52.Shanmugam A, Rajoria S, George AL, Mittelman A, Suriano R, Tiwari RK. PLoS ONE. 2012;7:e30839. doi: 10.1371/journal.pone.0030839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ohashi K, Burkart V, Flohé S, Kolb H. J Immunol. 2000;164:558. doi: 10.4049/jimmunol.164.2.558. [DOI] [PubMed] [Google Scholar]
- 54.Asea A. Journal of Biological Chemistry. 2002;277:15028–15034. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]
- 55.Smiley ST, King JA, Hancock WW. J Immunol. 2001;167:2887. doi: 10.4049/jimmunol.167.5.2887. [DOI] [PubMed] [Google Scholar]
- 56.Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, Chow JC, Stauss JF., III Journal of Biological Chemistry. 2001;276:10229–10233. doi: 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- 57.Semeraro F, Ammollo CT, Morrissey JH, Dale GL, Friese P, Esmon NL, Esmon CT. Blood. 2011;118:1952–1961. doi: 10.1182/blood-2011-03-343061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chan M, Hayashi T, Mathewson RD, Nour A, Hayashi Y, Yao S, Tawatao RI, Crain B, Tsigelny IF, Kouznetsova VL, Messer K, Pu M, Corr M, Carson DA, Cottam HB. J Med Chem. 2013;56:4206–4223. doi: 10.1021/jm301694x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Latz E, Verma A, Visintin A, Gong M, Sirois C, Klein D, Monks B, McKnight CJ, Lamphier M, Duprex P, Espevik T, Golenbock D. Nature. 2007;8:772–779. doi: 10.1038/ni1479. [DOI] [PubMed] [Google Scholar]
- 60.Wei T, Gong J, Jamitzky F, Heckl W, Stark R, Rossle S. Protein Sci. 2009;18:1684–1691. doi: 10.1002/pro.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tanji H, Ohto U, Shibata T, Miyake K, Shimizu T. Science. 2013;339:1426–1429. doi: 10.1126/science.1229159. [DOI] [PubMed] [Google Scholar]
- 62.Gibbard RJ, Morley PJ, Gay NJ. J Biol Chem. 2006;281:27503–27511. doi: 10.1074/jbc.M605003200. [DOI] [PubMed] [Google Scholar]
- 63.Chan M, Hayashi T, Mathewson RD, Yao S, Gray C, Tawatao RI, Kalenian K, Zhang Y, Hayashi Y, Lao FS, Cottam HB, Carson DA. Bioconjugate Chem. 2011;22:445–454. doi: 10.1021/bc1004813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chan M, Hayashi T, Kuy CS, Gray CS, Wu CCN, Corr M, Wrasidlo W, Cottam HB, Carson DA. Bioconjugate Chem. 2009;20:1194–1200. doi: 10.1021/bc900054q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kokatla HP, Sil D, Malladi SS, Balakrishna R, Hermanson AR, Fox LM, Wang X, Dixit A, David SA. J Med Chem. 2013;56:6871–6885. doi: 10.1021/jm400694d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shukla NM, Salunke DB, Balakrishna R, Mutz CA, Malladi SS, David SA. PLoS ONE. 2012;7:e43612. doi: 10.1371/journal.pone.0043612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cusson-Hermance N, Khurana S, Lee TH, Fitzgerald KA, Kelliher MA. J Biol Chem. 2005;280:36560–36566. doi: 10.1074/jbc.M506831200. [DOI] [PubMed] [Google Scholar]
- 68.Leonard JN, Ghirlando R, Askins J, Bell JK, Margulies DH, Davies DR, Segal DM. PNAS. 2008;105:258–263. doi: 10.1073/pnas.0710779105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Choe J, Kelker MS, Wilson IA. Science. 2005;309:581–585. doi: 10.1126/science.1115253. [DOI] [PubMed] [Google Scholar]
- 70.Toscano F, Estornes Y, Virard F, Garcia-Cattaneo A, Pierrot A, Vanbervliet B, Bonnin M, Ciancanelli MJ, Zhang SY, Funami K, Seya T, Matsumoto M, Pin JJ, Casanova JL, Renno T, Lebecque S. J Immunol. 2013;190:764–773. doi: 10.4049/jimmunol.1202173. [DOI] [PubMed] [Google Scholar]
- 71.Zhou Y, Guo M, Wang X, Li J, Wang Y, Ye L, Dai M, Zhou L, Persidsky Y, Ho W. Innate Immun. 2013;19:184–192. doi: 10.1177/1753425912459975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Barbuto S, Idoyaga J, Vila-Perelló M, Longhi MP, Breton G, Steinman RM, Muir TW. Nature Chemical Biology. 2013 doi: 10.1038/nchembio.1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ivičak-Kocjan K, Panter G, Benčina M, Jerala R. Biochemical and Biophysical Research Communications. 2013;435:40–45. doi: 10.1016/j.bbrc.2013.04.030. [DOI] [PubMed] [Google Scholar]
- 74.Mizel SB, West AP, Hantgan RR. J Biol Chem. 2003;278:23624–23629. doi: 10.1074/jbc.M303481200. [DOI] [PubMed] [Google Scholar]
- 75.Yoon S, Kurnasov O, Natarajan V, Hong M, Gudkov AV, Osterman AL, Wilson IA. Science. 2012;335:859–864. doi: 10.1126/science.1215584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Smith KD, Andersen-Nissen E, Hayashi F, Strobe K, Bergman MA, Barrett SLR, Cookson BT, Aderem A. Nat Immunol. 2003;4:1247–1253. doi: 10.1038/ni1011. [DOI] [PubMed] [Google Scholar]
- 77.Andersen-Nissen E, Smith KD, Bonneau R, Strong RK, Aderem A. J Exp Med. 2007;204:393–403. doi: 10.1084/jem.20061400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Barton GM, Kagan JC, Medzhitov R. Nat Immunol. 2006;7:49–56. doi: 10.1038/ni1280. [DOI] [PubMed] [Google Scholar]
- 79.Rutz M, Metzger J, Gellert T, Luppa P, Lipford GB, Wagner H, Bauer S. European Journal of Immunology. 2004;34:2541–2550. doi: 10.1002/eji.200425218. [DOI] [PubMed] [Google Scholar]
- 80.Vollmer J, Weeratna R, Payette P, Jurk M, Schetter C, Laucht M, Wader T, Tluk S, Liu M, Davis HL, Krieg AM. European Journal of Immunology. 2004;34:251–262. doi: 10.1002/eji.200324032. [DOI] [PubMed] [Google Scholar]
- 81.Krug A, Rothenfusser S, Hornung V, Jahrsdörfer B, Blackwell S, Ballas ZK, Endres S, Krieg AM, Hartmann G. European Journal of Immunology. 2001;31:2154–2163. doi: 10.1002/1521-4141(200107)31:7<2154::aid-immu2154>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 82.Krieg AM, Wagner H. Immunology Today. 2000;21:521–526. doi: 10.1016/s0167-5699(00)01719-9. [DOI] [PubMed] [Google Scholar]
- 83.Krieg AM, Yi AK, Matson S, Waldschmidt TJ, Bishop GA, Teasdale R, Koretzky GA, Klinman DM. Nature. 1995;374:546–549. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- 84.Krieg AM. Current Opinion in Immunology. 2000;12:35–43. doi: 10.1016/s0952-7915(99)00048-5. [DOI] [PubMed] [Google Scholar]
- 85.Krieg AM. Annual Review of Immunology. 2002;20:709–760. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- 86.Haas T, Metzger J, Schmitz F, Heit A, Müller T, Latz E, Wagner H. Immunity. 2008;28:315–323. doi: 10.1016/j.immuni.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 87.Li Y, Berke IC, Modis Y. EMBO J. 2012;31:919–931. doi: 10.1038/emboj.2011.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ewald SE, Engel A, Lee J, Wang M, Bogyo M, Barton GM. J Exp Med. 2011;208:643–651. doi: 10.1084/jem.20100682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Park B, Brinkmann MM, Spooner E, Lee CC, Kim YM, Ploegh HL. Nat Immunol. 2008;9:1407–1414. doi: 10.1038/ni.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Li Y, Berke IC, Modis Y. EMBO J. 2012;31:919–931. doi: 10.1038/emboj.2011.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Krummen M, Balkow S, Shen L, Heinz S, Loquai C, Probst HC, Grabbe S. J Leukoc Biol. 2010;88:189–199. doi: 10.1189/jlb.0408228. [DOI] [PubMed] [Google Scholar]
- 92.Mäkelä SM, Strengell M, Pietilä TE, Osterlund P, Julkunen I. J Leukoc Biol. 2009;85:664–672. doi: 10.1189/jlb.0808503. [DOI] [PubMed] [Google Scholar]
- 93.Wu H. Cell. 2013;153:287–292. doi: 10.1016/j.cell.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Trinchieri G, Sher A. Nat Rev Immunol. 2007;7:179–190. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- 95.Garcia-Cordero JL, Nembrini C, Stano A, Hubbell JA, Maerkl SJ. Integr Biol (Camb) 2013;5:650–658. doi: 10.1039/c3ib20263a. [DOI] [PubMed] [Google Scholar]
- 96.Timmermans K, Plantinga TS, Kox M, Vaneker M, Scheffer GJ, Adema GJ, Joosten LAB, Netea MG. Clin Vaccine Immunol. 2013;20:427–432. doi: 10.1128/CVI.00703-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Napetschnig J, Wu H. Annual Review of Biophysics. 2013;42:443–468. doi: 10.1146/annurev-biophys-083012-130338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lin SC, Lo YC, Wu H. Nature. 2010;465:885–890. doi: 10.1038/nature09121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Barton GM, Kagan JC. Nat Rev Immunol. 2009;9:535–542. doi: 10.1038/nri2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Medzhitov R, Janeway CA., Jr Current Opinion in Immunology. 1997;9:4–9. doi: 10.1016/s0952-7915(97)80152-5. [DOI] [PubMed] [Google Scholar]