

Abstract
Drug-induced liver injury (DILI) is a leading cause of drug failure in clinical trials and a major reason for drug withdrawals. DILI has been shown to be dependent on both daily dose and extent of hepatic metabolism. Yet, early in drug development daily dose is unknown. Here, we perform a comprehensive analysis of the published hypotheses that attempt to predict DILI, including a new analysis of the Biopharmaceutics Drug Disposition Classification System (BDDCS) in evaluating the severity of DILI warning in drug labels approved by the FDA and the withdrawal status due to ADRs. Our analysis confirms that higher doses ≥ 50mg/day lead to increased DILI potential but this property alone is not sufficient to predict DILI potential. We evaluate prior attempts to categorize DILI such as Rule of 2, BSEP inhibition, and measures of key mechanisms of toxicity compared to BDDCS classification. Our results show that BDDCS Class 2 drugs exhibit the highest DILI severity, and that all of the published methodologies evaluated here, except when daily dose is known, do not yield markedly better prediction than BDDCS. The assertion that extensive metabolized compounds are at higher risk of developing DILI is confirmed, but can be enhanced by differentiating BDDCS Class 2 from Class 1 drugs. We do not propose that BDDCS classification, which does not require knowledge of the clinical dose, is sufficiently predictive/accurate of DILI potential for new molecular entities, but suggest that comparison of proposed DILI prediction methodologies with BDDCS classification is a useful tool to evaluate the potential reliability of newly proposed algorithms.
Conclusion
The most successful approaches to predict DILI potential all include a measure of dose, yet there is a quantifiable uncertainty associated with the predicted dose early in drug development. Here we compare the possibility of predicting DILI potential using BDDCS classification versus previously published methods, and suggest that comparison of predictive metrics versus the outcome by just avoiding BDDCS Class 2 drugs may serve as a useful baseline in evaluating these metrics.
Keywords: BDDCS, BSEP inhibition, DILI assessment, FDA label warnings, Hepatotoxicity
INTRODUCTION
Drug-induced liver injury (DILI) is a leading cause of drug failure in clinical trials and a major reason for drug withdrawals from the market. Idiosyncratic DILI (IDILI) is very complex. Most IDILI appears to be immune mediated, and reactive metabolites appear to be involved in most, but not all IDILI. In addition, there are probably several mechanisms by which a drug or reactive metabolite can induce an immune response. Numerous compound- and/or patient-specific risk factors can contribute to the susceptibility to DILI. IDILI has been shown to be dependent on both daily dose and extent of hepatic metabolism of a drug 1–4.
Dose appears to be a key component in the risk assessment of toxicity. While there is not a clear dose-response relationship for idiosyncratic adverse drug reactions, epidemiological DILI studies have shown that dose of a compound is an important parameter in determining the likelihood that an individual drug will cause an idiosyncratic adverse drug reaction in the human population2. At the same time, numerous studies have shown that dose alone is not an adequate discriminator between high and low risk compounds5. There are a number of preclinical strategies where dose has been combined with other parameters directly or indirectly related to key measures of toxicity endpoints to help assess the potential DILI risk such as the formation of reactive metabolites, inhibition of the bile salt export pump, BSEP, resulting in the intracellular accumulation of bile salts and high covalent body burden6,7, mitochondrial dysfunction (resulting in the depletion of cellular energy supply and the generation of damaging reactive oxygen species), cell damage from oxidative stress (caused by reactive oxygen or reactive nitrogen species), and local inflammatory effects8. All of these mechanisms are often interconnected and have, under various circumstances, been associated with the formation of chemically reactive metabolites. Recently, Chen et al. reported that high lipophilicity in combination with high daily dose increases DILI risk potential in humans9. However, one would like to have a predictive DILI methodology early in drug development, long before the clinical dose is known.
Here we consider the possibility of using the Biopharmaceutics Drug Disposition Classification System (BDDCS), which can be determined prior to dosing a drug to humans or animals, as a potential baseline tool to be compared with presently proposed predictive procedures in evaluating DILI toxicity. The BDDCS was developed in 2005 after Wu and Benet recognized that highly permeable compounds, as outlined by the Biopharmaceutics Classification System (BCS), were extensively metabolized, while poorly permeable drugs were primarily eliminated unchanged in the urine or bile10. Furthermore, BDDCS demonstrated that simple passive membrane permeability measures were highly selective in differentiating extensively vs. poorly metabolized drugs in humans. Drugs in the BDDCS are classified according to the membrane permeability rate and aqueous solubility. These characteristics have helped BDDCS define whether metabolic enzymes and/or transporters are clinically important. BDDCS features are demarcated by high and low values, classifying drugs into four categories. These classes are each associated with specific predictions regarding route of elimination and which interactions may be a clinical concern.
Since its inception, the BDDCS has been useful in drug discovery for predicting routes of elimination, oral drug disposition, food effects on drug absorption, transporter effects on drug absorption, and potentially clinically significant drug interactions that may arise in the intestine, liver and brain11. Most recently we have shown that the BDDCS can be useful in predicting the potential for antiepileptic drugs to cause cutaneous adverse reactions12. A goal of this perspective was to explore the extent to which BDDCS defining characteristics, independent of knowing actual drug pharmacokinetics/pharmacodynamics and dose can be used as a comparison baseline matrix of potential DILI adverse events with prior published predictive proposals9,13–16
Here, we perform a comprehensive analysis to examine the clinical impact of BDDCS in evaluating the severity of DILI warning in drug labels approved by the Food and Drug Administration (FDA)17, the withdrawal status due to ADRs, the role of BSEP inhibition, and proposed models including: the Rule of 2 (Ro2), Ro2 and reactive metabolite formation, maximum daily dosages prescribed, and assays applied to cover various mechanisms and endpoints associated with human DILI. (These assays included the generation of reactive metabolites, namely time-dependent inhibition (TDI) of Cytochrome P450 3A4 and glutathione (GSH) adduct formation, inhibition of the human bile salt export pump (BSEP), mitochondrial toxicity and cytotoxicity)14. Recently, Zhang et al.16 evaluated specific metabolic pathways predictive of DILI and Chen et al.13 added the measurement of known reactive metabolites, both reporting a marked improvement in the previous methodologies employed to predict DILI; we have also included these studies in our analysis.
Because one of the strongest determinant hypotheses with respect to DILI is reactive metabolite formation, we expect that drugs that are extensively metabolized/highly permeable (BDDCS Class 1 and 2) will have heightened susceptibility to DILI. Conversely, drugs that are poorly metabolized/poorly permeable (BDDCS Class 3 and 4) will be at a lower risk for causing DILI because they are primarily eliminated unchanged into the urine and bile. The strong relationship between dose, metabolic susceptibility, solubility and idiosyncratic DILI highlights the potential benefits of BDDCS as a comparison matrix for DILI prediction.
Relationship between FDA Drug Label Section, DILI Assessment and BDDCS Classification
In our current comparative analysis, we leveraged the unique information contained in FDA drug labels and DILI severity assessment with respect to the BDDCS classification system. The DILI potential of the drugs in the data set was classified on the basis of the information on hepatic ADRs extracted from FDA drug labels; we note that only drugs that have been on the market for a minimum of ten years were chosen for review18. Briefly, depending on the ADR severity, off market status and FDA drug labels, ADRs may be classified in different categories (“Discontinued”, “Withdrawn”, “Boxed Warning”, “Warning and Precautions”, “Adverse Reactions” and “No Mention”, ordered by decreasing severity)19–21. The DILI severity assessment is categorized as follows: “Severe DILI”, “Moderate DILI”, “Mild DILI”, and “No DILI”, ordered by decreasing severity as described by Chen et al.18. However, with the recent publication of prediction based on metabolic pathways16, “Moderate DILI” and “Mild DILI” were combined into a category designated “Non-severe DILI,” which we have utilized here.
BDDCS Classification
The BDDCS Class of each drug was initially evaluated based on the available solubility data, maximum dose strength (mg), and extent of metabolism22. Recently we expanded the list of BDDCS drug classification to more than 1100 drugs, including many drugs that have been removed from the market as a result of toxic manifestations23. Expansion of the BDDCS classification list was particularly challenging since for many drugs that came onto the market a number of years ago, and then removed because of toxicity, little reliable information both in terms of metabolism and solubility can be found in the literature. Therefore, when a drug is on the border of two classes, the BDDCS class is selected based on expected or known drug interactions.
There is a marked distinction between extensively and poorly metabolized compounds and this can be well predicted based on an in vitro measure of drug permeability24. Recently, Dave and Morris showed that the solubility classification could be evaluated using a 0.3 mg/mL cut-off, 25, thus not requiring knowledge of the clinical dose, thereby allowing BDDCS classification to be made without knowledge of clinical dose (See Supplementary Figure 1).
We examined the BDDCS class relationship of hepatotoxicity between the different ADR categories by calculating the proportion of drugs in each FDA hepatic liability category, and DILI severity category. Categorical variables were tested for statistically significant differences using the chi-square tests (test for trends in proportions and test of equal or given proportions), p < 0.05 was considered statistically significant. Analyses and plots were carried out using R (http://cran.r-project.org)26,27 and GraphPad Prism software version 7.0 (GraphPad Software, Inc., San Diego, CA). Furthermore, The p-values for evaluating BDDCS class trends of FDA hepatic liability category and DILI severity are computed by the implemented functions in R for testing for trends in proportions. The test of equal or given proportions was used for testing the null hypothesis that the proportions in several groups are the same as the “No Mention” or “No DILI” where applicable. Out of 287 eligible compounds from the NCTR dataset, 19 compounds could not be classified due to limited available data (See legend of Supplementary Figure 2).
We observe that as the hepatic warning severity increases, the proportion of BDDCS Class 2 drugs increases and the proportions of both BDDCS Class 1 and 3 drug decrease as depicted in Figure 1A, all with highly significant trends. The “No Mention” category is significantly different from all other categories, except for “Adverse Reactions.” BDDCS Class 2 drugs were incriminated with the highest proportions in the following drug label sections: “Warning and Precautions” (45.6%, 36/79), “Boxed Warning” (47.2%, 17/36), “Withdrawn” (62.5%, 25/40) and “Discontinued” (83.3%, 5/6). Obviously, the number of drugs designated as exhibiting severe DILI drugs increases as the ADR severity increases. That is, 15.9% (7/44) in the “Adverse Reactions” category, 36.7% (29/79) in the “Warning and Precautions” and 81.6% (31/38) of the drugs in the “Black Box Warning” are assessed to exhibit severe DILI (See Supplementary Figure 2). In Figure 1B and 1C the two BDDCS determinants (extent of metabolism and solubility) are examined. The percentage of poorly metabolized and of highly soluble drugs decrease, while low solubility drugs increase with hepatic liability. The percent of extensively metabolized drugs also increases with hepatic liability, but since almost 2/3 of “No Mention” drugs are metabolized, it is apparent that extent of metabolism itself is not a discriminating parameter. Although greater extent of metabolism has been reported to significantly increase the potential of a compound to cause DILI 1, this property alone is not able to distinguish compounds that are “No Mention” of hepatic liability from those compounds exhibiting hepatic liability (See Figure 1B).
Figure 1.
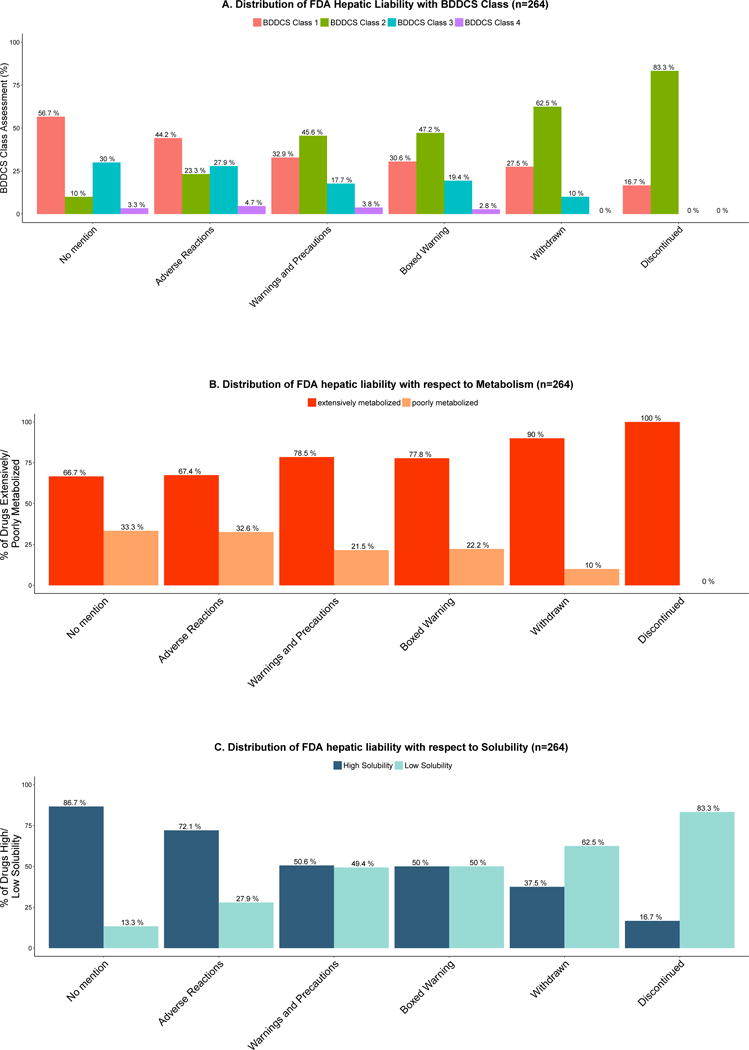
A. Distribution of FDA hepatic liability with BDDCS Class
Drugs were assigned according to the most severe drug label section reporting a hepatic ADR or withdrawn and discontinued, and to the “No mention” class if no hepatic ADRs were reported. Bars show the percentage of all compounds in the same category that are associated with each FDA hepatic liability. BDDCS Class 2 drugs are shown to significantly increase the frequency of hepatic ADRs reported in the “Boxed Warning”, “Warning and Precautions”, “Withdrawn” and “Discontinued” categories. There was a significant difference between BDDCS Classes when the proportionality trend test was calculated: BDDCS Class 1 trend p-value = 0.0003842; BDDCS Class 2 trend p-value = 2.014e-10; BDDCS Class 3 trend p-value = 0.003928; BDDCS Class 4 trend NS, p-value = 0.2963. Differences in the BDDCS Class distributions were evaluated among the following groups: “No mention” vs. “Adverse Reactions”, NS, p-value = 0.2908; “No mention” vs. “Warning and Precautions”, p-value=0.0001058; “No mention” vs. “Boxed Warning”, p-value=0.0006222; “No mention” vs. “Withdrawn”, p-value= 6.439e-07“; No mention” vs. “Discontinued”, p-value= 9.36e-05
B. Distribution of FDA hepatic liability with respect to extensively metabolized vs. poorly metabolized drugs
The extensive hepatic metabolism group consisted of 102 BDDCS Class 1 and 99 BDDCS Class 2 drugs; the poor hepatic metabolism group consisted of 55 BDDCS Class 3 and 8 BDDCS Class 4 drugs. There was a significant difference between extensively metabolized vs. poorly metabolized drugs when the proportionality test was calculated p-value = 0.001536).
C. Distribution of FDA hepatic liability with respect to high solubility vs. low solubility drugs
The high solubility group consisted of 102 BDDCS Class 1 and 55 BDDCS Class 3 drugs; the low solubility group consisted of 99 BDDCS Class 2 and 8 BDDCS Class 4 drugs. There was a significant difference between extensively high solubility vs. low solubility drugs when the proportionality test was calculated p-value = 3.481e-09.
When assessing DILI severity using the FDA DILI severity assignment (but combining “Mild” and “Moderate” DILI as “Non-severe DILI”) with BDDCS Class (See Figure 2A), we also observe statistically significant trends for the increase in BDDCS Class 2 and decreases for BDDCS Classes 1 and 3. BDDCS Class 2 represents 53.6% (60/112) of the drugs in the “Severe DILI” category vs. 10% (6/60) in the “No DILI” category. BDDCS Class 1 represents 28.6% (32/112) of drugs in the “Severe DILI” vs. 56.7% (34/60) in the “No DILI” category. BDDCS Class 3 represents 14.3% (16/112) of drugs in the “Severe DILI” vs. 30% (18/60) in the “No DILI” category. The “Severe DILI” category comprises the following endpoints: acute liver failure, fatal hepatotoxicity, and “Discontinued and Withdrawn” drugs as defined by Chen et al.18. The “Non-severe DILI” category comprises compounds exhibiting hyperbilirubinemia, jaundice, and/or liver necrosis (“Moderate DILI”) and compounds exhibiting liver aminotransferases increase (“Mild DILI”). In the “No DILI” category, we observe that most drugs are BDDCS Classes 1 (56.7%, 34/60) and 3 (30%, 18/60). Here again, we observe a significant trend of high DILI liability for extensively metabolized compounds (Figure 2B). Drugs exhibiting high solubility (Figure 2C) show a trend of lower DILI severity.
Figure 2.
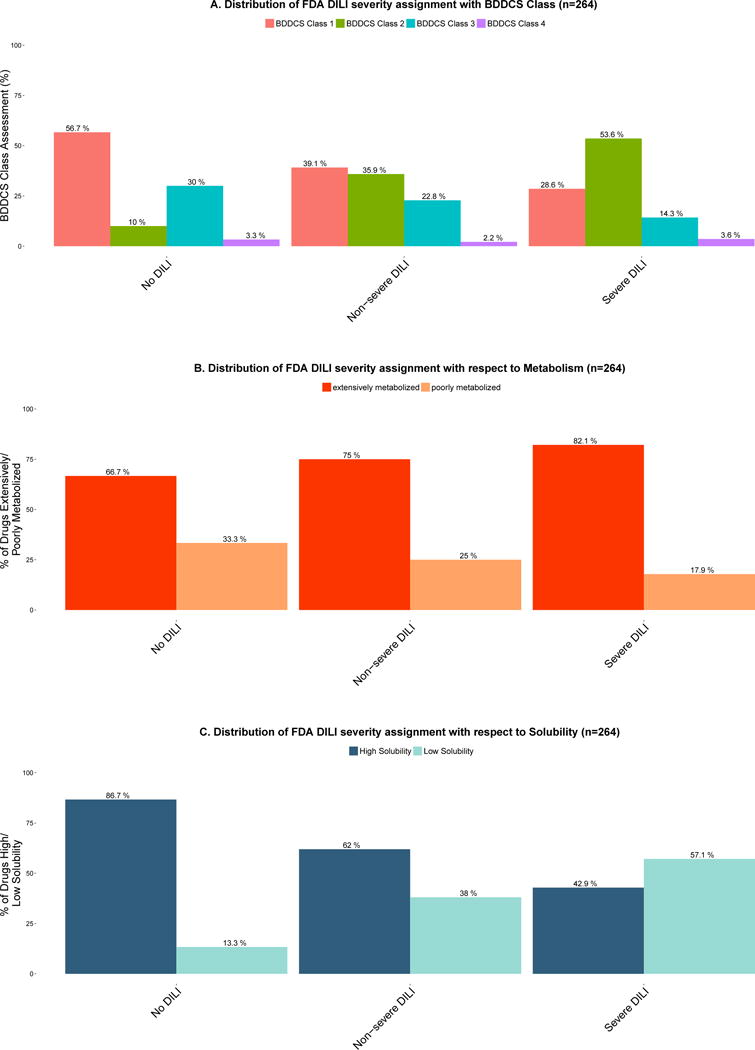
A. Distribution of FDA DILI severity assignment with BDDCS Class
FDA Drugs Labels were assigned to a DILI severity class according to the most severe reporting of a hepatic ADR or to the No DILI class if no hepatic ADRs were reported (6). Bars show the percentage of all compounds in the same DILI severity class (“No DILI”, “Non-severe DILI”, and “Severe DILI”). BDDCS Class 2 shows the highest frequency in the “Severe DILI” class assessment. BDDCS Class 1 and 3 drugs show the highest frequency in the “No DILI” class assessment. There was a significant difference between BDDCS classes when the proportionality trend test was calculated: BDDCS Class 1 trend p-value = 0.0003608; BDDCS Class 2 trend p-value = 2.105e-08; BDDCS Class 3 trend NS, p-value = 0.01297; BDDCS Class 4 trend NS, p-value = 0.8457. There was also significant differences in the BDDCS Class distributions among the following groups: “No DILI” vs. “Non-severe DILI”, p-value = 0.005063; “No DILI” vs. “Severe DILI”, p-value = 4.627e-07.
B. Distribution of FDA DILI severity assignment with respect to extensively metabolized vs. poorly metabolized drugs. There was a significant difference between extensively metabolized vs. poorly metabolized drugs when the proportionality test was calculated p-value = 0.02208.
C. Distribution of FDA DILI severity assignment with respect to high vs. low solubility drugs. There was a significant different between high vs. low solubility drugs when the proportionality test was calculated p-value = 2.23e-08.
Our examination of the relationship between the BDDCS’s determinant properties: solubility and extent of metabolism led to some novel observations. Drugs belonging to BDDCS Class 1 and 3 exhibited a lower proportion of DILI severity. Drugs that are extensively metabolized and have low aqueous solubility, i.e., BDDCS Class 2 drugs have the highest rates of DILI risk. BDDCS Class 2 drugs exhibited the highest proportions among the ”Warning and Precautions”, “Black Box Warning”, “Withdrawn” and “Discontinued” categories. These are notably considered the most serious DILI risk categories (See Figure 1A). These findings demonstrate the importance of the intrinsic drug properties as a potential factor for the development of a DILI event.
Relationship between Daily Dosage, FDA Drug Label and DILI Assessment Score
Lammert and coworkers1,2 have attributed hepatic adverse events to compounds with significant hepatic metabolism and daily dose ≥ 50mg. We have also evaluated the relationship between daily dosages ≥ 50mg against the already assessed FDA hepatic liability categories and DILI severity assessment9. Our analysis concurs with the association of drugs being given at dosages ≥ 50mg/day having more adverse hepatic events. We have further evaluated this observation by examining the FDA hepatic liability distribution and DILI severity assessment. Drugs with a daily dose ≥ 50mg had a much higher frequency of toxicity as evidenced by the higher percentages in the “Warning and Precautions”, “Boxed Warning” and “Withdrawn” label sections. For the DILI assessment in Figure 3B we also observe a higher frequency in DILI severity for compounds that are dosed at ≥ 50mg/day.
Figure 3.
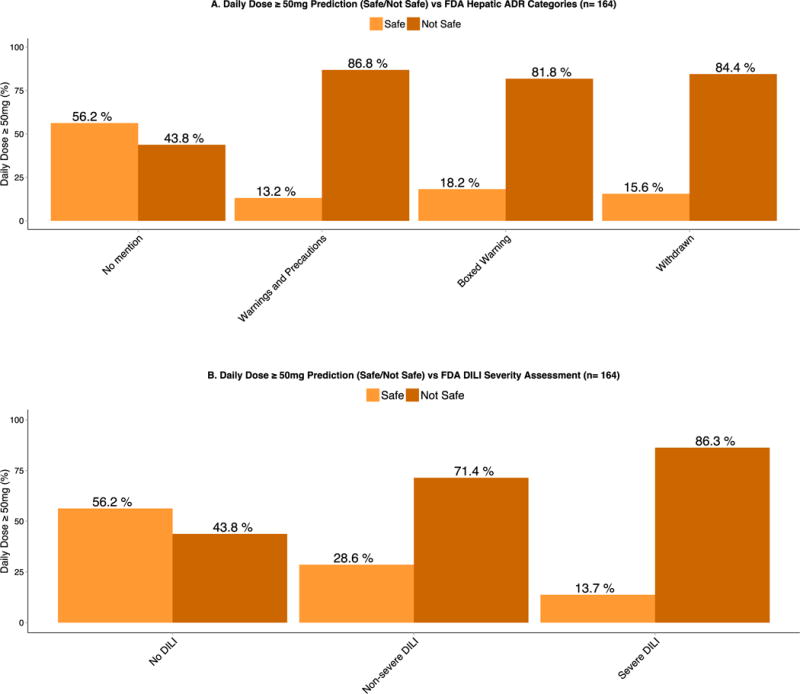
A. Daily Dose ≥ 50mg prediction (Safe/Not Safe) vs. FDA Hepatic ADR Categories.
There is a marked increase in the proportion of compounds that are dosed at greater than 50mg/day and have FDA drug label warnings associated with DILI adverse effects as illustrated in the “Warning and Precautions”, “Boxed Warning” and “Withdrawn” categories.
B. Daily Dose ≥ 50mg prediction (Safe/Not Safe) vs. FDA DILI severity assessment.
Similarly, there is a marked increase in the proportion of compounds that are dosed at greater than 50mg/day and have some type of DILI toxicity as illustrated in the “Non-severe DILI” and “Severe DILI” categories.
Although, there is strong evidence that dosages ≥ 50mg/day are associated with increased risk for hepatotoxicity, many drugs are safe at such dosages. For instance, the 50mg/day dosage cut off would predict that 44% of “No Mention” and/or “No DILI” drugs (See Figure 3A and 3B) exhibit “Not Safe” potential in terms of hepatotoxicity. Thus, supporting that daily dosage alone is not a reliable means of guiding the drug development process, regulatory application, and clinical practice.
Comparison of DILI – No DILI Predictive Metrics
In our comparative analysis we believe that positive predictive value (PPV, i.e., those drugs predicted to cause DILI that actually do so, or the true positive rate) is the most important value, since a high percentage will indicate the ability of the method to identify drugs that cause DILI. We believe that the false negative rate (FNR, i.e. those drugs causing DILI that are not identified by the metric, the type 2 error of the metric) is the second most important criteria, since a low number indicates that we do not incorrectly predict DILI when it occurs. The third parameter that we list, accuracy of the metric (ACC, i.e., the true positive and true negative predictions of the metric divided by the total number of compounds evaluated), represents the total % predicted correctly. Many other predictive metrics can be calculated, as has been done. However, we believe that PPV and FNR are the most relevant in evaluating an analysis of toxicity potential.
Chen and coworkers9 have proposed that drugs with high lipophilicity (LogP) given at high doses likely become hepatotoxic as expressed in the Ro2. Using the same data set and the same annotations that Chen and coworkers9 used for the proposed Ro2 (log P ≥ 3 and daily dosages ≥ 100mg (n=164)), we have reviewed the relationship between DILI hepatic adverse events and daily dose ≥ 50mg, daily dose ≥ 100mg, BDDCS Class, cLogP ≥ 3 and combinations of these characteristics. The data set was classified into two categories: “Most DILI Concern” and “No DILI Concern.” We believe that the use of these standardized annotations allows for a more direct comparison of the models. Of the 164 drugs, BDDCS classification could be assigned for 151 drugs. Our evaluation includes a comparison of the different predictive metrics against the first proposed Ro2 Chen et al. data set9 and the most recent report adding generation of reactive metabolites to the Ro213.
Chen and coworkers9 claim that the Ro2 is the best method for identifying drugs that cause DILI (PPV=95.3%), but we maintain that it is not a good method in terms of its FNR (61.7%), and therefore its ACC is low (55.0%). We can see in Table 1 that comparing the Ro2 with cLogP ≥ 3 or cLogP ≥ 3 + Dose ≥ 50mg (rather than 100mg), that similar ACC values are achieved, but PPV for cLogP ≥ 3 alone is markedly decreased. BDDCS Class 2 identification yields a slightly higher ACC than Ro2 due to the bigger decrease in FNR vs. PPV. We observe that accuracy of DILI is best predicted by “Dose ≥ 50mg”, followed by “Dose ≥ 100mg”. The next best predictive model was “Metabolism (BDDCS Class 1 and 2) + Dose ≥ 50mg” together. Additionally, when we compared the Ro2 with “cLogP ≥ 3” or “cLogP ≥ 3 + Dose ≥ 50mg” (rather than 100mg), we observe similar ACC values, but PPV for cLogP ≥ 3 alone is markedly decreased. Thus showing that dose alone is a stronger contributing factor to DILI risk than cLogP. Although BDDCS Class 2 and Ro2 show relatively high PPV, their ACC is decreased due to FNR outcomes. We also found that poor solubility “(BDDCS Class 2 + 4)” has a correlation with DILI toxicity, but this characteristic alone was not able to distinguish accurately DILI vs. No DILI events. Our comparison in Table 1 suggests that BDDCS classification alone is not sufficiently predictive of DILI potential, but that the Ro2, which includes a dose parameter may be no better a predictor and possibly even poorer than just looking at BDDCS Class 2, which does not require knowledge of dose.
Table 1.
Comparison of Different Predictive Metrics for the First Published Chen et al. Data Set9
| Criteria | % Correct (Positive Predictive Value, PPV) | % DILI Missing (False Negative Rate, FNR) | % Accuracy (ACC) (True Positive + True Negative)/151 |
|---|---|---|---|
| Rule of Two | 95.3% | 61.7% | 55.0% |
|
| |||
| BDDCS Class 2 | 90.2% | 48.6% | 61.6% |
| BDDCS Class 2 + Dose ≥ 50mg | 94.1% | 55.1% | 58.9% |
| BDDCS Class 2+ Dose ≥ 100 mg | 93.8% | 57.9% | 57.0% |
|
| |||
| Dose ≥ 50mg | 83.5% | 15.0% | 77.5% |
| Dose ≥ 100mg | 85.6% | 22.4% | 74.8% |
|
| |||
| BDDCS Class (1 + 2) | 74.8% | 16.8% | 68.2% |
| BDDCS Class (1 + 2) + Dose ≥ 50mg | 87.4% | 29.0% | 72.2% |
| BDDCS Class (1 + 2) + Dose ≥ 100mg | 90.8% | 35.5% | 70.2% |
|
| |||
| BDDCS Class (2 + 4) | 89.4% | 44.9% | 63.6% |
| BDDCS Class (2 + 4) + Dose ≥ 50mg | 92.9% | 51.4% | 60.9% |
| BDDCS Class (2 + 4) + Dose ≥ 100mg | 92.5% | 54.2% | 58.9% |
|
| |||
| CLogP ≥ 3 | 76.1% | 52.3% | 52.3% |
| CLogP ≥ 3 +Dose ≥50mg | 91.7% | 58.9% | 55.6% |
|
| |||
| BDDCS Class 1 | 58.6% | 68.2% | 35.8% |
| BDDCS Class 3 | 51.9% | 86.9% | 29.8% |
| BDDCS Class 4 | 80.0% | 96.3% | 31.1% |
A more recent report on the Ro213 includes the addition of reactive metabolites formation13. Of the 192 drugs used in their follow up analysis, BDDCS classification could be assigned to 166 drugs. This comparative analysis is depicted in Table 2. This “Ro2 + Reactive Metabolites” shows an increase in PPV, but only has a marginal improvement in the overall ACC and, in fact, appears to be less useful than “BDDCS Class 2 + Reactive Metabolites”. BDDCS Class 2 compounds show higher DILI predictability as compared to the other BDDCS classes. Furthermore, BDDCS Class 2 alone in this dataset performed better in terms of ACC than the first proposed “Ro2” and most recently proposed model for “Ro2 + Reactive Metabolite formation”. We also observe that reactive metabolite formation alone, followed by “Dose ≥ 50mg + Reactive Metabolite Formation” and “Dose ≥ 100mg + Reactive Metabolite Formation” had the best performance in terms of ACC and PPV. However, these conditions also have an increase in FNR. The next best predictive model was “Metabolism (BDDCS Class 1 and 2) + Reactive Metabolite Formation” together, which does not require any knowledge of the dose taken, performed similarly to “Metabolism (BDDCS Class 1 and 2) + Dose ≥ 50mg or Dose ≥ 100mg”, which has showed the best predictability in our initial analysis. Moreover, taking into account reactive metabolite formation or having better methods to account for reactive metabolite formation together with high permeability compounds can potentially lead to an improvement in DILI prediction without the need to rely on dose.
Table 2.
Comparison of Different Predictive Metrics for the Chen et al. Data Set (Filtered for only BDDCS Classifiable Drugs)13
| Criteria | % Correct (Positive Predictive Value, PPV) | % DILI Missing (False Negative Rate, FNR) | % Accuracy (ACC) (True Positive + True Negative)/166 |
|---|---|---|---|
| Rule of Two | 92.2% | 58.0% | 58.4% |
| Rule of Two + Reactive Metabolite Formation | 100.0% | 60.7% | 59.0% |
|
| |||
| BDDCS Class 2 | 92.4% | 45.5% | 66.3% |
| BDDCS Class 2 + Reactive Metabolite Formation | 98.2% | 50.9% | 65.1% |
|
| |||
| BDDCS Class (1 + 2) | 71.4% | 15.2% | 66.9% |
| BDDCS Class (1 + 2)+ Reactive Metabolite Formation | 91.2% | 25.9% | 77.7% |
| Dose ≥50 | 78.3% | 9.8% | 76.5% |
| Dose ≥100 | 82.6% | 15.2% | 77.7% |
|
| |||
| Dose ≥50 + Reactive Metabolite Formation | 93.4% | 24.1% | 80.1% |
| Dose ≥100 + Reactive Metabolite Formation | 95.2% | 28.6% | 78.3% |
|
| |||
| Reactive Metabolite Formation | 88.7% | 16.1% | 81.9% |
Lammert and coworkers1 attributed hepatic adverse events to compounds exhibiting extensive metabolism. This attribute is represented by “BDDCS Class (1+ 2)” in Table 1 and Table 2 that shows better ACC than Ro2 and “BDDCS Class 2” because of the marked decrease in FNR, but the lower PPV value is probably higher than is acceptable for DILI predictions. Lammert et al.2 had previously suggested that significant hepatic metabolism and daily dose ≥ 50mg was potentially predictive of hepatic adverse events. However, addition of dose to BDDCS Class (1 + 2) shows an increase in PPV, but with a corresponding increase in FNR, yielding negligible ACC changes. It is noteworthy that the best ACC in Table 1 is achieved with dose alone (i.e. Dose ≥ 50mg and Dose ≥ 100mg) with slightly lower PPV but the lowest FNRs as compared to Ro2 and BDDCS Class 2. In Table 2 what we find illuminating for this data set is that BDDCS Class 2 by itself performed better than “Ro2” and “Ro2 and reactive metabolite formation.” We also observe a comparable performance of “BDDCS Class (1+2) + reactive metabolite formation” vs. dose alone. But we also note that just considering only reactive metabolite formation yields the highest ACC of all the other methodologies, even when dose is added to reactive metabolite formation. (In Supplementary Tables S1A and S1B we show that selection of only BDDCS Class drugs vs. all drugs in the new Chen et al. 13 data set does not bias the outcome.)
Relationship between BSEP Inhibition and BDDCS Classification
Another model we used to evaluate DILI toxicity has been the supposition that BSEP inhibitors lead to DILI causation. FDA drug labels for 182 registered drugs have been evaluated for their BSEP inhibition by Pedersen et al.15. Assignment to BSEP inhibition categories was based on the ATP dependent taurocholate transport rate. Compounds that inhibited BSEP more than 50% at concentration of 50uM were considered “BSEP Inhibitors”; compounds in the 50%–72.5% range were considered “Weak BSEP Inhibitors”; compounds that inhibited less that 27.5% were considered “BSEP Non-Inhibitors”. All compounds but L-carnitine (“No mention”, “No DILI”) could be classified. For BDDCS classification, only active species were considered. The distribution of BSEP inhibition in each FDA hepatic liability category and BDDCS class were evaluated. 73/181 drugs were assigned to the “Adverse Reactions” category, 61/181 to the “Warning and Precautions”, 12/181 to the “Boxed Warning”, 2/181 in the “Withdrawn” category and 33/181 to the “No Mention” category.
When BSEP inhibition data were correlated with FDA drug labels of registered drugs15, we observed no discernible pattern between BSEP inhibition and ADR categories (See Figure 4A). For the BDDCS classification, we observe that the great majority of strong BSEP inhibitors are BDDCS Class 2 drugs, with concomitant decreases in the percentages of BDDCS class 1 and 3 drugs as BSEP inhibition increases, as depicted by Figure 4B. Here we point out that because we are able to make similar predictions just based on simple physicochemical parameters, this leads us to dismiss the predictive ability of the mechanistic association of BSEP and DILI. We suspect that previous analyses predicting that BSEP inhibition leads to DILI may have been confounded by the observation that most BSEP inhibitors are BDDCS Class 2 drugs, which show a high prevalence for DILI. In Table 3, we observe that in the condition of a positive GSH + BDDCS Class 2 or BSEP +BDDCS Class 2 we observe a marked improvement in the PPV. However, the predictability of these assays is still very limited as noted by their high FNR outcomes. Consideration of Cmax of drugs in relation to IC50 of BSEP inhibition could possibly improve the prediction of DILI based on BSEP inhibition.
Figure 4.
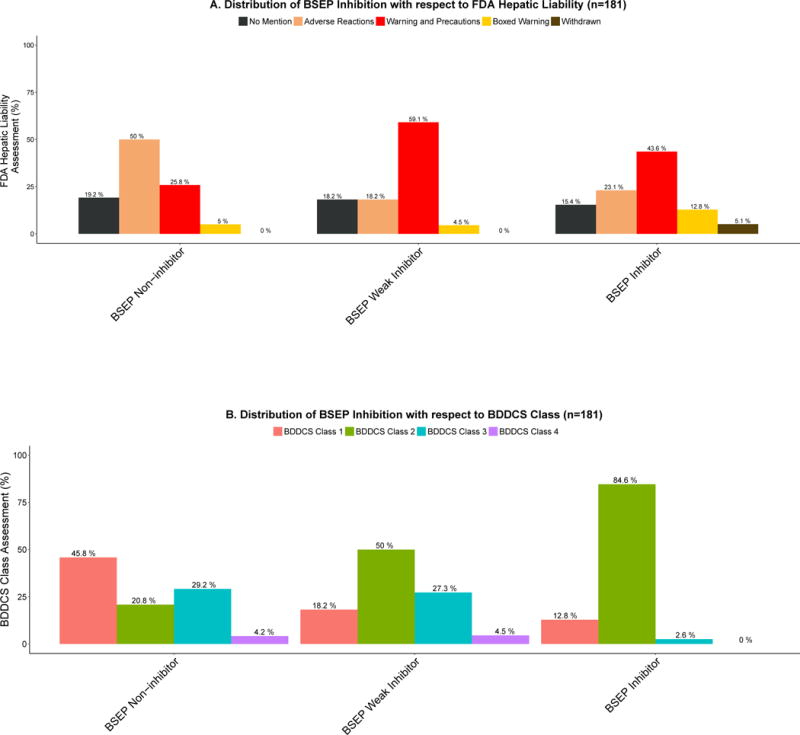
A. Distribution of BSEP inhibition with respect to FDA hepatic liability assignment
Assignment to BSEP inhibition categories was based on the ATP dependent taurocholate transport rate. Compounds that inhibited more than 50% were considered BSEP Inhibitors; compounds in the 50%–72.5% range were considered Weak BSEP Inhibitors; compounds less than 27.5% were considered BSEP Non-inhibitors. Bars show the percentage of all compounds in the same BSEP inhibition class (BSEP Non-Inhibitor, BSEP Weak Inhibitor, BSEP Inhibitor). There was a significant difference between the FDA hepatic liability categories when the proportionality trend test was calculated: “No mention” trend, NS, p-value = 0.6018, “Adverse Reactions” trend, p-value = 0.0007441, “Warning and Precautions” trend, p-value = 0.01111, “Boxed Warning” trend, NS, p-value = 0.1129, “Withdrawn” trend, p-value = 0.01243.
B. Distribution of BSEP inhibition assignment with respect to BDDCS Class.
BSEP inhibitors are overwhelmingly composed of BDDCS Class 2 drugs (84.6%).
There was a significant difference between BDDCS Classes when the proportionality trend test was calculated: BDDCS Class 1 trend, p-value = 5.521e-05; BDDCS Class 2 trend, p-value = 5.009e-13; BDDCS Class 3 trend, p-value = 0.00115; BDDCS Class 4 trend, NS, p-value = 0.2432. There was also significant differences in the BDDCS Class distributions among the following BSEP inhibition groups: “BSEP Non-inhibitor” vs. “BSEP Weak Inhibitor”, p-value = 0.0214; “BSEP Non-inhibitor” vs. “BSEP Inhibitor”, p-value = 2.78e-11.
Table 3.
Comparison of Various Assays Measuring Key Mechanisms of Toxicity Endpoints associated with DILI14
| Criteria | % Correct (Positive Predictive Value, PPV) | % DILI Missing (False Negative Rate, FNR) | % Accuracy (ACC) (True Positive + True Negative)/106 |
|---|---|---|---|
| GSH | 71.9% | 52.1% | 69.1% |
| TDI | 75.0% | 81.3% | 61.8% |
| Cytotoxicity (3T3 cells) | 48.3% | 70.8% | 55.5% |
| Mitotox | 71.4% | 79.2% | 61.8% |
| BSEP | 69.2% | 62.5% | 65.5% |
| All assays | 65.1% | 14.6% | 73.6% |
|
| |||
| BDDCS Class 1 | 33.3% | 75.0% | 45.5% |
| BDDCS Class 2 | 64.6% | 35.4% | 69.1% |
| BDDCS Class 1 and 2 | 51.2% | 10.4% | 58.2% |
|
| |||
| GSH and BDDCS Class 1 | 46.2% | 87.5% | 55.5% |
| GSH and BDDCS Class 2 | 89.5% | 64.6% | 70.0% |
| GSH and BDDCS Class 1 and 2 | 71.9% | 52.1% | 69.1% |
|
| |||
| BSEP and BDDCS Class 1 | 37.5% | 93.8% | 54.5% |
| BSEP and BDDCS Class 2 | 87.5% | 70.8% | 67.3% |
| BSEP and BDDCS Class 1 and 2 | 70.8% | 64.6% | 65.5% |
Comparison of Mechanism Based Toxicity Endpoints
Although, a number of compound–specific liability factors have been linked with DILI susceptibility, it is difficult to understand which risk factors are more important in patient-specific responses and/or environmental stimuli. One approach followed by many research groups to assess and reduce some of the more common, drug-specific factors in a set of targeted in vitro assays. The most common mechanisms covered in such in vitro panels or hazard matrices include formation of reactive metabolites, inhibitions of drug transporters involved in hepatobiliary elimination of bile acids and other metabolic endogenous products (BSEP, MRPs), mitochondrial toxicity and different cellular toxicity assays covering the formation of drug-metabolites28,29. Various approaches are used in the pharmaceutical industry for hazard identification and risk assessment of reactive metabolites and more integrated strategies that include measures of the initial mechanism of toxicity have been highlighted in our analysis.
Zhang et al.16 evaluated the in vitro hepatic toxicity of 152 drugs from the Chen and coworkers9 data set using four mechanistically relevant endpoints. They reported that the ratio of the measured reactive oxygen species to cellular ATP depletion (ROS/ATP) was able to not only differentiate compounds exhibiting severe DILI (65 compounds) from no DILI (35 compounds) but also severe DILI from non-severe DILI (52 compounds). Of the 152 drugs, streptozocin could not be BDDCS classified and chlorpropamide as a class 0 drug (extent of metabolism highly dependent upon urinary pH) was not included in our analysis. For the 152 drugs from the Chen dataset evaluated by Zhang et al.16, 134 drugs were BDDCS Class known. When we carried out an analysis for this data set in terms of BDDCS Class as was done for the entire Chen et al. 9 data set as shown in Fig. 2 a similar trend was observed (data not shown) but the trends were not to the same degree of significance. What would be more illuminating would be individual drug results with respect to the specific mechanisms based outcomes, which were not presented. However, the accuracy of this characterization remains in question. In the manuscript by Zhang et al. ibuprofen and atorvastatin were characterized as “severe DILI”, felbamate, methimazole, and pyrazinamide were characterized as “non-severe DILI” and streptozocin and penicillamine were characterized as “no DILI”, which we believe are inaccurate classifications. In addition, Zhang et al. include in their study directly cytotoxic anticancer drugs such as cisplatin, dacarbazine, bleomycin, etc., which should be evaluated separately from other drugs.
We also include here a comparison of the different predictive metrics in the various assays measuring key mechanisms of toxicity endpoints associated with DILI from the Schadt et al. data set14. Schadt et al.14 evaluated 120 marketed or withdrawn drugs, which were analyzed independent of FDA classification. These workers categorized severe and moderate DILI as “high DILI concern” and mild and no DILI as “low DILI concern.” The “high DILI concern” category was a merger of moderate and severe risk compounds based on the FDA categorization. Generation of reactive metabolites was tested via GSH adduct formation and P450 3A4 time-dependent inhibition (TDI). Further key measures of initial mechanism of toxicity were monitored in a panel consisting of assays assessing BSEP inhibition, mitochondrial toxicity and cytotoxicity. In the Schadt et al. data set of 120 compounds, 14 compounds had not been BDDCS classified. As depicted in Table 3 we evaluated 106/120 drugs that were screened based on different in vitro mechanism endpoints and BDDCS class, which has been previously published. The assays that performed the best were GSH and BSEP assays; this was determined based on the balance of the lowest FNR and highest PPV. However the FNR rates of these two assays are also very high, and the accuracy of these tests is comparable to the analysis of BDDCS class 2 alone. When GSH or BSEP measurements are added to BDDCS Class 2 PPV and FNR both increase but accuracy is no better. The highest ACC is obtained when all of the mechanisms of toxicity endpoints are confirmed, due to the low FNR. However, having a PPV of only 65.1% does not give much confidence.
Some of these risk factors can be mitigated during the drug design/development process to identify drugs with better chemical attributes with reduced potential to cause human DILI. The strengths and weaknesses have been highlighted in our analysis.
Although there may be some general trends between simple physical parameters, it is unlikely that such considerations could accurately predict risk. This problem could potentially be alleviated by the new in vitro approaches and utilization of state of the art instrumentation currently being evaluated. The development of improved physiological test systems based on information gained from studies with model hepatotoxins are required to encompass both chemical and biological factors associated with hepatotoxicity to try to screen for rare but often fatal idiosyncratic hepatotoxicities earlier in drug development.
BDDCS Classification Prior to Dosing in Humans
Although the finding of Uetrecht shows that idiosyncratic drug reactions were rare among individuals given drug doses <10mg/day and more likely among individuals given drug doses ≥ 1000mg/day30, the dose relationships can only be determined for a new molecular entity after the drug has been administered to human subjects/patients. In contrast, BDDCS Class can be predicted prior to ever dosing the compounds to humans as we have proposed previously31. Hosey and Benet24 showed that based on in vitro permeability measurements, PPV for prediction of extensive metabolism were all 90% or greater. And most recently Dave and Morris25 showed that they were able to correctly predict highly soluble vs. poorly soluble drugs using measured solubility parameters with greater than 85% probability. Thus as seen in Table 1, just knowing if a compound is BDDCS Class 2 prior to drug dosing has the ability to identify DILI potential with 90.2% PPV and 61.6% ACC. And in Table 2, the incorporation of better assays to the assignment of BDDCS Class and reactive metabolite formation can perform as well as dose without knowing the actual dose. As noted above, negligible improvements in PPV and decrements in ACC are observed when dose size is added to BDDCS categorization. BDDCS Class (2 + 4) gave comparable results for BDDCS Class 2 alone, but since there are so few Class 4 drugs, it is difficult to conclude if this is relevant.
Relationships between BDDCS and Toxicity
The hypothesis that compounds with significant hepatic metabolism may potentially be more hepatotoxic due to the generation of reactive intermediates and subsequent metabolic idiosyncrasies was first uncovered in an epidemiological survey by Lammert and coworkers1 who reported in their analysis that compounds exhibiting a significant hepatic metabolism resulted in ALT > 3 times ULN, liver failure, liver transplantation, and fatal DILI versus compounds with lesser degrees of hepatic metabolism. Our results show that DILI toxicity is most apparent in BDDCS Class 2 drugs, exhibiting the highest proportions among the ”Warning and Precautions”, “Black Box Warning”, “Withdrawn” and “Discontinued” categories. The great majority of approved drugs that cause acute liver failure, fatal hepatotoxicity, discontinued and or withdrawn are BDDCS Class 2 drugs. BDDCS Class 3 and 4 drugs show little risk of liver aminotransferases increase and hyperbilirubinemia. Lammert’s assertion that extensively metabolized compounds are at an increased risk to develop DILI is limited since we show in our data analysis that BDDCS Class 1 compounds, which are extensively metabolized, represent the majority of the compounds in the “No Mention” and “No DILI” groups (See Figure 1A and Figure 2A). The compounds that show the most toxicity are the extensively metabolized, low solubility compounds, i.e. BDDCS Class 2. Overall BDDCS classification appears to have an association with drug toxicity potential to lead to DILI adverse events.
Drugs belonging to BDDCS Class 3 and 4 exhibited much lower proportions in the FDA hepatic liability and DILI severity assessment categories (See Figures 1 and 2). However, we note the underrepresentation of BDDCS Class 4 drugs in the overall scheme of marketed approved drugs. Compounds with poor hepatic metabolism had been previously noted to be significantly less likely to cause hepatotoxicity1. In support of this observation, we also observe the increasing trend of BDDCS Class 3 drugs as the DILI severity decreases as depicted by Figure 2. Although a lack of hepatic metabolism does not assure total lack of hepatotoxicity, it indeed appears that BDDCS Class 3 and 4 drugs lead to a lower DILI severity.
We are not the first to investigate the BDDCS Class relationship and DILI. Previously Vuppalanchi et al.32 have analyzed 383 cases of DILI caused by a single orally administered prescription agent from the DILI Network Prospective Study. The relationship of daily dosage (≥ 50 mg vs. ≤ 49mg), preponderance of hepatic metabolism (≥50% vs. <50%), and BDDCS class were compared with clinical characteristics and outcomes. A total of 99 drugs belonging to BDDCS Classes 1 through 4 were responsible for the DILI episodes. In concordance with daily dosage relationship previously reported, there are a much smaller number of cases of DILI in the ≤ 49 mg/day group (n=50) than those with daily dosages ≥ 50mg/day (n=324). There is also a higher number of cases of DILI from drugs that underwent significant hepatic metabolism (n=305) compared to those without hepatic metabolism (n=71). However, in their BDDCS case analysis breakdown, they report 118 cases with BDDCS Class 1, 96 cases with BDDCS Class 2, 112 cases with BDDCS Class 3, and 38 cases with BDDCS Class 4, which shows that the actual number of extensively metabolized drugs is 214, while it is 150 for poorly metabolized drugs. Vuppalanchi et al. concludes that there is no DILI difference between BDDCS Class 1 and BDDCS Class 2 drugs. Patients with DILI caused by medications with or without preponderant hepatic metabolism did not differ in clinical characteristics or outcomes. There was also no significant difference between BDDCS 1, 2, 3 classes in terms of DILI cases. BDDCS Class 1 compounds were reported to have a longer latency and exhibit a greater proportion of hepatocellular injury. However, in our current analysis we observe that the majority of drugs in the “No DILI” group are composed of BDDCS Class 1 and BDDCS Class 3 and there is a much greater risk of BDDCS Class 2 leading to idiosyncratic DILI than BDDCS Class 1 or 3 compounds.
In this work and in our previous study predicting the prevalence of cutaneous adverse reactions with antiepileptic drug 12, BDDCS Class 2 drugs appear to present the most toxic liability. Why should this be true? A major finding in the development of the BDDCS classification system was the recognition that extensively metabolized, high permeability, high solubility Class 1 drugs may be shown in vitro to be substrates of both uptake and efflux transporters, but that effects of transporters on BDDCS Class 1 drugs are essentially clinically insignificant in the liver and intestine, as well as the brain. Thus, for BDDCS Class 1 drugs unbound concentrations in the systemic circulation will reflect unbound concentrations in the liver as well as in the rest of the body. However, this will not be true for BDDCS Classes 2, 3 and 4 drugs where transporter effects will lead to different unbound concentrations in the liver and throughout the body. That is, Class 1 drugs will follow the long held assumption in deriving pharmacologic/toxicologic relationships that free drug concentrations are the same throughout the body. But this assumption in pharmacology was made prior to any recognition of the importance of drug transporters in controlling permeability. Thus, according to BDDCS classification22,33 approximately 40% of marketed drugs (i.e., those that are Class 1) will still follow the equivalent free drug concentration hypothesis. It is important to recognize that the compounds evaluated here are drugs that reach the market where sponsors were able to convince the regulatory agencies based on in vitro and preclinical animal studies that toxicity potential, particularly DILI, would be manageable or at least acceptable when the drugs reached the market and were taken by large patient populations as compared to those limited number of patients studied during drug development. Thus, according to our hypothesis, drug company sponsors in their preclinical and clinical studies of Class 1 drugs would be able to reasonably predict drug concentrations in the liver and throughout the body. In contrast, for BDDCS Class 2 drugs, where metabolism is the significant process of elimination, drug concentration measurements in the systemic circulation for these compounds both in the preclinical and clinical studies would poorly predict what occurs in the liver and in other organs of the body. And since it is obvious that DILI occurs more frequently with metabolized drugs, studies in drug development with Class 2 drugs would be poorer predictors of toxicity potential due to the challenges to estimate intracellular concentrations and metabolic processes. Thus, the prevalence of DILI with BDDCS Class 2 drugs could just be circumstantial in that sponsors would be unable to properly evaluate hepatic toxicity for these compounds in designing their clinical studies. This problem could potentially be alleviated by new in vitro approaches and utilization of state of the art instrumentation currently being evaluated.
CONCLUSION
In our analysis we confirm previous reports that the best predictor of DILI requires knowledge of the daily dose, an unknown quantity early in drug development. We show here that the BDDCS methodology, where assignment can be made prior to ever dosing a drug to animals or man, yields similar and in a number of cases better than the DILI predictive potential of other methodologies such as Ro2. Although we observe strong trends of BDDCS Class 2 increasing toxicity as DILI severity increases, overall, BDDCS Classification only marginally improves the prediction of DILI toxicity potential. However, we observe that “BDDCS Class 2” alone have performed better than “Ro2” and “Ro2 + reactive metabolite formation.” As seen in Figs. 1 and 2, the BDDCS Class 2 versus Class 1 differentiation only becomes evident with the most severe hepatic toxicities, and then only a 2:1 differentiation between BDDCS Class 2 versus Class 1 is found.
Similarly, we demonstrate that those previous proposed models to predict DILI potential such as the “Ro2” and “Ro2 + reactive metabolite formation”, daily dosage ≥ 50mg, and the supposition that BSEP inhibitors lead to DILI causation are still not sufficiently predictive. Lammert et al.’s1 assertion that extensive metabolized compounds are at higher risk of developing DILI can be much improved by differentiating BDDCS Class 2 from BDDCS Class 1 drugs. Ro2 shows a high FNR missing significant cases of DILI assignment when “DILI” occurs and that the daily dosage ≥ 50mg alone can only depict a clear relationship with dose with compounds that have been previous associated with DILI, but very limited predictability in differentiating compounds with “No DILI” assignment. We also suspect that previous analyses predicting that BSEP inhibition leads to DILI may be confounded by our finding that most BSEP inhibitors are BDDCS Class 2 drugs. Thus, our BDDCS analysis and previous DILI toxicity potential are not sufficiently accurate in allowing early identification of new molecular molecules that will be DILI free. But we believe that comparison of proposed DILI predictive methodology with BDDCS assignment offers a useful tool by which new DILI predictive hypotheses can be evaluated. Hopefully, development of mechanism based toxicity endpoints, such as those proposed by Chen et al.13, Zhang et al.16 and Schadt et al.14, as discussed above, will greatly improve future predictability.
Toxicologists, medicinal chemists and drug development scientists will most likely conclude that no one in the drug development process will discontinue a drug candidate based on the predictive DILI potential using BDDCS class. We agree. The purpose of this analysis was to point out that many of the published “predictive DILI” hypotheses do no better than just avoiding BDDCS Class 2 drugs. We propose that comparison of predictive DILI hypotheses with BDDCS class assignment is a useful exercise in determining the relevance of predictive metrics.
Supplementary Material
Acknowledgments
Financial Support:
Rosa Chan was supported by the American Association of Pharmaceutical Scientists Graduate Fellowship, American Foundation for Pharmaceutical Education Pre-Doctoral Fellowship and NIGMS grant R25 GM56847. Dr. Benet is a member of the UCSF Liver Center supported by NIH Grant P30 DK026743.
List of Abbreviations
- DILI
Drug-Induced-Liver-Injury
- BDDCS
Biopharmaceutics Drug Disposition Classification System
- ADR
Adverse Drug Reaction
- AR
Adverse Reaction
- PPV
Positive Predictive Value
- FNR
False Negative Rate
- ACC
Accuracy
- Ro2
Rule of 2
Biographies

Dr. Leslie Z. Benet, Professor and former Chair (1978–1998) of Bioengineering and Therapeutic Sciences, UCSF, received his AB, BS and MS from the University of Michigan, Ph.D. from UCSF. He is the recipient of nine honorary doctorates. His recent honors include the 2015 ISSX North American Scientific Achievement Award and 2016 Remington Honor Medal of the APhA. Dr. Benet has published over 550 scientific articles and book chapters, holds 12 patents and served as editor of 7 books. He is listed by Thomson Reuters among the most highly cited pharmacologists worldwide with his peer-reviewed publications cited more than 24,000 times.
Photo credit: Christine Krieg, Photographer

Rosa Chan received her B.S. in Molecular Toxicology from UC Berkeley in 2011. She is currently a 5th year graduate student at the University of California San Francisco’s Pharmaceutical Sciences and Pharmacogenomics Ph.D. Program, where she is completing her thesis work in Dr. Benet’s laboratory. Her thesis work is focused on evaluating the application of the Biopharmaceutics Drug Disposition Classification System (BDDCS) for the risk assessment of skin and liver toxicity potential of small molecules using in vitro and human clinical data.
Footnotes
Conflict of Interest Disclosure
“The authors declare no competing financial interest.”
References
- 1.Lammert C, Bjornsson E, Niklasson A, Chalasani N. Oral medications with significant hepatic metabolism at higher risk for hepatic adverse events. Hepatology. 2010;51:615–620. doi: 10.1002/hep.23317. [DOI] [PubMed] [Google Scholar]
- 2.Lammert C, Einarsson S, Saha C, Niklasson A, Bjornsson E, Chalasani N. Relationship between daily dose of oral medications and idiosyncratic drug-induced liver injury: search for signals. Hepatology. 2008;47:2003–2009. doi: 10.1002/hep.22272. [DOI] [PubMed] [Google Scholar]
- 3.Lewis JH. Drug-induced liver injury, dosage, and drug disposition: is idiosyncrasy really unpredictable? Clin Gastroenterol Hepatol. 2014;12:1556–1561. doi: 10.1016/j.cgh.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 4.Chen M, Suzuki A, Borlak J, Andrade RJ, Isabel Lucena M. Drug-induced liver injury: interactions between drug properties and host factors. J Hepatol. 2015;63:503–514. doi: 10.1016/j.jhep.2015.04.016. [DOI] [PubMed] [Google Scholar]
- 5.Thompson RA, Isin EM, Ogese MO, Mettetal JT, Williams DP. Reactive Metabolites: Current and Emerging Risk and Hazard Assessments. Chem Res Toxicol. 2016;29:505–533. doi: 10.1021/acs.chemrestox.5b00410. [DOI] [PubMed] [Google Scholar]
- 6.Srivastava A, Maggs JL, Antoine DJ, Williams DP, Smith DA, Park BK. Role of reactive metabolites in drug-induced hepatotoxicity. Handb Exp Pharmacol. 2010;196:165–194. doi: 10.1007/978-3-642-00663-0_7. [DOI] [PubMed] [Google Scholar]
- 7.Knowles SR, Uetrecht J, Shear NH. Idiosyncratic drug reactions: the reactive metabolite syndromes. Lancet. 2000;356:1587–1591. doi: 10.1016/S0140-6736(00)03137-8. [DOI] [PubMed] [Google Scholar]
- 8.Jaeschke H, Gores GJ, Cederbaum AI, Hinson JA, Pessayre D, Lemasters JJ. Mechanisms of hepatotoxicity. Toxicol Sci. 2002;65:166–176. doi: 10.1093/toxsci/65.2.166. [DOI] [PubMed] [Google Scholar]
- 9.Chen M, Borlak J, Tong W. High lipophilicity and high daily dose of oral medications are associated with significant risk for drug-induced liver injury. Hepatology. 2013;58:388–396. doi: 10.1002/hep.26208. [DOI] [PubMed] [Google Scholar]
- 10.Wu CY, Benet LZ. Predicting drug disposition via application of BCS: transport/absorption/elimination interplay and development of a Biopharmaceutics Drug Disposition Classification System. Pharm Res. 2005;22:11–23. doi: 10.1007/s11095-004-9004-4. [DOI] [PubMed] [Google Scholar]
- 11.Benet LZ. Predicting drug disposition via application of a biopharmaceutics drug disposition classification system. Basic Clin Pharmacol Toxicol. 2010;106:162–167. doi: 10.1111/j.1742-7843.2009.00498.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chan R, Wei C, Chen Y, Benet LZ. Use of the biopharmaceutics drug disposition classification system (BDDCS) to help predict the occurrence of idiosyncratic cutaneous adverse drug reactions associated with antiepileptic drug usage. AAPS J. 2016;18:757–766. doi: 10.1208/s12248-016-9898-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen M, Borlak J, Tong W. A Model to predict severity of drug-induced liver injury in humans. Hepatology. 2016;64:931–940. doi: 10.1002/hep.28678. [DOI] [PubMed] [Google Scholar]
- 14.Schadt S, Simon S, Kustermann S, Boess F, McGinnis C, Brink A, Lieven R, Fowler S, Youdim K, Ullah M, Marschmann M, Zihlmann C, Siegrist YM, Cascais AC, Di Lenarda E, Durr E, Schaub N, Ang X, Starke V, Singer T, Alvarez-Sanchez R, Roth AB, Schuler F, Funk C. Minimizing DILI risk in drug discovery – A screening tool for drug candidates. Toxicol Vitr. 2015;30:429–437. doi: 10.1016/j.tiv.2015.09.019. [DOI] [PubMed] [Google Scholar]
- 15.Pedersen JM, Matsson P, Bergström CaS, Hoogstraate J, Norén A, LeCluyse EL, Artursson P. Early identification of clinically relevant drug interactions with the human bile salt export pump (BSEP/ABCB11) Toxicol Sci. 2013;136:328–343. doi: 10.1093/toxsci/kft197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang J, Doshi U, Suzuki A, Chang CW, Borlak J, Li AP, Tong W. Evaluation of multiple mechanism-based toxicity endpoints in primary cultured human hepatocytes for the identification of drugs with clinical hepatotoxicity: Results from 152 marketed drugs with known liver injury profiles. Chem Biol Interact. 2016;255:3–11. doi: 10.1016/j.cbi.2015.11.008. [DOI] [PubMed] [Google Scholar]
- 17.Trontell AE. How the US food and drug administration defines and detects adverse drug events. Curr Ther Res. 2001;62:641–649. [Google Scholar]
- 18.Chen M, Vijay V, Shi Q, Liu Z, Fang H, Tong W. FDA-approved drug labeling for the study of drug-induced liver injury. Drug Discov Today. 2011;16:697–703. doi: 10.1016/j.drudis.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 19.United States Food and Drug Administration. Guidance for Industry Warning and precautions, contraindications and boxed warning sections of labeling for human prescription drug and biological products-content and format. http://www.fda.gov/downloads/Drugs/…/Guidances/ucm075096.pdf (accessed Jun 08, 2016)
- 20.United States Food and Drug Administration. Guidance for Industry Adverse reactions section of labeling for human prescription drug and biological products — content and format. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm075057.pdf (accessed Jun 08, 2016)
- 21.United States Food and Drug Administration. Guidance for Industry Labeling for human prescription drug and biological products — implementing the new content and format requirements. http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm075082.pdf (accessed Jun 08, 2016)
- 22.Benet LZ, Broccatelli F, Oprea TI. BDDCS applied to over 900 drugs. AAPS J. 2011;13:519–547. doi: 10.1208/s12248-011-9290-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hosey CM, Chan R, Benet LZ. BDDCS predictions, self-correcting aspects of BDDCS assignments, BDDCS assignment corrections, and classification for more than 175 additional drugs. AAPS J. 2016;18:251–260. doi: 10.1208/s12248-015-9845-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hosey CM, Benet LZ. Predicting the extent of metabolism using in vitro permeability rate measurements and in silico permeability rate predictions. Mol Pharm. 2015;12:1456–1466. doi: 10.1021/mp500783g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dave RA, Morris ME. Novel high/low solubility classification methods for new molecular entities. Int J Pharm. 2016;511:111–126. doi: 10.1016/j.ijpharm.2016.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.R Core Team. R: A language and environment for statistical computing. Vienna, Austria: 2015. [Google Scholar]
- 27.Wickham H. ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag; New York: 2009. [Google Scholar]
- 28.Aleo MD, Luo Y, Swiss R, Bonin PD, Potter DM, Will Y. Human drug-induced liver injury severity is highly associated with dual inhibition of liver mitochondrial function and bile salt export pump. Hepatology. 2014;60:1015–1022. doi: 10.1002/hep.27206. [DOI] [PubMed] [Google Scholar]
- 29.Russmann S, Kullak-Ublick Ga, Grattagliano I. Current concepts of mechanisms in drug-induced hepatotoxicity. Curr Med Chem. 2009;16:3041–3053. doi: 10.2174/092986709788803097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uetrecht J, Dan L. Idiosyncratic drug eeactions: current understanding. Annu Rev Pharmacol Toxicol. 2007;47:513–539. doi: 10.1146/annurev.pharmtox.47.120505.105150. [DOI] [PubMed] [Google Scholar]
- 31.Broccatelli F, Cruciani G, Benet LZ, Oprea TI. BDDCS class prediction for new molecular entities. Mol Pharm. 2012;9:570–580. doi: 10.1021/mp2004302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vuppalanchi R, Gotur R, Reddy KR, Fontana RJ, Ghabril M, Kosinski AS, Gu J, Serrano J, Chalasani N. Relationship between characteristics of medications and drug-induced liver disease phenotype and outcome. Clin Gastroenterol Hepatol. 2014;12:1550–1555. doi: 10.1016/j.cgh.2013.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benet LZ. The role of BCS (Biopharmaceutics Classification System) and BDDCS (Biopharmaceutics Drug Disposition Classification System) in drug development. J Pharm Sci. 2013;102:34–42. doi: 10.1002/jps.23359. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.