

SUMMARY
Smoothened (SMO) inhibitors are under clinical investigation for the treatment of several cancers. Vismodegib is approved for the treatment of locally advanced and metastatic basal cell carcinoma (BCC). Most BCC patients experience significant clinical benefit on vismodegib, however, some develop resistance. Genomic analysis of tumor biopsies revealed vismodegib resistance is associated with Hedgehog (Hh) pathway reactivation, predominantly through mutation of the drug target SMO and to a lesser extent through concurrent copy number changes in SUFU and GLI2. SMO mutations either directly impaired drug binding or activated SMO to varying levels. Furthermore, we found evidence for intra-tumor heterogeneity, suggesting that a combination of therapies targeting components at multiple levels of the Hh pathway is required to overcome resistance.
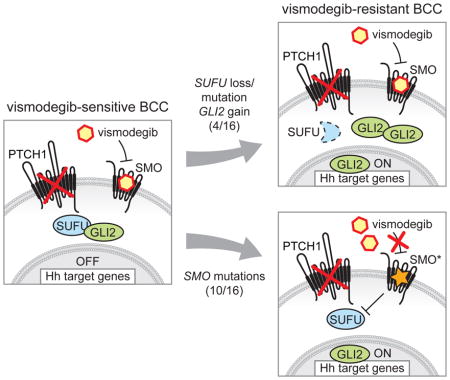
Graphical Abstract
INTRODUCTION
BCC is the most common human cancer and is predominantly driven by hyperactivation of the Hh pathway (Oro et al., 1997; Xie et al., 1998). The association between Hh signaling and cancer was first discovered in patients with Gorlin or basal cell nevus syndrome (BCNS), who are highly susceptible to medulloblastoma (MB) and BCC. These patients generally possess heterozygous germline mutations in the Patched 1 gene (PTCH1), which encodes a receptor for Hh ligands (Hahn et al., 1996; Johnson et al., 1996). Hh ligand binding relieves PTCH1 suppression of the serpentine transmembrane (TM) signal transducer Smoothened (SMO; see Figure 1A for a schematic of the Hh pathway). The vast majority of sporadic BCCs are driven by inactivating mutations and loss of heterozygosity (LOH) in PTCH1, with most of the remainder harboring activating mutations in SMO (Reifenberger et al., 2005). SMO promotes the activation and nuclear localization of GLI transcription factors by inhibition of Suppressor of fused (SUFU) and Protein kinase A (PKA). SUFU negatively regulates the Hh pathway by binding and sequestering GLI transcription factors in the cytoplasm (Stone et al., 1999). Loss-of-function mutations in SUFU are also associated with Gorlin Syndrome (Pastorino et al., 2009; Smith et al., 2014; Taylor et al., 2002). Approximately 50% of sporadic BCCs also have TP53 mutations (Jayaraman et al., 2014).
Figure 1. Genomic analysis of vismodegib-resistant BCC.

(A) Schematic of the Hh pathway; see Introduction section in main text for details.
(B) Initial response and disease progression of a sporadic BCC from PT12 that metastasized to lung. A red arrow indicates the target lesion in computerized tomography (CT) scans of the chest before treatment (PreRx) and after 4 (showing a decrease in lesion size) and 37 (revealing disease progression) months of vismodegib treatment.
(C) Photographs of two locally advanced BCCs from Gorlin syndrome patient PT10 that initially responded to vismodegib but subsequently relapsed (black arrow) after the indicated length of treatment.
(D) Hematoxylin and Eosin (H&E) stained sections of a locally advanced sporadic BCC from PT09.1 before and after 11 months of vismodegib treatment. Note that the relapsed lesion maintains the histology of the untreated tumor. The scale bar represents 50 μm.
(E) GLI1 and MKI67 expression levels in vismodegib-resistant and normal skin biopsies. Pearson’s correlation coefficient (R) = 0.96. Normalized read counts are shown.
(F) Overview of genetic alterations in Hh pathway genes and TP53 identified in 12 relapsed BCC patients. Germline PTCH1 variants are reported for Gorlin BCCs, whereas only somatic mutations are shown for sporadic BCCs. Two regionally distinct biopsies were obtained upon regrowth of the same initial tumor for PT06, PT08 and PT09. Two separate BCCs developed resistance in PT10. LOH was determined by minor allele frequencies from SNP arrays. Green boxes highlight LOH events followed by copy number gain of the mutant allele. Allele-specific expression was determined by RNA-seq.
Several Hh pathway inhibitors (HPIs) are currently under clinical investigation for treatment of BCC and MB (Amakye et al., 2013). Vismodegib, previously known as GDC-0449, is a SMO inhibitor approved for the treatment of metastatic and locally advanced BCC (Sekulic et al., 2012). The majority of BCC patients treated with vismodegib experience a clinical benefit, including both complete and partial responses (Sekulic et al., 2012). However, a preliminary estimate suggests that up to 20% of advanced BCC patients develop resistance to vismodegib within the first year of treatment (Chang and Oro, 2012).
To date, the only functionally characterized mechanism of acquired resistance to vismodegib in the clinic came from a patient with metastatic MB. A SMO-D473H mutation was detected in a biopsy from a relapsed metastatic tumor and was shown to abrogate drug binding in vitro (Yauch et al., 2009). Four other clinical SMO mutations were recently reported in vismodegib-resistant BCC, but were not examined functionally (Brinkhuizen et al., 2014; Pricl et al., 2014). Several resistance mechanisms to SMO inhibitors have been delineated from preclinical models, including additional SMO mutations, amplification of downstream Hh pathway components such as GLI2, and activation of bypass signaling pathways including phosphatidylinositol 3-kinase (PI3K) kinase and atypical protein kinase C ι/λ (aPKC-ι/λ) (Atwood et al., 2013; Buonamici et al., 2010; Dijkgraaf et al., 2011). However, it remains unclear which mechanisms drive resistance in patients. Here we use genomic and functional approaches to investigate mechanisms of vismodegib resistance in the clinic.
RESULTS
Genomic analysis of vismodegib-resistant and untreated BCCs
To identify mutations associated with vismodegib resistance we performed whole exome sequencing (WES) of BCCs from Gorlin syndrome (n=5) and sporadic (n=6) patients, and targeted SMO sequencing of a formalin-fixed paraffin-embedded (FFPE) sample from a further Gorlin patient (Tables S1 and S2). All patients initially experienced a clinical benefit on vismodegib but subsequently progressed while undergoing treatment. We collected two distinct biopsies from each of four of the patients such that a total of sixteen biopsies from vismodegib-resistant BCCs were analyzed (Table S1). Patients were initially diagnosed with metastatic (Figure 1B) or locally advanced BCC (Figure 1C) and we confirmed histologically that the drug-resistant lesions were BCCs (Figure 1D). For comparison we subjected tumors from untreated Gorlin syndrome (n=16) and sporadic (n= 27) BCC patients to WES (Table S1). Two distinct biopsies were obtained from each of five of the Gorlin patients giving 48 untreated BCC biopsies in total (Tables S1 and S3). The mean somatic mutation rate of untreated BCC samples from Gorlin patients was 33.5/megabase (Mb), varying from 6.2–68.9/Mb, and for sporadic patients was 50.5/Mb with a range of 2.4–162.2/Mb (Table S1). These rates are high in comparison to other cancers, including melanoma (Lawrence et al., 2013). Global analysis of the somatic mutation spectrum revealed a predominance of cytosine to thymine (C>T) transition mutations in both cohorts (Figures S1A and S1B), indicative of ultraviolet light-induced mutagenesis (Miller, 1985).
Transcriptional analysis of relapsed BCC biopsies (n=11) using RNA-seq, revealed that the Hh target gene GLI1 was expressed 10-fold higher (DESeq2, p < 0.003) than in a collection of normal skin samples (Figure 1E). Additionally, GLI1 expression levels were highly correlated (R=0.96) with expression levels of the proliferation marker MKI67, consistent with reactivation of Hh signaling driving BCC regrowth. Therefore, we focused our analysis on identifying genetic mechanisms that reactivate Hh signaling to bypass SMO inhibition by vismodegib. To this end, we identified mutations in selected cancer genes (Kandoth et al., 2013) and canonical Hh pathway components (Tables S4 and S5). Next, we determined genome-wide copy number alterations and LOH in vismodegib-resistant BCCs using single nucleotide polymorphism (SNP; n=11) and comparative genomic hybridization (CGH; n=4) arrays (Table S6). Figure 1F summarizes the genetic alterations identified in vismodegib-resistant tumors for the genes that encode canonical Hh pathway components as well as TP53, which is also commonly mutated in BCCs (Jayaraman et al., 2014).
PTCH1 and SMO mutations in BCC initiation
First, we aimed to identify the genetic alterations that were responsible for tumor initiation in vismodegib-resistant BCCs. Consistent with previous reports on BCC genetics (Jayaraman et al., 2014; Reifenberger et al., 2005), all of the relapsed Gorlin (100%) and the majority of sporadic (75%) BCCs displayed mutations in the tumor suppressor PTCH1 (Figures 1F and S1C), which occur throughout the length of the gene (Figure S1D) and are probably deleterious: seven are truncating, four are likely to affect exon splicing (Figure S1E) and two are predicted to be deleterious by the Condel algorithm (Table S4; Gonzalez-Perez and Lopez-Bigas, 2011). The Gorlin patient BCC (PT12; Figure 1F) for which we had only an FFPE sample and performed targeted SMO sequencing, was also likely to have initiated by alterations in PTCH1. The relapsed sporadic tumors without PTCH1 alterations (n=2) harbored the known oncogenic mutation SMO-W535L (Xie et al., 1998). These PTCH1 and SMO variants are likely to be the initiating events in the BCCs that first responded and subsequently displayed vismodegib resistance.
We observed a similar trend for the frequency of PTCH1 variants in the untreated Gorlin (90%) and sporadic (78%) BCCs (Figure S1F), and identified known oncogenic SMO mutations in three sporadic cases (Figures S1F and S1G). Relapsed BCCs showed a similar frequency of TP53 variants between Gorlin (50%) and sporadic (57%) cases (Figure 1F), whereas in the untreated cohort TP53 variants were observed more frequently in sporadic BCCs (59%) than Gorlin BCCs (24%) (Figure S1F), which could reflect the higher mutation rates observed in the untreated sporadic BCCs (Table S1).
Vismodegib-dependent selection of SMO variants
Strikingly, the majority of relapsed tumor biopsies harbored mutations in SMO encoding the drug target (11/16; 69%) and most co-occurred with PTCH1 variants (Figure 1F). By comparison, SMO variants were absent from untreated Gorlin BCCs and present in only 4/27 (15%) untreated sporadic BCCs (Figure S1F and Table S3). The SMO mutations identified in relapsed BCCs are outlined in Figure 2A. SMO-L412F, SMO-W535L and SMO-S533N mutations were previously reported as oncogenic drivers (Reifenberger et al., 1998; Sweeney et al., 2014; Xie et al., 1998), while SMO-W281C and SMO-V321M were recently identified in vismodegib-resistant BCCs (Brinkhuizen et al., 2014). We discovered four SMO mutations including, SMO-T241M, SMO-I408V, SMO-A459V and SMO-C469Y that were not observed in our untreated BCC cohort (Figure S1G) or in previous genomic analyses of Hh-driven cancers (Brastianos et al., 2013; Clark et al., 2013; Jayaraman et al., 2014; Kool et al., 2014; Reifenberger et al., 1998), strongly implicating them in vismodegib resistance. All SMO mutations from this study are situated within the TM region (Figures 2B and S2A) and confer amino acid substitutions in residues that are highly conserved among SMO proteins from several species (Figure S2B), likely reflecting their importance in SMO function.
Figure 2. SMO mutations in vismodegib-resistant BCC.

(A) Overview of SMO mutations identified in this study. All mutations were somatic in nature, as they were not detected in either blood or other tissue from the same patient.
(B) Computational model of vismodegib (yellow) docked onto the crystal structure of the SMO TM region (grey helices; Wang et al., 2013). Previously uncharacterized mutant residues are highlighted in green.
(C–F) Prevalence of SMO mutations in pre- and post-treatment biopsies. Bar graphs show the incorporation frequency of either wild-type (blue) or mutant (red) nucleotides at positions corresponding to SMO-A459V for PT03, PT04 and PT12 (C), SMO-V321M for PT09 (D), SMO-C469Y and SMO-T241M for PT10 (E) and SMO-L412F for PT11 (F) as determined by pyrosequencing. Note that SMO mutations are expected to be heterozygous and that SMO copy number determines the maximum Y-axis value, which is 50% for PT03, PT04, PT12, PT10 and PT11 (SMO copy number is 2) and 25% for PT09 (SMO copy number is 4). Incorporation of mutant nucleotides was considered to be within the background levels (<5%) of the pyrosequencing assay in all pre-treatment samples.
(G) PTCH1 copy number in pre- and post-treatment biopsies from PT11. Data plotted are mean and the range of quadruplicates.
(H) Photographs of a locally advanced BCC (white arrow) from PT11 that initially responded to vismodegib, but subsequently relapsed after the indicated length of time.
Resistance mechanisms can be acquired de novo or more likely by selection of minor subclones present in the pre-treatment tumor. In both scenarios we expected to observe enrichment of alterations responsible for drug-resistance with treatment. To assess drug-dependent selection of SMO mutants, we examined whether mutations were detectable in pre-treatment tumors and what proportion of tumor cells harbored SMO mutations after treatment. To this end, we sequenced pre-treatment FFPE tumor samples that were available from six patients and analyzed post-treatment tumor clonality. SMO-A459V was detected in post-treatment biopsies from three patients, but was not detectable above background levels in corresponding pre-treatment biopsies (Figure 2C). Similarly, the nucleotide changes corresponding to SMO-V321M, SMO-T241M and SMO-C469Y were only detectable above background levels in post-treatment samples, consistent with drug-induced selection of SMO mutant cells that arose de novo or were initially present at levels below the detection limit of our assay (Figures 2D and 2E). Interestingly, we readily detected the previously reported SMO-L412F mutation in both pre- and post-treatment samples from patient PT11, suggesting that this variant was likely to be the oncogenic driver for this tumor (Figure 2F). Note that the frequency of mutant nucleotides appears to decrease upon treatment; this is due to a higher level of contaminating normal tissue in the post-treatment sample. Copy number and SNP array analysis revealed that this tumor was initially diploid for PTCH1 and acquired PTCH1 copy number loss after treatment (Figure 2G and Table S6). The fact that this patient initially responded to vismodegib (Figure 2H) raises the intriguing possibility that reduced PTCH1 levels (through copy loss), in the context of this oncogenic mutation, might promote tumor regrowth while on drug.
To address whether SMO mutations were present in dominant clones in the relapsed BCCs, we calculated the tumor cell fractions of PTCH1 and SMO variants using allele frequencies from WES, as well as copy number and tumor content information derived from SNP arrays (See Supplemental Experimental Procedures and Table S7; Greenman et al., 2010; Nik-Zainal et al., 2012; Stjernqvist et al., 2011). We accounted for heterozygous germline PTCH1 mutations in contaminating normal skin in biopsies from Gorlin patients and, where observed, subsequent LOH in tumor cells. Except for PT09, SMO was diploid in relapsed BCCs (Table S6), therefore, the expected allele frequencies of fully clonal heterozygous SMO variants was 50% of the tumor content, which we then compared with the observed allele frequency. PTCH1 mutations were present in >80% of tumor cells (Table S7), consistent with deleterious events in PTCH1 being the oncogenic drivers in these tumors. Based on normal contamination and observed allele frequencies, all S MO mutations were estimated to be present in >60% of tumor cells in our vismodegib-resistant BCCs (Table S7), consistent with their selection upon drug treatment.
Mutations in the drug-binding pocket of SMO confer resistance to vismodegib
To gain insight into the properties of the SMO mutations discovered in this study, we took advantage of the recently solved crystal structure of the SMO TM region (Wang et al., 2013). Computational docking of vismodegib onto the SMO structure revealed that SMO-W281, SMO-V321, SMO-I408 and SMO-C469 are located in proximity of the drug-binding pocket (DBP; Figure 3A). The aromatic indole of SMO-W281 forms an edge-to-face pi-stacking interaction with the pyridine ring of vismodegib and helps to form a narrow and hydrophobic pocket, which is disrupted by substitution for the less bulky sulfur of the SMO-W281C mutant (Figure 3B, middle panel). Furthermore, mutation of valine 321 to methionine is likely to interfere with the positioning of W281, exerting a secondary effect on drug binding (Figure 3B, right panel). Unlike W281, residue I408 does not directly contact the drug in our computational model; instead it packs against the binding pocket residues H470 and V404 with its delta methyl group, which when lost is expected to affect binding by changing the conformations of these residues (Figure 3C). This mutation may cause even greater changes in the overall protein backbone structure and hence affect drug binding via a second-shell effect. Finally, we predict that substitution of C469 to a bulky tyrosine would elicit steric effects on the binding pocket, disrupting its conformation.
Figure 3. Structure-function modeling of SMO drug-binding pocket mutants.

(A) Computational docking model showing a top down view of vismodegib (yellow) binding to SMO (grey) and revealing the proximity of W281, V321, I408 and C469 (all green) to the drug-binding pocket.
(B) Left: The position of V321 and W281 (both green) relative to vismodegib (yellow). Middle: The C281 mutant from PT02. Right: The M321 mutant from PT09 is expected to impact the conformation of W281.
(C) Positioning of I408 (left) and the mutant V408 (right) relative to vismodegib. In all panels mutant residues are highlighted in red text.
To test the functional impact of mutations in the DBP, we used a Gli-luciferase based Hh reporter assay. The DBP mutations increased the IC50 of vismodegib 12 to 49-fold over that of SMO-WT, which had an IC50 of 80 nM (Figure 4A). It should be noted that these IC50 values are over-estimates due to overexpression of SMO in this assay (Dijkgraaf et al., 2011). Although each DBP mutant displayed a small (<1.5 fold) increase in basal activity compared to SMO-WT (Figure S3A), all but SMO-I408V were readily inhibited by PTCH1 overexpression (Figure S3B). We next tested [3H]-labeled vismodegib binding to SMO-I408V and SMO-W281C, which respectively exhibited the smallest and largest increases in IC50 (Figure 4A). Both mutants were expressed at cell surface levels similar to SMO-WT, but displayed impaired vismodegib binding (Figures 4B and S3C).
Figure 4. Functional analyses of SMO drug-binding pocket mutants.

(A) Normalized Gli-luciferase reporter activity in C3H10T½ cells transfected with constructs expressing indicated SMO variants, following a dose response with vismodegib. Values were normalized to untreated activity and data plotted are mean +/− standard deviation (SD) of triplicates. IC50 values were calculated after non-linear regression fitting.
(B) Binding of [3H]-vismodegib to HEK-293 cells transfected with constructs expressing indicated SMO variants. EV stands for empty vector and drug binding was measured in counts per minute (cpm). Specific binding was calculated after competition with an excess of unlabeled vismodegib by subtracting non-specific binding from total binding. Data shown are the mean +/− SD.
(C) Viral transduction scheme of primary CGNPs. Only transduced CGNPs proliferate in the absence of SHH, allowing us to specifically test the ability of our SMO variants to promote proliferation in the presence of vismodegib.
(D) Overlay of a representative bright field and red fluorescent image from a PPT CGNP culture after infection with a Cre-expressing lentivirus. The scale bar represents 50 μm.
(E) Normalized methyl-[3H]-thymidine incorporation of PPT CGNPs transduced with indicated viruses, following a dose response with vismodegib after removal of SHH ligand. Each graph shows the same control data. Data plotted are mean +/− SD of triplicates.
See also Figure S3.
It has been demonstrated in preclinical tumor models that the Hh pathway must be inhibited >90% at the transcriptional level to induce tumor regression (Wong et al., 2011). To better understand the impact of these SMO mutations on cell proliferation in the presence of vismodegib, we developed an assay for viral transduction of cerebellar granule neuron precursor (CGNP) cells. It has previously been noted that Hh-driven tumor cells rapidly lose their Hh pathway dependence during culturing (Sasai et al., 2006). However, CGNPs proliferate in vivo in a Hh dependent manner and maintain their Hh pathway dependence in culture for a finite period (Wechsler-Reya and Scott, 1999). CGNPs isolated from Ptch1loxp/loxp;Trp53loxp/loxp;Rosa26LSL-tdTomato (PPT) pups were infected with lentiviral constructs expressing a SMO variant together with an enhanced green fluorescent protein (eGFP)-Cre fusion protein (Figure 4C). The Cre recombinase induces loss of Trp53 and Ptch1, and thus ensures that only transduced CGNPs can proliferate in the absence of exogenous Sonic hedgehog ligand (SHH; Figure S3D). This allowed us to test the ability of the various SMO mutants to promote proliferation in the presence of vismodegib and other inhibitors, after removal of SHH ligand. We monitored proliferation by methyl-[3H]-thymidine incorporation, while Cre-dependent tandem dimer (td) Tomato expression enabled visualization and quantification of infected cells (Figure 4D). This system also enabled us to better model patient genetics because most of the SMO mutations were identified in tumors that harbored TP53 mutations and were driven by loss of PTCH1.
PPT CGNPs infected with SMO-WT and Cre had an IC50 of ~22 nM and proliferation was maximally inhibited at 100 nM vismodegib. In contrast, all DBP mutations had a dramatic effect on vismodegib sensitivity, with infected cells continuing to proliferate at high levels of vismodegib (>1 μM; Figure 4E). Strikingly, cells infected with either SMO-W281C or SMO-C469Y continued to proliferate at near untreated levels even in the presence of 5 μM vismodegib possibly reflecting the direct role of these residues in drug binding. We confirmed that CGNPs were infected at similar frequencies by fluorescence-activated cell sorting (FACS) analysis for Cre-dependent tdTomato reporter expression (Figure S3E), and that the SMO variants were expressed at equivalent levels by quantitative reverse transcription (qRT) PCR (Figure S3F).
Predicting resistance to vismodegib through mutation of the SMO drug binding pocket
To investigate whether other DBP mutations could promote drug resistance, we used our computational model to identify the 21 SMO residues with atoms located within 4.5Å of vismodegib (Figure 5A). We used an algorithm to identify 160 different single nucleotide variants that resulted in non-synonymous changes to these DBP residues, including SMO-W281C and SMO-I408V from this study (Table S8). SMO-D473 was not identified with this method, but the SMO crystal structure revealed that D473 forms a hydrogen-bonding network with several residues that do directly contact vismodegib including R400, H470, E518 and N521 (Wang et al., 2013; Yauch et al., 2009). SMO-E518 was previously identified by alanine scan mutagenesis as a residue that affects vismodegib sensitivity when mutated (Dijkgraaf et al., 2011). Our approach also identified residues that were previously implicated in preclinical models of resistance to the SMO inhibitor sonidegib (LDE225) including N219 and D384 (Table S8; Buonamici et al., 2010), which are predicted to stabilize the SMO conformation through a hydrogen-bonding network (Figure 5B). Strikingly, SMO-N219D, SMO-D384N and SMO-S387N all displayed reduced sensitivity to vismodegib compared to SMO-WT in our Gli-luciferase based Hh reporter assay (Figure 5C). Moreover, we found that the SMO inhibitor LY2940680 and vismodegib share 14 contact residues (Table S8). This suggests that chemically distinct inhibitors interact with overlapping SMO residues and that cross-resistance between inhibitors might occur in the clinic.
Figure 5. Predicting resistance by mutation of the SMO drug-binding pocket.

(A) Illustration of the 21 residues (green) predicted to have atoms within 4.5Å of vismodegib (yellow) bound to the SMO TM structure (grey helices).
(B) Illustration of a hydrogen-bonding network (dashed lines) formed by N219, D384 and S387. Mutation of any of these residues is likely to change the shape of the vismodegib-binding pocket.
(C) Gli-luciferase reporter activity in C3H10T½ cells transfected with indicated SMO- expressing constructs and treated with 1 μM vismodegib. Values were normalized to untreated activity levels for each construct and data plotted are mean +/− SD of triplicates. See also Table S8.
SMO mutations beyond the drug-binding pocket confer vismodegib resistance
SMO mutations located distally with respect to the vismodegib-binding pocket were also associated with vismodegib resistance (Figure 6A). Interestingly, both SMO-T241M and SMO-A459V display increased basal activity over SMO-WT, albeit to a lesser extent than the established oncogenic mutations (Figure 6B). This elevated activity correlated with reduced sensitivity to inhibition by both vismodegib (Figures 6C and S4A) and PTCH1 overexpression (Figure S4B), with SMO-T241M and SMO-A459V shifting the IC50 of vismodegib approximately 3- and 9-fold, respectively. Additionally, all activating mutants tested displayed impaired vismodegib binding despite comparable levels of cell surface expression to SMO-WT (Figures S4C and S4D).
Figure 6. Characterization of SMO mutants outside the drug-binding pocket.

(A) Computational model of vismodegib (yellow) docked onto the crystal structure of the SMO TM region (grey helices; Wang et al., 2013). Mutant residues distal to the drug-binding pocket are highlighted in green.
(B) Gli-luciferase reporter activity in C3H10T½ cells transfected with indicated SMO-expressing constructs. Values were normalized to activity levels of SMO-WT and data plotted are mean +/− SD of triplicates.
(C) Normalized Gli-luciferase reporter activity in C3H10T½ cells transfected with indicated SMO-expressing constructs, following a dose response with vismodegib. Data plotted are mean +/− SD of triplicates.
(D) Normalized methyl-[3H]-thymidine incorporation of PPT CGNPs transduced with indicated viruses, following a dose response with vismodegib after removal of SHH ligand. Data plotted are mean +/− SD of triplicates.
See also Figure S4.
We used our PPT CGNP assay to investigate the impact of non-DBP SMO mutations on proliferation in the presence of vismodegib. SMO-T241M, SMO-A459V and SMO-W535L expressing CGNPs continued to proliferate at high concentrations of vismodegib (Figures 6D and S4E). These data are consistent with mutations outside the DBP destabilizing the SMO architecture to promote activation and reduce affinity for antagonists, as has been observed for GPCRs (Gether et al., 1997). However, we cannot rule out potential allosteric effects on the DBP by these mutations, for example, in the case of SMO-T241M, which only slightly increased basal activity (Figure 2B). Several BCCs (from PT01, PT07 and PT11) that were PTCH1 wild-type and harbored oncogenic SMO mutations initially responded to treatment despite the fact that these SMO mutations reduce sensitivity to vismodegib inhibition (Figure S4A). This could suggest a role for PTCH1 loss-of-function in the sensitivity of SMO mutants to vismodegib.
Intra-tumor heterogeneity and downstream resistance mechanisms
We next examined intra-tumor heterogeneity of resistance mechanisms in relapsed BCC samples. Two distinct SMO mutations were observed in a single biopsy from a Gorlin patient (PT03); SMO-A459V was present in ~70% of tumor cells, whereas the known oncogenic SMO-S533N mutant was present in ~25% of tumor cells (Table S7). This is consistent with outgrowth of two distinct resistant subclones and supports the notion of genetic heterogeneity in drug resistance. To further assess whether genetic heterogeneity plays a role in resistance, we analyzed adjacent biopsies from the same progressing lesion for PT08 and PT09, and biopsies from two distinct relapses originating from the same tumor bed for PT06.
Using the analogy of a tree for tumor evolution, mutations common between regions were assigned to the trunk and unique mutations were considered private to a given branch. We identified respectively 284 and 548 private mutations in PT08.1 and PT08.2, indicating significant genetic divergence between these locations (Figure 7A). Among the 1240 trunk mutations were multiple PTCH1 mutations, which are likely to be oncogenic drivers (Figure S1D). A chromosome 10 duplication and a focal loss in 10q containing the tumor suppressors SUFU and PTEN (Figures 7B and S5A) were also common to the biopsies. This 10q deletion was associated with slightly reduced SUFU expression compared to normal skin (Figure S5B). Whereas one biopsy displayed a large-scale gain of chromosome 2, including GLI2, the other exhibited a focal GLI2 amplification (Figure 7B). Thus, despite significant mutational heterogeneity, these two tumor regions appear to have converged upon the same putative resistance mechanism involving components downstream of the drug target SMO.
Figure 7. Intra-tumor heterogeneity and downstream resistance mechanisms.

(A) Schematic depicting genetic heterogeneity between two biopsies from PT08. Common (Trunk; blue) and unique (Private; orange or green) genetic events are shown with their respective number of somatic mutations. Putative resistance mechanisms are highlighted in green text. Cartoon on right shows spatially separated biopsy sites from the same progressing lesion.
(B) CNV plots for PT08.1 and PT08.2 highlighting key genes that likely contributed to tumor initiation and vismodegib resistance. Copy numbers were derived from array CGH and are shown relative to control tissue.
(C) Schematic depicting genetic heterogeneity between two biopsies from PT06 (same labeling scheme as in (A)). Mutational analysis of a pre-defined list of cancer-associated genes was used to compare the two sites, as germline calls were not available. Cartoon on right shows two separate sites of regrowth from the same regressed lesion.
(D) CNV plots for PT06.1 and PT06.2 highlighting key genes that likely contributed to tumor initiation and vismodegib resistance. Absolute copy number values were derived from SNP arrays.
See also Figure S5.
The two relapsed lesions of PT06 (see schematic in Figure 7C) shared nine trunk mutations from a panel of cancer genes, including TP53-G244D and PTCH1-S616G, consistent with this Gorlin patient harboring a germline PTCH1 mutation (Figure 7C). A chromosome 9 alteration resulting in PTCH1 LOH, and a chromosome 10 duplication were shared between the two biopsies further supporting shared tumor evolution (Figure 7D). One site harbored a SMO-I408V mutation, which was the only private cancer gene mutation detectable at >10% exome sequencing reads. Conversely, this variant was not observed in >160 reads at the corresponding genomic locus from the other biopsy (PT06.2). Similar to PT08.2, PT06.2 harbored a gain of chromosome 2 covering GLI2 (Figure 7D), and a loss in chromosome 10 encompassing SUFU and PTEN (Figure S5C), which correlates with reduced SUFU expression compared to PT06.1 (Figure S5B). These findings suggest recurrent copy number variants could drive vismodegib resistance downstream of SMO.
PT09.1 and PT09.2 had just 75 and 48 private mutations, respectively, and shared 376 trunk mutations including PTCH1-E380* and drug-resistant SMO-V321M, indicating similar evolutionary origins of resistance driven by the same SMO variant (Figure S5D). In conclusion, analysis of progressed lesions from the same initial tumor site revealed common genetic origins and distinct evolutionary paths resulting in mutual and heterogeneous resistance mechanisms, which include alterations in the Hh pathway components SMO, SUFU and GLI2.
Therapeutic options to overcome vismodegib resistance
Having established that multiple SMO mutations can confer resistance to vismodegib we asked whether chemically distinct SMO inhibitors could overcome vismodegib resistance. LY2940680 and LDE225 are currently in clinical trials for various cancers (Clinicaltrials.gov) and compound 5 is a SMO inhibitor that showed preclinical efficacy against SMO-D473H (Dijkgraaf et al., 2011). While all compounds similarly inhibited the proliferation of SMO-WT expressing PPT CGNPs, SMO-mutant expressing cells continued to proliferate, albeit to differing extents (Figure 8A). This observed cross-resistance between the various SMO inhibitors is consistent with our structural predictions (Table S8), and suggests that combining SMO antagonists is not a suitable therapeutic option to overcome acquired resistance. Moreover, the identification of recurrent SUFU and GLI2 variants in relapsed tumors argues for targeting Hh pathway components downstream of SMO. While GLI inhibitors developed so far lack potency and bioavailability, recent studies found that the bromodomain-containing protein BRD4 occupies GLI promoters and is required for transcriptional output of the Hh pathway (Long et al., 2014; Tang et al., 2014). Therefore, the current generation of bromodomain inhibitors could hold promise for Hh-driven cancers. Consistent with this report, PPT CGNPs expressing vismodegib-resistant SMO mutants showed reduced proliferation in the presence of the bromodomain inhibitor JQ1 (Figure 8B).
Figure 8. Therapeutic options for vismodegib resistant BCC.

(A) Normalized methyl-[3H]-thymidine incorporation of PPT CGNPs transduced with various SMO variants and treated with 500 nM of indicated compounds. Values were normalized to proliferation levels without drug and data plotted are mean +/− SD of triplicates. Note that the residual proliferation of SMO-WT in the presence of drug is due to fibroblast and glial contamination of these primary CGNP cultures.
(B) Same as in (A), but transduced CGNPs were treated with 1 μM of either vismodegib or JQ1.
DISCUSSION
Our genomic analysis of vismodegib resistant BCCs revealed recurrent mechanisms for reactivation of the Hh signaling pathway at the level, or downstream, of SMO. Intriguingly, secondary, or acquired, resistance in BCC is relatively rare, with an estimated incidence of ~20% per year (Chang and Oro, 2012). This differs from the situation for other targeted therapies, such as BRAF inhibitors in melanoma and EGFR inhibitors in lung cancer, where secondary resistance is almost inevitable within the first 12 months of treatment (Lackner et al., 2012). Although we could not detect SMO mutations in pre-treatment biopsies, other examples in the literature, such as the detection of gefitinib-resistant EGFR-T790M in pre-treatment samples from non-small cell lung cancer patients, support the idea that resistance arises through selection of minor resistant clones (Inukai et al., 2006). The somatic mutation rate in our samples and in BCC in general is very high when compared to other tumor types and would be expected to correlate with a high number of pre-existing resistance mutations, and an increased probability of resistance. Moreover, we observed a high proportion of TP53 mutations in vismodegib-resistant BCCs, which would be predicted to increase genomic instability (Negrini et al., 2010). However, unlike other cancers where pathway switching can be observed (Lackner et al., 2012), our genomic analysis suggests that BCC is addicted to Hh signaling, which could limit the range of possible resistance variants. Alternatively, the relatively low incidence of resistance to date may simply be an underestimate, as vismodegib has only recently entered clinical practice and BCC is a slow-growing cancer (Wang et al., 2011). Thus, it could be that patients have not yet been followed long enough on treatment to accurately gauge resistance rates and durability of response.
We found that the vast majority of vismodegib resistance in PTCH1 mutation-driven BCC is caused by acquired mutations in the drug target, SMO. These mutations fall into two classes: those in proximity of the SMO DBP, and those located distally, including known oncogenic variants. The DBP mutants were not detectable in untreated samples and all conferred resistance to vismodegib, consistent with their key role in drug binding. Interestingly, all DBP mutants except SMO-I408V were as sensitive as SMO-WT to inhibition by PTCH1 overexpression. The DBP has been proposed as a possible binding site for a putative endogenous, PTCH-regulated, SMO ligand (Nedelcu et al., 2013). Our observations suggest that such a ligand would bind SMO in a manner distinct from clinical inhibitors.
SMO mutations located outside the DBP included two novel mutants (T241M and A459V) and three previously described oncogenic mutants (L412F, S533N and W535L). Interestingly, the recurrent SMO-A459V mutant displayed elevated basal activity and is located on TM helix 6, which plays a key role in the activation of class A GPCRs (Katritch et al., 2013). This conservative mutation might disrupt helix-packing leading to increased conformational flexibility of SMO, and thereby reduce the affinity for antagonists (Gether et al., 1997). SMO-A459V and the oncogenic mutations displayed a similar reduction in sensitivity to vismodegib. Yet, SMO mutant-driven BCCs initially responded to vismodegib prior to relapse. We propose that the difference between sensitive and resistant SMO mutant tumors is the mutational status of PTCH1 because SMO-A459V was identified in PTCH1 mutant relapsed BCCs. In another case, an oncogenic SMO-L412F mutation was present before and after treatment, and PTCH1 copy number loss was acquired upon treatment. Partial or total PTCH1 loss could affect drug sensitivity by increasing overall pathway activity, reducing PTCH1-mediated negative feedback, or promoting a competitive interaction between a PTCH1-regulated SMO ligand and inhibitors. Through such mechanisms PTCH1 loss may increase the amount of drug required to block the Hh pathway output >90%, the level of inhibition needed to achieve tumor regression (Wong et al., 2011). Thus, small changes in sensitivity, through combinations of SMO and PTCH1 mutations or copy number alterations, could have dramatic effects on drug response.
In two patients we detected deletion of chromosome 10q, covering SUFU and PTEN. This alteration has not been observed previously in untreated BCCs (Pesz et al., 2013) and therefore might only provide a selective advantage in the presence of a SMO inhibitor. Strikingly, both copies of PTEN were lost in one case, suggesting this may not be a passenger CNV. PI3K signaling has been implicated in the response of preclinical MB models to SMO inhibition (Buonamici et al., 2010; Metcalfe et al., 2013). However, despite lessening the response to vismodegib treatment, PTEN loss did not result in progression of MB allografts (Metcalfe et al., 2013). Loss of PTEN might provide a selective advantage in treated Hh-driven tumors, but further hits in the Hh pathway are likely to be required for tumor progression in the presence of a SMO inhibitor.
SUFU is a highly conserved negative regulator of the Hh pathway that functions as a tumor suppressor in MB and BCC, and has been associated with Gorlin syndrome (Pastorino et al., 2009; Smith et al., 2014). Mice heterozygous for Sufu display enhanced basaloid proliferation in the epidermis, consistent with haploinsufficiency of Sufu in the skin (Svard et al., 2006). Our observation of co-occurring alterations, such as GLI2 amplification, suggests that partial loss of SUFU function may not be sufficient to drive vismodegib resistance. However, homozygous mutation of SUFU likely confers resistance in one of our relapsed sporadic BCCs (PT05) because SUFU mutant patient MB xenografts or mouse models of Sufu−/− MB tumors do not respond to SMO inhibition (Kool et al., 2014; Lee et al., 2007).
GLI2 amplification has previously been associated with HPI resistance in preclinical MB models (Buonamici et al., 2010; Dijkgraaf et al., 2011). This raises the question: Why does SUFU loss co-occur with GLI2 gain in vismodegib resistant BCCs? One explanation could be the extent of amplification; GLI2 was amplified 17-fold in a resistant MB allograft (Dijkgraaf et al., 2011), while we only see evidence for one or two copies gained. Thus, SUFU loss might alleviate the need for high copy number gains by reducing negative regulation of GLI transcription factors.
Identification of resistance mechanisms is essential for improving therapeutic strategies to obtain durable responses to targeted therapies. Our analysis has revealed divergent mechanisms of resistance to SMO inhibition in BCC, even within the same tumor. We found significant cross-resistance between clinical SMO inhibitors for all SMO mutants identified in this study. Combinatorial treatment with BRAF and MEK inhibitors has recently been implemented in melanoma, where resistance to BRAF inhibitors is primarily associated with reactivation of the MAP kinase pathway (Das Thakur and Stuart, 2013). Similarly, mechanisms of SMO inhibitor resistance in BCC converge on the Hh pathway. Therefore, we anticipate combination with agents acting downstream in the Hh pathway, including those targeting epigenetic regulators such as BRD4 (Long et al., 2014; Tang et al., 2014), will be required to circumvent acquired resistance to SMO inhibitors in BCC and other Hh-driven cancers.
EXPERIMENTAL PROCEDURES
Patient and tissue specimens
We characterized primary tissue samples from 12 vismodegib-treated patients with locally advanced or metastatic BCC, who experienced a prior, investigator-assessed, clinical benefit on therapy (LoRusso et al., 2011; Sekulic et al., 2012) followed by tumor regrowth. Tissue samples were collected after receiving written informed consent according to protocols approved by the institutional review boards (IRB) of centers participating in clinical trials NCT00607724, NCT00833417 and NCT01367665, including Hôpital Saint-Louis, Stanford University, Sint-Augustinus Cancer Center, Dana Farber Cancer Institute, Memorial Sloan Kettering Cancer Center, UCLA School of Medicine and MD Anderson Cancer Center. For comparison, we analyzed BCC biopsies and matched blood from 43 untreated BCC patients. Samples from untreated sporadic BCC patients were obtained according to University of Michigan IRB-approved protocols HUM00069052 and HUM00050085. Untreated Gorlin patient samples were obtained according to the Stanford University IRB-approved protocol 2012–029. 5 normal skin RNA samples were procured from ProteoGenex, the use of which was approved by relevant IRBs. Detailed information is available from ProteoGenex. All samples used had informed consent from study participants. Patient and biopsy characteristics are summarized in Table S1.
Genomic analyses
DNA from 15 vismodegib-resistant BCC samples, 48 untreated BCCs and 52 matched blood samples were subjected to WES. WES of tumor biopsies was achieved with a minimum average coverage of more than 67-fold (Table S1). Copy number changes were assessed for vismodegib-resistant BCCs by SNP or CGH arrays. RNA from 11 resistant BCC samples was subjected to RNA-seq. DNA from 7 FFPE samples was analyzed by pyrosequencing. RNA-seq data from 5 normal skin samples were used as baseline gene expression for comparisons with BCC patient samples.
Animals
All mouse studies were performed according to protocols approved by the Genentech Inc. institutional animal care and use committee, which conformed to the animal-use guidelines of Genentech Inc. and to California State legal and ethical practices.
Functional analyses
SMO mutants were generated in pRK5-SMO vectors as described (Dijkgraaf et al., 2011; Yauch et al., 2009) and were either utilized in Gli-luciferase reporter assays as described (Dijkgraaf et al., 2011) or cloned into lentiviral vectors for transduction of primary CGNP cultures. Proliferation was assayed using methyl-[3H]-thymidine incorporation (Kool et al., 2014). Binding of [3H]-vismodegib to SMO mutants was carried out in HEK-293 cells as described (Dijkgraaf et al., 2011).
Supplementary Material
Significance.
Acquired resistance represents a major challenge to the success of targeted cancer therapies. Therefore, understanding resistance mechanisms will help guide future therapeutic strategies. Vismodegib was approved for treatment of locally advanced and metastatic BCC, and is under clinical investigation for several other cancers. We performed molecular analyses of tumor biopsies from BCC patients who initially responded, but subsequently progressed on treatment. We found that vismodegib resistance is invariably linked to Hh pathway reactivation, indicating that BCC is addicted to Hh signaling. Variants occurred either in or downstream of the drug target SMO, with intra-tumor heterogeneity being observed for both resistance mechanisms, suggesting combination of SMO and downstream targeting agents will be required to overcome vismodegib resistance in Hh-driven cancers.
Acknowledgments
We thank A Durlach (Pathology) and F Grange (Dermatology) from the Centre Hospitalier Universitaire de Reims, France. We thank N Meireles from the University of Michigan Multidisciplinary Cutaneous Oncology Program for skin cancer biobank and data management. We thank A Dupati and U Amadi for assistance during sample collection. We thank K Robarge for small molecule support and H Chen for FACS help. We thank C Metcalfe and J Hoeck for critical reading of the manuscript. We thank A Bruce for assistance with the graphical abstract. HJS, GP, GJD, ZM, TJ, VT, PMH, RB, RLY and FJdS are employees of Genentech Inc. and own Roche shares. NBS was a visiting scientist at Genentech Inc. JYT was a consultant for Genentech Inc.
Footnotes
Accession numbers
All datasets generated in this study using exome and RNA sequencing as well as aCGH and SNP arrays are accessible at the European Genome-phenome Archive (EGA) under EGAS00001000845.
Supplemental information includes Supplemental Experimental Procedures, five figures and eight tables and can be found with this article online at:
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Amakye D, Jagani Z, Dorsch M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nature medicine. 2013;19:1410–1422. doi: 10.1038/nm.3389. [DOI] [PubMed] [Google Scholar]
- Atwood SX, Li M, Lee A, Tang JY, Oro AE. GLI activation by atypical protein kinase C iota/lambda regulates the growth of basal cell carcinomas. Nature. 2013;494:484–488. doi: 10.1038/nature11889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brastianos PK, Horowitz PM, Santagata S, Jones RT, McKenna A, Getz G, Ligon KL, Palescandolo E, Van Hummelen P, Ducar MD, et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nature genetics. 2013;45:285–289. doi: 10.1038/ng.2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkhuizen T, Reinders MG, van Geel M, Hendriksen AJ, Paulussen AD, Winnepenninckx VJ, Keymeulen KB, Soetekouw PM, van Steensel MA, Mosterd K. Acquired resistance to the Hedgehog pathway inhibitor vismodegib due to smoothened mutations in treatment of locally advanced basal cell carcinoma. Journal of the American Academy of Dermatology. 2014 doi: 10.1016/j.jaad.2014.08.001. [DOI] [PubMed] [Google Scholar]
- Buonamici S, Williams J, Morrissey M, Wang A, Guo R, Vattay A, Hsiao K, Yuan J, Green J, Ospina B, et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci Transl Med. 2010;2:51ra70. doi: 10.1126/scitranslmed.3001599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang AL, Oro AE. Initial assessment of tumor regrowth after vismodegib in advanced Basal cell carcinoma. Archives of dermatology. 2012;148:1324–1325. doi: 10.1001/archdermatol.2012.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark VE, Erson-Omay EZ, Serin A, Yin J, Cotney J, Ozduman K, Avsar T, Li J, Murray PB, Henegariu O, et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science. 2013;339:1077–1080. doi: 10.1126/science.1233009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das Thakur M, Stuart DD. The evolution of melanoma resistance reveals therapeutic opportunities. Cancer research. 2013;73:6106–6110. doi: 10.1158/0008-5472.CAN-13-1633. [DOI] [PubMed] [Google Scholar]
- Dijkgraaf GJ, Alicke B, Weinmann L, Januario T, West K, Modrusan Z, Burdick D, Goldsmith R, Robarge K, Sutherlin D, et al. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer research. 2011;71:435–444. doi: 10.1158/0008-5472.CAN-10-2876. [DOI] [PubMed] [Google Scholar]
- Gether U, Ballesteros JA, Seifert R, Sanders-Bush E, Weinstein H, Kobilka BK. Structural instability of a constitutively active G protein-coupled receptor. Agonist-independent activation due to conformational flexibility. The Journal of biological chemistry. 1997;272:2587–2590. doi: 10.1074/jbc.272.5.2587. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Perez A, Lopez-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. American journal of human genetics. 2011;88:440–449. doi: 10.1016/j.ajhg.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenman CD, Bignell G, Butler A, Edkins S, Hinton J, Beare D, Swamy S, Santarius T, Chen L, Widaa S, et al. PICNIC: an algorithm to predict absolute allelic copy number variation with microarray cancer data. Biostatistics. 2010;11:164–175. doi: 10.1093/biostatistics/kxp045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, Vorechovsky I, Holmberg E, Unden AB, Gillies S, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85:841–851. doi: 10.1016/s0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- Inukai M, Toyooka S, Ito S, Asano H, Ichihara S, Soh J, Suehisa H, Ouchida M, Aoe K, Aoe M, et al. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non-small cell lung cancer. Cancer research. 2006;66:7854–7858. doi: 10.1158/0008-5472.CAN-06-1951. [DOI] [PubMed] [Google Scholar]
- Jayaraman SS, Rayhan DJ, Hazany S, Kolodney MS. Mutational landscape of basal cell carcinomas by whole-exome sequencing. The Journal of investigative dermatology. 2014;134:213–220. doi: 10.1038/jid.2013.276. [DOI] [PubMed] [Google Scholar]
- Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, Quinn AG, Myers RM, Cox DR, Epstein EH, Jr, Scott MP. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272:1668–1671. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333–339. doi: 10.1038/nature12634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katritch V, Cherezov V, Stevens RC. Structure-function of the G protein-coupled receptor superfamily. Annual review of pharmacology and toxicology. 2013;53:531–556. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kijima C, Miyashita T, Suzuki M, Oka H, Fujii K. Two cases of nevoid basal cell carcinoma syndrome associated with meningioma caused by a PTCH1 or SUFU germline mutation. Fam Cancer. 2012;11:565–570. doi: 10.1007/s10689-012-9548-0. [DOI] [PubMed] [Google Scholar]
- Kool M, Jones DT, Jager N, Northcott PA, Pugh TJ, Hovestadt V, Piro RM, Esparza LA, Markant SL, Remke M, et al. Genome Sequencing of SHH Medulloblastoma Predicts Genotype-Related Response to Smoothened Inhibition. Cancer cell. 2014;25:393–405. doi: 10.1016/j.ccr.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackner MR, Wilson TR, Settleman J. Mechanisms of acquired resistance to targeted cancer therapies. Future Oncol. 2012;8:999–1014. doi: 10.2217/fon.12.86. [DOI] [PubMed] [Google Scholar]
- Lee Y, Kawagoe R, Sasai K, Li Y, Russell HR, Curran T, McKinnon PJ. Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene. 2007;26:6442–6447. doi: 10.1038/sj.onc.1210467. [DOI] [PubMed] [Google Scholar]
- Long J, Li B, Rodriguez-Blanco J, Pastori C, Volmar CH, Wahlestedt C, Capobianco A, Bai F, Pei XH, Ayad NG, Robbins DJ. The BET bromodomain inhibitor I-BET151 acts downstream of Smoothened to abrogate the growth of Hedgehog driven cancers. The Journal of biological chemistry. 2014 doi: 10.1074/jbc.M114.595348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoRusso PM, Rudin CM, Reddy JC, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR, Chang I, Darbonne WC, et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17:2502–2511. doi: 10.1158/1078-0432.CCR-10-2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalfe C, Alicke B, Crow A, Lamoureux M, Dijkgraaf GJ, Peale F, Gould SE, de Sauvage FJ. PTEN loss mitigates the response of medulloblastoma to Hedgehog pathway inhibition. Cancer research. 2013;73:7034–7042. doi: 10.1158/0008-5472.CAN-13-1222. [DOI] [PubMed] [Google Scholar]
- Miller JH. Mutagenic specificity of ultraviolet light. J Mol Biol. 1985;182:45–65. doi: 10.1016/0022-2836(85)90026-9. [DOI] [PubMed] [Google Scholar]
- Nedelcu D, Liu J, Xu Y, Jao C, Salic A. Oxysterol binding to the extracellular domain of Smoothened in Hedgehog signaling. Nature chemical biology. 2013;9:557–564. doi: 10.1038/nchembio.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability--an evolving hallmark of cancer. Nature reviews Molecular cell biology. 2010;11:220–228. doi: 10.1038/nrm2858. [DOI] [PubMed] [Google Scholar]
- Nik-Zainal S, Van Loo P, Wedge DC, Alexandrov LB, Greenman CD, Lau KW, Raine K, Jones D, Marshall J, Ramakrishna M, et al. The life history of 21 breast cancers. Cell. 2012;149:994–1007. doi: 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oro AE, Higgins KM, Hu Z, Bonifas JM, Epstein EH, Jr, Scott MP. Basal cell carcinomas in mice overexpressing sonic hedgehog. Science. 1997;276:817–821. doi: 10.1126/science.276.5313.817. [DOI] [PubMed] [Google Scholar]
- Pastorino L, Ghiorzo P, Nasti S, Battistuzzi L, Cusano R, Marzocchi C, Garre ML, Clementi M, Scarra GB. Identification of a SUFU germline mutation in a family with Gorlin syndrome. Am J Med Genet A. 2009;149A:1539–1543. doi: 10.1002/ajmg.a.32944. [DOI] [PubMed] [Google Scholar]
- Pesz KA, Bieniek A, Makowska I, Sasiadek MM. Basal cell carcinoma of the skin: whole genome screening by comparative genome hybridization revisited. Journal of cutaneous pathology. 2013;40:25–29. doi: 10.1111/cup.12033. [DOI] [PubMed] [Google Scholar]
- Pricl S, Cortelazzi B, Dal Col V, Marson D, Laurini E, Fermeglia M, Licitra L, Pilotti S, Bossi P, Perrone F. Smoothened (SMO) receptor mutations dictate resistance to vismodegib in basal cell carcinoma. Molecular oncology. 2014 doi: 10.1016/j.molonc.2014.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reifenberger J, Wolter M, Knobbe CB, Kohler B, Schonicke A, Scharwachter C, Kumar K, Blaschke B, Ruzicka T, Reifenberger G. Somatic mutations in the PTCH, SMOH, SUFUH and TP53 genes in sporadic basal cell carcinomas. Br J Dermatol. 2005;152:43–51. doi: 10.1111/j.1365-2133.2005.06353.x. [DOI] [PubMed] [Google Scholar]
- Reifenberger J, Wolter M, Weber RG, Megahed M, Ruzicka T, Lichter P, Reifenberger G. Missense mutations in SMOH in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer research. 1998;58:1798–1803. [PubMed] [Google Scholar]
- Sasai K, Romer JT, Lee Y, Finkelstein D, Fuller C, McKinnon PJ, Curran T. Shh pathway activity is down-regulated in cultured medulloblastoma cells: implications for preclinical studies. Cancer research. 2006;66:4215–4222. doi: 10.1158/0008-5472.CAN-05-4505. [DOI] [PubMed] [Google Scholar]
- Sekulic A, Migden MR, Oro AE, Dirix L, Lewis KD, Hainsworth JD, Solomon JA, Yoo S, Arron ST, Friedlander PA, et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. The New England journal of medicine. 2012;366:2171–2179. doi: 10.1056/NEJMoa1113713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MJ, Beetz C, Williams SG, Bhaskar SS, O'Sullivan J, Anderson B, Daly SB, Urquhart JE, Bholah Z, Oudit D, et al. Germline Mutations in SUFU Cause Gorlin Syndrome-Associated Childhood Medulloblastoma and Redefine the Risk Associated With PTCH1 Mutations. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2014 doi: 10.1200/JCO.2014.58.2569. [DOI] [PubMed] [Google Scholar]
- Stjernqvist S, Ryden T, Greenman CD. Model-integrated estimation of normal tissue contamination for cancer SNP allelic copy number data. Cancer informatics. 2011;10:159–173. doi: 10.4137/CIN.S6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone DM, Murone M, Luoh S, Ye W, Armanini MP, Gurney A, Phillips H, Brush J, Goddard A, de Sauvage FJ, Rosenthal A. Characterization of the human suppressor of fused, a negative regulator of the zinc-finger transcription factor Gli. Journal of cell science. 1999;112(Pt 23):4437–4448. doi: 10.1242/jcs.112.23.4437. [DOI] [PubMed] [Google Scholar]
- Svard J, Heby-Henricson K, Persson-Lek M, Rozell B, Lauth M, Bergstrom A, Ericson J, Toftgard R, Teglund S. Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Developmental cell. 2006;10:187–197. doi: 10.1016/j.devcel.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Sweeney RT, McClary AC, Myers BR, Biscocho J, Neahring L, Kwei KA, Qu K, Gong X, Ng T, Jones CD, et al. Identification of recurrent SMO and BRAF mutations in ameloblastomas. Nature genetics. 2014;46:722–725. doi: 10.1038/ng.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Gholamin S, Schubert S, Willardson MI, Lee A, Bandopadhayay P, Bergthold G, Masoud S, Nguyen B, Vue N, et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nature medicine. 2014;20:732–740. doi: 10.1038/nm.3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, Zhang X, Agatep R, Chiappa S, Gao L, Lowrance A, et al. Mutations in SUFU predispose to medulloblastoma. Nature genetics. 2002;31:306–310. doi: 10.1038/ng916. [DOI] [PubMed] [Google Scholar]
- Wang C, Wu H, Katritch V, Han GW, Huang XP, Liu W, Siu FY, Roth BL, Cherezov V, Stevens RC. Structure of the human smoothened receptor bound to an antitumour agent. Nature. 2013;497:338–343. doi: 10.1038/nature12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GY, So PL, Wang L, Libove E, Wang J, Epstein EH., Jr Establishment of murine basal cell carcinoma allografts: a potential model for preclinical drug testing and for molecular analysis. The Journal of investigative dermatology. 2011;131:2298–2305. doi: 10.1038/jid.2011.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron. 1999;22:103–114. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- Wong H, Alicke B, West KA, Pacheco P, La H, Januario T, Yauch RL, de Sauvage FJ, Gould SE. Pharmacokinetic-pharmacodynamic analysis of vismodegib in preclinical models of mutational and ligand-dependent Hedgehog pathway activation. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17:4682–4692. doi: 10.1158/1078-0432.CCR-11-0975. [DOI] [PubMed] [Google Scholar]
- Xie J, Murone M, Luoh SM, Ryan A, Gu Q, Zhang C, Bonifas JM, Lam CW, Hynes M, Goddard A, et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391:90–92. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
- Yauch RL, Dijkgraaf GJ, Alicke B, Januario T, Ahn CP, Holcomb T, Pujara K, Stinson J, Callahan CA, Tang T, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science. 2009;326:572–574. doi: 10.1126/science.1179386. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.