

Abstract
The lymph node periphery is an important site for many immune functions, from pathogen containment to T helper differentiation, yet the cues that position cells in this region are largely undefined. Here, using a sphingosine 1-phosphate (S1P) reporter, we found that cells sensed higher concentrations of S1P in the medullary cords than in the T zone, and the S1P transporter SPNS2 on lymphatic endothelial cells generated this gradient. Natural killer (NK) cells are located at the periphery of the lymph node, predominantly in the medulla, and we found that expression of SPNS2, S1P receptor 5 on NK cells, and CXCR4 were all required for NK cell localization during homeostasis and rapid interferon-γ production by NK cells after challenge. Our findings elucidate the spatial cues for NK cell organization, and reveal a novel role of S1P in positioning cells within the medulla.
Introduction
The lymph node (LN) periphery surrounding the T zone and B follicles is increasingly recognized to be an important site for many immune functions. Lymph-borne pathogens drain into the LN subcapsular and medullary sinuses, where infection must be contained and antigen must be routed to lymphocytes surveying the LN parenchyma1–7. Lymphocytes are initially activated near the subcapsular sinus in many models of tissue infection, and CD4+ T cell access to the interfollicular regions and medulla is important for differentiation of both TH1 and TH2 cells8–17. Yet, we are only beginning to define the signals that direct cell movement through the LN periphery.
An important set of cells that resides in the LN periphery and helps contain infection is a group of “innate-like” lymphocytes, including natural killer (NK) cells, NKT cells, γδ T cells, and some memory CD8+ T cells5. During homeostasis, NK cells are positioned predominantly in the medulla, as well as in the interfollicular regions neighboring the subcapsular sinus, and at the boundary of the T zone and B cell follicles, where cortical lymphatic sinuses are concentrated18–24. When a pathogen drains into the LN, inflammasome activation in macrophages that line the subcapsular and medullary sinuses induces those macrophages to secrete cytokines including interleukin 18 (IL-18), which activate NK cells and other innate-like lymphocytes to secrete interferon-γ (IFN-γ)5. IFN-γ in turn stimulates macrophage microbicidal activity, and blocking IFN-γ allows pathogens to exit the draining LN via efferent lymph and spread systemically5,25. The IFN-γ response is rapid, within two hours of infection in some cases5. The speed with which the NK cell IFN-γ response occurs suggests that the close proximity between NK cells and macrophages prior to infection is important, but it is not known what chemotactic cues position NK cells near the macrophage-filled lymphatic sinuses5.
We hypothesized that lymphatic endothelial cells (LECs) may secrete a chemoattractant that brings immune cells near the lymphatic sinuses. We have previously shown that lymphatic endothelial cells supply sphingosine 1-phosphate (S1P) to lymph using the S1P transporter SPNS2, and that lymphocytes require this source of S1P to egress from lymph nodes26. The shape of the S1P gradient within the LN parenchyma is unknown, but we hypothesized that S1P concentrations may be high not only in the lymph but also within the lymph node parenchyma near lymphatics, and that this S1P may position cells in the LN periphery.
Here, using a mouse that expresses an S1P sensor, we found that cells within the medullary cords sensed higher concentrations of S1P than cells in the T zone, and SPNS2 expressed on lymphatic endothelial cells generated this gradient. Moreover, NK cells were displaced from the medulla into the T zone in the absence of Spns2 and when NK cells lacked S1P receptor 5 (S1PR5). Seeking cues that may act with S1P, we found that CXCR4 inhibition resulted in NK cell mislocalization. In each of these cases, the defect in positioning attenuated IFN-γ production by NK cells just after infection, demonstrating that precise localization of innate cells within the LN facilitates the early response to infection.
Results
High S1P concentrations in the medullary cords
The concentration of S1P in lymph is higher than in the interstitial fluid of LN, when averaged over the whole LN, and this differential guides lymphocyte exit from LN into lymph27. We hypothesized, however, that S1P concentrations may not be uniformly low across the LN parenchyma, and that gradients may exist within the LN —higher near lymphatics than deep in the T zone (Supp. Fig. 1a).
To test this hypothesis, we used a mouse expressing a reporter of extracellular, “signaling available” S1P (Supp. Fig. 1b)28. The core of the reporter is an S1P receptor 1 (S1PR1)-eGFP fusion. Because S1PR1 is internalized upon binding its ligand S1P, S1PR1-eGFP remains on the plasma membrane in the absence of extracellular S1P, and is lost from the plasma membrane (relocalized into endosomes and partially degraded) in the presence of extracellular S1P29. As an internal control, the reporter also encodes a transmembrane glycoprotein human CD2 (hCD2)-tRFP fusion, lacking the 100 amino acids in the hCD2 cytoplasmic tail responsible for signaling. hCD2-tRFP marks the plasma membrane of reporting cells, and allows for ratiometric calculation of surface expression of S1PR1-GFP. The two proteins are separated by a P2A ribosomal skip sequence so that they are translated at a 1:1 ratio, knocked into the Rosa26 locus with the strong synthetic CAG promoter, and preceded by a floxed transcriptional stop28. To induce reporter expression, we bred animals expressing the reporter and Cre driven by the interferon-responsive Mx1 promoter (Mx1-Cre30), treated 3–5 day old pups with the double-stranded RNA mimic poly(I:C) to induce expression of Cre, and analyzed the mice as adults. We also bred animals expressing the reporter and tamoxifen-inducible CreERT2 driven by the widely expressed Ubiquitin C promoter (UBC-CreERT231), and induced Cre activity in adult animals. In the LN, using either Cre, the reporter was well-expressed by high endothelial venules and a subset of myeloid cells. We limited our reporter analysis to CD11b+ myeloid cells, which are relatively stationary in the LN in homeostasis and should therefore report on S1P in their current location32.
In LN sections, we could clearly identify the subcapsular sinus (SCS), the T zone, and the medulla by staining with antibodies to B220 (expressed by B cells) and Lyve1 (expressed by lymphatic endothelial cells and a subset of lymphatic sinus-lining macrophages33) (Supp. Fig. 1a, Fig. 1a). The SCS is identified by a rim of Lyve1 staining underneath the LN capsule, marking the sinus floor. The medulla as a whole can be identified as Lyve1-rich regions. The medullary cords (the LN parenchyma) and medullary sinuses (the lymphatic vessels) can be delineated using Lyve1 staining34, 35 (Fig. 1b), and we verified that FITC-dextran injected subcutaneously drained quickly into the Lyve1+ areas of the medulla (not shown). The T zone is at the center of the LN, circumscribed by B follicles and the medulla.
Figure 1. Cells in the medullary cords sense higher concentrations of extracellular S1P than cells in the T zone.
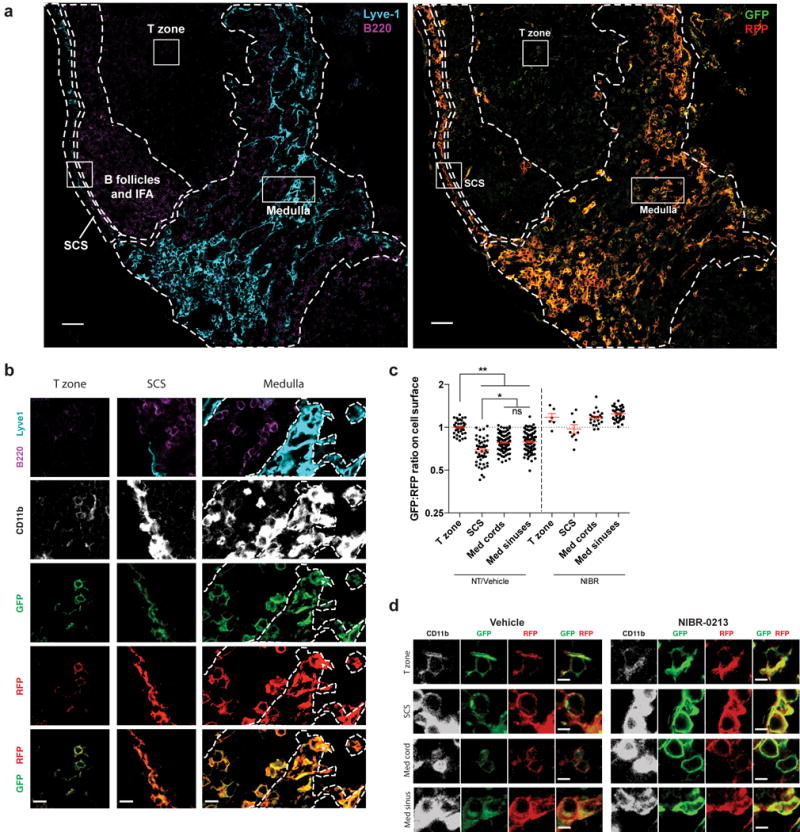
(a) LN section from an S1P reporter mouse (Mx1-Cre) stained with antibodies against CD11b (not shown), Lyve1 (cyan), B220 (purple), GFP (green), and RFP (red). Left: B220 and Lyve1 staining was used to identify the subcapsular sinus (SCS), medulla, T zone, and B follicles/interfollicular areas (IFA). Boundaries between regions are indicated by dashed lines. The medulla was defined as Lyve1-rich regions. Within the medulla, cords were defined as Lyve1-negative areas that contained relatively dense B cells. Medullary sinuses were defined as Lyve1 positive areas with scarce B cells. Medullary cords and sinuses were identified conservatively, and ambiguous areas were excluded. Right: Reporter staining. (high resolution file: https://figshare.com/s/a88afb6d06b90b3af860). Boxed areas are shown in greater detail in Fig. 1b. Bright reporting cells at the T/B boundary are high endothelial venules. Scale bar, 50 μm. Representative of 16 images from 5 mice in 5 experiments.
(b) Insets of cells from the T zone, SCS, and medulla from the reporter LN shown in Fig. 1a. Lymphatic sinuses are indicated by dotted lines in the medulla inset. Scale bars, 10 μm.
(c) Quantification of S1P reporting in the T zone, subcapsular sinus (SCS), medullary cords, and medullary sinuses. Pixels positive for both RFP and CD11b were identified, marking the surface of reporter-expressing myeloid cells. The ratio of GFP to RFP for each CD11b+RFP+ pixel was calculated, and the ratio was averaged over a defined area. Each point on the graph represents the average ratio over an area of at least 1.8×103 square microns for the SCS; 8×102 square microns for medullary cords or sinuses; and 5.5×102 square microns for the T zone. Horizontal lines indicate the mean and SEM. To account for different microscope settings on different days, for each experiment all ratios were normalized such that the average ratio for the T zone in non-treated (NT) or vehicle-treated animals was 1.0. For non-treated and vehicle-treated animals, graph compiles 16 sections from 5 mice. For NIBR-0213 treated animals (see Fig. 1d legend), graph compiles 4 sections from 2 mice. Data compile 3 experiments analyzing non-treated mice and 2 experiments analyzing NIBR-0213 and vehicle-treated animals. *, p<0.01; **, p<0.0001; (ANOVA with post-hoc Tukey HSD test for multiple comparisons).
(d) Littermate pairs of S1P reporter mice (UBC-CreERT2 or Mx1-Cre) were treated i.p. with 30 mg/kg of the S1PR1 antagonist NIBR-0213 or vehicle once daily for 8 days. LN were harvested 3–5 hours after the last dose. LN sections were stained with antibodies against GFP (green), RFP (red), CD11b (white), and Lyve1 and B220 (to define LN regions, not shown). Representative cells are shown from the T zone, SCS, medullary cords, and medullary sinuses of NIBR-0213 or vehicle-treated S1P reporter mice. Scale bar, 5 μm. Representative of 3 (vehicle-treated) or 4 (NIBR-0213-treated) images from 2 pairs of mice in 2 experiments (image files: https://figshare.com/s/fd985aa726576b6cb9e7 and https://figshare.com/s/bc2ada688bc9ba074cfe).
As expected, CD11b+ cells within the SCS, which is filled with lymph fluid, reported high concentrations of S1P, while CD11b+ cells in the T zone reported little S1P (Fig. 1a, b). Furthermore, reporter imaging revealed that CD11b+ cells in the medullary cords sensed more S1P than those in the T zone (Fig. 1a, b). Quantification confirmed these observations, with a consistently lower GFP:RFP ratio on the surface of reporting CD11b+ cells within the medullary cords, medullary sinuses, and SCS compared to their counterparts in the T zone (Fig. 1c).
Interestingly, we saw little difference in S1P sensing between medullary cord and medullary sinus macrophages (Fig. 1a–c). We had expected more S1P sensing by cells in the sinuses than the cords, reflecting the S1P gradient that guides lymphocyte egress. The reporter was not saturated, as we detected increased S1P sensing by medullary sinus macrophages in mice treated with the S1PR1 agonist FTY720 compared to control-treated animals (Supp. Fig. 1c), and increased S1P sensing by subcapsular sinus macrophages compared to medullary sinus macrophages (Fig. 1b, c). It is possible that the reporter is not sufficiently sensitive to detect a small difference in S1P sensing across the sinus wall. However, we speculate that the dense network of cells on both sides of the medullary lymphatic sinus wall in macrophage-packed regions degrades S1P secreted in either direction, and that S1P concentrations may be higher in macrophage-sparse areas of the medullary sinuses and in the cortical sinuses (which lack reporting cells).
To verify that medullary cord cells were faithfully reporting elevated S1P in the cords, we treated mice with the specific S1PR1 antagonist NIBR-0213, which prevents internalization of the receptor36. After 7 days of treatment, reporting CD11b+ cells within the medullary cords had recovered S1PR1-GFP on the plasma membrane, reflected in higher surface GFP:RFP ratios compared to reporting cells in the medullary cords of vehicle treated animals (Fig. 1c, d). It is possible that medullary cord cells sense S1P only because they extend processes into the lymphatic sinus lumen, where they may be exposed to lymph S1P. We have not observed such processes on reporting cells in our system, however, as many medullary cord macrophages have a rounded morphology (Fig. 1b). We predicted that if S1P sensing by cells in the cords were due only to extension of processes into the lymphatic sinuses, sensing by cells in the cords would be substantially less than cells in the sinuses because only a small portion of the cord-resident cells would be exposed to lymph S1P, in contrast to the sinus-resident cells bathed in S1P. Because we did not detect a substantial difference in S1P sensing across the lymphatic sinus wall, at least in areas of medullary lymphatic sinuses packed with reporting macrophages, our data suggest that S1P sensing by medullary cord macrophages is attributable to S1P within the LN parenchyma. Finally, we validated the pattern using a second cell type. Although expression of the S1P reporter transgene by lymphocytes was below our limit of detection, we were able to visualize the reporter in lymphocytes after transducing cells with retrovirus encoding the reporter construct. We transduced primary T cells with the S1P reporter and transferred them to wild-type hosts, and observed that T cells in the medullary cords internalized a larger proportion of S1PR1-GFP than those in the T zone (Supp. Fig. 1d).
We hypothesized that the S1P gradient between the medullary cords and the T zone was generated by SPNS2, an S1P transporter required in lymphatic endothelial cells to supply S1P to lymph26. To determine whether SPNS2 also generates S1P gradients within the LN, we used reporter bone marrow (BM) to generate BM chimeras in wild-type and their littermate Spns2f/f Lyve1-Cre (Spns2ΔLyve1) or Spns2f/fUBC-CreERT2 (Spns2ΔUBC-CreERT2) hosts. In Spns2ΔLyve1 chimeras, Spns2 was deleted primarily in lymphatic endothelial cells33. In tamoxifen-treated Spns2ΔUBC-CreERT2 chimeras, Spns2 was deleted more globally in radiation-resistant cells. While reporter cells in wild-type hosts reported comparatively high concentrations of S1P in the medullary cords, they reported low concentrations of S1P in the medullary cords of Spns2Δ hosts (Fig. 2a). Quantification revealed that the S1P gradient between the T zone and the medulla seen in wild-type animals was flattened (Fig. 2b). Taken together, our results suggest that cells are able to sense substantial S1P within the medullary cords, either due to direct secretion of S1P into the cords by lymphatic endothelial cells using SPNS2 or due to leak of S1P-containing lymph from the sinuses into the cords.
Figure 2. NK cells are displaced into the T zone of LN in Spns2ΔLyve1 mice.
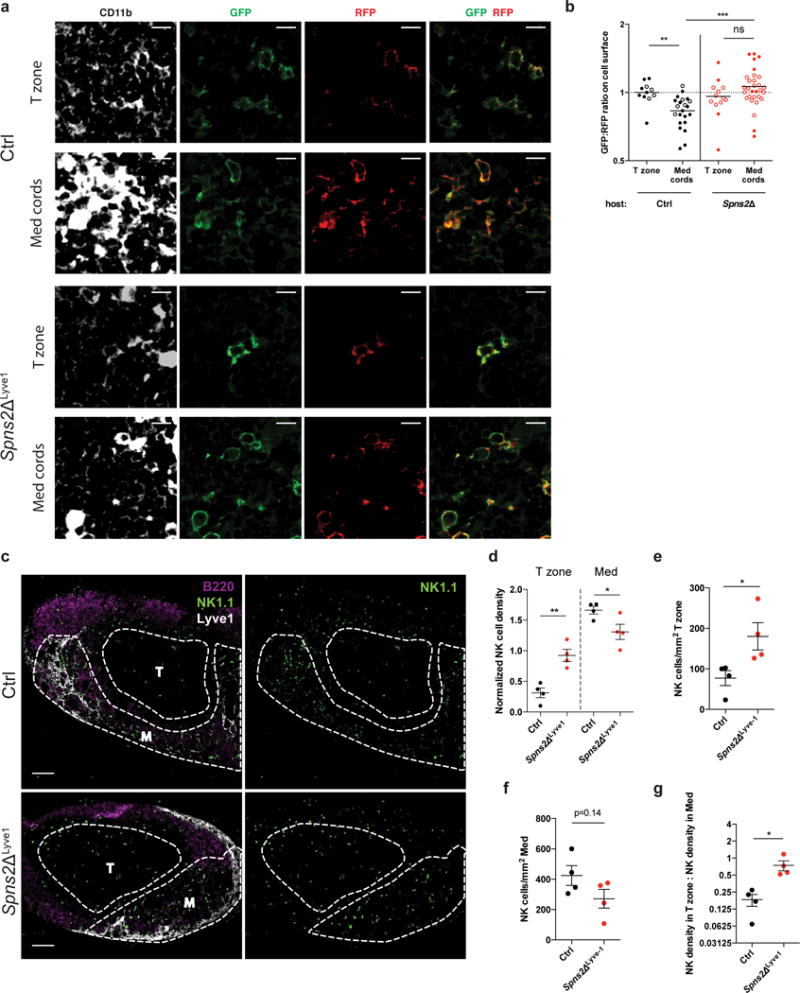
(a, b) Bone marrow chimeras were generated in Spns2ΔLyve1, Spns2ΔUBC-CreERT2, and their respective littermate control hosts using S1P reporter (Mx1-Cre) bone marrow. LN were analyzed 10 weeks after reconstitution.
(a) Representative images from Spns2ΔLyve1 LN sections stained with antibodies against GFP (green), RFP (red), CD11b (white), and Lyve1 and B220 (to define LN regions, not shown). Scale bar, 10 μm. Representative of 6 (littermate control) or 11 (Spns2Δ) images from 3 (littermate control) or 4 (Spns2Δ – 2 Spns2ΔLyve1 and 2 Spns2ΔUBC-CreERT2) chimeras in 2 experiments (image files: https://figshare.com/s/6d3bce9419b2829cb459).
(b) Quantification of S1P reporting in the T zone and medullary cords of Spns2ΔLyve1 (filled circles), Spns2ΔUBC-CreERT2 (open circles), or control reporter chimeras. Pixels positive for both RFP and CD11b were identified, marking the surface of reporter-expressing myeloid cells. The ratio of GFP to RFP for each CD11b+RFP+ pixel was calculated, and the ratio was averaged over a defined area. Each point on the graph represents the average ratio over an area of at least 6.9×102 square microns for the T zone and 3.2×103 square microns for the medullary cords. Horizontal lines indicate the mean. All ratios were normalized such that the average ratio for the T zone in control animals was 1.0.
(c) Representative confocal immunofluorescence images of LN sections from a Spns2ΔLyve1 mouse (bottom) and littermate control (top) showing localization of NK cells. M indicates medulla, T indicates T zone. NK cells were stained with anti-NK1.1 (green), B cells with anti-B220 (magenta), and lymphatic endothelial cells (LECs) and some sinus-lining macrophages with anti-Lyve1 (white). Single channel images of the NK1.1 stain are shown to the right. Scale bar, 100 μm. Representative of 4 Ctrl and 4 Spns2ΔLyve1 images from 4 pairs of mice in 4 experiments.
(d) NK cell densities in the T zone and medulla of Spns2ΔLyve1 and littermate control LN; for each section, densities were normalized by setting the NK density over the entire section (total number of NK cells in section/total section area) as 1. Each symbol represents one section; lines indicate the mean and SEM.
(e) Absolute NK cell density in T zone of LN sections from Spns2ΔLyve1 mice and littermate controls. Each symbol represents one section; lines indicate the mean and SEM.
(f) Absolute NK cell density in medulla of LN sections from Spns2ΔLyve1 mice and littermate controls. Each symbol represents one section; lines indicate the mean and SEM.
(g) Ratio of NK cell density in T zone to NK cell density in medulla. Each symbol represents the ratio for one LN section; lines indicate the mean and SEM.
Graphs compile 6–11 sections from 3–4 mice per condition analyzed in 2 experiments (b); 4 pairs of images from 4 pairs of mice in 4 experiments (d-g). *, p<0.05; **, p<0.001; ***, p<0.0001; ns, p>0.05, not significant. Fisher’s LSD test (b, d); unpaired Student’s 2-tailed t-test (e-g).
SPNS2 regulates NK cell positioning and IFN-γ response
Because the majority of LN NK cells are positioned in proximity to medullary sinus lymphatic endothelial cells, we hypothesized that S1P secreted by LEC draws NK cells near lymphatics (Supp. Fig. 1a). We evaluated NK cell positioning in LN of Spns2ΔLyve1 and littermate control animals. As expected, NK cells in control LNs were primarily in the medulla, outside the T zone. In Spns2ΔLyve1 LNs, by contrast, many NK cells could be seen mislocalized in the T zone (Fig. 2c, Supp. Fig. 2). Quantification confirmed an increased fraction of NK cells in the T zone, and a decreased fraction in the medulla of Spns2ΔLyve1 LN (Fig. 2d). This observation reflected an increase in the absolute density of NK cells in the T zone (Fig. 2e). There was a trend towards a decrease in the absolute density of NK cells in the medulla, which did not reach statistical significance (Fig. 2f). It is known that NK cells exit LN in response to lymph S1P using S1PR523, 37. In Spns2ΔLyve1 mice, both lymph S1P26 and medullary cord S1P were lost (Fig. 2a, b). Hence it is likely that the medulla simultaneously gains some NK cells that would otherwise have exited the LN and loses some NK cells to the T zone. Overall, the ratio of NK cell density in the T zone to NK cell density in the medulla increased substantially as compared to littermate control mice (Fig. 2g). These data suggest that S1P secretion by cells targeted by Lyve1-Cre, most likely LEC, plays an important role positioning NK cells in the LN periphery.
We next asked whether mislocalization of NK cells in Spns2ΔLyve1 mice affects the ability of NK cells to respond to pathogens entering the LN via afferent lymphatics. To address this, we assessed the early NK cell IFN-γ response, in which effects of mis-positioning within the LN could be isolated from effects of blocked exit from the LN due to loss of lymph S1P23, 37. We used a model of subcutaneous injection of Salmonella enterica Typhimurium, which induced a strong and reproducible NK cell response in wild-type mice. 2–4 hours after infection, the fraction of NK cells producing IFN-γ in Spns2ΔLyve1 mice was half that in littermate controls (Fig. 3a, b). Although we hypothesized that the reduced IFN-γ response was due to mislocalization, other possible explanations remained. We did not observe gross displacement of macrophages from the SCS or the medulla (Supp. Fig. 3a). We predicted that if the defect were due to NK cell mislocalization, NK cells isolated from LN of Spns2ΔLyve1 mice and stimulated in vitro should have a similar response to LN NK cells from littermate controls, and this was indeed the case (Fig. 3c, d). Furthermore, we predicted that the NK cells in Spns2ΔLyve1 mice that remain positioned in the medulla should respond similarly to NK cells in the medulla of controls. Using immunofluorescence staining of LN sections, we found that the fraction of NK cells producing IFN-γ was comparable in the medulla of Spns2ΔLyve1 mice and littermate controls, while NK cells in the T zone did not secrete IFN-γ in either Spns2ΔLyve1 or control animals (Fig. 3e, f). Thus, proper pre-positioning of NK cells within the medulla enables their early production of IFN-γ after infection.
Figure 3. Mislocalization causes defective IFN-γ production by LN NK cells after infection in Spns2ΔLyve1 mice.
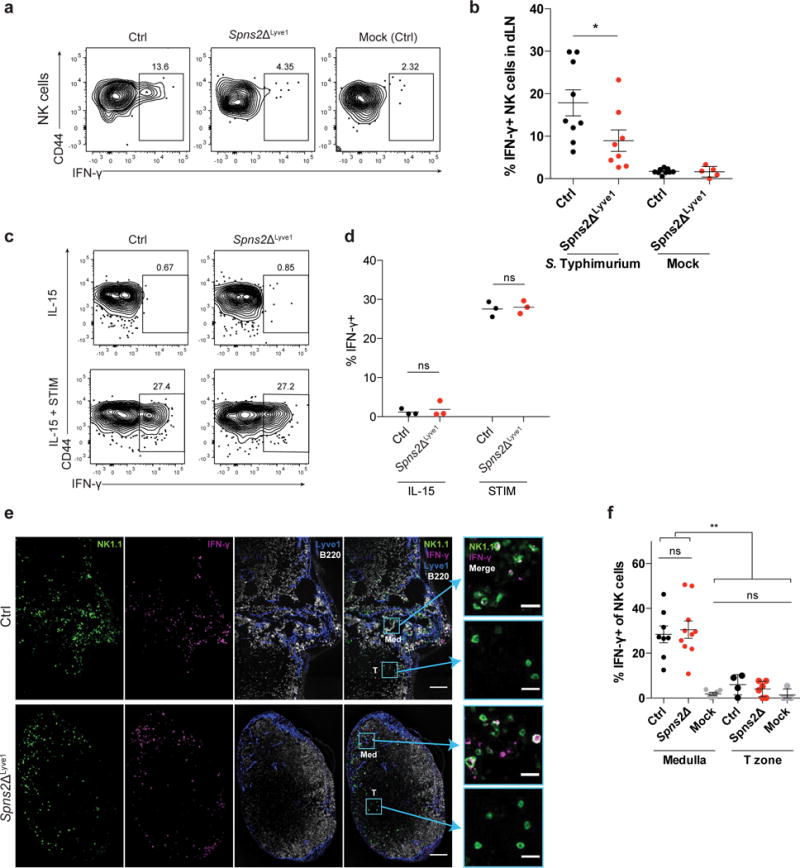
(a, b) 108 CFU Salmonella Typhimurium or PBS (mock) were injected subcutaneously into footpads of Spns2ΔLyve1 and littermate control mice. Draining LN were harvested 2–4h later and IFN-γ production was assessed by flow cytometry.
(a) Representative FACS plots of IFN-γ production, gated on NK1.1+CD3– cells.
(b) Percent IFN-γ positive cells among total NK1.1+CD3– cells. Each symbol represents one mouse; lines indicate the mean and SEM.
(c, d) NK cells isolated from LN of Spns2ΔLyve1 or littermate control animals were treated in vitro with IL-15 alone or IL-15, IL-12, and IL-18 (STIM) for 3 hours, and assessed for IFN-γ production by flow cytometry.
(c) Representative FACS plots of IFN-γ production, gated on NK1.1+CD3– cells.
(d) Percent IFN-γ positive cells among total NK1.1+CD3– cells. Each symbol represents the average of duplicate samples from one mouse; lines indicate the mean.
(e, f) 108 CFU Salmonella Typhimurium or PBS (mock) were injected subcutaneously in the footpad or intradermally into the ear of Spns2ΔLyve1 and littermate control mice. (Mock injections were in control mice.) Draining LN were harvested 2h later.
(e) Representative sections from draining LN stained with anti-NK1.1 (NK cells, green), anti- IFN-γ (magenta), anti-Lyve1 (LEC and some sinus-lining macrophages, blue), and anti-B220 (B cells, white). Med indicates medulla, T indicates T zone. Scale bars, 100 μm. Insets show NK1.1 and IFN-γ stains in the boxed areas of medulla and T zone. Scale bars, 20 μm. Representative of 8 (littermate control) or 10 (Spns2ΔLyve1) images from 4 sets of mice in 2 experiments.
(f) Percent IFN-γ+ cells among total NK cells within the medulla or T zone of draining LN. Mock-treated mice were controls. For the T zone, sections containing fewer than 10 NK cells within the T zone were excluded from analysis. Each symbol represents one section; lines indicate the mean and SEM.
Graphs compile 5–9 mice for each condition, analyzed in 6 experiments (b); 3 pairs of mice, analyzed in 2 experiments (d); or 6–10 sections from 4 mice for each condition, analyzed in 2 experiments (f). *, p<0.05; **, p<0.001; ns, not significant. Unpaired Student’s 2-tailed t-test (b, d); Fisher’s LSD test (f).
S1PR5 regulates NK cell positioning and IFN-γ response
Next, we asked which receptor enables NK cells to sense S1P secreted by LECs. NK cells use S1PR5 to exit LN23, 37, and by analyzing S1pr5−/− mice we addressed whether S1PR5 also regulates NK cell migration within the LN. As in Spns2ΔLyve1 animals, many S1pr5−/− NK cells were displaced into the T zone from the medulla (Fig. 4a–e). The finding that S1PR5 regulates NK cell positioning enabled us to ask whether the requirement for S1P signaling was cell-intrinsic, as it was also possible that NK cell displacement was due to the increased frequency of NK cells in LN of Spns2ΔLyve1 (Supp. Fig. 4a) and S1pr5−/− mice (23, 37, and Supp. Fig. 4b) or due to a requirement for S1P signaling in an intermediate cell type. We made mixed BM chimeras in CD45.1+ wild-type hosts using BM from GFP+ CD45.2+ wild-type mice and BM from GFPneg CD45.2+ S1pr5−/− mice or littermate controls. We found that a greater percentage of S1pr5−/− NK cells were mislocalized in the T zone of the mixed chimeras compared to GFP+ wild-type NK cells in the same mice, while control NK cells were positioned similarly to GFP+ wild-type NK cells in the same mice (Fig. 4f, g, Supp. Fig. 4c). These data demonstrate that NK cells have a cell-intrinsic requirement for S1PR5 for proper positioning within LNs.
Figure 4. NK cells are displaced into the T zone of LN in S1pr5−/− mice.
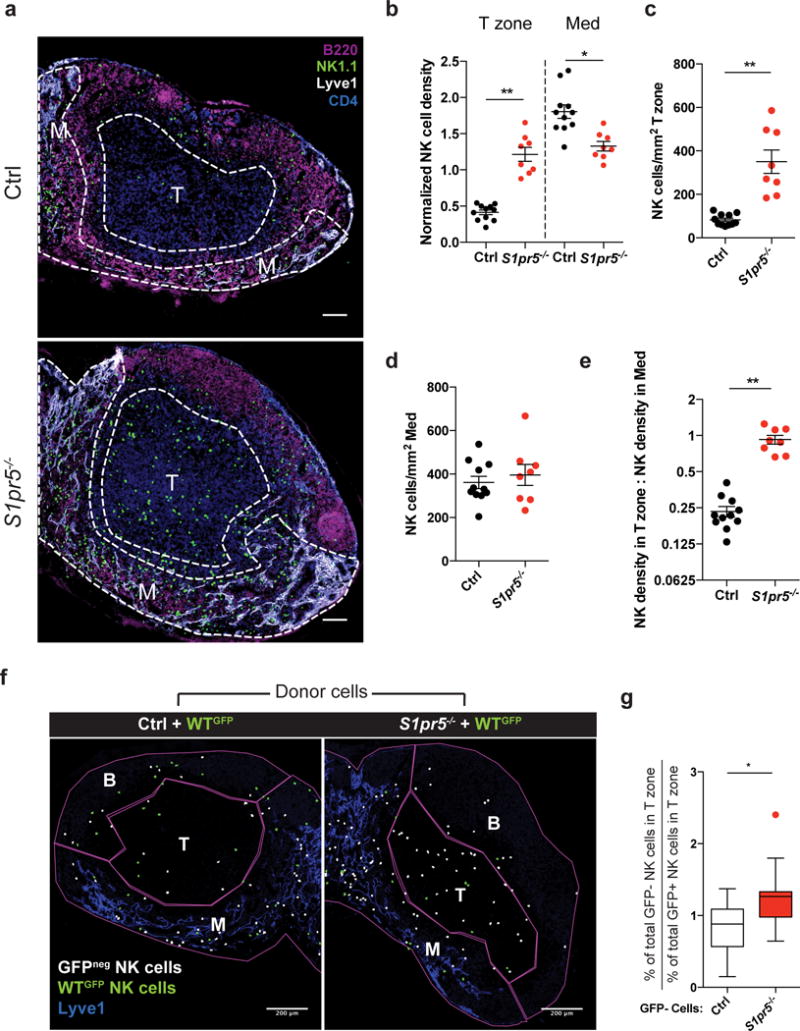
(a) Representative confocal immunofluorescence images of LN sections from an S1pr5−/− mouse (bottom) and littermate control (top) showing localization of NK cells. M indicates medulla, T indicates T zone. NK cells were stained with anti-NK1.1 (green), B cells with anti-B220 (magenta), LEC and some sinus-lining macrophages with anti-Lyve1 (white), and CD4 T cells with anti-CD4 (blue). Scale bars, 100 μm. Representative of 11 (littermate control) or 8 (S1pr5−/−) images from 4 (littermate control) or 3 (S1pr5−/−) mice in 3 experiments.
(b) NK cell densities in the T zone and medulla of S1pr5−/− and littermate control LN; for each section, densities were normalized by setting the NK density over the entire section (total number of NK cells in section/total section area) as 1. Each symbol represents one section; lines indicate the mean and SEM.
(c) Absolute NK cell density in T zone of LN sections from S1pr5−/− mice and littermate controls. Each symbol represents one section; lines indicate the mean and SEM.
(d) Absolute NK cell density in medulla of LN sections from S1pr5−/− mice and littermate controls. Each symbol represents one section; lines indicate the mean and SEM.
(e) Ratio of NK cell density in T zone to NK cell density in medulla. Each symbol represents the ratio for one LN section; lines indicate the mean and SEM.
(f–h) Mixed bone marrow chimeras (mBMCs) were generated in WT CD45.1+ hosts using a 1:1 ratio of S1pr5−/− GFPneg CD45.2+ bone marrow and WT GFP+ CD45.2+ bone marrow (average LN NK cell chimerism: 50% CD45.2+ and 34% GFP+). Control mBMCs were generated using a 1:1 ratio of S1pr5+/+ GFPneg CD45.2+ littermate control bone marrow and WT GFP+ CD45.2+ bone marrow (average LN NK cell chimerism: 56% CD45.2+ and 25% GFP+).
(f) ImageJ analysis was used to identify and pseudocolor GFPneg (white) and GFP+ (green) NK cells in LN sections from chimeric mice; see original images in Supp. Fig. 4c. LN sections were stained with antibodies against Lyve1, B220, and NK1.1. (The analysis did not distinguish between donor GFPneg NK cells and any remaining host GFPneg NK cells, biasing against detecting an effect of S1PR5-deficiency.) M, Medulla; T, T zone; B, includes B cell follicles, interfollicular regions, and T-B boundary. Scale bars, 200 μm. Representative of 17 (littermate control) or 12 (S1pr5−/−) images from 3 pairs of mice in 3 experiments.
(g) Ratio of (the percent GFPneg NK cells in the T zone of total GFPneg NK cells) to (the percent WT GFP+ NK cells in the T zone of total WT GFP+ NK cells) in S1pr5−/− mBMCs and S1pr5+/+ mBMCs. Center line represents the median, box limits extend from the 25th to 75th percentile, Tukey whiskers extend to 1.5 × IQR beyond the box, and symbol represents an outlier.
Graphs compile 8–11 sections from 3–4 mice per genotype, analyzed in 3 experiments (b-e); and 12–17 sections from 3 pairs of chimeras with 2 sets of donors, analyzed in 3 experiments (g). *, p<0.01; **, p<0.0001. Fisher’s LSD test (b); unpaired Student’s 2-tailed t-test (c-e, g).
We then addressed whether S1PR5 expression by NK cells regulates the IFN-γ response to bacterial infection, using the same model of subcutaneous Salmonella infection. We found that, similar to NK cells in Spns2ΔLyve1 mice, a reduced fraction of S1pr5−/− NK cells secreted IFN-γ 2 hours after bacterial challenge (Fig. 5a, b). Infection of mixed BM chimeras demonstrated that the requirement for S1PR5 for rapid IFN-γ secretion is cell-intrinsic. The ratio of the percent IFN-γ-producing S1pr5−/− NK cells of total S1pr5−/− NK cells, to the percent IFN-γ-producing wild-type NK cells of total wild-type NK cells in the same mouse was consistently lower than the ratio of the percent IFN-γ-producing S1pr5+/+ to wild-type NK cells (Fig. 5c). The ratio of the percent IFN-γ-producing S1pr5+/+ to wild-type NK cells was reproducibly higher than 1:1. This observation held in mixed chimeras made with bone marrow from 7 pairs of S1pr5−/− and S1pr5+/+ littermate donors and three different B6-congenic wild-type donor mouse lines. This finding suggests that the background of the S1pr5 mouse strain favors slightly increased IFN-γ secretion compared to the B6-congenic lines.
Figure 5. Mislocalization causes defective IFN-γ production by S1pr5−/− LN NK cells after infection.
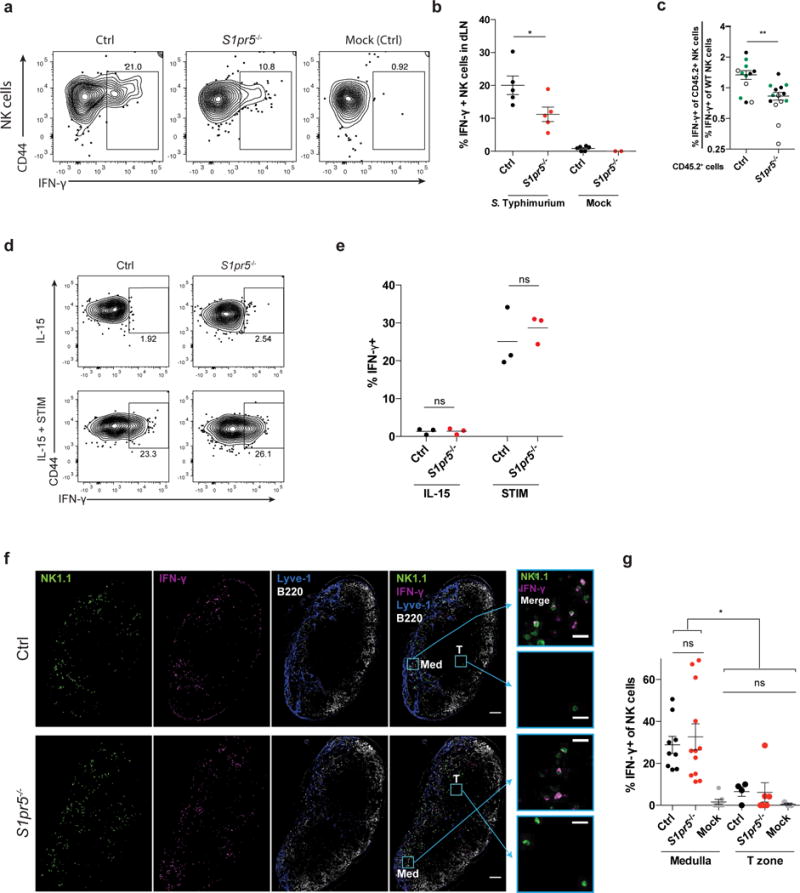
(a, b) 108 CFU Salmonella Typhimurium or PBS (mock) were injected subcutaneously into footpads of S1pr5−/− and littermate control mice. Draining LN were harvested 2h later and IFN-γ production was assessed by flow cytometry.
(a) Representative FACS plots of IFN-γ production, gated on NK1.1+CD3– cells.
(b) Percent IFN-γ positive cells among total NK1.1+CD3– cells. Each symbol represents one mouse; lines indicate the mean.
(c) Mixed bone marrow chimeras (mBMCs) were generated as in Fig. 4f using GFP+ (
 ), CD45.1+ (○), or CD45.1+CD45.2+ (●) mice as WT donors. Mixed chimeras were injected subcutaneously with 108 CFU Salmonella Typhimurium in the footpad. Draining LN were harvested 2 hours later for analysis by flow cytometry. Graph shows the ratio of the [percent IFN-γ+ of CD45.2+ experimental NK cells] to the [percent IFN-γ+ of WT NK cells] in S1pr5−/− mBMCs and S1pr5+/+ control mBMCs. Each symbol represents one mouse; lines indicate the mean and SEM.
), CD45.1+ (○), or CD45.1+CD45.2+ (●) mice as WT donors. Mixed chimeras were injected subcutaneously with 108 CFU Salmonella Typhimurium in the footpad. Draining LN were harvested 2 hours later for analysis by flow cytometry. Graph shows the ratio of the [percent IFN-γ+ of CD45.2+ experimental NK cells] to the [percent IFN-γ+ of WT NK cells] in S1pr5−/− mBMCs and S1pr5+/+ control mBMCs. Each symbol represents one mouse; lines indicate the mean and SEM.
(d, e) NK cells isolated from LN of S1pr5−/− or littermate control animals were treated in vitro with IL-15 alone or IL-15, IL-12, and IL-18 (STIM) for 3 hours, and assessed for IFN-γ production by flow cytometry.
(d) Representative FACS plots of IFN-γ production, gated on NK1.1+CD3– cells.
(e) Percent IFN-γ positive cells among total NK1.1+CD3– cells. Each symbol represents the average of duplicate samples from one mouse; lines indicate the mean.
(f, g) 108 CFU Salmonella Typhimurium (ST) or PBS (mock) were injected subcutaneously in footpads of S1pr5−/− and littermate control mice. Draining LN were harvested 2h later for analysis.
(f) Representative sections from draining LN stained with anti-NK1.1 (NK cells, green), anti- IFN-γ (magenta), anti-Lyve1 (LEC and some sinus-lining macrophages, blue), and anti-B220 (B cells, white). Med indicates medulla, T indicates T zone. Scale bars, 100 μm. Insets show NK1.1 and IFN-γ stains in the boxed areas of medulla and T zone. Scale bars, 20 μm. Representative of 9 (littermate control) or 12 (S1pr5−/−) images from 3 pairs of mice in 2 experiments.
(g) Percent IFN-γ+ among total NK cells within the medulla or T zone of draining LN. Mock-treated mice were controls. For the T zone, sections containing fewer than 10 NK cells within the T zone were excluded from analysis. Each symbol represents one section; lines indicate the mean and SEM.
Graphs compile 2–5 mice per condition, analyzed in 4 experiments (b); 12–14 mice per genotype from 7 sets of donors, analyzed in 9 experiments (c); 3 pairs of mice, analyzed in 2 experiments (e); or 3–12 sections from 3 mice per condition analyzed in 2 experiments (g). *, p<0.05; **, p<0.01; ns, p>0.05, not significant. Unpaired Student’s 2-tailed t-test (b, c, e); Fisher’s LSD test (g).
We then asked whether the defect in IFN-γ secretion could be attributed to mislocalization. We found that, like NK cells from Spns2ΔLyve1 mice, S1pr5−/− NK cells secreted IFN-γ similarly to littermate control NK cells in response to stimulation in vitro (Fig. 5d, e). Furthermore, the S1pr5−/− NK cells that remained properly localized in the medulla secreted IFN-γ similarly to littermate control NK cells (Fig. 5f, g). These data indicated that displacement into the T zone is responsible for the reduced IFN-γ response by S1pr5−/− NK cells.
CXCR4 regulates NK cell positioning and IFN-γ response
While NK cell distribution shifted from the medulla to the T zone upon loss of SPNS2-S1P-S1PR5 signaling, a substantial fraction of NK cells remained in the medulla. This finding led us to ask whether other cues are required to position NK cells in the medulla. The chemokine CXCL12 is abundant in the LN medullary cords, and the chemokine receptor CXCR4, which binds CXCL12, positions plasma cells in this area38, 39. Mice expressing a mutant CXCR4 that cannot be internalized have elevated numbers of LN NK cells40, suggesting that CXCR4 may antagonize NK cell exit from LN, and we hypothesized that CXCR4 signaling may also contribute to NK cell positioning within the LN.
We confirmed that LN NK cells express CXCR4 (Fig. 6a) and found that treatment with the specific CXCR4 antagonist AMD310041, 42 for two hours caused NK cells to move into the T zone (Fig. 6b–f). The increases in the fraction of NK cells in the T zone and the ratio of T zone to medulla NK cell density (Fig. 6c, f) were similar to what we observed upon loss of S1pr5 signaling. However, the increase in the absolute number of NK cells in the T zone was smaller and there was a significant reduction of NK cells in the medulla (Fig. 6d, e). This finding is consistent with our finding that unlike loss of SPNS2 or S1PR5, inhibition of CXCR4 signaling by AMD3100 treatment did not cause NK cells to accumulate in LN (Supp. Fig. 5a). We found the same mislocalization in Cxcr4f/fMx1-Cre (Cxcr4ΔMx1) animals treated with poly I:C to induce deletion of Cxcr4 (Fig. 6g–k)43. These data demonstrated that CXCR4 is required for proper NK cell positioning within the medulla.
Figure 6. NK cells are displaced into the T zone of LN upon CXCR4 antagonism.
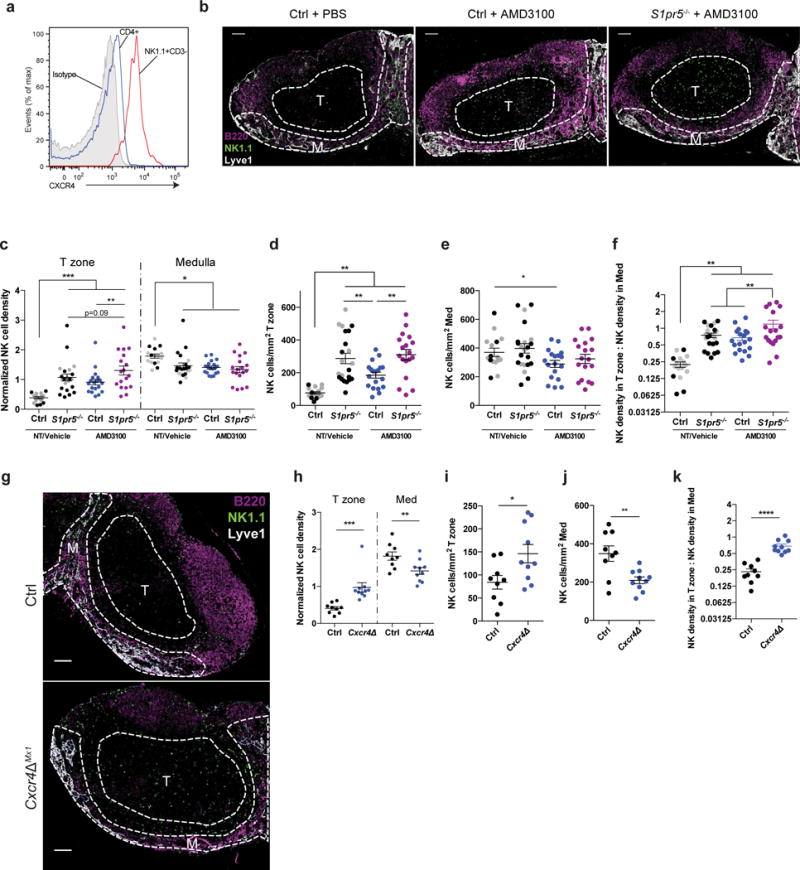
(a) Surface CXCR4 staining on WT LN NK cells (NK1.1+CD3–) and CD4+ T cells. Representative of 3 mice in 3 experiments.
(b–f) Mice were injected i.p. with 5 mg/kg of the CXCR4 inhibitor AMD3100 or PBS, and analyzed 2 hours later.
(b) Representative confocal immunofluorescence images of LN sections from a PBS-treated littermate control, an AMD3100-treated littermate control, and an AMD3100-treated S1pr5−/− mouse showing localization of NK cells. M indicates medulla, T indicates T zone. NK cells were stained with anti-NK1.1 (green), B cells with anti-B220 (magenta), LEC and some sinus-lining macrophages with anti-Lyve1 (white). Scale bars, 100 μm. Representative of 16 (PBS-treated control), 19 (AMD3100-treated control), or 18 (AMD3100-treated S1pr5−/−) images from 5 sets of mice in 4 experiments.
(c) NK cell densities in the T zone and medulla of LN from S1pr5−/− and littermate control mice treated with AMD3100 or PBS; for each section, densities were normalized by setting the NK density over the entire section (total number of NK cells in section/total section area) as 1. Gray symbols were from untreated control and S1pr5−/− mice previously shown in Fig. 4b, and are shown here again for completeness. Each symbol represents one section; lines indicate the mean and SEM.
(d) Absolute NK cell density in T zone of LN sections. Gray symbols were from untreated control and S1pr5−/− mice previously shown in Fig. 4c, and are shown here again for completeness. Each symbol represents one section; lines indicate the mean and SEM.
(e) Absolute NK cell density in medulla of LN sections. Gray symbols were from untreated control and S1pr5−/− mice previously shown in Fig. 4d, and are shown here again for completeness. Each symbol represents one section; lines indicate the mean and SEM.
(f) Ratio of NK cell density in T zone to NK cell density in medulla. Gray symbols were from untreated control and S1pr5−/− mice previously shown in Fig. 4e, and are shown here again for completeness. Each symbol represents one LN section; lines indicate the mean and SEM.
(g) Representative confocal immunofluorescence images of LN sections from a Cxcr4ΔMx1 mouse and littermate control showing localization of NK cells. M indicates medulla, T indicates T zone. NK cells were stained with anti-NK1.1 (green), B cells with anti-B220 (magenta), LEC and some sinus-lining macrophages with anti-Lyve1 (blue). Scale bars, 100 μm. Representative of 9 (littermate control) or 10 (Cxcr4ΔMx1) images from 2 pairs of mice in 2 experiments.
(h) NK cell densities in the T zone and medulla of Cxcr4ΔMx1 and littermate control LN; for each section, densities were normalized by setting the NK density over the entire section (total number of NK cells in section/total section area) as 1. Each symbol represents one section; lines indicate the mean and SEM.
(i) Absolute NK cell density in T zone of LN sections from Cxcr4ΔMx1 mice and littermate controls. Each symbol represents one section; lines indicate the mean and SEM.
(j) Absolute NK cell density in medulla of LN sections from Cxcr4ΔMx1 mice and littermate controls. Each symbol represents one section; lines indicate the mean and SEM.
(k) Ratio of NK cell density in T zone to NK cell density in medulla. Each symbol represents one LN section; lines indicate the mean and SEM.
Graphs compile 16–22 sections from 5–7 mice per genotype, analyzed in 6 experiments (c-f); or 9–10 sections from 2 mice per genotype, analyzed in 2 experiments (h-k). *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001. Fisher’s LSD test (c-f, h); unpaired Student’s 2-tailed t-test (i-k).
As in Spns2ΔLyve1 mice and S1pr5−/− mice, a substantial fraction of NK cells remained in the medulla after CXCR4 inhibition. We therefore asked how concomitant loss of CXCR4 and S1PR5 signaling would affect NK cell distribution, by treating S1pr5−/− mice with AMD3100. We found a small increase in mislocalization into the T zone compared to either PBS-treated S1pr5−/− or AMD3100-treated control mice (Fig. 6b–f), reflected in an increased fraction of NK cells in the T zone (Fig. 6c) and an increased ratio of T zone to medulla NK cell density (Fig. 6f). These results demonstrated that NK cells became evenly distributed between the T zone and medulla upon loss of both receptors; they never fully left the medulla.
Like the loss of SPNS2 and/or S1PR5, CXCR4 antagonism reduced the early NK cell IFN-γ response to Salmonella infection (Fig. 7a). Again, our data suggest that the defect is attributable to NK cell mislocalization, as AMD3100 did not impact NK cells’ ability to secrete IFN-γ in vitro (Fig. 7b), and the NK cells that remained in the medulla of AMD3100-treated animals produced IFN-γ comparably to NK cells in vehicle-treated controls (Fig. 7c, d). We also saw a trend towards further reduced IFN-γ secretion upon loss of both S1PR5 and CXCR4 (Fig. 7a). We observed that a subset of NK cells expressed CXCR3 even in homeostasis, which we speculate may also contribute to positioning NK cells in the LN periphery upon upregulation of CXCR3 ligands after infection (Supp. Fig. 5b).
Figure 7. Mislocalization causes defective IFN-γ production by LN NK cells after infection in mice treated with a CXCR4 antagonist.
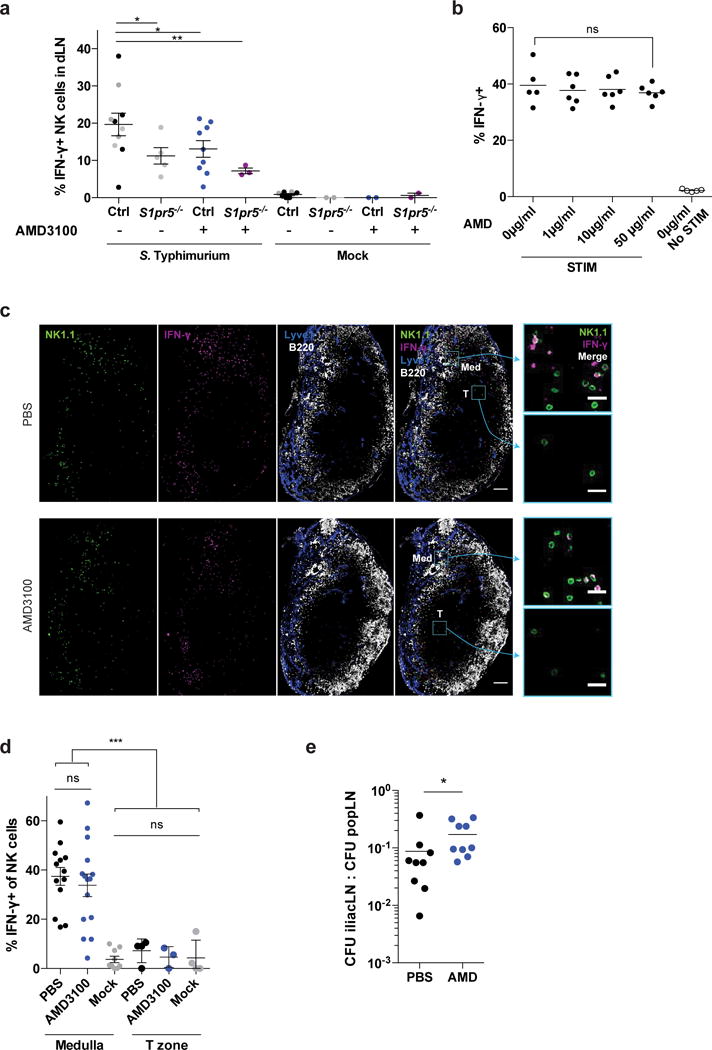
(a) 108 CFU Salmonella Typhimurium or PBS (mock) were injected subcutaneously in footpads of AMD3100-treated or PBS-treated S1pr5−/− or littermate control mice. Animals were treated i.p. with 5 mg/kg AMD3100 or PBS 2h prior to and at the time of infection. Draining LN were harvested 2h after infection and IFN-γ production was assessed by flow cytometry. Percent IFN-γ positive cells among total NK1.1+CD3– cells is shown. Gray symbols were from untreated S1pr5−/− or littermate control mice previously shown in Fig. 5b and are shown here again for completeness. Each symbol represents one mouse; lines indicate the mean and SEM.
(b) NK cells isolated from LN of WT mice were incubated in media with the indicated concentrations of AMD3100 for 45m at 37°C. These concentrations of AMD3100 inhibited NK cell migration towards CXCL12 in chemotaxis assays in a dose-dependent manner (not shown). IL-15 alone or IL-15, IL-12, and IL-18 (STIM) were then added to the media. 3 hours later, cells were analyzed by flow cytometry. Percent IFN-γ positive cells among total NK1.1+CD3– cells is shown. Each symbol represents the average of duplicate samples from one mouse; lines indicate the mean.
(c, d) 108 CFU Salmonella Typhimurium (ST) or PBS (mock) were injected subcutaneously in footpads of AMD3100-treated and PBS-treated (control) WT mice. Animals were treated with 5 mg/kg AMD3100 i.p. or PBS 2h prior to and at the time of infection. Draining LN were harvested 2h after infection.
(c) Representative sections from draining LN stained with anti-NK1.1 (NK cells, green), anti- IFN-γ (magenta), anti-Lyve1 (LEC and some sinus-lining macrophages, blue), and anti-B220 (B cells, white). Med indicates medulla, T indicates T zone. Scale bar, 100 μm. Insets show NK1.1 and IFN-γ stains in the boxed areas of medulla and T zone. Scale bar, 20 μm. Representative of 13 (PBS control) or 15 (AMD3100-treated) images from 4 pairs of mice in 2 experiments.
(d) Percent IFN-γ+ among total NK cells within the medulla or T zone of draining LN. Mock-treated mice were controls. For the T zone, sections containing fewer than 10 NK cells within the T zone were excluded from analysis. Each symbol represents one section; lines indicate the mean and SEM.
(e) 108 CFU Salmonella Typhimurium (ST) were injected subcutaneously into footpads of AMD3100-treated and PBS-treated mice. 0.125mg AMD3100 or PBS was injected 2h before, at the time of, and 3h after ST injection. The popliteal LN (primary draining LN) and iliac LN (downstream of the popliteal LN) were harvested 4h after ST injection, and bacterial colony forming units (CFU) were counted.
Graphs compile 2–9 mice per condition analyzed in 5 experiments (a); 5–6 mice analyzed in 2 experiments (b); 9–15 sections from 4 mice of each condition analyzed in 2 experiments (d), or 9 mice per condition analyzed in 3 experiments (e). *, p<0.05; **, p<0.01; ***, p<0.001; ns, not significant. Fisher’s LSD test (a, d); unpaired Student’s 2-tailed t-test (b), paired Student’s 2-tailed t-test (e).
Finally, we addressed whether NK cell redistribution had an effect on bacterial control. We expected that reduced IFN-γ secretion by NK cells near the medullary sinuses would impair the ability of sinus-lining macrophages to contain infection in the draining LN (in this case the popliteal LN), and enable infection to spread to the downstream LN (in this case the iliac LN). We saw little effect on the spread of Salmonella after 4 hours in Spns2ΔLyve1 mice and S1pr5−/− mice (not shown), as expected because there was little change in the density of NK cells in the medulla. In AMD3100-treated mice, however, where there was a statistically significant decrease in density of NK cells in the medulla, we saw a slight increase in the ratio of Salmonella CFU’s in the iliac to the popliteal LN (Fig. 7e). This result suggested that positioning of NK cells in the medulla contributes to bacterial containment within the draining LN.
Discussion
Precise positioning of immune cells within the LN is critical for host defense, but the factors required are still being defined. Using S1P reporter mice, we mapped LN gradients of S1P and found that cells sensed higher levels of S1P in the medullary cords than the T zone. In mice deficient for SPNS2 in lymphatic endothelial cells, however, extracellular S1P concentrations in the medullary cords were decreased, flattening the gradient. Notably, our findings are in contrast to suggestions that Spns2−/− mice have increased levels of S1P in lymph node interstitial fluid44, highlighting the importance of measuring signaling-available S1P as opposed to total tissue lipid. Consistent with our reporter data, both SPNS2 on cells targeted by Lyve1-Cre, most likely lymphatic endothelial cells, and S1PR5 on NK cells were required to maintain proper NK cell localization in the medulla. Additionally, CXCR4 was required for NK cell positioning. Our work suggests that mislocalization was directly responsible for the early NK cell defect in responding to bacterial infection in Spns2ΔLyve1, S1pr5−/−, and CXCR4 antagonist-treated animals.
Surprisingly, we found little difference in S1P sensing by macrophages in the medullary cords versus macrophages in regions of the medullary sinuses packed with cells. We hypothesize that the dense network of macrophages within the medullary sinuses rapidly degrades S1P. We further speculate that S1P concentrations may be higher in the macrophage-sparse cortical sinuses and macrophage-sparse areas of the medullary sinuses, and that “hotspots” that have been observed for T cell exit in the medulla may be in areas of the sinuses with lower macrophage density45. Recent studies have suggested the hypothesis that, in addition to the medulla, other areas of the LN contain fine gradients of S1P that regulate cell positioning. For example, S1PR2, which antagonizes migration toward S1P, is required to retain germinal center B cells and follicular T helper cells in LN germinal centers. The edges of B cell follicles are thus posited to have higher concentrations of S1P than germinal centers46, 47. Here, we have begun to map LN S1P gradients, and it will be important to further dissect the gradients within this tissue.
S1PR5 has previously been shown to direct NK cell exit from LN into lymph, an observation which raises the question of how S1P signaling can simultaneously promote NK cell egress and localization in the medulla23, 37. Unlike S1PR1, which guides T and B cell egress, S1PR5 is resistant to ligand-induced internalization37. Hence S1PR5 may position NK cells in the medulla, yet remain sensitive to the S1P in lymph and enable NK cells to exit the LN when they encounter a permissive site in the medullary or cortical sinuses. Alternatively, S1PR5’s principal role in egress may be to guide NK cells to areas near lymphatics, where they patrol the LN parenchyma and ultimately exit via a random walk.
Upon tissue infection, NK cells are recruited to LN of wild-type mice and become concentrated near the subcapsular sinus within 16–24 hours19, 20. Interaction with collagen fibers, perhaps exposed initially due to inflammation-induced damage and later to LN expansion, is one cue that pulls NK cells to the subcapsular sinus19. We have observed that even in the absence of S1PR5 or CXCR4 signaling, NK cells began to move towards the edges of the T zone within 2 hours of challenge. It is not clear what cues induce this early relocalization, although we speculate that CXCR3 ligands are involved. Upon infection, CXCR3 ligands are upregulated in the LN periphery10, 11, 13, and we found that a subset of LN NK cells expressed CXCR3 at homeostasis. It will be important to delineate the sequential signals that regulate NK cell recruitment to lymphatics in inflammation.
More generally, the LN periphery is increasingly recognized to be an important site of many immune functions, from pathogen containment to helper T cell differentiation. Yet we know little about the organization of cells within this region, and whether and how different types of responses may be spatially segregated from each other. A recent study showed that CCR6, which is regulated by RORγt, directed the localization of a network of IL17-producing innate-like lymphocytes to the subcapsular sinus49. We found that S1PR5, which is regulated by T-bet, directed the localization of IFN-γ-producing NK cells to the medullary sinuses. These studies suggest that there may be different territories for different types of response, at least early after infection. It will be important to investigate whether these differences contribute to functional differences between macrophages in distinct locations4, and whether these early differences regulate the localization of differentiating helper T cell subsets. The knowledge that S1P can position cells in the medulla will be valuable as the LN periphery is better defined spatially and functionally to determine how different regions contribute to host defense.
METHODS
Mice
Mx1-Cre30, UBC-CreERT231, Cd69−/−50, UBC-GFP51, S1P reporter28, Spns2f/f26, Lyve1-Cre33, S1pr5−/−37 and Cxcr4f/f43 mice have been described. CD45.1+ and CD45.2+ C57BL/6 congenic mice were obtained from Charles River Laboratories. All mice were either C57BL/6 or a B6-129 mix. Mice were used at 6–40 weeks of age, and female and male mice were used depending on availability (we observed no sex differences in any of the parameters measured). Mice were housed in specific pathogen–free conditions at the Skirball Institute Animal Facility. All animal experiments were performed in accordance with protocols approved by the New York University Institutional Animal Care and Use Committee.
Mouse treatments
For induction of Cre-mediated deletion of floxed gene alleles in Mx1-Cre S1P reporter mice, 3- to 5-day-old mice received a single intraperitoneal injection of 70 μl polyI:C (GE Healthcare) at a concentration of 2 mg/ml in PBS. All mice in each litter were treated identically. For induction of Mx1-Cre in Cxcr4f/fMx1-Cre mice, adult mice received intraperitoneal injections of 100 μl polyI:C at a concentration of 2 mg/ml in PBS once every other day for a total of 3 injections. LNs were harvested at least at least 9 days after the last treatment. For induction of UBC-CreERT2, adult mice received daily intraperitoneal injections of 200 μg tamoxifen for 5 days, and mice were analyzed at least 5 days after the final tamoxifen dose.
NIBR-0213, provided by Novartis, was used to inhibit S1PR1 internalization in S1P reporter mice. Dessicated powder was resuspended at 3 mg/ml in 30% polyethylene glycol 200 (TCI America) in PBS36. Mixture was heated to 45° C for 1 h to dissolve the powder. Animals were injected once daily with 250 μl (30 mg/kg) for 8 days. LNs were collected 3–5 h after the last treatment.
FTY720 (Cayman Chemical) was used to induce S1PR1 internalization in S1P reporter mice. Mice were treated with FTY720 at a dose of 1 mg per kg body weight (mg/kg) intraperitoneally 5 h before harvest of LNs.
Bone marrow chimeras
Recipients were lethally irradiated by two doses of 6.6 Gray separated by 3 h, followed by intravenous transfer of 2 × 106 donor bone marrow cells. Chimeras were analyzed at least 6 weeks after transplantation.
Infections
Salmonella enterica Typhimurium (ST, SL1344) was provided by K. Cadwell. 108 CFU ST was diluted in PBS and injected into either the footpad (30 μl) or the ear (10 μl). LN were harvested 2–4 h after infection for flow cytometry, confocal imaging, or CFU measurement.
Retroviral transduction of primary T cells
The coding region of the S1P reporter was cloned into the pMXs retroviral vector. pCL-Eco Retrovirus packaging vector (Novus Biologicals) and HEK293T cells were used for generation of retrovirus. T cells were purified from lymph nodes and spleen by negative selection with magnetic beads (EasySep™ Mouse Biotin Positive Selection Kit from StemCell technologies or MagniSort™ Streptavidin Negative Selection Beads from eBioscience) using biotinylated antibodies against CD19 (6D5), CD11c (N418), and NKp46 (29A1.4). Purified T cells were resuspended in activation media containing 1 μg/ml anti-CD28 (37.51) and 0.25 μg/ml anti-CD3 (145-2C11), and plated on anti-hamster IgG (MP Biomedicals) coated plates overnight. T cells were transduced the following day with the retrovirus encoding the S1P reporter by spinning at 1258g (no brake), 90 min, 32° C with polybrene (8 μg/ml); incubated at 37°C overnight with new activation media; and transduced a second time the following day. T cells were rested for two days without anti-CD28 and anti-CD3, and transferred i.v. into wild-type mice. LN from recipient mice were analyzed the following day by confocal microscopy.
In vitro stimulation of NK cells
A single-cell suspension was obtained from pooled peripheral and mesenteric LNs by mechanical disruption and filtration through a 70 μm nylon cell strainer. NK cells were enriched by negative selection using the EasySep Mouse Biotin Positive Selection Kit (Stem Cell 18556) with biotinylated antibodies against CD4 (RM4-5), CD8 (53-6.7), and CD19 (6D5). Enriched cells were resuspended at 106/ml in media containing IL-15 (10 ng/ml, Peprotech) or stimulation media containing IL-15 (10 ng/ml, Peprotech), IL-18 (50 ng/ml, Medical and Biological Labs Co), and IL-12 (10 ng/ml, Peptrotech). All media contained GolgiPlug (BD Biosciences). Cells were incubated for 3 h at 37° C before staining and analysis by flow cytometry. In experiments utilizing AMD3100 (Fig. 7b), cells were first incubated in the indicated concentrations of AMD3100 for 45 min before stimulation.
Antibodies
The following fluorochrome conjugated antibodies were used: anti-CD4 (RMA 4–5), anti-CD8 (53-6.7), anti-CD19 (6D5), anti-CD45.1 (A20), anti-CD45.2 (104), anti-B220/CD45RO (RA3-6B2), anti-CD169 (3D6.112), anti-NK1.1 (PK136), anti-CD3 (145-2C11), anti-NKp46 (29A1.4), anti-IFN-γ (XMG1.2), anti-CXCR4 (L276F12), anti-CXCR3 (CXCR3-173), and streptavidin (all from BioLegend). Fluorochrome-conjugated anti-Lyve1 (ALY7) was from eBioscience. Reporter antibodies are detailed below. Zombie Aqua™ fixable viability kit (Biolegend) was used to exclude dead cells in flow cytometry experiments.
Confocal microscopy
Lymph nodes were harvested, fixed in 4% PFA for 1 h at 22–25° C with gentle shaking, dehydrated by sucrose gradient (15% sucrose in PBS >1h at 4° C, then 30% sucrose at 4° C overnight), embedded in OCT (Sakura), and snap-frozen in dry ice-cooled 2-methylbutane. 8–12 μm sections were cut, fixed with ice cold acetone for 10 min at −20°C, and air dried. All staining was performed at 22–25 °C. Sections were permeabilized for 10–30 min with 0.5% Triton X-100 in PBS, then incubated for 10–30 min in block buffer consisting of 0.1% Triton X-100 in PBS with 4% normal rat serum, 4% normal mouse serum, and 10 μg/ml anti-CD16/32 (Biolegend, clone 93). Stains were done with antibodies diluted in the block buffer; an Avidin/Biotin blocking kit (Vector Laboratories) was used with biotinylated antibodies. Slides were mounted with G-Fluoromount (Southern Biotech) and visualized using a Zeiss 710 inverted confocal microscope with a 25× or 40× oil-immersion objective and ZEN 2010 software. Images were processed with ImageJ v1.49. For all direct comparisons samples were stained and imaged the same day with the same settings.
For reporter LN analysis (Fig. 1, 2, Supp. Fig. 1), the above methods were utilized with the following changes: Mice were lethally anesthetized with ketamine and xylazine and fix-perfused with 1% PFA in PBS prior to downstream processing. Sections were blocked with 4% normal goat serum, 4% normal donkey serum, and 10 μg/ml anti-CD16/32 (Biolegend, clone 93) instead of normal mouse and normal rat serum. Slides were incubated for 1 h with polyclonal chicken anti-GFP (Abcam, ab13970, diluted 1:250) and polyclonal rabbit anti-TagRFP (Evrogen, AB234, diluted 1:250), washed in PBS, then incubated with polyclonal Alexa Fluor 488–conjugated goat anti-chicken (Jackson Immunoresearch, 103-545-155, diluted 1:1000) and polyclonal Alexa Fluor 647–conjugated donkey anti-rabbit (Jackson Immunoresearch, 711-605-152, diluted 1:400) for 1 h.
Flow cytometry
A single-cell suspension was obtained from lymphoid organs by mechanical disruption and filtration through a 70 μm nylon cell strainer. Cells were stained and analyzed on an LSRII (BD). Data were analyzed with FlowJo software (TreeStar).
CFU counting
LN were disaggregated with frosted glass slides in 0.1% Triton-X in PBS. Serial dilutions of the sample were made (undiluted to 1/10,000) and 10 μl of each dilution was plated in duplicate on LB plates with 100 mg/ml streptomycin.
Statistics
Samples were compared by Student’s two-tailed t-test paired where appropriate, Tukey multiple comparisons test, or uncorected Fisher’s LSD test, as noted in figure legends.
Code availability
All image analysis and programming was done with ImageJ software v1.49 (NIH). The colocalization programs are provided as Supplementary Text.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supplementary Material
Acknowledgments
We thank members of the Schwab lab for valuable discussions; K. Cadwell (New York University School of Medicine) for Salmonella enterica Typhimurium; K. Manova, S. Fujisawa, and Y. Romin (Memorial Sloan Kettering Cancer Center Molecular Cytology Core Facility) for assistance with microscopy and image analysis; and B. Breart (Institut Pasteur, Paris, France) and J. Cyster (University of California, San Francisco) for critical reading of the manuscript. This work was funded by NIH R01 AI085166 to S.R.S.; NIH T32 AI100853 to V.F. and A.M.; and NIH R01 NIH DA019674 and NS084398 to J.C.
Footnotes
Author Contributions
V.F. designed and conducted experiments, wrote ImageJ programs, analyzed data, and wrote the manuscript; V.S.C. conducted experiments and analyzed data; W.D.R.-P., A.M., and A.B. conducted experiments; R.R. and J.C. provided S1pr5−/− mice; M.C. wrote ImageJ programs; S.R.S. designed experiments, interpreted data, and wrote the manuscript.
Competing Financial Interests
The authors declare no competing financial interests.
References
- 1.Phan TG, Grigorova I, Okada T, Cyster JG. Subcapsular encounter and complement-dependent transport of immune complexes by lymph node B cells. Nat Immunol. 2007;8:992–1000. doi: 10.1038/ni1494. [DOI] [PubMed] [Google Scholar]
- 2.Phan TG, Green JA, Gray EE, Xu Y, Cyster JG. Immune complex relay by subcapsular sinus macrophages and noncognate B cells drives antibody affinity maturation. Nat Immunol. 2009;10:786–793. doi: 10.1038/ni.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Junt T, et al. Subcapsular sinus macrophages in lymph nodes clear lymph-borne viruses and present them to antiviral B cells. Nature. 2007;450:110–114. doi: 10.1038/nature06287. [DOI] [PubMed] [Google Scholar]
- 4.Iannacone M, et al. Subcapsular sinus macrophages prevent CNS invasion on peripheral infection with a neurotropic virus. Nature. 2010;465:1079–1083. doi: 10.1038/nature09118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kastenmuller W, Torabi-Parizi P, Subramanian N, Lammermann T, Germain RN. A spatially-organized multicellular innate immune response in lymph nodes limits systemic pathogen spread. Cell. 2012;150:1235–1248. doi: 10.1016/j.cell.2012.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carrasco YR, Batista FD. B cells acquire particulate antigen in a macrophage-rich area at the boundary between the follicle and the subcapsular sinus of the lymph node. Immunity. 2007;27:160–171. doi: 10.1016/j.immuni.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 7.Gonzalez SF, et al. Capture of influenza by medullary dendritic cells via SIGN-R1 is essential for humoral immunity in draining lymph nodes. Nat Immunol. 2010;11:427–434. doi: 10.1038/ni.1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gerner MY, Torabi-Parizi P, Germain RN. Strategically localized dendritic cells promote rapid T cell responses to lymph-borne particulate antigens. Immunity. 2015;42:172–185. doi: 10.1016/j.immuni.2014.12.024. [DOI] [PubMed] [Google Scholar]
- 9.Suan D, et al. T follicular helper cells have distinct modes of migration and molecular signatures in naive and memory immune responses. Immunity. 2015;42:704–718. doi: 10.1016/j.immuni.2015.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Kastenmuller W, et al. Peripheral prepositioning and local CXCL9 chemokine-mediated guidance orchestrate rapid memory CD8+ T cell responses in the lymph node. Immunity. 2013;38:502–513. doi: 10.1016/j.immuni.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Groom JR, et al. CXCR3 chemokine receptor-ligand interactions in the lymph node optimize CD4+ T helper 1 cell differentiation. Immunity. 2012;37:1091–1103. doi: 10.1016/j.immuni.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hickman HD, et al. Direct priming of antiviral CD8+ T cells in the peripheral interfollicular region of lymph nodes. Nat Immunol. 2008;9:155–165. doi: 10.1038/ni1557. [DOI] [PubMed] [Google Scholar]
- 13.Sung JH, et al. Chemokine guidance of central memory T cells is critical for antiviral recall responses in lymph nodes. Cell. 2012;150:1249–1263. doi: 10.1016/j.cell.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chtanova T, et al. Dynamics of T cell, antigen-presenting cell, and pathogen interactions during recall responses in the lymph node. Immunity. 2009;31:342–355. doi: 10.1016/j.immuni.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leon B, et al. Regulation of T(H)2 development by CXCR5+ dendritic cells and lymphotoxin-expressing B cells. Nat Immunol. 2012;13:681–690. doi: 10.1038/ni.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Woodruff MC, et al. Trans-nodal migration of resident dendritic cells into medullary interfollicular regions initiates immunity to influenza vaccine. J Exp Med. 2014;211:1611–1621. doi: 10.1084/jem.20132327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barral P, et al. CD169(+) macrophages present lipid antigens to mediate early activation of iNKT cells in lymph nodes. Nat Immunol. 2010;11:303–312. doi: 10.1038/ni.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dokun AO, Chu DT, Yang L, Bendelac AS, Yokoyama WM. Analysis of in situ NK cell responses during viral infection. J Immunol. 2001;167:5286–5293. doi: 10.4049/jimmunol.167.9.5286. [DOI] [PubMed] [Google Scholar]
- 19.Coombes JL, Han SJ, van Rooijen N, Raulet DH, Robey EA. Infection-induced regulation of natural killer cells by macrophages and collagen at the lymph node subcapsular sinus. Cell Rep. 2012;2:124–135. doi: 10.1016/j.celrep.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia Z, et al. Subcapsular sinus macrophages promote NK cell accumulation and activation in response to lymph-borne viral particles. Blood. 2012;120:4744–4750. doi: 10.1182/blood-2012-02-408179. [DOI] [PubMed] [Google Scholar]
- 21.Beuneu H, et al. Dynamic behavior of NK cells during activation in lymph nodes. Blood. 2009;114:3227–3234. doi: 10.1182/blood-2009-06-228759. [DOI] [PubMed] [Google Scholar]
- 22.Bajenoff M, et al. Natural killer cell behavior in lymph nodes revealed by static and real-time imaging. J Exp Med. 2006;203:619–631. doi: 10.1084/jem.20051474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walzer T, et al. Natural killer cell trafficking in vivo requires a dedicated sphingosine 1-phosphate receptor. Nat Immunol. 2007;8:1337–1344. doi: 10.1038/ni1523. [DOI] [PubMed] [Google Scholar]
- 24.Garrod KR, Wei SH, Parker I, Cahalan MD. Natural killer cells actively patrol peripheral lymph nodes forming stable conjugates to eliminate MHC-mismatched targets. Proc Natl Acad Sci USA. 2007;104:12081–12086. doi: 10.1073/pnas.0702867104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Decker T, Stockinger S, Karaghiosoff M, Muller M, Kovarik P. IFNs and STATs in innate immunity to microorganisms. J Clin Invest. 2002;109:1271–1277. doi: 10.1172/JCI15770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mendoza A, et al. The transporter Spns2 is required for secretion of lymph but not plasma sphingosine-1-phosphate. Cell Rep. 2012;2:1104–1110. doi: 10.1016/j.celrep.2012.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cyster JG, Schwab SR. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol. 2012;30:69–94. doi: 10.1146/annurev-immunol-020711-075011. [DOI] [PubMed] [Google Scholar]
- 28.Ramos-Perez WD, Fang V, Escalante-Alcalde D, Cammer M, Schwab SR. A map of the distribution of sphingosine 1-phosphate in the spleen. Nat Immunol. 2015;16:1245–1252. doi: 10.1038/ni.3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu CH, et al. Ligand-induced trafficking of the sphingosine-1-phosphate receptor EDG-1. Mol Biol Cell. 1999;10:1179–1190. doi: 10.1091/mbc.10.4.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269:1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
- 31.Ruzankina Y, et al. Deletion of the developmentally essential gene ATR in adult mice leads to age-related phenotypes and stem cell loss. Cell stem cell. 2007;1:113–126. doi: 10.1016/j.stem.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gray EE, Cyster JG. Lymph Node Macrophages. Innate Immun. 2012;4:424–36. doi: 10.1159/000337007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pham TH, et al. Lymphatic endothelial cell sphingosine kinase activity is required for lymphocyte egress and lymphatic patterning. J Exp Med. 2010;207(17–27):S11–14. doi: 10.1084/jem.20091619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Braun A, et al. Afferent lymph-derived T cells and DCs use different chemokine receptor CCR7-dependent routes for entry into the lymph node and intranodal migration. Nat Immunol. 2011;12:879–887. doi: 10.1038/ni.2085. [DOI] [PubMed] [Google Scholar]
- 35.Grigorova IL, Panteleev M, Cyster JG. Lymph node cortical sinus organization and relationship to lymphocyte egress dynamics and antigen exposure. Proc Natl Acad Sci USA. 2010;107:20447–20452. doi: 10.1073/pnas.1009968107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quancard J, et al. A potent and selective S1P(1) antagonist with efficacy in experimental autoimmune encephalomyelitis. Chemistry & biology. 2012;19:1142–1151. doi: 10.1016/j.chembiol.2012.07.016. [DOI] [PubMed] [Google Scholar]
- 37.Jenne CN, et al. T-bet-dependent S1P5 expression in NK cells promotes egress from lymph nodes and bone marrow. J Exp Med. 2009;206:2469–2481. doi: 10.1084/jem.20090525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cyster JG. Homing of antibody secreting cells. Immunol Rev. 2003;194:48–60. doi: 10.1034/j.1600-065x.2003.00041.x. [DOI] [PubMed] [Google Scholar]
- 39.Schmidt TH, Bannard O, Gray EE, Cyster JG. CXCR4 promotes B cell egress from Peyer’s patches. J Exp Med. 2013;210:1099–1107. doi: 10.1084/jem.20122574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mayol K, Biajoux V, Marvel J, Balabanian K, Walzer T. Sequential desensitization of CXCR4 and S1P5 controls natural killer cell trafficking. Blood. 2011;118:4863–4871. doi: 10.1182/blood-2011-06-362574. [DOI] [PubMed] [Google Scholar]
- 41.Schols D, Este JA, Henson G, De Clercq E. Bicyclams, a class of potent anti-HIV agents, are targeted at the HIV coreceptor fusin/CXCR-4. Antiviral Res. 1997;35:147–156. doi: 10.1016/s0166-3542(97)00025-9. [DOI] [PubMed] [Google Scholar]
- 42.Uy GL, et al. A phase 1/2 study of chemosensitization with the CXCR4 antagonist plerixafor in relapsed or refractory acute myeloid leukemia. Blood. 2012;119:3917–3924. doi: 10.1182/blood-2011-10-383406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nie Y, et al. The role of CXCR4 in maintaining peripheral B cell compartments and humoral immunity. J Exp Med. 2004;200:1145–1156. doi: 10.1084/jem.20041185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagahashi M, et al. Spns2, a transporter of phosphorylated sphingoid bases, regulates their blood and lymph levels, and the lymphatic network. FASEB J. 2013;27:1001–1011. doi: 10.1096/fj.12-219618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wei SH, et al. Sphingosine 1-phosphate type 1 receptor agonism inhibits transendothelial migration of medullary T cells to lymphatic sinuses. Nat Immunol. 2005;6:1228–1235. doi: 10.1038/ni1269. [DOI] [PubMed] [Google Scholar]
- 46.Green JA, et al. The sphingosine 1-phosphate receptor S1P(2) maintains the homeostasis of germinal center B cells and promotes niche confinement. Nat Immunol. 2011;12:672–680. doi: 10.1038/ni.2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moriyama S, et al. Sphingosine-1-phosphate receptor 2 is critical for follicular helper T cell retention in germinal centers. J Exp Med. 2014;211:1297–1305. doi: 10.1084/jem.20131666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fukuhara S, et al. The sphingosine-1-phosphate transporter Spns2 expressed on endothelial cells regulates lymphocyte trafficking in mice. J Clin Invest. 2012;122:1416–1426. doi: 10.1172/JCI60746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Y, et al. Migratory and adhesive cues controlling innate-like lymphocyte surveillance of the pathogen-exposed surface of the lymph node. eLife. 2016;5 doi: 10.7554/eLife.18156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Murata K, et al. CD69-null mice protected from arthritis induced with anti-type II collagen antibodies. Int Immunol. 2003;15:987–992. doi: 10.1093/intimm/dxg102. [DOI] [PubMed] [Google Scholar]
- 51.Schaefer BC, Schaefer ML, Kappler JW, Marrack P, Kedl RM. Observation of antigen-dependent CD8+ T-cell/dendritic cell interactions in vivo. Cell Immunol. 2001;214:110–122. doi: 10.1006/cimm.2001.1895. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.