

Abstract
Common methods for the generation of human embryonic-derived neural stem cells (hNSCs) result in cells with potentially compromised safety profiles due to maintenance of cells in conditions containing non-human proteins (e.g. in bovine serum or on mouse fibroblast feeders). Additionally, sufficient expansion of resulting hNSCs for scaling out or up in a clinically relevant time frame has proven to be difficult. Here, we report a strategy that produces hNSCs in completely “Xeno-Free” culture conditions. Furthermore, we have enriched the hNSCs for the cell surface marker CD133 via magnetic sorting, which has led to an increase in the expansion rate and neuronal fate specification of the hNSCs in vitro. Critically, we have also confirmed neural lineage specificity upon sorted hNSC transplantation into the immunodeficient NOD-scid mouse brain. The future use or adaptation of these protocols has the potential to better facilitate the advancement of pre-clinical strategies from the bench to the bedside.
Introduction
Regenerative medicine strategies for central nervous system (CNS) injury and disease represent a major unmet clinical need. One approach will likely include the transplantation of human neural stem cells (hNSCs). Indeed, fetal- and embryonic-derived hNSCs are currently in phase I clinical trials for multiple neurological disorders, including spinal cord injury (Cummings et al., 2005; Salazar et al., 2010), Pelizaeus Merzbacher disease (Uchida et al., 2012), and dry age-related macular degeneration (Schwartz et al., 2012). However, despite the promise afforded by these trials, obstacles (including a complicated FDA approval process for cell lines, difficulties expanding cell lines sufficiently for human transplantation, and tumorigenicity concerns (Germain et al., 2012) resulting from residual, non-differentiated pluripotent cells) still remain.
Future cell-based strategies using new cell lines will benefit from the use of protocols designed to produce readily expandable cell lines with robust safety profiles during the initial pre-clinical phases of research that address FDA concerns for clinical compliance. Here, we report feasible methodologies to generate highly expandable multipotent hNSCs from human embryonic stem cells (hESCs) under completely Xeno-Free (XF) and feeder-free culture conditions. Additionally, we have magnetically sorted the XF hNSCs to further enrich for a highly proliferative neural stem population (CD133+) and reduce the potential for non-neural tumor formation (Tamaki et al., 2002). Together, XF cell culture methods and population enrichment via cell sorting may offer a streamlined approach to generate more readily approvable, expandable, and potentially safer cell populations for CNS transplantation.
Materials and methods
Human embryonic and neural stem cell culture and differentiation
Culture of hESC lines Shef3, Shef4, and Shef6 (University of Sheffield, UK) was established at UC Irvine in accordance with all appropriate hSCRO and IBC protocols on mitotically-inactivated mouse embryonic fibroblasts (MEFs, EMD Millipore) and in defined media consisting of KO DMEM/ F12, 20% KO Serum Replacement (KO SR), 0.1 mM NEAA, 2 mM GlutaMAX, 0.1 mM β-Mercaptoethanol, and 20 ng/mL bFGF (All from Life Technologies). To transition cells to Xeno-Free (XF) culture conditions, all non-human animal-based components (MEFs, KOSR) were removed and replaced with human-based or recombinant alternatives including CELLstart CTS, KO SR Xeno-Free CTS, and KO SR GF Cocktail CTS (All from Life Technologies). XF hESC culture media consisted of KO DMEM/F12, 15% KO SR Xeno-Free CTS, 2 mM GlutaMAX, 1× KO SR GF Cocktail CTS, 0.1 mM β-Mercaptoethanol, and 20 ng/mL bFGF. Cells were manually split every 4–7 days upon reaching ~90% confluence.
For neuralization, an adapted version of a previously published “EZ-sphere” based neuralization protocol (Ebert et al., 2013) was utilized where hESC colonies were manually detached and cultured as floating spheres in Ultra Low Cell Culture Flasks (Corning Inc.) and in media consisting of X-Vivo 15 (Lonza Group Ltd.; Basel, Switzerland), 1× N2, 100 ng/mL bFGF, and 100 ng/mL EGF (Life Technologies). Spheres were split approximately every 2 weeks via mechanical trituration using a wide-end P1000 pipette tip with care taken to avoid dissociation to single cells. 5 days prior to adherent monolayer culture, 10 ng/mL LIF (EMD Millipore) was added to the sphere culture media (Xeno-Free Neural Stem Media, or XF-NSM). To begin adherent monolayer culture, spheres were plated onto CELLstart coated plates in XF-NSM. Within 1–2 days following sphere attachment, single cells began migrating away from the large sphere and upon reaching 80–90% confluence were dissociated using TrypLE Select (Life Technologies) and replated onto CELLstart coated plates in XF-NSM. Cells were then split in this manner every 4–6 days. All karyotype analyses of cell lines were performed off-site (Cell Line Genetics Inc.; Madison, WI).
For neural differentiation, TrypLE Select dissociated single cells were plated onto CELLstart coated Lab-Tek Permanox chamber slides (Thermo Fisher Scientific/Nunc) in XF-NSM. 24 h after attachment, the media was changed to differentiation media (DM) consisting of X-Vivo 15, 10 ng/mL BDNF (Peprotech), 10 ng/mL GDNF (Peprotech), 1× N2, 1× B27 (Life Technologies), 2 ng/mL Heparin (Sigma-Aldrich; St. Louis, MO), 63 μg/mL NAC (Sigma-Aldrich), 0.1 ng/mL bFGF, and 10 μg/mL Ciprofloxacin (Mediatech, Inc.). The media was changed every 3 days with half being removed and replaced with fresh DM. Differentiation was carried out for a total of 2–4 weeks before cells were permeabilized and immunostained.
Magnetic-activated cell sorting and flow cytometric analysis
Magnetic-Activated Cell Sorting (MACS, Miltenyi Biotec) was performed using an autoMACS Pro Separator (Miltenyi Biotec) according to manufacturer-provided protocols via a two-step process: 1) positive selection of CD133+ cells (retain), followed one passage later by 2) negative selection for CD34+ cells (remove), to obtain a CD133+/CD34‒ enriched cell population. Human serum albumin (HSA, Octapharma USA Inc.) was used in place of bovine serum albumin. Magnetic microbead kits human CD133 microbead kit (130-050-801, Miltenyi Biotec) and human CD34 microbead kit (130-046-702, Miltenyi Biotec) were used. All cells were grown prior to sorting as well as post-sorting on CELLstart coated plates in XF-NSM. TrypLE was used to dissociate cells prior to sorting.
For flow cytometric analysis, antibodies used were human CD133/2 (293C3)-PE (130-090-853, Miltenyi Biotec) and human CD34-FITC (130-081-001, Miltenyi Biotec). Surface marker staining was performed according to supplied antibody protocols. Briefly, pelleted cells were resuspended in 80 μL MACS buffer. 20 μL of FcR Blocking Reagent and 10 μL of each respective antibody (either alone, or in combination) were then added to the cell suspension. The suspension was then mixed and incubated at 4 °C for 10 min. Following antibody incubation the cells were washed then incubated with Annexin V according to manufacturer protocols (Life Technologies) for 15 min at room temperature. Human IgG beads (BD Biosciences) were used as fluorescent antibody binding controls. All flow cytometry analyses were performed using a BD FACSAria II and FACS Diva and FlowJo (ver. 10.0.6) software.
In vitro immunocytochemistry
Immunocytochemistry procedures were as described previously (Piltti et al., 2011) with minor modifications. Briefly, cells were fixed with 4% paraformaldehyde for 15 min and then permeabilized in PBS containing 0.5% Triton X-100 (Sigma-Aldrich) for 20 min. The cells were then blocked in PBS containing 5% donkey serum (Jackson ImmunoResearch), and 1% BSA (Sigma-Aldrich) for 10 min, followed by primary antibody incubation in blocking buffer for 2 h at room temperature. Primary antibodies included monoclonal mouse anti-SSEA4 (1:75, ab16287, Abcam), polyclonal rabbit anti-Oct4 (1:200, ab19857, Abcam), polyclonal rabbit anti-Sox2 Alexa Fluor 488-Conjugated (1:100, AB5603A4, EMD Millipore), monoclonal mouse anti-hNestin (1:200, MAB1259, R&D Systems), monoclonal mouse anti-βIII Tubulin (1:500, MMS-435P, Covance), polyclonal rabbit anti-GFAP (1:500, Z0334, DakoCytomation), and polyclonal goat anti-hOlig2 (1:100, AF2418, R&D Systems). Following primary antibody incubation and washes, cells were incubated in appropriate secondary antibodies for 1 h at room temperature and then mounted with Flouromount-G (Southern-Biotech) for imaging. Secondary antibodies included: Alexa Fluor 488 Donkey Anti-Rabbit, Alexa Fluor 555 Donkey Anti-Mouse, Alexa Fluor 488 Donkey Anti-Mouse, Alexa Fluor 568 Donkey Anti-Rabbit, and Alexa Fluor 647 Donkey Anti-Goat. Hoechst 33342 (H1399) was used for nuclear identification. All secondary antibodies and Hoechst were from MolecularProbes/Life Technologies and used at a 1:1000 dilution. All secondary antibodies were tested for cross-reactivity and non-specific binding. Imaging was performed using an Olympus FluoView FV10i (Olympus America Inc.) and a Zeiss Axio Imager.M2 Apotome System (Carl Zeiss).
RNA extraction and PCR analysis
Total RNA was isolated from harvested cells from 2 wells of a 6-well culture plate by scraping with lysis buffer from an RNeasy mini kit (Qiagen). Extracted total RNA was treated with RNase free DNase I (DNA free; Ambion), and synthesis of cDNA was performed by oligo(dT)/random primer mediated reverse transcription with a minimum of 300 ng of total RNA input using a high capacity RNA-to-cDNA kit (Applied Biosystems). For quantitative PCR experiments, each reaction was performed using 100 ng of cDNA with TaqMan® probe-based gene expression primers that were analyzed per gene (technical duplicates) in biological triplicate (three independent experiments) for all samples for each cell stage in the study. For non-quantitative PCR experiments, GoTaq Green Master Mix (Promega Corp.) was combined with specifically designed forward and reverse primers and 200 ng cDNA. PCR conditions used were 95 °C denaturation, 62 °C annealing, and 72 °C extension for either 35 or 40 cycles. Due to the number of comparisons, samples of the same cycle number were run in parallel across multiple agarose gels on the same day, and identical exposure settings were used to capture all gel band images. Images were then arranged side-by-side in the figure for ease of visual comparison. All kits were used according to manufacturer’s instructions. For primers used see Table S1.
In vitro cell growth and proliferation analysis
To assess cell growth and obtain mean doubling times of both non-sorted and CD133+/CD34− hNSCs, cells were seeded at an initial density of 5 × 105 cells per well onto CELLstart-coated 6-well plates. Cells were harvested and dissociated into single cells using TrypLE and counted manually via Trypan Blue exclusion at 24-, 48-, 72-, 96-, and 120-h post-plating. All cell growth assays were performed in biological triplicates. To assess proliferation via 5-ethynyl-2’-deoxyuridine (EdU, C10337, Life Technologies) incorporation, cells were plated onto CELLstart coated Permanox chamber slides and allowed to grow to approximately 80–90% confluence before adding 10 μM EdU and incubating for 24 h. Post-EdU incubation, cells were fixed and EdU incorporation was detected via a Click-iT® reaction. The quantification of EdU-positive cells to total number of Hoechst-positve nuclei was performed by analyzing fluorescent images of at least 8 randomly chosen fields from 3 independent experiments using Volocity 3D Image Analysis Software (PerkinElmer) (Piltti et al., 2011).
Human stem cell preparation for transplantation
All animal housing conditions, surgical procedures, and postoperative care were approved by and conducted according to the Institutional Animal Care and Use Committee (IACUC) guidelines at the University of California, Irvine. All hESC and hNSC preparations took place during the day of transplantation/injection. All cells were maintained under normal culture conditions prior to harvesting and collection for hippocampal/subcutaneous injection. For hippocampal hNSC injections, cells were dissociated to single cells using TrypLE and resuspended in X-Vivo 15 to yield 7.5 × 104 cells/μl. hESCs were harvested in a similar manner for hippocampal injection but care was taken to avoid single-cell dissociation by using a cell scraper (BD Falcon) rather than TrypLE. Hippocampal cell preparations were kept at room temperature.
For subcutaneous leg injections of both hESC and hNSC, cells were prepared similarly to hippocampal preparations but were resuspended in Matrigel rather than X-Vivo 15 and kept on ice to prevent Matrigel from polymerizing prematurely. Dilutions ranged from 1.25 to 2.5 × 106 cells per 50 μl.
Intracranial and subcutaneous cell transplantations
For intracranial injections, both male and female NOD-scid mice (12–20 weeks old; The Jackson Laboratory) were anesthetized with 2% isoflurane and positioned in a stereotaxic holder (Leica Microsystems Inc.). A midline incision was made exposing the skull. A 5 μl Hamilton syringe (Cat #87930) with a custom 1” 30G blunt needle was mounted into an UMP-3 (World Precision Instruments) injector connected to a SYS-Micro4 Controller. The needle was then moved to 1.94 mm posterior to bregma and 2.00 mm lateral to midline, and a mark was made using a mechanical pencil. The needle was moved out of the way, and a burr hole was drilled using a Dremel rotary tool at ~5,000 rpm. Cells were first triturated 5 times with a 10 μl pipette then pulled up into the syringe over approximately 10 s. The needle was first lowered to 2.15 mm ventral relative to bregma to create a pocket in the brain, then raised to 2.10 mm. Cells were then injected at doses ranging from 7.5 × 104 to 1.5 × 105 (at 7.5 × 104 cells/μl) over 2 min. Speeds varied relative to the total volume, but time of injection remained constant. The needle was raised out of the brain 4 min after injection. The syringe was tested to make sure it was not clogged by ejecting some of the solution into the air manually through the Micro4 controller. Bone wax was applied to seal the burr hole, and the midline incision was stapled shut.
For subcutaneous injections, anesthetized mice received 50 μl injections of either cells (hESC or hNSC) or vehicle control (Matrigel, BD Biosciences) via injection through a 23G needle attached to a 1 ml syringe that was inserted underneath the skin, between the knee and ankle. The needle was held in place for approximately 10 s before retraction, and all cell/vehicle preparations, and injection equipment, were kept on ice to prevent Matrigel from polymerizing prematurely.
Animals were randomly allocated to different cell type and injection location groups as described in Table 1 and were furthermore processed blind to injection cohort. Animals received lactated ringers (50 ml/kg) subcutaneously immediately after surgery as well as Buprenorphine (0.5 mg/kg) immediately after surgery and for 2 days thereafter. Additionally, due to the immunodeficient nature of NOD-scid mice, an antibiotic (Baytril, 2.5 mg/kg) was administered immediately after surgery and daily for 5 days thereafter. No immunosuppressant drugs were administered to any animal.
Table 1.
Engraftment and tumorigenesis potential of XF Shef6 hESCs and hNSCs.
| Cell line/type | Leg injection dose | # leg grafts | % with leg tumors | CNS injection dose | # CNS grafts | % with CNS engraftment | % with CNS tumors |
|---|---|---|---|---|---|---|---|
| Shef6 hESC | 1,250,000 | 14 | 79% (11/14) | 75,000 | 8 | 63% (5/8) | 63% (5/8) |
| Shef6 hNSC (unsorted) | 1,250,000 | 10 | 0% (0/10) | 75,000 | 8 | 0% (0/8) | 0% (0/8) |
| Shef6 hNSC (unsorted) | 150,000 | 2 | 0% (0/2) | 0% (0/2) | |||
| Shef6 hNSC (sorted) | 1,250,000 | 23 | 0% (0/23) | 75,000 | 15 | 93% (14/15) | 0% (0/15) |
| Shef6 hNSC (sorted) | 2,500,000 | 17 | 0% (0/17) | 150,000 | 4 | 100% (4/4) | 0% (0/4) |
Engraftment and tumor forming potential of subcutaneously (leg) and intracranially (hippocampus) injected XF Shef6 hESCs as well as both non-sorted and low passage (P7) CD133+/CD34‒ sorted hNSCs at multiple doses. Shef6 hESC transplants resulted in tumors in 79% (leg) and 63% (CNS) of transplants at 8–12 weeks post-transplant. All animals with successful CNS hESC engraftment exhibited tumors. No non-sorted hNSCs were detected 12 weeks post-transplant. CD133+/CD34‒ sorted Shef6 hNSCs engrafted in the CNS at a rate of 93–100% and no CD133+/CD34‒ sorted Shef6 hNSC transplants resulted in tumors. See also Figure S2.
Tissue collection and immunohistochemistry
Tissue collection and immunohistochemistry were performed as previously described (Hooshmand et al., 2009) with minor modifications. Briefly, all animals, ranging from 8 to 20 weeks post-transplant, were anesthetized with a lethal dose of Euthasol (100 mg/kg, i.p.) and transcardially perfused with 30 ml of PBS, followed by 100 ml of 4% paraformaldehyde. Brains were carefully dissected and post-fixed overnight in a solution of 4% paraformaldehyde and 20% sucrose in PBS at 4 °C, flash frozen at ‒65 °C in isopentane (2-methyl butane), and stored at ‒80 °C.
For cryosectioning and immunohistochemistry, frozen brains were embedded in Optimal Cutting temperature (O.C.T.) compound (Sakura Finetek USA, Inc.). 35 μm thick coronal sections were cut on a sliding microtome, collected in 96-well plates containing 0.1 M Tris and 0.02% Sodium Azide, and kept at 4 °C until processed for immunostaining. All immunostaining procedures were conducted at room temperature. Briefly, sections were washed in 0.1 M Tris followed by a 15 minute incubation in 0.3% hydrogen peroxide/methanol. After a brief 0.1% Triton X-100 wash, sections were blocked for 1 h with bovine serum albumin (BSA) and normal serum from the species in which the secondary antibody was raised. Sections were then exposed overnight to the appropriate primary antibody. The primary antibodies used were: monoclonal mouse anti-SC121 (1:10, 000), monoclonal mouse anti-SC101 (1:2,000), mouse monoclonal anti-SC123 (1:3,000) (all from StemCells Inc.), monoclonal mouse anti-hNestin (1:1,000) (MAB1259, R&D Systems), polyclonal rabbit anti-PDX1 (1:2,000) (ab47267, Abcam), polyclonal rabbit anti-αSMA (1:100) (ab5694, Abcam), mono-clonal rabbit anti-βIII Tubulin (1:500) (MRB-435P, Covance), polyclonal rabbit anti-VGlut1 (1:50, ab72311, Abcam), and polyclonal rabbit anti-GAD65 + 67 (1:1000, ab49832, Abcam). The next day, sections were incubated with either fluorescent or biotin-conjugated, purified IgG secondary antibody (1:500, Jackson Immunoresearch) pre-adsorbed against the species in which the primary was raised, followed by avidin–biotinylated peroxidase complex (ABC) using a Vectastain Elite ABC kit (Vector Laboratories, USA) prepared according to the manufacturer’s recommendations (for DAB staining). After several washes, the signal was visualized with diaminobenzidine (DAB) (Vector Laboratories, USA). Sections were mounted onto slides and allowed to dry overnight at 37 °C. DAB stained sections were lightly counter-stained with the nuclear marker, methyl green. Fluorescent stained sections were counterstained with Hoechst 33342 (1:1000, Life Technologies). All slides were coverslipped using Depex or Fluoromount-G (SouthernBiotech) mounting medium. Imaging was performed using an Olympus FluoView FV10i (Olympus America Inc.) and also a Zeiss Imager.M2 Apotome System (Carl Zeiss).
Statistical analysis
All qPCR and EdU/growth kinetics data are representative of three or more individual independent experiments. Grubb’s test (ESD method) was used to eliminate qPCR replicate statistical outliers. Errors are the standard error (SEM) of averaged results. EdU data analyzed by one-way ANOVA (Bonferroni post-hoc) with P < 0.001 using GraphPad Prism software.
Results
Xeno-Free transition and neuralization of three human embryonic stem cell lines
Culture of the human embryonic stem cell lines Shef3, Shef4, and Shef6 was initially established in “standard” conditions that contain both bovine and murine components (Fig. 1B–D, non-XF hESC). To enhance the clinical applicability of these human stem cell lines, all hESC lines were transitioned to “Xeno-Free” conditions in a 1-step simultaneous conversion of both media (to a human serum-based media) and substrate (to a human vitronectin-based substrate, CELLstart) (Fig. 1A). XF transitioned hESCs typically required an adaptation or a stabilization period of 1–2 passages post-transition in which spontaneously differentiating cells were removed via manual dissection. Following XF transition, all three XF hESC lines maintained circular, hESC-like colony morphologies and expressed the pluripotency markers Oct4 and SSEA4 at both 9–11 passages in XF conditions (Fig. 1B–D, XF hESC) as well as after prolonged XF hESC culture (Figure S1D).
Figure 1.
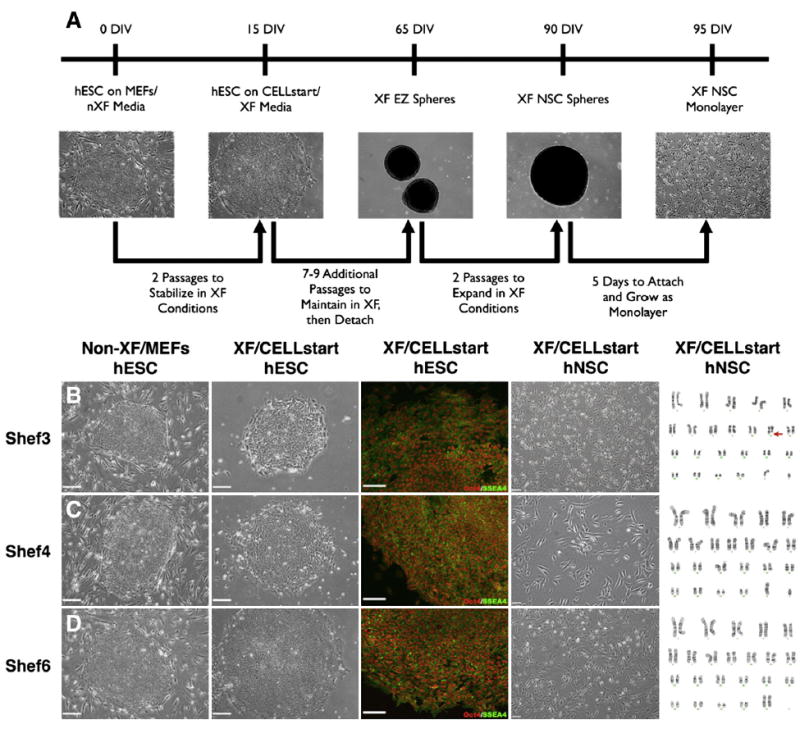
hESCs transitioned to XF conditions maintain pluripotency and can be further neuralized to form XF hNSCs. (A) Schematic for the XF transition and subsequent neuralization of hESCs into bi- and multi-polar neural stem/progenitor-like cells via a modified EZ sphere-based protocol. (B–D) Shef3 (B) undifferentiated hESCs were originally maintained on MEFs and in standard non-XF hESC culture media containing BSA. hESCs were then transitioned to XF conditions utilizing the XF substrate CELLstart and KnockOut SR XF/ KO SR GF Cocktail. Following XF transition, hESCs continued to express the pluripotency markers Oct4 (red) and SSEA4 (green). Finally, XF transitioned hESCs were further neuralized to form adherent XF hNSCs that exhibited characteristic neural morphologies. Subsequent karyotype analysis revealed one chromosomal abnormality (Shef3, Chromosome 11) and two karyotypically normal (Shef4, Shef6) hNSC lines. (C) Shef4 and (D) Shef6 hESCs were similarly transitioned to XF conditions and neuralized. Scale: 200 μm (hESCs, phase), 100 μm (hESCs, fluorescent) and 50 μm (hNSCs). See also Figure S1.
After 9 to 11 passages in XF hESC conditions (40–60 days in culture), transitioned XF hESCs were subsequently subjected to neural induction via EGF/FGF supplementation to generate intermediate neuralized spheres (Ebert et al., 2013). Further neuralization of spheres with LIF supplementation and cell attachment on CELLstart generated an adherent monolayer culture system (Fig. 1A) that produced cells with bi- and multi-polar neural morphologies (Fig. 1B–D, XF hNSC). One cell line (Shef3) was determined to be karyotypically abnormal after XF transition and neuralization (Fig. 1B, karyotype; Figure S1). The other two hNSC lines (Shef4, Shef6) were karyotypically normal (Fig. 1C–D, karyotype). Prior to any subsequent in vitro or in vivo studies, XF adherent hNSCs had, at a minimum, been cultured in XF conditions for over 100 days (40–60 days as XF hESC, followed by 60+ days as XF hNSC spheres/monolayer), a time period sufficient to eliminate detectable non-human molecules (Ludwig et al., 2006; Heiskanen et al., 2007).
Magnetically sorted (CD133+/CD34‒) Xeno-Free Shef6 hNSCs exhibit increased neuronal differentiation in vitro
Following XF neuralization, magnetic sorting was used to further enrich one of the hNSC lines, Shef6, for a stem/ progenitor population via positive selection of hNSCs expressing CD133 and depletion of cells expressing CD34. Flow cytometric analysis of non-sorted XF Shef6 hNSCs demonstrated that ~17% expressed CD133, and less than 2% expressed CD34 (Fig. 2A). Post-sorting, the CD133 + proportion increased to ~ 82%, while CD34 remained at less than 2% (Fig. 2B). The increase in the CD133 + proportion of cells was maintained over long-term culture as greater than 70% of sorted XF Shef6 hNSCs still expressed CD133 after 20 additional passages (P27, Fig. 2C).
Figure 2.
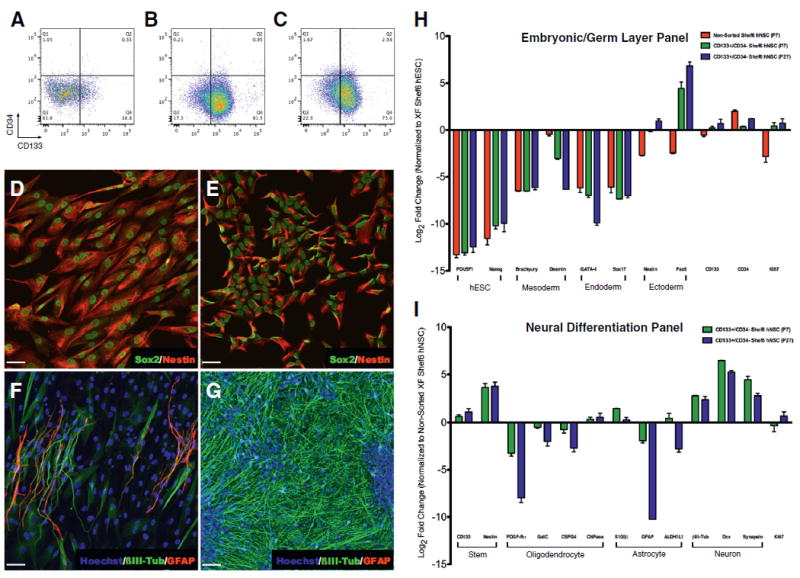
Undifferentiated CD133+/CD34‒ sorted XF Shef6 hNSCs have reduced levels of embryonic/germ layer expression, and generate fewer glial cells but more neurons after in vitro differentiation. (A–C) Flow cytometric analysis of (A) non-sorted (P7), (B) low passage (P7) CD133+/CD34‒ sorted, and (C) high passage (P27) CD133+/CD34‒ sorted XF Shef6 hNSCs displayed an increase in CD133+ proportion post-sorting (16.8% to 81.5%, non-sorted to low passage sorted) that was maintained at higher passages (73.0%, high passage sorted). The CD34+ proportion remained under 5% for all samples. (D-G) Immunocytochemistry of both (D) non-sorted and (E) low passage CD133+/CD34‒ sorted XF Shef6 hNSCs expressed neural stem/progenitor markers Sox2 (D–E, green) and Nestin (D–E, red) in vitro under non-differentiating conditions. Upon differentiation, both (F) non-sorted and (G) low passage CD133+/ CD34‒ sorted XF Shef6 hNSCs expressed more mature neural lineage markers βIII-Tub (F–G, green) and GFAP (F, red; Figure S1C, red). Nuclei were counter-stained with Hoechst 33342 (F–G, blue). Scale: 50 μm (D–G). (H–I) qPCR analysis of (H) embryonic and germ layer/early differentiation markers expressed in both non-sorted and CD133+/CD34‒ sorted undifferentiated XF Shef6 hNSCs normalized to XF Shef6 hESCs and (I) neural lineage differentiation markers expressed in CD133+/CD34‒ sorted XF Shef6 hNSCs after 28DIV neural differentiation normalized to non-sorted XF Shef6 hNSCs. All data were normalized to internal 18s rRNA and expressed as mean log2 fold change ± SEM (n = 3 independent experiments with technical duplicates of each independent biological experiment).
Interestingly, while both XF undifferentiated non-sorted and CD133+/CD34- sorted Shef6 hNSCs expressed the neural stem and progenitor markers Sox2 and Nestin (Fig. 2D–E), morphological differences were observed between the two neuralized populations. Notably, non-sorted cells appeared larger and more cytoplasmic (Fig. 2D) than sorted cells (Fig. 2E). Additionally, analysis via qPCR (Fig. 2H) demonstrated a reduction of pluripotent (Oct4, Nanog) and embryonic germ layer (Brachyury, Desmin, GATA-4, Sox17) mRNA expression in the neuralized populations compared to XF Shef6 hESCs. Furthermore, the sorted XF Shef6 hNSCs also expressed higher levels of neural lineage markers Nestin (6-fold upregulation in P7 CD133+/CD34− sorted hNSCs, 12.5-fold in P27 sorted hNSCs vs. non-sorted Shef6 hNSCs) and Pax6 (115-fold upregulation in P7 CD133+/CD34− sorted hNSCs, 600-fold in P27 sorted hNSCs) as well as smaller increases in both CD133 and Ki67 expression and a decrease in CD34 expression in comparison to non-sorted XF Shef6 hNSCs.
Differences were also observed between the two populations in cells undergoing neural differentiation, with non-sorted cells expressing neuronal Class III β-Tubulin (βIII-Tub) and GFAP in elongated, bipolar cells (Fig. 2F) while CD133+/ CD34‒ cells expressed primarily βIII-Tub in densely populated areas (Fig. 2G) as well as GFAP in sparse but clustered regions (Figure S1C). Morphologically, in the sorted population, βIII-Tub immunostaining was observed in numerous spineous bipolar and multipolar neuronal cells and GFAP in larger, flatter astrocytic cell types. qPCR was used to further probe the differentiation potential of the two populations. As observed via ICC, there was an increase in neuronal differentiation (βIII-Tub 7-fold upregulation, Dcx 89-fold, Synapsin 22-fold) in the sorted XF Shef6 hNSCs (P7) in comparison to non-sorted hNSCs (Fig. 2I). Interestingly, sorted XF Shef6 hNSCs also exhibited a decrease in glial fate potential (PDGF-Ra, GalC, CSPG, GFAP) compared to non-sorted cells, suggesting that CD133+ enrichment at this developmental stage may bias cells toward a neuronal fate. Importantly, even though the sorted populations exhibited enhanced proliferation as detected by Ki67 gene expression under non-differentiating conditions in comparison to non-sorted XF Shef6 hNSCs (Fig. 2H), there do not appear to be significant differences once the hNSCs have undergone differentiation (Fig. 2I), suggesting a downregulation of proliferative potential upon differentiation.
Furthermore, regional identity of both the non-sorted and CD133+/CD34‒ sorted Shef6 hNSCs was evaluated via RT-PCR (Fig. 3A–B). Non-sorted Shef6 hNSCs expressed hindbrain markers GBX2 and KROX20, and lacked forebrain and forebrain/midbrain marker expression (FOXG1 and OTX2, respectively). Non-sorted Shef6 hNSCs also lacked expression or expressed very low levels of the neural stem cell markers PAX6 and ASCL1 as well as neural rosette markers PLZF and DACH1. Similarly, both low passage (P7) and high passage (P27) CD133+/CD34‒ sorted Shef6 hNSCs expressed the hindbrain markers GBX2 and KROX20, and also lacked or had very low expression of forebrain markers FOXG1 and OTX2. However, the sorted hNSCs exhibited expression of both neural stem (Pax6, ASCL1) and rosette-specific (PLZF, DACH1) markers, suggesting that the CD133+/CD34‒ Shef6 hNSCs are most likely an expanding neural stem-like cell population possessing a hindbrain regional specification preference.
Figure 3.
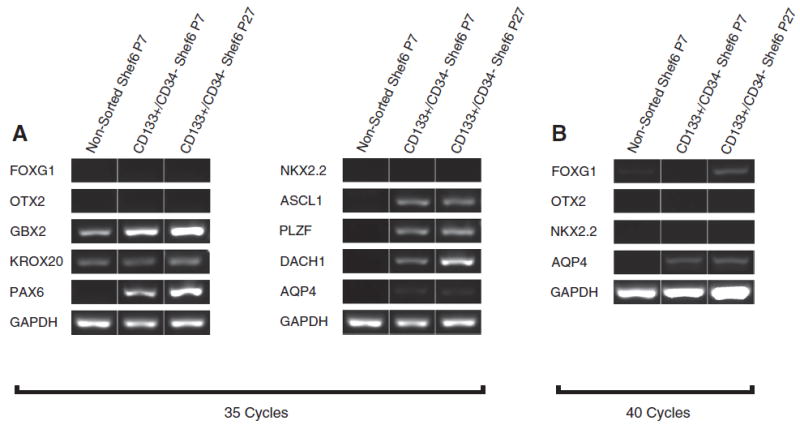
CD133+/CD34‒ sorted Shef6 hNSCs express hindbrain as well as neural stem and rosette-specific transcription factors in vitro. (A–B) RT-PCR analysis of non-sorted XF Shef6 (P7), low passage (P7) CD133+/CD34‒ sorted, and high passage (P27) CD133+/ CD34‒ sorted XF Shef6 hNSCs at (A) 35 cycles of PCR amplification exhibited expression of hindbrain transcription factors GBX2 and KROX20, and lacked expression of forebrain and forebrain/midbrain factors FOXG1 and OTX2, respectively, as well as the ventral marker NKX2.2 and the more mature FGF/EGF-expanded marker AQP4. CD133+/CD34‒ sorted Shef6 hNSCs demonstrated expression of neural stem cell markers PAX6 and ASCL1, as well as neural rosette markers PLZF and DACH1, whereas non-sorted Shef6 hNSCs exhibited very low to no expression of these neural stem/rosette markers. (B) At 40 cycles of PCR amplification, CD133+/CD34‒sorted Shef6 hNSCs exhibited very low levels of FOXG1 (P27 hNSCs) and AQP4 expression (P7 and P27), but no or extremely low expression of OTX2 and NKX2.2.
Magnetically sorted (CD133+/CD34‒) Xeno-Free Shef6 hNSCs proliferate extensively in vitro
In addition to differences in morphology and differentiation propensity, differences in proliferation and cellular growth kinetics were also observed between non-sorted and sorted XF Shef6 hNSCs. EdU incorporation over a 24-hour period was used to gauge proliferation of undifferentiated hNSCs, with 21% of non-sorted XF Shef6 hNSCs (Fig. 4A, C) incorporating EdU compared to 91% of CD133+/CD34‒ sorted XF Shef6 hNSCs (Fig. 4B, C). EdU incorporation remained consistent after 20 additional passages of prolonged culture (95%, Fig. 4C). Additionally, non-sorted Shef6 hNSCs expanded to ~850,000 cells from an initial starting density of 500,000 cells over the course of 5 days in vitro (Fig. 4D), giving a doubling time of 5.1–6.4 days. Conversely, over the same 5 day period, CD133+/CD34‒ sorted Shef6 hNSCs expanded to over 14 million cells (Fig. 4D) from the same initial density of 500,000 cells (a doubling time of 0.9–1.4 days). The overall increase in cell expansion of sorted XF Shef6 hNSCs in comparison to non-sorted XF Shef6 hNSCs was maintained after 20 additional passages (Fig. 4D), although a minor decrease in expansion capability was observed between low (P7, 0.9–1.4 days doubling) and high passage (P27, 1.0–1.4 day doubling) sorted hNSCs. Interestingly, non-sorted and CD133+/CD34‒ sorted Shef4 hNSCs exhibited similar in vitro expansion rates to each other, but both were slower than the non-sorted Shef6 hNSCs (data not shown). Due to the slow proliferative nature of the Shef4 hNSCs, as well as the karyotypic abnormality in the Shef3 hNSCs, further in vivo studies utilized Shef6 hNSCs exclusively.
Figure 4.
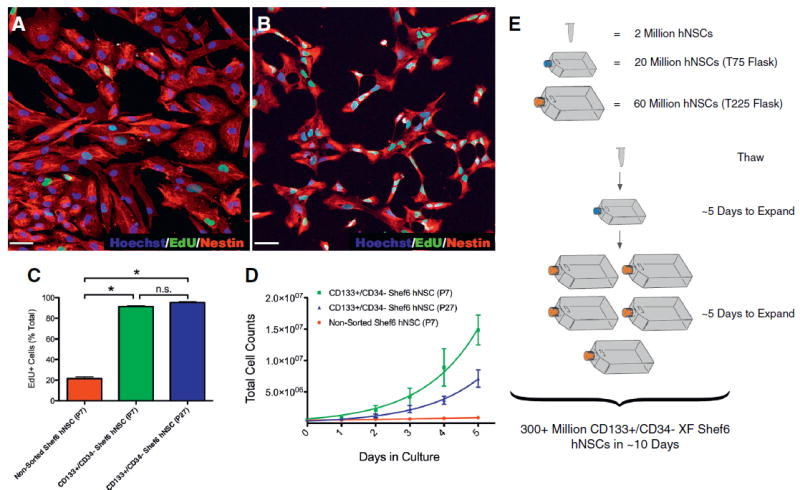
CD133+/CD34‒ XF Shef6 hNSCs proliferate extensively in vitro. (A–B) Undifferentiated (A) non-sorted (P7) and (B) low passage (P7) CD133+/CD34‒ sorted XF Shef6 hNSCs incorporated EdU over a 24-hour pulse period. EdU (A–B, green) and Nestin (A–B, red). Nuclei were counterstained with Hoechst 33342 (A–B, blue). Scale: 50 μm (A–B). (C) Quantification of EdU incorporation as a percentage of total cells (EdU + Hoechst+/Hoechst+). Error bars are mean percentage ± SEM (n = 3 independent experiments) analyzed by ANOVA (Bonferroni), * P < 0.001. (D) Growth kinetics of non-sorted (P7) as well as both low (P7) and high passage (P27) CD133+/CD34‒ sorted XF Shef6 hNSCs plated on CELLstart at an initial density of 500,000 cells and cultured for 5 subsequent days. Cells were counted daily at the same time. Error bars are mean cell counts ± SEM (n = 3 independent experiments). (E) Schematic example of CD133+/CD34− sorted XF Shef6 hNSC expansion capability to generate in excess of 300 million hNSCs from one thawed vial (containing 2 million hNSCs) over the course of ~10 days.
Taken together, the EdU and growth kinetics data suggest a dramatic increase in proliferative potential of the sorted XF Shef6 hNSCs in vitro such that a starting population of 2 million hNSCs could be expanded to over 300 million hNSCs in ~10 days (Fig. 4E).
Xeno-Free CD133+/CD34‒ sorted hNSCs do not form teratomas in vivo and differentiate primarily into neurons
To examine tumorigenicity potential, intrahippocampal transplanted non-sorted and CD133+/CD34‒ sorted Shef6 hNSCs were compared to similarly transplanted undifferentiated Shef6 hESCs. As expected from a pluripotent cell population, transplanted Shef6 hESCs exhibited graft overgrowth with widespread intrahippocampal migration, that in many cases caused the endogenous tissue architecture to become deformed (Fig. 5A). Transplanted Shef6 hESCs also expressed both neural and non-neural (Fig. 5B–D) germ layer-specific markers, including hNestin (ectoderm), α-SMA (mesoderm), and PDX1 (endoderm) at 8 weeks post-transplantation.
Figure 5.
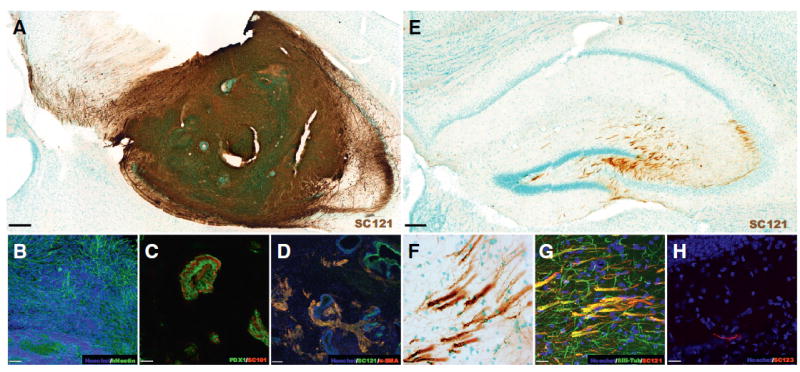
Evaluation of tumorigenesis potential – in vivo engraftment and fate of hippocampal transplanted XF Shef6 hESC and CD133+/CD34‒ sorted XF hNSCs. (A–D) Transplanted XF Shef6 hESCs (dose: 75,000 cells) survive and engraft in the immunodeficient NOD-scid hippocampus at 8-weeks post-transplantation. (A) Formation of large, dense human-positive (SC121, brown) masses that engulf the Shef6 hESC transplanted hippocampus. Within the transplanted region, cells of each embryonic germ layer lineage were detected, including (B) ectoderm (hNestin, green), (C) endoderm (PDX1, green; SC101, red), and (D) mesoderm (α-SMA, red; SC121, green). Scale: 200 μm (A), 50 μm (B, C), and 80 μm (D). (E–H) Transplanted low passage (P7) CD133+/CD34‒ sorted XF Shef6 hNSCs (Dose: 150,000 cells) survive and engraft in the immunodeficient NOD-scid hippocampus at 12-weeks post-transplantation. (E) Engraftment of Shef6 hNSCs (SC121, brown) and migration into the molecular layer as well as dentate hilar and CA3 hippocampal regions. (F) Transplanted Shef6 hNSCs often form fibrous bundles that (G) stain positive for the neuronal marker βIII-Tubulin (βIII-Tub, green; SC121, red). (H) Human astrocytes (SC123, red) were also visualized occasionally. For (A, E, and F) methyl green was used to counterstain nuclei. For (B, D, G, and H) Hoechst 33342 was used to counterstain nuclei (blue). Scale: 200 μm (E), and 20 μm (F–H). See also Figure S2.
Additionally, subcutaneous leg injections of both Shef6 hESC and hNSC populations were performed at doses ranging from 1.25 million to 2.5 million cells per injection. Only the Shef6 hESC population displayed gross anatomical abnormalities, with 79% of injected animals exhibiting large masses (Table 1, Figure S2).
Conversely, intrahippocampal transplanted Shef6 hNSCs (as opposed to Shef6 hESCs) exhibited markedly different engraftment dynamics. We saw no evidence of cell engraftment in any of the 10 animals transplanted with non-sorted Shef6 hNSCs (Table 1) at 12 weeks post-transplantation. Conversely, a high level of engraftment was observed in CD133+/CD34‒ Shef6 hNSC transplanted animals (18/19, 95%; Table 1). Engrafted cells migrated laterally from the single injection site, spanning from the dentate gyrus to CA3 (Fig. 5E). The sorted Shef6 hNSCs extended cell projections that often displayed bundled or fasciculated phenotypes (Fig. 5F). Occasionally, cells appeared to migrate out from hippocampus and were located adjacent to the walls of the lateral ventricle. Engrafted CD133+/CD34‒ Shef6 hNSCs exhibited primarily neuronal differentiation at 12 weeks post-transplantation, determined via co-expression of the human-specific cytoplasmic marker SC121 and βIII-Tub (Fig. 5G). Rare astroglial differentiation was also observed (SC123; Fig. 5H). Additionally, SC121 + human cells were nearly always found to be associated with vesicular glutamate transporter 1 (VGlut1) immunopositive neurons (Fig. 6A–D), suggesting that the majority of the transplanted CD133+/CD34‒ Shef6 hNSCs differentiated into glutamatergic neurons in vivo. Glutamic acid decarboxyl-ase (GAD67) positive puncta were also occasionally associated with SC121 + human cells, although the pattern of GAD67 immunoreactivity appeared more isolated, sparse, and on the surface of SC121 + cells (Fig. 6E–H), suggesting that the human, glutamatergic neurons were receiving GABAergic innervation rather than being GABAergic themselves. In no cases examined was non-neural cell type staining found to be co-localized with transplanted human hNSCs (e.g. α-SMA, PDX1).
Figure 6.
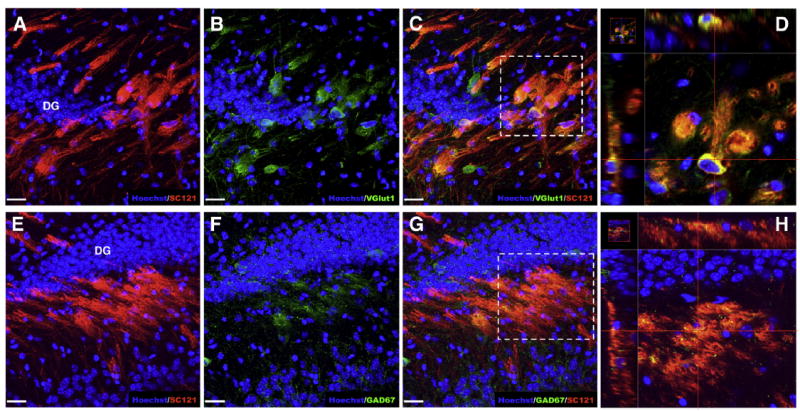
Hippocampal transplanted CD133+/CD34‒ sorted Shef6 hNSCs predominantly differentiate into glutamatergic neurons and receive GABAergic inputs. (A–D) Nearly all engrafted CD133+/CD34‒ Shef6 hNSCs (A, SC121, red) were immunopositive for vesicular glutamate transporter 1, a glutamatergic neuron marker (B, VGlut1, green), throughout the cytoplasm of the transplanted cells (C, co-expression; D, magnified inset from C). (E–H) A subset of engrafted CD133+/CD34‒ Shef6 hNSCs (E, SC121, red) also appeared to receive glutamic acid decarboxylase (GAD) positive GABAergic inputs (F, GAD67, green), visualized as small GAD67 immunopositive puncta contacting regions of human SC121+ cytoplasm (G, co-expression; H, magnified inset from G). Hoechst 33342 was used to counterstain nuclei (blue). Scale: 20 μm.
Discussion
The data presented here establish protocols to transition and maintain multiple hESC and hNSC lines long-term in completely XF culture conditions, thus providing straightforward methodologies to produce clinically compliant cell lines. Additionally, we have used magnetic sorting to enrich these XF Shef6 hNSCs for cells expressing the surface marker CD133. Sorting for CD133+ cells dramatically increased both the neuronal fate preference as well as the proliferative potential of the Shef6 hNSCs, to the extent that enough cells could potentially be generated within ~10 days for a theoretical human brain transplant (Gupta et al., 2012). Importantly, we have also addressed safety concerns inherent with embryonic stem cell-derived hNSCs. We have demonstrated that while these cells expand rapidly in culture, once they are transplanted into the brain there was no evidence of non-neural lineage tumors. Both XF human cell types (embryonic and neural) generated via the methods presented here exhibited good stability. After more than 10 passages in completely XF conditions, XF hESCs remained karyotypically normal, expressed key pluripotency markers, exhibited minimal spontaneous differentiation, and were able to generate teratomas upon transplantation into NOD-scid mice. XF transitioned hESCs were subsequently able to be differentiated into hNSCs in completely XF conditions as well. CD133+/CD34‒ XF Shef6 hNSCs were able to be maintained for over 25 passages in XF neural conditions, maintained a normal karyotype, exhibited similar differentiation and proliferation profiles to earlier passage hNSCs, and engrafted after intrahippocampal transplantation.
Positive selection via magnetic sorting for CD133 increased the CD133+ proportion of the XF Shef6 hNSCs from 17% to 82%. Interestingly, the CD34+ proportion of the Shef6 hNSCs was measured at less than 5%, as detected by flow cytometry, in both the non-sorted and CD133+/CD34‒ sorted hNSCs, which may obviate the need for CD34 depletion in the cell populations discussed herein. However, CD34 is expressed on multiple cell types, including hematopoetic stem cells (Civin et al., 1984; Baum et al., 1992; Goodell et al., 1997), endothelial cells (Pusztaszeri et al., 2006), macrophages (Ryncarz and Anasetti, 1998), and microglia (Asheuer et al., 2004; Ladeby et al., 2005), and therefore warrants depletion consideration in future pluripotent-derived neural populations. In this regard, we have found that CD133+/CD34‒ sorting has altered the in vitro fate profile of the hNSCs. Both the non-sorted and CD133+/CD34‒ populations similarly expressed neural stem/ progenitor markers Sox2 and Nestin under non-differentiating conditions. However, the sorted population, under differentiating conditions, appeared to be highly enriched for neurons. One possible explanation could relate to the developmental stage of the hNSCs at the time of cell sorting. Recent evidence has suggested that the neuralization method utilized in this study generates very early stage, pre-rosette hNSCs (Ebert et al., 2013). By sorting for the stemness marker CD133 at this early stage, we may have enriched the cell population for a more mitotically active early neural stem cell subtype that readily generates neuronal lineage cells (Tropepe et al., 2001; Pruszak et al., 2007; Elkabetz et al., 2008; Koch et al., 2009; Li et al., 2011; Falk et al., 2012; Yan et al., 2013). Supporting evidence exists for the derivation of early lineage neural stem or neuroepithelial-like stem cells that are continuously expandable in culture, express over 90% CD133 on the cell surface, and generate predominantly neurons (Koch et al., 2009; Falk et al., 2012). These cells, termed long-term self-renewing neuroepithelial-like stem cells or lt-NES cells, exhibit hindbrain specification but are amenable to regional patterning in vitro, engraft after intracranial transplantation and fire action potentials in vivo, and differentiate predominantly into GABAergic neurons (Steinbeck et al., 2012). Similarly, the early stage hNSCs generated via the protocols described herein appeared to be largely hindbrain specified, though a small glial population was present at least early in culture (up to passage 7). However, upon intrahippocampal transplantation, CD133+/CD34‒ XF Shef6 hNSCs generate predominantly glutamatergic neurons. Regardless, the CD133+/CD34‒ hNSCs described here represent a population of stable, self-renewing, and neurogenic early stage neural stem-like cells that may be of benefit for researchers studying human neural development.
A second interesting finding was the increased in vitro proliferation rate of the sorted XF Shef6 hNSCs. CD133+/ CD34‒ hNSCs have previously been shown to have enhanced neurosphere forming capabilities (Uchida et al., 2000). Additionally, some proliferative neuroepithelial cell populations express CD133 (Pruszak et al., 2007; Falk et al., 2012), and CD133+ fractions of cancer biopsies and cancer cell lines have been shown to have increased proliferative rates (Singh et al., 2003; Kelly et al., 2010). Here, we have demonstrated that CD133 enrichment of XF Shef6 hNSCs has dramatically increased their expansion rate, effectively shortening their doubling time from ~6 days (non-sorted XF Shef6 hNSCs) to ~1 day (CD133+/CD34‒ XF Shef6 hNSCs). One possible explanation for the noted sub-optimal degree of cell growth and expansion in the non-sorted Shef6 hNSCs could be heterogeneity in the proportion of slower-cycling or quiescent neural stem/progenitor-like cells to faster-cycling neural stem- or transit amplifying-like cells. Additionally, non-neural epithelial or multipotent stromal cell types could be present in the non-sorted population, as evidenced by the increased level of Desmin mRNA present in the non-sorted Shef6 hNSCs compared to the CD133+/CD34‒ sorted hNSCs. The possibility exists then that these slower-cycling, possibly non-neural cells could be maintaining an overall slower rate of proliferation via secretion of factors such as BMPs, which have been shown to decrease NSPC proliferation and induce cell cycle exit (Liu and Niswander, 2005; Mathieu et al., 2008). By either enriching for faster-cycling cells or by eliminating slower-cycling cells (or both) via CD133+/CD34‒cell sorting, we have potentially disengaged the brakes, at least partially, on this proliferation and cell cycle governing system in vitro. Moving forward, additional replication of the above Xeno-Free transition and CD133+/CD34‒ sorting studies with multiple different cell lines and cell types, including human induced pluripotent stem cells, will be critical to fully evaluate the utility of the methods presented here across a wide range of human pluripotent and neural cell types.
However, unabated in vivo cell proliferation can be problematic and often has undesired consequences such as tumor formation. CNS transplantation of highly proliferative hESCs and immature neural progenitors (Brederlau et al., 2006; Seminatore et al., 2010), as well as known neural tumor-forming cell lines (Fogh et al., 1977; Pollard et al., 2009), result in severe over-growth of the host tissue transplant region. In this study, we transplanted XF Shef6 hESCs into the hippocampus of immunodeficient NOD-scid mice and find similar results to previous studies – namely, the hESCs proliferate extensively, differentiate into cell types of all three embryonic germ layers (form teratomas), and engulf nearly the entire transplant region (hippocampus). Conversely, CD133+/CD34‒ XF Shef6 hNSCs do not exhibit transplant region over-growth or non-neural lineage differentiation. Rather, hippocampal engrafted sorted XF Shef6 hNSCs tend to distribute more sparsely throughout the molecular layer, dentate gyrus, and CA3 hippocampal regions and project fasciculated axon bundles. Interestingly, using similar cell preparation and transplantation methods, hippocampal transplanted non-sorted XF Shef6 hNSCs failed to survive when examined at 12-weeks post-transplant. This failure could be the result of a combination of different factors, including, but not limited to, poor cell survival immediately post-transplant (Okada et al., 2005), insufficient proliferation of engrafted cells (as opposed to CD133+/ CD34‒ XF Shef6 hNSCs that are more proliferative in vitro), and/or delayed cell death (Steinbeck et al., 2012). Future investigation of both the interactions between the transplanted non-sorted cell population and the host transplant niche, as well as the mechanisms responsible for the observed differences in gene expression and proliferation rate between the non-sorted and CD133+/CD34‒ XF Shef6 hNSCs, will be necessary to enhance our understanding of neural stem cell biology and cell transplantation, and critically, additional replication of these findings and long-term survival studies (i.e. 12 months or greater) in the injured or diseased niche must be performed before final conclusions can be drawn in regards to cell safety for future human use.
Supplementary Material
Acknowledgments
We would like to thank the technical staff at the Christopher and Dana Reeve Foundation Animal Injury Core and other members of the Anderson lab: Rebecca Nishi, Zhe Xu, Oscar Mendez, Jovanny Lucero, and Diane Su for their help with animal surgeries and animal care. We would also like to thank Usha Nekanti for assistance with qPCR procedures, Denisse Moreno for technical support in maintenance and preparation of human cells for transplantation, Kelly Cunningham for assistance with histology, and Vanessa Scarfone for assisting with flow cytometry experiments. This work was supported by California Institute for Regenerative Medicine (CIRM) Early Translational Award TR2-01767 (to B.J.C.), Basic Biology RB2-01496 (to H.X.N. and A.J.A.) and Pre-Doctoral Training Grant TG2-01152 (to D.L.H.), and also a UK-US Collaboration Development Award from the Foreign and Commonwealth Office Award (to B.J.C.). H.D.M. is supported by grant award G0801059 from the Medical Research Council, UK.
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.scr.2014.06.008.
References
- Asheuer M, Pflumio F, Benhamida S, Dubart-Kupperschmitt A, Fouquet F, Imai Y, Aubourg P, Cartier N. Human CD34+ cells differentiate into microglia and express recombinant therapeutic protein. Proc Natl Acad Sci U S A. 2004;101:3557–3562. doi: 10.1073/pnas.0306431101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum CM, Weissman IL, Tsukamoto AS, Buckle AM, Peault B. Isolation of a candidate human hematopoietic stem-cell population. Proc Natl Acad Sci U S A. 1992;89:2804–2808. doi: 10.1073/pnas.89.7.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brederlau A, Correia AS, Anisimov SV, Elmi M, Paul G, Roybon L, Morizane A, Bergquist F, Riebe I, Nannmark U, Carta M, Hanse E, Takahashi J, Sasai Y, Funa K, Brundin P, Eriksson PS, Li JY. Transplantation of human embryonic stem cell-derived cells to a rat model of Parkinson’s disease: effect of in vitro differentiation on graft survival and teratoma formation. Stem Cells. 2006;24:1433–1440. doi: 10.1634/stemcells.2005-0393. [DOI] [PubMed] [Google Scholar]
- Civin CI, Strauss LC, Brovall C, Fackler MJ, Schwartz JF, Shaper JH. Antigenic analysis of hematopoiesis. III. A hematopoietic progenitor cell surface antigen defined by a monoclonal antibody raised against KG-1a cells. J Immunol. 1984;133:157–165. [PubMed] [Google Scholar]
- Cummings BJ, Uchida N, Tamaki SJ, Salazar DL, Hooshmand M, Summers R, Gage FH, Anderson AJ. Human neural stem cells differentiate and promote locomotor recovery in spinal cord-injured mice. Proc Natl Acad Sci U S A. 2005;102:14069–14074. doi: 10.1073/pnas.0507063102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert AD, Shelley BC, Hurley AM, Onorati M, Castiglioni V, Patitucci TN, Svendsen SP, Mattis VB, McGivern JV, Schwab AJ, Sareen D, Kim HW, Cattaneo E, Svendsen CN. EZ spheres: a stable and expandable culture system for the generation of pre-rosette multipotent stem cells from human ESCs and iPSCs. Stem Cell Res. 2013;10:417–427. doi: 10.1016/j.scr.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkabetz Y, Panagiotakos G, Al Shamy G, Socci ND, Tabar V, Studer L. Human ES cell-derived neural rosettes reveal a functionally distinct early neural stem cell stage. Genes Dev. 2008;22:152–165. doi: 10.1101/gad.1616208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk A, Koch P, Kesavan J, Takashima Y, Ladewig J, Alexander M, Wiskow O, Tailor J, Trotter M, Pollard S, Smith A, Brustle O. Capture of neuroepithelial-like stem cells from pluripotent stem cells provides a versatile system for in vitro production of human neurons. PLoS One. 2012;7:e29597. doi: 10.1371/journal.pone.0029597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogh J, Fogh JM, Orfeo T. One hundred and twenty-seven cultured human tumor cell lines producing tumors in nude mice. J Natl Cancer Inst. 1977;59:221–226. doi: 10.1093/jnci/59.1.221. [DOI] [PubMed] [Google Scholar]
- Germain ND, Hartman NW, Cai C, Becker S, Naegele JR, Grabel LB. Teratocarcinoma formation in embryonic stem cell-derived neural progenitor hippocampal transplants. Cell Transplant. 2012;21:1603–1611. doi: 10.3727/096368912X647243. [DOI] [PubMed] [Google Scholar]
- Goodell MA, Rosenzweig M, Kim H, Marks DF, DeMaria M, Paradis G, Grupp SA, Sieff CA, Mulligan RC, Johnson RP. Dye efflux studies suggest that hematopoietic stem cells expressing low or undetectable levels of CD34 antigen exist in multiple species. Nat Med. 1997;3:1337–1345. doi: 10.1038/nm1297-1337. [DOI] [PubMed] [Google Scholar]
- Gupta N, Henry RG, Strober J, Kang SM, Lim DA, Bucci M, Caverzasi E, Gaetano L, Mandelli ML, Ryan T, Perry R, Farrell J, Jeremy RJ, Ulman M, Huhn SL, Barkovich AJ, Rowitch DH. Neural stem cell engraftment and myelination in the human brain. Sci Transl Med. 2012;4:155ra137. doi: 10.1126/scitranslmed.3004373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiskanen A, Satomaa T, Tiitinen S, Laitinen A, Mannelin S, Impola U, Mikkola M, Olsson C, Miller-Podraza H, Blomqvist M, Olonen A, Salo H, Lehenkari P, Tuuri T, Otonkoski T, Natunen J, Saarinen J, Laine J. N-glycolylneuraminic acid xenoantigen contamination of human embryonic and mesenchymal stem cells is substantially reversible. Stem Cells. 2007;25:197–202. doi: 10.1634/stemcells.2006-0444. [DOI] [PubMed] [Google Scholar]
- Hooshmand MJ, Sontag CJ, Uchida N, Tamaki S, Anderson AJ, Cummings BJ. Analysis of host-mediated repair mechanisms after human CNS-stem cell transplantation for spinal cord injury: correlation of engraftment with recovery. PLoS One. 2009;4:e5871. doi: 10.1371/journal.pone.0005871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly SE, Di Benedetto A, Greco A, Howard CM, Sollars VE, Primerano DA, Valluri JV, Claudio PP. Rapid selection and proliferation of CD133+ cells from cancer cell lines: chemotherapeutic implications. PLoS One. 2010;5:e10035. doi: 10.1371/journal.pone.0010035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch P, Opitz T, Steinbeck JA, Ladewig J, Brustle O. A rosette-type, self-renewing human ES cell-derived neural stem cell with potential for in vitro instruction and synaptic integration. Proc Natl Acad Sci U S A. 2009;106:3225–3230. doi: 10.1073/pnas.0808387106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladeby R, Wirenfeldt M, Dalmau I, Gregersen R, Garcia-Ovejero D, Babcock A, Owens T, Finsen B. Proliferating resident microglia express the stem cell antigen CD34 in response to acute neural injury. Glia. 2005;50:121–131. doi: 10.1002/glia.20159. [DOI] [PubMed] [Google Scholar]
- Li W, Sun W, Zhang Y, Wei W, Ambasudhan R, Xia P, Talantova M, Lin T, Kim J, Wang X, Kim WR, Lipton SA, Zhang K, Ding S. Rapid induction and long-term self-renewal of primitive neural precursors from human embryonic stem cells by small molecule inhibitors. Proc Natl Acad Sci U S A. 2011;108:8299–8304. doi: 10.1073/pnas.1014041108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A, Niswander LA. Bone morphogenetic protein signalling and vertebrate nervous system development. Nat Rev Neurosci. 2005;6:945–954. doi: 10.1038/nrn1805. [DOI] [PubMed] [Google Scholar]
- Ludwig TE, Levenstein ME, Jones JM, Berggren WT, Mitchen ER, Frane JL, Crandall LJ, Daigh CA, Conard KR, Piekarczyk MS, Llanas RA, Thomson JA. Derivation of human embryonic stem cells in defined conditions. Nat Biotechnol. 2006;24:185–187. doi: 10.1038/nbt1177. [DOI] [PubMed] [Google Scholar]
- Mathieu C, Sii-Felice K, Fouchet P, Etienne O, Haton C, Mabondzo A, Boussin FD, Mouthon MA. Endothelial cell-derived bone morphogenetic proteins control proliferation of neural stem/progenitor cells. Mol Cell Neurosci. 2008;38:569–577. doi: 10.1016/j.mcn.2008.05.005. [DOI] [PubMed] [Google Scholar]
- Okada S, Ishii K, Yamane J, Iwanami A, Ikegami T, Katoh H, Iwamoto Y, Nakamura M, Miyoshi H, Okano HJ, Contag CH, Toyama Y, Okano H. In vivo imaging of engrafted neural stem cells: its application in evaluating the optimal timing of transplantation for spinal cord injury. FASEB J. 2005;19:1839–1841. doi: 10.1096/fj.05-4082fje. [DOI] [PubMed] [Google Scholar]
- Piltti KM, Haus DL, Do E, Perez H, Anderson AJ, Cummings BJ. Computer-aided 2D and 3D quantification of human stem cell fate from in vitro samples using Volocity high performance image analysis software. Stem Cell Res. 2011;7:256–263. doi: 10.1016/j.scr.2011.05.005. [DOI] [PubMed] [Google Scholar]
- Pollard SM, Yoshikawa K, Clarke ID, Danovi D, Stricker S, Russell R, Bayani J, Head R, Lee M, Bernstein M, Squire JA, Smith A, Dirks P. Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell. 2009;4:568–580. doi: 10.1016/j.stem.2009.03.014. [DOI] [PubMed] [Google Scholar]
- Pruszak J, Sonntag KC, Aung MH, Sanchez-Pernaute R, Isacson O. Markers and methods for cell sorting of human embryonic stem cell-derived neural cell populations. Stem Cells. 2007;25:2257–2268. doi: 10.1634/stemcells.2006-0744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusztaszeri MP, Seelentag W, Bosman FT. Immunohis-tochemical expression of endothelial markers CD31, CD34, von Willebrand factor, and Fli-1 in normal human tissues. J Histochem Cytochem. 2006;54:385–395. doi: 10.1369/jhc.4A6514.2005. [DOI] [PubMed] [Google Scholar]
- Ryncarz RE, Anasetti C. Expression of CD86 on human marrow CD34(+) cells identifies immunocompetent committed precursors of macrophages and dendritic cells. Blood. 1998;91:3892–3900. [PubMed] [Google Scholar]
- Salazar DL, Uchida N, Hamers FP, Cummings BJ, Anderson AJ. Human neural stem cells differentiate and promote locomotor recovery in an early chronic spinal cord injury NOD-scid mouse model. PLoS One. 2010;5:e12272. doi: 10.1371/journal.pone.0012272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz SD, Hubschman JP, Heilwell G, Franco-Cardenas V, Pan CK, Ostrick RM, Mickunas E, Gay R, Klimanskaya I, Lanza R. Embryonic stem cell trials for macular degeneration: a preliminary report. Lancet. 2012;379:713–720. doi: 10.1016/S0140-6736(12)60028-2. [DOI] [PubMed] [Google Scholar]
- Seminatore C, Polentes J, Ellman D, Kozubenko N, Itier V, Tine S, Tritschler L, Brenot M, Guidou E, Blondeau J, Lhuillier M, Bugi A, Aubry L, Jendelova P, Sykova E, Perrier AL, Finsen B, Onteniente B. The postischemic environment differentially impacts teratoma or tumor formation after transplantation of human embryonic stem cell-derived neural progenitors. Stroke. 2010;41:153–159. doi: 10.1161/STROKEAHA.109.563015. [DOI] [PubMed] [Google Scholar]
- Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- Steinbeck JA, Koch P, Derouiche A, Brustle O. Human embryonic stem cell-derived neurons establish region-specific, long-range projections in the adult brain. Cell Mol Life Sci. 2012;69:461–470. doi: 10.1007/s00018-011-0759-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamaki S, Eckert K, He D, Sutton R, Doshe M, Jain G, Tushinski R, Reitsma M, Harris B, Tsukamoto A, Gage F, Weissman I, Uchida N. Engraftment of sorted/expanded human central nervous system stem cells from fetal brain. J Neurosci Res. 2002;69:976–986. doi: 10.1002/jnr.10412. [DOI] [PubMed] [Google Scholar]
- Tropepe V, Hitoshi S, Sirard C, Mak TW, Rossant J, van der Kooy D. Direct neural fate specification from embryonic stem cells: a primitive mammalian neural stem cell stage acquired through a default mechanism. Neuron. 2001;30:65–78. doi: 10.1016/s0896-6273(01)00263-x. [DOI] [PubMed] [Google Scholar]
- Uchida N, Buck DW, He D, Reitsma MJ, Masek M, Phan TV, Tsukamoto AS, Gage FH, Weissman IL. Direct isolation of human central nervous system stem cells. Proc Natl Acad Sci U S A. 2000;97:14720–14725. doi: 10.1073/pnas.97.26.14720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida N, Chen K, Dohse M, Hansen KD, Dean J, Buser JR, Riddle A, Beardsley DJ, Wan Y, Gong X, Nguyen T, Cummings BJ, Anderson AJ, Tamaki SJ, Tsukamoto A, Weissman IL, Matsumoto SG, Sherman LS, Kroenke CD, Back SA. Human neural stem cells induce functional myelination in mice with severe dysmyelination. Sci Transl Med. 2012;4:155ra136. doi: 10.1126/scitranslmed.3004371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Shin S, Jha BS, Liu Q, Sheng J, Li F, Zhan M, Davis J, Bharti K, Zeng X, Rao M, Malik N, Vemuri MC. Efficient and rapid derivation of primitive neural stem cells and generation of brain subtype neurons from human pluripotent stem cells. Stem Cells Transl Med. 2013;2:862–870. doi: 10.5966/sctm.2013-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.