

Abstract
Epigenetic enzymes including histone modifying enzymes are key regulators of gene expression in normal and disease processes. Many drug development strategies to target histone modifying enzymes have focused on ligands that bind to enzyme active sites, but allosteric pockets offer potentially attractive opportunities for therapeutic development. Recent biochemical studies have revealed roles for small molecule and peptide ligands binding outside of the active sites in modulating the catalytic activities of histone modifying enzymes. Here we highlight several examples of allosteric regulation of epigenetic enzymes and discuss the biological significance of these findings.
Graphical Abstract
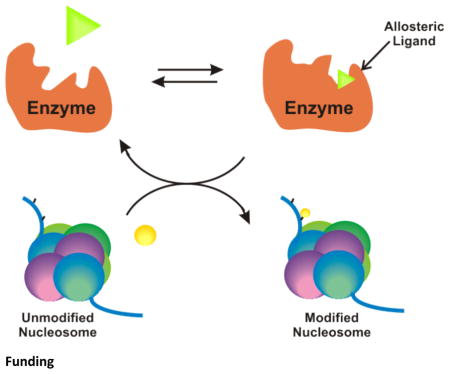
Introduction
Epigenetic enzymes are pivotal determinants of cell fate by regulating chromatin modifications on both nucleosomal proteins and DNA. These modifications result in changes in the “timing” and “volume” of gene expression; and when occurring on histone residues, constitute the proposed histone code. This code of histone modifications is generated by “writers,” interpreted by “readers,” and removed by “erasers.” The most intensively studied writer enzymes include the Lys acetyltransferases and the Lys and Arg methyltransferases. The best understood family of acetyl-Lys readers is the bromodomain containing proteins. There are several classes of methyl-Lys readers including chromodomains, PHD fingers, tudor domains, and MBT proteins. Eraser enzymes for acetyl-Lys belong to two major families, the classical HDACs which are Zn hydrolases and the more chemically unusual NAD-dependent sirtuins. Two major families of Lys demethylases have been identified including the flavin-dependent demethylases and the non-heme iron monoxygenase Jumonji enzymes [1–5].
Within each of these writer, reader, and eraser families are multiple well-characterized examples making the epigenetic machinery intricate and complex [4,6,7]. Moreover, a wide array of acyl chain modifications have been identified recently including propionylation, butyrylation, 2-hydroxyisobutyrylation, succinylation, malonylation, glutarylation, crotonylation and β-hydroxybutyrylation [4,8,9]. Specific modifications on particular histone residues are generally associated with “open” or transcriptionally active gene states while others are associated with “closed” or transcriptionally silent chromatin [7,10,11]. Aberrant activity or mutation of histone modifying enzymes can alter the chromatin structure and gene expression profile contributing to cancer, developmental abnormalities, and other diseases [1,6,7,12].
Understanding how these enzymes are regulated in both normal physiology and disease is of great fundamental importance and may offer therapeutic opportunities. The broad significance of epigenetic writers and readers as factors in disease processes has stimulated researchers to identify and design small molecule modulators of these protein activities. Targeting the enzyme active sites of the writers and erasers has been the primary focus of drug discovery programs. However, given the conserved active sites of many epigenetic enzyme families, achieving specificity for particular enzyme family members can prove challenging [13–15]. In contrast, allosteric modulators of their activities pave the way to unique and specific pharmacologic agents. In addition, dissecting allosteric mechanisms within epigenetic enzymes can facilitate a fundamental understanding of the principles of their biological regulation. Accordingly, the past six years has seen the budding of allosteric regulation of epigenetic enzymes. Lessons from cell signaling enzymes such as protein kinases indicate how various domains and structural features can dramatically impact the activity of phosphoryl transfer. The protein tyrosine kinase Src has served as a paradigm in this regard. In Src, engagement of its SH2 and SH3 adaptor domains by phosphotyrosine and proline-rich ligands can relieve autoinhibition of its catalytic activity [16–18]. Related themes are beginning to emerge in epigenetic modifying enzymes. Below, we describe several examples of epigenetic enzyme allosteric mechanisms and their connection to opportunities in pharmacology.
Allosteric regulation of histone demethylase KDM5A
The retinoblastoma binding protein KDM5A (RBP2, JARID1A) is a histone demethylase that catalyzes the removal of methyl groups from histone H3K4me3 and H3K4me2 [11,19]. KDM5A has been shown to have a role in adipocyte development, osteogenesis, and immunoactivation [20–22]. It has been implicated in numerous cancers, including multiple myeloma, gastric, lung, and breast [23–28]. Like many histone demethylases, the protein KDM5A contains both reader and eraser domains within a single polypeptide. KDM5A contains both a Jumonji (Jmj) catalytic domain and three plant homeodomain (PHD) reader domains. The Jmj enzymes require iron(II) and α-ketoglutarate as cofactors [1,2,29]. In KDM5A as in other KDM5 enzymes, the JmjC domain is preceded indirectly by a JmjN domain which folds with the JmjC domain to form a stable, catalytic core [29–32]. Inserted between the JmjN and JmjC domains in this subclass of Jmj enzymes is the first PHD finger and ARID DNA binding domain (Figure 1). In general, PHD domains recruit methyltransferases and demethylases to chromatin in a sequence/modification specific paradigm. Seemingly promiscuous, PHD domains can bind acetylated, methylated and unmethylated lysines depending on the context [1,33,34]. PHD1 of KDM5A can bind unmodified H3K4 peptide with low micromolar affinity [35], and deletion of PHD1 leads to increased cellular H3K4me3 [36].
Figure 1.

Protein domains of each of the epigenetic enzymes discussed. Catalytic sites in shades of blue, allosteric ligand interacting domains in shades of green or purple, and DNA interacting regions in yellow. All other domains as labeled.
Studies with an unmodified H3(1-18) tail peptide demonstrate that the affinity of PHD1 of KDM5A is dependent upon the first four residues of the H3 tail. By studying intact KDM5A protein, it was revealed that the binding of unmodified H3(1-18) by PHD1 stimulates the demethylation of H3K4me3 by the JmjC catalytic domain of KDM5A (Figure 2 and Table 1). This enhancement of catalysis was even more dramatic on homogenously modified H3K4me3 nucleosomes, where the kobs of demethylation increased over 20-fold in the presence of unmodified H3(1-18) peptide. In the current model, binding of the PHD1 of KDM5A to the unmodified tail of H3 on one nucleosome may perform the role of feedback activation through allosteric changes to dramatically enhance demethylation of adjacent nucleosomes by the JmjC domain and propagate spreading of nucleosome demethylation [37]. Such propagation may explain gene cluster silencing as KDM5A is known to silence Hox gene clusters which are marked by H3K4 methylation [38,39]. This catalytic link between a reader domain and nucleosome demethylation suggests that these domains structurally communicate although the molecular details remain to be elucidated. This mechanism also raises the possibility that small molecule ligands for PHD1 may provide a tool to inhibit gene expression for genes under the control of KDM5A.
Figure 2.

Allosteric ligands discussed in this review. A) Histone peptides interacting with GCN5 and KDM5A, (un)modified as listed, respectively. B) I-CBP112 is a bromodomain ligand for p300 and CBP. C) MAML1 binds the CH3 domain of p300 to activate enzyme activity. D) SGC707 binds to the dimerization interface of PRMT3 to disengage the activation helix and inhibit activity.
Table 1.
Epigenetic enzymes, their allosteric ligands, and functional effects.
| Enzyme | Active Site | Ligand | Ligand Binding Site | Ligand Action |
|---|---|---|---|---|
| KDM5A | JmjC/N | H3(1-18) unmodified | PHD1 | activate H3K4me3 demethylation |
| p300/CBP | HAT | MAML1 Proline-rich region | CH3 | increase H4 acetylation and p300 target gene expression |
| p300/CBP | HAT | CBP112 | Bromodomain | increase H3K18ac |
| GCN5 | HAT | H3K14ac(1-18) | Bromodomain | increase H3K18ac |
| PRMT3 | SAM-Dependent MTase | SGC707 | Distal interface between SAM-Dependent MTase, α-Helix, and Dimerization Arm | decrease H4R3me2a |
Multi-domain regulation of p300/CBP
p300 (EP300), as well as its paralog CREB-binding protein (CBP), is a promiscuous and prolific lysine acetyltransferase (KAT). p300 and CBP have been shown to play key roles in regulating a myriad of biological processes and diseases [8]. Consistent with its name, p300 is a large protein (264 kDa) with multiple domains (Figure 1). Acetyl transfer from a tightly-bound acetyl-CoA cofactor to the substrate Lys-containing protein or peptide substrate is catalyzed by p300's histone acetyltransferase (HAT) domain via a hit and run catalytic mechanism [8]. For p300, several mechanisms of allosteric regulation have been described. The HAT domain is preceded by a bromodomain which “reads” PTMs by binding acetylated lysine side chains. Between the bromodomain and the HAT domains are the RING and PHD domains, also known together as the CH2 domain [40]. These have been shown to have inhibitory effects on p300 activity by an unidentified mechanism [40,41]. Also the HAT domain contains an autoacetylation loop (AL) that binds to the substrate binding pocket of the HAT domain, blocking substrate binding. However, upon acetylation of the AL by the HAT active site, the loop is released and
p300 acetylation of target proteins increases [42]. p300 has over 400 known protein-protein interactions and over 100 known protein substrates [8]. One of these is the transcriptional coregulator Mastermind like-1 (MAML1) [43]. Not only does p300 acetylate MAML1, but also the presence of MAML1 increases p300 autoacetylation. This effect is independent of the p300 AL but is dependent upon the subsequent CH3 domain with which MAML1 interacts. The MAML1-p300 interaction also increases the acetylation of the histone H4 tail peptide and the colocalization of p300 and H4-ac in nuclear bodies. Full-length MAML1 dramatically increases the expression of p300 target genes (Figure 2 and Table 1). The potential exists that a portion of the CH3 domain may interact with the HAT domain to inhibit its activity, and MAML1-CH3 binding abrogates this inhibition [44]. This would pose the CH3 domain as a potential drug target in an attempt to make specific activators of p300 acetylation.
The bromodomain of p300/CBP has also been investigated as a regulator of p300/CBP function. For example, after being acetylated by p300/CBP, the tumor suppressor p53-K382ac peptide can bind to the CBP bromodomain and this can enhance p300/CBP recruitment to transcriptional loci [45]. Blocking this interaction with a cyclic p53-K382 peptide, blocks p53 induction of p21 expression [46]. Relatively recently, several high affinity small molecule ligands for the p300/CBP bromodomains have been developed [47–52]. Each of these was shown to induce some cellular phenotype, and several were crystallized bound to the p300/CBP [50–52]. It was further shown that one of the p300/CBP bromodomain ligands, I-CBP112, specifically activates acetylation of H3K18ac in the context of intact nucleosomes (Figure 2 and Table 1). This activation of acetylation is seen both in vitro and in cell culture. The requirement of p300/CBP domains beyond the HAT domain and the intact nucleosome substrate points toward a multifaceted interaction shift propagated by I-CBP112, potentially unique to this bromodomain ligand among other small molecules [53]. The importance of the bromodomain to activate p300 acetylation was consistent with an earlier study in which deletion of the bromodomain abolished p300 acetylation of an entire library of nucleosome substrates [54]. Even weakening the bromodomain-Ac-Lys interaction by bromodomain point mutations [55] diminished p300’s propensity to acetylate “primed” nucleosomes (with preexisting H4ac) consistent with a “cooperative acetylation model” [54]. These and other data point to allosteric binding ligands regulating the acetylation activity of the HAT domain. The precise mechanisms of these actions are largely unknown but are areas of active investigation.
Catalytic regulation of Gcn5 by its bromodomain
Gcn5 is a histone acetyltransferase with much more narrow substrate selectivity compared with p300/CBP. Gcn5 has been shown to be especially important for hepatic metabolism and may also be an anti-cancer target [12,56,57]. Like p300/CBP, Gcn5 also possesses a bromodomain (Figure 1), and the role of its bromodomain has been explored in the context of histone site selectivity. As a part of the catalytic ADA complex (Gcn5 + Ada2 + Ada3), Gcn5 acetylates H3 tail lysines with the following specificity: H3K14 > H3K23 > H3K9 ≈ H3K18 > H3K27 > H3K36. However, if the Gcn5 bromodomain is mutated to make it incompetent to bind Ac-Lys, H3K18ac was most severely diminished. Accordingly, the same result was found with H3K14R mutants and wild type Gcn5. Therefore, a model is proposed in which Gcn5 acetylates H3K14 and then this Ac-Lys is bound by the bromodomain of Gcn5. H3K14Ac-Gcn5-bromodomain association then enhances H3K18ac by the Gcn5 HAT domain (Figure 2 and Table 1) [58]. Reminiscent of p300/CBP regulation by I-CBP112 or of KDM5A by unmethylated peptide, the acetylation of H3K18 seems to be susceptible to allosteric regulation. The short distance between H3K14 and H3K18 makes it unlikely that the Gcn5-K14ac association results from orienting the K18 of the same histone tail into the HAT active site [59]. More likely the activation occurs in trans. This trans acetylation activation model is similar to a proposal for the activation of H3 acetylation when p300 is incubated with nucleosomes already containing penta-acetylated H4 tails or at least one H4 tail acetylation site [54]. The recent reports of potent Gcn5 bromodomain selective ligands [60,61] offer promise to more deeply probe this allostery.
Allosteric inhibitors of PRMT3 arginine methyltransferase
PRMT3 is an example of a protein methyltransferase than methylates the Arg guanidinium sidechain. In general, protein methyltransferases catalyze the transfer of 1–3 methyl groups to either arginine or lysine side chains. Methylation of the terminal (ε) nitrogen of lysine side chains can be mono-, di-, or tri-methylated. Methylation of the guanidinium arginine group can occur as mono-, symmetrical di- or asymmetrical di-methylation [1,13,15]. Beyond the multiple sites on histone tails, over one-thousand cellular proteins have been identified as containing methylated residues (www.phosphosite.org) [62]. The transcriptionally repressed state of heterochromatin and polycomb genes is marked by histone H3 trimethylated at lysine 9 or 27 (H3K9me3 or H3K27me3); whereas H3K4me marks active chromatin regions including enhancers and promoters [2,3,63,64]. The mutation or dysregulation of many protein methyltransferases gives rise to disease, especially cancer and neurological disorders [6,13,15] and therefore poses an enticing pharmacological target.
All protein methyltransferases use S-adenosylmethionine (SAM) as the methyl donor. Thus, generating agents that specifically target the SAM binding site of a particular methyltransferase such as PRMT3 may prove challenging. PRMT3 can catalyze mono- or asymmetrical di-methylation. PRMT3 methylates the 40S ribosomal protein S2 (rpS2) which is mandatory for mature 80S formation [65]. Additionally, PRMT3 is associated with oculopharyngeal muscular dystrophy likely via PAPN1 methylation, with cardiomyocyte excitability via sodium voltage channel expression, and methylates the H4 tail in vitro among many other substrates [66–69]. Therefore, inhibitors of PRMT3 have therapeutic potential in disorders involving these pathways.
Like other Type 1 PRMTs, PRMT3 is active as a homodimer (Figure 1) [70]. Using high throughput screening, moderately potent allosteric inhibitors of PRMT3 were discovered. Siaheyeva et.al. discovered a urethane compound from a library screen of over 16,000 compounds as a molecule that inhibited PRMT3 methylation of H4 tail peptide (1-24) [71]. After synthetic optimization, SGC707 (Figure 2 and Table 1) was developed as a highly potent and selective PRMT3 inhibitor probe. SGC707 was demonstrated to inhibit the methyltransferase activity of PRMT3. Consistent with earlier generation compounds, PRMT3 was bound over 15 A away from the active site and non-overlapping with the substrate peptide and SAM cofactor binding pockets. SGC707 binds in the large cavity of an inactive PRMT3 conformation between the β-barrel and the homodimerization arm near the dimer interface and likely forces the catalytically regulatory N-terminal α-helix into a conformationally inactive state. Enzymology confirmed the non-competitive inhibition mode. In contrast to parent compounds, SGC707 shows inhibition of H4R3me2a (asymmetric demethylation) in cells with an IC50 that is submicromolar [72]. This model of a small molecule binding to a conformationally dynamic and catalytically regulatory protein domain may be applicable to other epigenetic allosteric ligands.
Summary
KDM5A, p300, Gcn5, and PRMT3 are recent examples of histone modifying enzymes subject to allosteric regulation. In fact, many epigenetic enzymes possess non-catalytic domains whose biochemical functions are not well-characterized and may well participate in allosteric regulation. Emerging chemical tools to investigate allosteric mechanisms offer promise for clarifying biological function and opportunities for therapeutics development. As the catalytic functions and mechanisms of the wider array of histone modifying enzymes are increasingly defined, we believe that a rich tapestry of allosteric modulation will be identified and found actionable for pharmacologic application. Beyond small molecule probes, chemical techniques that allow for post-translational modifications to be introduced site-specifically into proteins [73,74] have the potential to greatly deepen our understanding of epigenetic regulation.
Highlights.
Allosteric ligands of histone modifying enzymes pose a new drug targeting strategy.
KDM5A, p300/CBP, Gcn5, and PRMT3 have allosteric ligands that modulate function.
SGC707, an allosteric ligand of PRMT3, functions by inducing conformational changes.
Acknowledgments
Funding: This work was supported by the National Institutes of Health [GM62437] and FAMRI Foundation and the Jane Coffin Childs Memorial Fund (B.E.Z.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
• Articles of special interest
- 1.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11:384–400. doi: 10.1038/nrd3674. [DOI] [PubMed] [Google Scholar]
- 2.Fierz B. Synthetic Chromatin Approaches To Probe the Writing and Erasing of Histone Modifications. ChemMedChem. 2014;9:495–504. doi: 10.1002/cmdc.201300487. [DOI] [PubMed] [Google Scholar]
- 3.Kouzarides T. Chromatin Modifications and Their Function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 4.Musselman CA, Lalonde M-E, Cote J, Kutateladze TG. Perceiving the epigenetic landscape through histone readers. Nat Struct Mol Biol. 2012;19:1218–1227. doi: 10.1038/nsmb.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith SG, Zhou M-M. The Bromodomain: A New Target in Emerging Epigenetic Medicine. ACS Chem Biol. 2016;11:598–608. doi: 10.1021/acschembio.5b00831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chi P, Allis CD, Wang GG. Covalent histone modifications — miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer. 2010;10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 8•.Dancy BM, Cole PA. Protein Lysine Acetylation by p300/CBP. Chem Rev. 2015;115:2419–2452. doi: 10.1021/cr500452k. A review of p300/CBP covering known ligands, both active site and allosteric, through the time of its publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sabari BR, Zhang D, Allis CD, Zhao Y. Metabolic regulation of gene expression through histone acylations. Nat Rev Mol Cell Biol. 2017;18:90–101. doi: 10.1038/nrm.2016.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hojfeldt JW, Agger K, Helin K. Histone lysine demethylases as targets for anticancer therapy. Nat Rev Drug Discov. 2013;12:917–930. doi: 10.1038/nrd4154. [DOI] [PubMed] [Google Scholar]
- 12.Sun X-J, Man N, Tan Y, Nimer SD, Wang L. The Role of Histone Acetyltransferases in Normal and Malignant Hematopoiesis. Front Oncol. 2015;5:108. doi: 10.3389/fonc.2015.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hu H, Qian K, Ho M-C, Zheng YG. Small Molecule Inhibitors of Protein Arginine Methyltransferases. Expert Opin Investig Drugs. 2016;25:335–358. doi: 10.1517/13543784.2016.1144747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marmorstein R, Zhou M-M. Writers and Readers of Histone Acetylation: Structure, Mechanism, and Inhibition. Cold Spring Harb Perspect Biol. 2014;6:a018762. doi: 10.1101/cshperspect.a018762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15•.Schapira M. Chemical Inhibition of Protein Methyltransferases. Cell Chem Biol. 2016;23:1067–1076. doi: 10.1016/j.chembiol.2016.07.014. A thorough review of all PRMT inhibitors describing their modes of action and binding sites. [DOI] [PubMed] [Google Scholar]
- 16.Fajer M, Meng Y, Roux B. The Activation of c-Src Tyrosine Kinase: Conformational Transition Pathway and Free Energy Landscape [Internet] J Phys Chem B. 2016 doi: 10.1021/acs.jpcb.6b08409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mayer BJ. The discovery of modular binding domains: building blocks of cell signalling. Nat Rev Mol Cell Biol. 2015;16:691–698. doi: 10.1038/nrm4068. [DOI] [PubMed] [Google Scholar]
- 18.Varkaris A, Katsiampoura AD, Araujo JC, Gallick GE, Corn PG. Src signaling pathways in prostate cancer. Cancer Metastasis Rev. 2014;33:595–606. doi: 10.1007/s10555-013-9481-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Christensen J, Agger K, Cloos PAC, Pasini D, Rose S, Sennels L, Rappsilber J, Hansen KH, Salcini AE, Helin K. RBP2 belongs to a family of demethylases, specific for tri-and dimethylated lysine 4 on histone 3. Cell. 2007;128:1063–1076. doi: 10.1016/j.cell.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 20.Brier A-SB, Loft A, Madsen JGS, Rosengren T, Nielsen R, Schmidt SF, Liu Z, Yan Q, Gronemeyer H, Mandrup S. The KDM5 family is required for activation of pro-proliferative cell cycle genes during adipocyte differentiation. Nucleic Acids Res. 2016 doi: 10.1093/nar/gkw1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang C, Wang J, Li J, Hu G, Shan S, Li Q, Zhang X. KDM5A controls bone morphogenic protein 2-induced osteogenic differentiation of bone mesenchymal stem cells during osteoporosis. Cell Death Dis. 2016;7:e2335. doi: 10.1038/cddis.2016.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao D, Zhang Q, Liu Y, Li X, Zhao K, Ding Y, Li Z, Shen Q, Wang C, Li N, et al. H3K4me3 Demethylase Kdm5a Is Required for NK Cell Activation by Associating with p50 to Suppress SOCS1. Cell Rep. 2016;15:288–299. doi: 10.1016/j.celrep.2016.03.035. [DOI] [PubMed] [Google Scholar]
- 23.Cao J, Liu Z, Cheung WKC, Zhao M, Chen SY, Chan SW, Booth CJ, Nguyen DX, Yan Q. Histone demethylase RBP2 is critical for breast cancer progression and metastasis. Cell Rep. 2014;6:868–877. doi: 10.1016/j.celrep.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li L, Wang L, Song P, Geng X, Liang X, Zhou M, Wang Y, Chen C, Jia J, Zeng J. Critical role of histone demethylase RBP2 in human gastric cancer angiogenesis. Mol Cancer. 2014;13:81. doi: 10.1186/1476-4598-13-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Teng Y-C, Lee C-F, Li Y-S, Chen Y-R, Hsiao P-W, Chan M-Y, Lin F-M, Huang H-D, Chen Y-T, Jeng Y-M, et al. Histone demethylase RBP2 promotes lung tumorigenesis and cancer metastasis. Cancer Res. 2013;73:4711–4721. doi: 10.1158/0008-5472.CAN-12-3165. [DOI] [PubMed] [Google Scholar]
- 26.Tumber A, Nuzzi A, Hookway ES, Hatch SB, Velupillai S, Johansson C, Kawamura A, Savitsky P, Yapp C, Szykowska A, et al. Potent and Selective KDM5 Inhibitor Stops Cellular Demethylation of H3K4me3 at Transcription Start Sites and Proliferation of MM1S Myeloma Cells. Cell Chem Biol. 2017;24:371–380. doi: 10.1016/j.chembiol.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang S, Wang Y, Wu H, Hu L. RBP2 induces epithelial-mesenchymal transition in non-small cell lung cancer. PloS One. 2013;8:e84735. doi: 10.1371/journal.pone.0084735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeng J, Ge Z, Wang L, Li Q, Wang N, Bjorkholm M, Jia J, Xu D. The histone demethylase RBP2 Is overexpressed in gastric cancer and its inhibition triggers senescence of cancer cells. Gastroenterology. 2010;138:981–992. doi: 10.1053/j.gastro.2009.10.004. [DOI] [PubMed] [Google Scholar]
- 29.Horton JR, Engstrom A, Zoeller EL, Liu X, Shanks JR, Zhang X, Johns MA, Vertino PM, Fu H, Cheng X. Characterization of a Linked Jumonji Domain of the KDM5/JARID1 Family of Histone H3 Lysine 4 Demethylases. J Biol Chem. 2016;291:2631–2646. doi: 10.1074/jbc.M115.698449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang F, Chandrasekharan MB, Chen Y-C, Bhaskara S, Hiebert SW, Sun Z-W. The JmjN domain of Jhd2 is important for its protein stability, and the plant homeodomain (PHD) finger mediates its chromatin association independent of H3K4 methylation. J Biol Chem. 2010;285:24548–24561. doi: 10.1074/jbc.M110.117333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pilka ES, James T, Lisztwan JH. Structural definitions of Jumonji family demethylase selectivity. Drug Discov Today. 2015;20:743–749. doi: 10.1016/j.drudis.2014.12.013. [DOI] [PubMed] [Google Scholar]
- 32.Shi Y, Whetstine JR. Dynamic Regulation of Histone Lysine Methylation by Demethylases. Mol Cell. 2007;25:1–14. doi: 10.1016/j.molcel.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 33.Musselman CA, Kutateladze TG. Handpicking epigenetic marks with PHD fingers. Nucleic Acids Res. 2011;39:9061–9071. doi: 10.1093/nar/gkr613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang GG, Song J, Wang Z, Dormann HL, Casadio F, Li H, Luo J-L, Patel DJ, Allis CD. Haematopoietic malignancies caused by dysregulation of a chromatin-binding PHD finger. Nature. 2009;459:847–851. doi: 10.1038/nature08036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li L, Greer C, Eisenman RN, Secombe J. Essential functions of the histone demethylase lid. PLoS Genet. 2010;6:e1001221. doi: 10.1371/journal.pgen.1001221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37•.Torres IO, Kuchenbecker KM, Nnadi CI, Fletterick RJ, Kelly MJS, Fujimori DG. Histone demethylase KDM5A is regulated by its reader domain through a positive-feedback mechanism. Nat Commun. 2015;6:6204. doi: 10.1038/ncomms7204. An elegant full description of the discovery of activation of KDM5A demethylation of H3 tail peptide and nucleosomes and by unmodified H3 peptide association with PHD1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.de Rooij JDE, Hollink IHIM, Arentsen-Peters STCJM, van Galen JF, Berna Beverloo H, Baruchel A, Trka J, Reinhardt D, Sonneveld E, Zimmermann M, et al. NUP98/JARID1A is a novel recurrent abnormality in pediatric acute megakaryoblastic leukemia with a distinct HOX gene expression pattern. Leukemia. 2013;27:2280–2288. doi: 10.1038/leu.2013.87. [DOI] [PubMed] [Google Scholar]
- 39.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A Chromatin Landmark and Transcription Initiation at Most Promoters in Human Cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Delvecchio M, Gaucher J, Aguilar-Gurrieri C, Ortega E, Panne D. Structure of the p300 catalytic core and implications for chromatin targeting and HAT regulation. Nat Struct Mol Biol. 2013;20:1040–1046. doi: 10.1038/nsmb.2642. [DOI] [PubMed] [Google Scholar]
- 41.Rack JGM, Lutter T, Kjareng Bjerga GE, Guder C, Ehrhardt C, Varv S, Ziegler M, Aasland R. The PHD finger of p300 Influences Its Ability to Acetylate Histone and Non-Histone Targets. J Mol Biol. 2014;426:3960–3972. doi: 10.1016/j.jmb.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 42.Thompson PR, Wang D, Wang L, Fulco M, Pediconi N, Zhang D, An W, Ge Q, Roeder RG, Wong J, et al. Regulation of the p300 HAT domain via a novel activation loop. Nat Struct Mol Biol. 2004;11:308–315. doi: 10.1038/nsmb740. [DOI] [PubMed] [Google Scholar]
- 43.Saint Just Ribeiro M, Hansson ML, Wallberg AE. A proline repeat domain in the Notch co-activator MAML1 is important for the p300-mediated acetylation of MAML1. Biochem J. 2007;404:289–298. doi: 10.1042/BJ20061900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44•.Hansson ML, Popko-Scibor AE, Saint Just Ribeiro M, Dancy BM, Lindberg MJ, Cole PA, Wallberg AE. The transcriptional coactivator MAML1 regulates p300 autoacetylation and HAT activity. Nucleic Acids Res. 2009;37:2996–3006. doi: 10.1093/nar/gkp163. This is the primary study showing the activation of p300 acetyltransferase activity by MAML1 including enzymology, ac-H3 co-localization, and reporter activation studies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mujtaba S, He Y, Zeng L, Yan S, Plotnikova O, Sachchidanand, Sanchez R, Zeleznik-Le NJ, Ronai Z’ev, Zhou M-M. Structural Mechanism of the Bromodomain of the Coactivator CBP in p53 Transcriptional Activation. Mol Cell. 2004;13:251–263. doi: 10.1016/s1097-2765(03)00528-8. [DOI] [PubMed] [Google Scholar]
- 46•.Gerona-Navarro G, Yoel-Rodriguez, Mujtaba S, Frasca A, Patel J, Zeng L, Plotnikov AN, Osman R, Zhou M-M. Rational Design of Cyclic Peptide Modulators of the Transcriptional Coactivator CBP: A New Class of p53 Inhibitors. J Am Chem Soc. 2011;133:2040–2043. doi: 10.1021/ja107761h. This research article describes the optimization of the p53 cyclic peptide and its effect on p53-CBP association and p53 reporter gene activation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chekler ELP, Pellegrino JA, Lanz TA, Denny RA, Flick AC, Coe J, Langille J, Basak A, Liu S, Stock IA, et al. Transcriptional Profiling of a Selective CREB Binding Protein Bromodomain Inhibitor Highlights Therapeutic Opportunities. Chem Biol. 2015;22:1588–1596. doi: 10.1016/j.chembiol.2015.10.013. [DOI] [PubMed] [Google Scholar]
- 48.Conery AR, Centore RC, Neiss A, Keller PJ, Joshi S, Spillane KL, Sandy P, Hatton C, Pardo E, Zawadzke L, et al. Bromodomain inhibition of the transcriptional coactivators CBP/EP300 as a therapeutic strategy to target the IRF4 network in multiple myeloma. eLife. 2016;5:e10483. doi: 10.7554/eLife.10483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ghosh S, Taylor A, Chin M, Huang H-R, Conery AR, Mertz JA, Salmeron A, Dakle PJ, Mele D, Cote A, et al. Regulatory T Cell Modulation by CBP/EP300 Bromodomain Inhibition. J Biol Chem. 2016;291:13014–13027. doi: 10.1074/jbc.M115.708560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hammitzsch A, Tallant C, Fedorov O, O’Mahony A, Brennan PE, Hay DA, Martinez FO, Al-Mossawi MH, de Wit J, Vecellio M, et al. CBP30, a selective CBP/p300 bromodomain inhibitor, suppresses human Th17 responses. Proc Natl Acad Sci. 2015;112:10768–10773. doi: 10.1073/pnas.1501956112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Picaud S, Fedorov O, Thanasopoulou A, Leonards K, Jones K, Meier J, Olzscha H, Monteiro O, Martin S, Philpott M, et al. Generation of a Selective Small Molecule Inhibitor of the CBP/p300 Bromodomain for Leukemia Therapy. Cancer Res. 2015;75:5106–5119. doi: 10.1158/0008-5472.CAN-15-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rooney TPC, Filippakopoulos P, Fedorov O, Picaud S, Cortopassi WA, Hay DA, Martin S, Tumber A, Rogers CM, Philpott M, et al. A Series of Potent CREBBP Bromodomain Ligands Reveals an Induced-Fit Pocket Stabilized by a Cation–π Interaction. Angew Chem Int Ed. 2014;53:6126–6130. doi: 10.1002/anie.201402750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53•.Zucconi BE, Luef B, Xu W, Henry RA, Nodelman IM, Bowman GD, Andrews AJ, Cole PA. Modulation of p300/CBP Acetylation of Nucleosomes by Bromodomain Ligand I-CBP112. Biochemistry. 2016;55:3727–3734. doi: 10.1021/acs.biochem.6b00480. This research article identifies and characterizes the activation of H3K18ac by p300/CBP induced by the I-CBP112 bromodomain ligand. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nguyen UTT, Bittova L, Muller MM, Fierz B, David Y, Houck-Loomis B, Feng V, Dann GP, Muir TW. Accelerated chromatin biochemistry using DNA-barcoded nucleosome libraries. Nat Methods. 2014;11:834–840. doi: 10.1038/nmeth.3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ragvin A, Valvatne H, Erdal S, Arskog V, Tufteland KR, Breen K, Oyan AM, Eberharter A, Gibson TJ, Becker PB, et al. Nucleosome Binding by the Bromodomain and PHD Finger of the Transcriptional Cofactor p300. J Mol Biol. 2004;337:773–788. doi: 10.1016/j.jmb.2004.01.051. [DOI] [PubMed] [Google Scholar]
- 56.Liu X, Tesfai J, Evrard YA, Dent SYR, Martinez E. c-Myc transformation domain recruits the human STAGA complex and requires TRRAP and GCN5 acetylase activity for transcription activation. J Biol Chem. 2003;278:20405–20412. doi: 10.1074/jbc.M211795200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sakai M, Tujimura-Hayakawa T, Yagi T, Yano H, Mitsushima M, Unoki-Kubota H, Kaburagi Y, Inoue H, Kido Y, Kasuga M, et al. The GCN5-CITED2-PKA signalling module controls hepatic glucose metabolism through a cAMP-induced substrate switch. Nat Commun. 2016;7:13147. doi: 10.1038/ncomms13147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58•.Cieniewicz AM, Moreland L, Ringel AE, Mackintosh SG, Raman A, Gilbert TM, Wolberger C, Tackett AJ, Taverna SD. The bromodomain of Gcn5 regulates site specificity of lysine acetylation on histone H3. Mol Cell Proteomics MCP. 2014;13:2896–2910. doi: 10.1074/mcp.M114.038174. This research quantifies the H3 acetylation preferences of Gcn5 and shows the key role of H3K14ac-bromodomain association in activating H3K18ac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trievel RC, Li F-Y, Marmorstein R. Application of a Fluorescent Histone Acetyltransferase Assay to Probe the Substrate Specificity of the Human p300/CBP-Associated Factor. Anal Biochem. 2000;287:319–328. doi: 10.1006/abio.2000.4855. [DOI] [PubMed] [Google Scholar]
- 60.Humphreys PG, Bamborough P, Chung C, Craggs PD, Gordon L, Grandi P, Hayhow TG, Hussain J, Jones KL, Lindon M, et al. Discovery of a Potent, Cell Penetrant, and Selective p300/CBP-Associated Factor (PCAF)/General Control Nonderepressible 5 (GCN5) Bromodomain Chemical Probe. J Med Chem. 2017;60:695–709. doi: 10.1021/acs.jmedchem.6b01566. [DOI] [PubMed] [Google Scholar]
- 61.Moustakim M, Clark PGK, Trulli L, Fuentes de Arriba AL, Ehebauer MT, Chaikuad A, Murphy EJ, Mendez-Johnson J, Daniels D, Hou C-FD, et al. Discovery of a PCAF Bromodomain Chemical Probe. Angew Chem Int Ed Engl. 2017;56:827–831. doi: 10.1002/anie.201610816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–520. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen T, Dent SYR. Chromatin modifiers and remodellers: regulators of cellular differentiation. Nat Rev Genet. 2014;15:93–106. doi: 10.1038/nrg3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Becker JS, Nicetto D, Zaret KS. H3K9me3-Dependent Heterochromatin: Barrier to Cell Fate Changes. Trends Genet. 2016;32:29–41. doi: 10.1016/j.tig.2015.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Swiercz R, Person MD, Bedford MT. Ribosomal protein S2 is a substrate for mammalian PRMT3 (protein arginine methyltransferase 3) Biochem J. 2005;386:85–91. doi: 10.1042/BJ20041466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Banerjee A, Apponi LH, Pavlath GK, Corbett AH. PABPN1: molecular function and muscle disease. FEBS J. 2013;280:4230–4250. doi: 10.1111/febs.12294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Beltran-Alvarez P, Espejo A, Schmauder R, Beltran C, Mrowka R, Linke T, Batlle M, Perez-Villa F, Perez GJ, Scornik FS, et al. Protein arginine methyl transferases-3 and -5 increase cell surface expression of cardiac sodium channel. FEBS Lett. 2013;587:3159–3165. doi: 10.1016/j.febslet.2013.07.043. [DOI] [PubMed] [Google Scholar]
- 68.Chen X, Niroomand F, Liu Z, Zankl A, Katus HA, Jahn L, Tiefenbacher CP. Expression of nitric oxide related enzymes in coronary heart disease. Basic Res Cardiol. 2006;101:346–353. doi: 10.1007/s00395-006-0592-5. [DOI] [PubMed] [Google Scholar]
- 69.Singh V, Miranda TB, Jiang W, Frankel A, Roemer ME, Robb VA, Gutmann DH, Herschman HR, Clarke S, Newsham IF. DAL-1/4. 1B tumor suppressor interacts with protein arginine N-methyltransferase 3 (PRMT3) and inhibits its ability to methylate substrates in vitro and in vivo. Oncogene. 2004;23:7761–7771. doi: 10.1038/sj.onc.1208057. [DOI] [PubMed] [Google Scholar]
- 70.Schapira M, Ferreira de Freitas R. Structural biology and chemistry of protein arginine methyltransferases. MedChemComm. 2014;5:1779–1788. doi: 10.1039/c4md00269e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71•.Siarheyeva A, Senisterra G, Allali-Hassani A, Dong A, Dobrovetsky E, Wasney GA, Chau I, Marcellus R, Hajian T, Liu F, et al. An Allosteric Inhibitor of Protein Arginine Methyltransferase 3. Structure. 2012;20:1425–1435. doi: 10.1016/j.str.2012.06.001. This research identifies the first allosteric PRMT3 inhibitor, crystallizes it bound to PRMT3 and shows its structure-activity relationship and its effect on enzyme kinetics. [DOI] [PubMed] [Google Scholar]
- 72•.Liu F, Li F, Ma A, Dobrovetsky E, Dong A, Gao C, Korboukh I, Liu J, Smil D, Brown PJ, et al. Exploiting an Allosteric Binding Site of PRMT3 Yields Potent and Selective Inhibitors. J Med Chem. 2013;56:2110–2124. doi: 10.1021/jm3018332. This research outlines the SAR in SGC707 development, divulges the crystal structure of SGC707 bound by PRMT3, and shows its specificity in inhibiting PRMT3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Muir TW, Sondhi D, Cole PA. Expressed protein ligation: A general method for protein engineering. Proc Natl Acad Sci. 1998;95:6705–6710. doi: 10.1073/pnas.95.12.6705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chin JW, Cropp TA, Anderson JC, Mukherji M, Zhang Z, Schultz PG. An Expanded Eukaryotic Genetic Code. Science. 2003;301:964–967. doi: 10.1126/science.1084772. [DOI] [PubMed] [Google Scholar]