


Abstract
We combine ion mobility spectrometry with cryogenic, messenger-tagging, infrared spectroscopy and mass spectrometry to identify different isomeric disaccharides of chondroitin sulfate (CS) and heparan sulfate (HS), which are representatives of two major subclasses of glycosaminoglycans. Our analysis shows that while CS and HS disaccharide isomers have similar drift times, they can be uniquely distinguished by their vibrational spectrum between ~3200 and 3700 cm−1 due to their different OH hydrogen-bonding patterns. We suggest that this combination of techniques is well suited to identify and characterize glycan isomers directly, which presents tremendous challenges for existing methods.
Graphical abstract
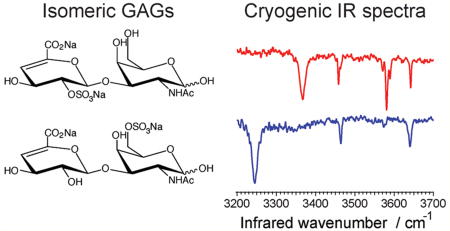
Glycosaminoglycans (GAGs) are linear polysaccharide chains of repeating disaccharide units of hexuronic acid and hexosamine that are found in extracellular matrices and on cell surfaces.1 Numerous factors contribute to the high structural diversity of GAGs, including the type of hexuronic acid or hexosamine moiety, variations in the glycosidic linkages between the disaccharide units, and chemical modifications of the monomeric units (e.g., sulfation).2 This isomeric heterogeneity inherent to GAGs is key to their biological function.3 However, the relationship between GAG structure and function remains elusive, in part because characterization of structural isomers remains a significant challenge for existing analytical techniques. Consequently, only a few function-specific sequences are currently known.4–8
Presently, GAG structures are characterized using a variety of tandem mass spectrometry (MS)-based techniques, including collision-induced dissociation (CID),9–15 electron detachment dissociation (EDD),16–20 infrared multiphoton dissociation (IRMPD),20 negative electron transfer dissociation (NETD),21,22 and photon-based methods, including ultraviolet photodissociation23 and activated-electron photodetachment.24 While these powerful approaches can provide important information about the structures of GAGs, limitations associated with each technique make their direct, unambiguous structural assignment elusive. For example, identification of sulfation sites with CID is difficult due to the lack of glycosidic and cross-ring cleavages as well as the loss of the labile sulfate group.25–27 In electron-based fragmentation methods such as EDD and ETD, the increase in the number of sulfate groups in the disaccharide unit reduces glycosidic and cross-ring cleavages, compromising the identification process.12,16 Since stereoisomers (e.g., glucose vs galactose vs mannose) generate similar fragments upon dissociation, MS-based methods cannot be used for their identification. Similarly, information regarding the connectivity of the glycosidic bond is not obtained easily and requires additional steps, such as chemical derivation, enzymatic digestion, and extensive MS/MS analysis.28
Perhaps the most important limitations encountered by analytical techniques in GAG analysis are that sample quantities are limited and sample workup often leads to a heterogeneous mixture of isomeric GAGs. To address such complex systems, MS-analysis is often preceded by a separation step. To this end, high-performance liquid chromatography (HPLC),29–31 capillary electrophoresis,32–35 hydrophilic interaction liquid chromatography,36 high-performance anion exchange chromatography,37 and porous graphite carbon liquid chromatography38,39 have all been used for the analysis of GAGs. Such methods improve the ability to characterize some isomers; however, coupling these strategies with MS is not straightforward and requires additional steps (e.g., removal of ion pairing reagents and salts). Consequently, novel analytical approaches are needed to separate and to identify isomeric GAG structures.
In the present study, we combine ion mobility spectrometry (IMS) with cryogenic, messenger-tagging, infrared (IR) spectroscopy and MS to elucidate the covalent structure of GAGs. Ion mobility is a gas-phase technique that separates ions based on the difference in their “shape”, which affects their mobility through a buffer gas. It can be easily combined with various types of MS, and IMS-MS techniques have been used to characterize structures for a range of biopolymers: folded and unfolded forms of proteins and protein complexes;40 peptides, including isomers that arise because of variation in amino acid sequence and composition;41 oligonucleotides;42 as well as oligosaccharides, glycans, and glycopeptides, including isomers arising from branching positions and linkage anomericity.43–50 While IMS has substantial potential for analyzing these biomolecules, with typical resolving power of 50–150, it falls short of unique characterization due to the lack of the structural resolution necessary to distinguish many closely related isomers. Infrared spectroscopy, on the other hand, can provide information on the distinct vibrational modes of the molecule, which are dependent on its structure. While a number of studies have been reported recently that combine IMS with IR spectroscopy for the study of various biomolecules,51–54 the broad bands in the IR spectrum decrease its ability to discern subtle differences in structure. One way to improve the resolution of the vibrational spectrum is by performing spectroscopic analysis on cryogenically cooled ions.55 We have recently combined ion mobility with cryogenic IR spectroscopy to probe the structure of peptides56,57 as well as a series of small glycans.58 Here, we report the use of a similar approach to distinguish five GAG isomers, three of which are chondroitin sulfate (CS) disaccharides and two of which are heparan sulfate (HS) disaccharides, as shown in Figure 1. CS and HS represent two major subclasses of GAGs that differ in their monosaccharide composition (N-acetylgalactosamine vs N-acetylglucosamine), glycosidic linkage (β(1–3) vs β(1–4)), and sulfate group location (6-O- vs 4-O- vs 2-O-sulfate). Recent work of Schindler et al. used IRMPD spectroscopy on some of these same GAG disaccharides in negative ion mode to establish spectroscopic markers for their sulfation patterns.59 While they clearly observe differences in spectra between the CS and HS GAGs, the vibrational spectra obtained at room temperature were relatively broad and not particularly diagnostic. We demonstrate here that cryogenic ion spectroscopy is sensitive to subtle structural differences and can easily distinguish all of the various types isomerisms. The ultimate goal of our multidimensional approach is to build a database that contains mass, collision cross sections, and IR spectra of various GAG standards with known structures, which would in turn be used to identify GAGs in a biological sample.
Figure 1.

Isomeric GAGs investigated in this work: (a–c) chondroitin sulfates; (d–e) heparan sulfates.
EXPERIMENTAL SECTION
We perform these experiments in a home-built instrument that combines an IMS drift tube with a cryogenic ion trap and a time-of-flight (TOF) mass spectrometer.57,60 This instrument provides a unique platform to measure the mass, collision cross section, and a cryogenic vibrational spectrum in a single experiment. Ions are generated using nanoelectrospray ionization (nESI) and pulsed into a 2-m long drift tube, which maintains a constant electric field gradient of ~10 V/cm. Under these weak-field conditions, ions traverse the drift tube with velocities proportional to their size-to-charge ratio. After exiting the drift tube, the ions are directed into a quadrupole mass filter and detected using a channeltron. The measured drift times (tD) can be converted into collision cross sections using the Mason–Schamp equation.61 The drift tube can also be operated in tandem IMS-IMS mode, with an ion funnel dividing it into two independent regions. In this case, the mobility-separated ions in the first section are selected with an ion gate and collisionally activated. Changes in the mobility distribution resulting from activation are then measured in the second section of the drift tube. For spectroscopic analysis, mobility- and mass-selected ions are sent o to a planar, cryogenic ion trap maintained at 13 K, where they are cooled through collisions with cold H2 buffer gas. Upon cooling, the H2 forms weakly bound adducts with the ions, which are later excited using infrared radiation. Resonant absorption of IR photons followed by infrared vibrational energy redistribution (IVR) leads to evaporation of the H2, which is measured as a depletion of that particular species in the mass spectrum. Infrared spectra are then obtained by plotting the depletion of selected adducts as a function of the laser wavenumber.62
Disodium salts of Δ-UA(6S)-GalNAc and Δ-UA(4S)-GalNAc (where ΔUA stands for 4,5 unsaturated uronic acid residue; GalNAc, N-acetyl galactosamine; 4S, 4-O-sulfate; and 6S, 6-O-sulfate) were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA) and used without further purification (minimum guaranteed purity ≥95%). Disodium salts of Δ-UA(2S)-GlcNAc and Δ-UA(6S)-GlcNAc (where GlcNAc stands for N-acetylglucosamine; 2S, 2-O-sulfate) were purchased from Dextra Laboratories (United Kingdom). The disodium salt of Δ-UA(2S)-GalNAc (minimum 90% purity) was purchased from Carbosynth Limited (United Kingdom). HPLC methanol and nuclease-free water were obtained from VWR International (Switzerland) and Ambion (Switzerland), respectively. Acetic acid (99%) was obtained from Sigma-Aldrich (Switzerland). Disaccharide salts were electrosprayed from 50:50 water:methanol solutions with 1% acetic acid. The concentration of disaccharide salt ranged from 200 to 600 µM. Metal-coated borosilicate emitters purchased from Thermo Scientific (Switzerland) were used for nanoelectrospray ionization.
In previous MS-based studies of HS disaccharides, considerable desalting was performed to remove sodium and potassium adducts that were detrimental to the identification process.27,63 However, desalting is not required in this study, and spectroscopic analysis is performed directly on the sodiated adduct. The use of a metal cation also simplifies the spectrum, as the interaction with the metal shifts some of the OH stretch bands outside the frequency region of our analysis. While the structure of the GAG complexed with a sodium cation will clearly be different than the uncomplexed GAG, our goal is to determine the covalent structure of the latter, and this is not altered by the presence of the cation. Because GAGs are acidic, one could also use our approach to study the negatively charged species, which we plan to do in the future.
RESULTS AND DISCUSSION
Chondroitin Sulfate Disaccharides
Figure 2a–c show the ion mobility distributions and vibrational spectra for [M − H + 2Na]+ ions (m/z 504) of the corresponding CS disaccharides shown in Figure 1a–c. Each CS disaccharide is composed of glucuronic acid attached to GalNAc through a β(1–3) linkage, but varies in its sulfation site (2S vs 4S vs 6S). Studies have shown that the precise position of the sulfate group in the monosaccharide unit of CS and HS is important for its biological function.64–67 It is thus crucial to obtain detailed information regarding the sulfation sites in CS and HS to fully understand how functional information is encoded in each GAG sequence. All three CS disaccharides display a single peak in the mobility distribution with drift times of 11.22, 10.45, and 10.72 ms for Δ-UA(2S)-GalNAc (Figure 2a), Δ-UA(4S)-GalNAc (Figure 2b), and Δ-UA(6S)-GalNAc (Figure 2c), respectively.
Figure 2.

Comparison of drift times and IR spectra of the isomeric CS and HS disaccharides. Key identifying bands in the IR spectra are indicated with dots.
The observed differences in drift times provide a way to distinguish CS isomers and suggest that the location of the sulfate group influences its overall shape, although the latter two are sufficiently close that it would be difficult to deconvolute them in a mixture. The mobility distributions do not change when ions corresponding to these isomers are selected and activated in the middle of the drift tube, which suggests that these conformers are stable, low-energy gas-phase conformers. We intentionally apply conditions that will activate the ions and anneal them into their lowest gas-phase conformation so that our measurements will be independent of electrospray conditions. The measured spectra are completely reproducible from day to day, despite slight differences in the harshness of the ion injection into the drift tube. The insensitivity of these annealed conformers to the source conditions allows us to make reproducible measurements that can be compared across different instruments. Numerous infrared active OH oscillators present throughout the backbone of CS and HS disaccharides make them excellent candidates to be probed with IR spectroscopy. The vibrational spectra of CS disaccharides (Figure 2a–c) show well-resolved peaks in the free (3580–3650 cm−1) and hydrogen-bonded (3200–3550 cm−1) OH stretch regions. These spectra are clearly distinct, consisting of unique bands that differ in both intensity and position, which is critical for using such data for unambiguous identification. The spectrum of Δ-UA(6S)-GalNAc (Figure 2c) is the simplest, containing three bands centered at 3244 cm−1, 3462 cm−1, and 3641 cm−1, whereas that of Δ-UA(4S)-GalNAc (Figure 2b) consists of five sharp transitions at 3407 cm−1, 3462 cm−1, 3568 cm−1, 3641 cm−1, and 3663 cm−1. In comparison to these two disaccharides, the spectrum of Δ-UA(2S)-GalNAc (Figure 2a) is fairly complex, but it still displays a series of sharp transitions centered at 3322 cm−1, 3459 cm−1, 3537 cm−1, 3582 cm−1, and 3643 cm−1. The spectral differences between these molecules result from a difference in the hydrogen-bonding patterns of the OH and NH oscillators, as hydrogen-bonding interactions cause vibrational bands to shift to lower energy and broaden. While the particular spectral pattern for a given GAG molecule depends upon its 3-dimensional structure, our purpose in measuring high-resolution vibrational spectra is to provide unique fingerprints for identification purposes (i.e., to determine the precise isomer). It is not our intention to extract a 3-dimensional structure by comparison with quantum chemical calculations, as is typically done for peptides. Instead, by developing a database that includes the mass, the cross section (from ion mobility), and the IR spectrum of standard GAGs, we have a method to identify an unknown GAG in a given mixture. For this purpose, it is critical that the spectral bands are sharp and distinctive for each molecule, which is clearly the case. Indeed in each spectrum, the full-width at half-maximum of the widest bands is ≤20 cm−1, with some being as narrow as 4 cm−1. The presence of these sharp peaks highlights the role of cryogenic cooling in increasing the resolution of the IR spectra by narrowing the absorption bands. This improved resolution increases the sensitivity of this technique to subtle structural differences and is essential for identification purposes.
Heparan Sulfate Disaccharides
Ion mobility distributions and vibrational spectra for [M − H + 2Na]+ ions (m/z 504) of isomeric HS disaccharides are shown in Figure 2d–e, with the corresponding structures shown in Figure 1d–e. Heparan sulfate disaccharide is composed of glucuronic acid attached to GlcNAc through an β(1–4) linkage with either 6S or 2S groups. Δ-UA(2S)-GlcNAc (Figure 2d) displays a single peak (10.98 ms) in the mobility distribution. Two different conformations, denoted as A (10.66 ms) and B (11.08 ms), are present in the mobility distribution of Δ-UA(6S)-GlcNAc (Figure 2e). Upon activation, these two conformers do not interconvert, indicating the presence of two structural isomers with a high barrier for interconversion that may arise from two competing metal binding sites on the HS backbone. The hydroxyl group at the C-1 position of GlcNAc, which can be in either the α- or β-position, may also influence the metal-binding site. Since the measured drift times of Δ-UA(2S)-GlcNAc and Δ-UA(6S)-GlcNAc are close to each other, distinguishing them based on IMS data is not possible.
Figure 2d–e also shows the IR spectra recorded for the HS disaccharides. In the case of Δ-UA(6S)-GlcNAc (Figure 2e), an IR spectrum of each conformer is obtained by selecting ions with a specific mobility in the middle of the drift tube before spectroscopic analysis. Similar to CS disaccharides, the spectra for isomeric HS disaccharides are distinctly different from each other, resulting from different hydrogen-bonding patterns of the OH and NH oscillators. Qualitative analysis of the high-frequency region shows the presence of two free OH stretches for Δ-UA(2S)-GlcNAc (Figure 2d; 3581 and 3641 cm−1), and for conformer B (Figure 2e) of Δ-UA(6S)-GlcNAc (3581 and 3661 cm−1). Conformer A of Δ-UA(6S)-GlcNAc contains only one free OH stretch (3575 cm−1).
In the lower frequency region, two sharp transitions (3440 and 3498 cm−1) are observed for conformer A of Δ-UA(6S)-GlcNAc (Figure 2e). On the other hand, conformer B of Δ-UA(6S)-GlcNAc contains only weak transitions in this region, occurring at 3440 cm−1, 3458 cm−1, and 3469 cm−1. These two conformers can be distinguished from each other based on the peaks at 3661 and 3498 cm−1. The broad feature in the hydrogen-bonded region of Δ-UA(2S)-GlcNAc (Figure 2d; 3367 cm−1) is absent for both conformers of Δ-UA(6S)-GlcNAc.
Comparative Study of CS and HS Disaccharides with the Same Sulfation Sites
The isomeric species Δ-UA(6S)-GalNAc and Δ-UA(6S)-GlcNAc, which have the same sulfation site (6-O-sulfate) but different linkage and monosaccharide composition, could not be distinguished using IMS alone due to overlapping drift times of 10.72 ms for Δ-UA(6S)-GalNAc (Figure 2c) as compared to 10.66 and 11.08 ms for the two conformers of Δ-UA(6S)-GlcNAc (Figure 2e). However, the IR spectra of these two species contain prominent peaks that can be used to distinguish them. The broad feature at 3244 cm−1 present in the spectrum of Δ-UA(6S)-GalNAc (Figure 2c) is absent for both conformers of Δ-UA(6S)-GlcNAc (Figure 2e). Moreover, the peaks at 3498 and 3661 cm−1 are unique to Δ-UA(6S)-GlcNAc, and that at 3641 cm−1 is unique to Δ-UA(6S)-GalNAc.
In contrast, CS and HS disaccharides with a 2-O-sulfate group exhibit different drift times: 11.22 ms for CS (Figure 2a) and 10.98 ms for HS (Figure 2d), which allows for identification based on IMS alone. When IR spectra of these isomers are compared, we find that the broad band in the low-frequency region appears at a different position: 3322 cm−1 for Δ-UA(2S)-GalNAc (Figure 2a) and 3367 cm−1 for Δ-UA(2S)-GlcNAc (Figure 2d), indicating the presence of stronger hydrogen-bonding interactions in the former. Moreover, the transitions at 3537 and 3388 cm−1 are unique to Δ-UA(2S)-GalNAc. Our findings show that in this case both IMS and IR spectroscopy can be effectively used to identify compositional and configurational isomers of CS and HS.
Comparing All Five Isomeric/Isobaric Disaccharides of CS and HS
With the exception of Δ-UA(2S)-GalNAc (Figure 2a) and Δ-UA(4S)-GalNAc (Figure 2b), the drift times of other CS and HS disaccharides are within 2% of each other, preventing IMS from being used to distinguish these isomers. While large differences in drift times are not expected from such small disaccharides with subtle structural differences, the use of sodium as a cationizing agent might even reduce the structural differences in isomers due to metal-induced compaction.45,68 Based on previous studies, possible ways to improve isomeric separation in IMS include the use of carbon dioxide as a drift gas,69,70 study of deprotonated species43 and different metal adducts,44,71 as well as application of alternative mobility approaches.72–74 Nevertheless, by comparing the IR spectra of all five isomeric disaccharides (Figure 2), one can see that each isomer gives rise to at least one unique band. Moreover, the entire spectrum of each disaccharide serves as a unique fingerprint by which one can identify it. This is an important finding for our ultimate goal of identifying GAG structures from a biological sample through a database search based on mass, collision cross section, and IR spectrum. If each GAG standard in our database displays such drastic differences in its vibrational signatures, this should allow for unambiguous identification of unknown GAGs in a mixture.
CONCLUSION
Considering the complexity faced by current analytical techniques in identifying isomeric GAGs, the rapid gas-phase separation provided by IMS together with the distinct vibrational patterns provided by cryogenic IR spectroscopy shows potential to become an important tool for comprehensive analysis of GAGs. In the present study, we have used this combined approach to unambiguously identify isomers of CS and HS disaccharides. While subtle structural differences of these isomeric disaccharides in their connectivity, composition, and sulfate group location do not lead to large differences in their drift times, the IR spectral fingerprint obtained for each disaccharide is clearly distinct, allowing for unique identification. The improved resolution of vibrational spectra due to the application of cryogenic IR spectroscopy makes this technique highly sensitive to subtle structural changes, which is of critical importance when closely related isomeric species are to be identified. Ultimately, the constitution of a database containing the mass, cross section, and IR spectrum of GAG standards with known chemical structural composition could prove to be an important tool for the identification of GAG structures in biological samples.
Acknowledgments
T.R.R. would like to thank the EPFL and the Swiss National Science Foundation (grant number 200020_165908) for their financial support of this work. D.E.C. would like to thank the National Institutes of Health for partial support of this work through grant (R01 GM117207-01). N.K. was supported by a Robert & Marjorie Mann Fellowship from Indiana University.
Footnotes
The authors declare no competing financial interest.
References
- 1.Kjellen L, Lindahl U. Annu. Rev. Biochem. 1991;60:443–475. doi: 10.1146/annurev.bi.60.070191.002303. [DOI] [PubMed] [Google Scholar]
- 2.Turnbull J, Powell A, Guimond S. Trends Cell Biol. 2001;11:75–82. doi: 10.1016/s0962-8924(00)01897-3. [DOI] [PubMed] [Google Scholar]
- 3.Hacker U, Nybakken K, Perrimon N. Nat. Rev. Mol. Cell Biol. 2005;6:530–541. doi: 10.1038/nrm1681. [DOI] [PubMed] [Google Scholar]
- 4.Gama CI, Tully SE, Sotogaku N, Clark PM, Rawat M, Vaidehi N, Goddard WA, 3rd, Nishi A, Hsieh-Wilson LC. Nat. Chem. Biol. 2006;2:467–473. doi: 10.1038/nchembio810. [DOI] [PubMed] [Google Scholar]
- 5.Kitagawa H, Tsutsumi K, Tone Y, Sugahara K. J. Biol. Chem. 1997;272:31377–31381. doi: 10.1074/jbc.272.50.31377. [DOI] [PubMed] [Google Scholar]
- 6.Plaas AH, West LA, Wong-Palms S, Nelson FR. J. Biol. Chem. 1998;273:12642–12649. doi: 10.1074/jbc.273.20.12642. [DOI] [PubMed] [Google Scholar]
- 7.Ly M, Leach FE, 3rd, Laremore TN, Toida T, Amster IJ, Linhardt RJ. Nat. Chem. Biol. 2011;7:827–833. doi: 10.1038/nchembio.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guerrini M, Naggi A, Guglieri S, Santarsiero R, Torri G. Anal. Biochem. 2005;337:35–47. doi: 10.1016/j.ab.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 9.Zaia J, McClellan JE, Costello CE. Anal. Chem. 2001;73:6030–6039. doi: 10.1021/ac015577t. [DOI] [PubMed] [Google Scholar]
- 10.Zaia J, Li XQ, Chan SY, Costello CE. J. Am. Soc. Mass Spectrom. 2003;14:1270–1281. doi: 10.1016/S1044-0305(03)00541-5. [DOI] [PubMed] [Google Scholar]
- 11.Zaia J, Costello CE. Anal. Chem. 2003;75:2445–2455. doi: 10.1021/ac0263418. [DOI] [PubMed] [Google Scholar]
- 12.Kailemia MJ, Li L, Ly M, Linhardt RJ, Amster IJ. Anal. Chem. 2012;84:5475–5478. doi: 10.1021/ac3015824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saad OM, Leary JA. Anal. Chem. 2005;77:5902–5911. doi: 10.1021/ac050793d. [DOI] [PubMed] [Google Scholar]
- 14.Meissen JK, Sweeney MD, Girardi M, Lawrence R, Esko JD, Leary JA. J. Am. Soc. Mass Spectrom. 2009;20:652–657. doi: 10.1016/j.jasms.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Desaire H, Leary JA. J. Am. Soc. Mass Spectrom. 2000;11:916–920. doi: 10.1016/S1044-0305(00)00168-9. [DOI] [PubMed] [Google Scholar]
- 16.Wolff JJ, Amster IJ, Chi L, Linhardt RJ. J. Am. Soc. Mass Spectrom. 2007;18:234–244. doi: 10.1016/j.jasms.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolff JJ, Chi L, Linhardt RJ, Amster IJ. Anal. Chem. 2007;79:2015–2022. doi: 10.1021/ac061636x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leach FE, Wolff JJ, Laremore TN, Linhardt RJ, Amster IJ. Int. J. Mass Spectrom. 2008;276:110–115. doi: 10.1016/j.ijms.2008.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wolff JJ, Laremore TN, Busch AM, Linhardt RJ, Amster IJ. J. Am. Soc. Mass Spectrom. 2008;19:294–304. doi: 10.1016/j.jasms.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leach FE, Xiao Z, Laremore TN, Linhardt RJ, Amster IJ. Int. J. Mass Spectrom. 2011;308:253–259. doi: 10.1016/j.ijms.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leach FE, III, Ly M, Laremore TN, Wolff JJ, Perlow J, Linhardt RJ, Amster IJ. J. Am. Soc. Mass Spectrom. 2012;23:1488–1497. doi: 10.1007/s13361-012-0428-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leach FE, III, Wolff JJ, Xiao Z, Ly M, Laremore TN, Arungundram S, Al-Mafraji K, Venot A, Boons G-J, Linhardt RJ. Eur. Mass Spectrom. 2011;17:167. doi: 10.1255/ejms.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Racaud A, Antoine R, Joly L, Mesplet N, Dugourd P, Lemoine J. J. Am. Soc. Mass Spectrom. 2009;20:1645–1651. doi: 10.1016/j.jasms.2009.04.022. [DOI] [PubMed] [Google Scholar]
- 24.Racaud A, Antoine R, Dugourd P, Lemoine J. J. Am. Soc. Mass Spectrom. 2010;21:2077–2084. doi: 10.1016/j.jasms.2010.08.021. [DOI] [PubMed] [Google Scholar]
- 25.Chi L, Amster J, Linhardt RJ. Curr. Anal. Chem. 2005;1:223–240. doi: 10.2174/157341105774573929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Naggar EF, Costello CE, Zaia J. J. Am. Soc. Mass Spectrom. 2004;15:1534–1544. doi: 10.1016/j.jasms.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 27.Jones CJ, Beni S, Limtiaco JF, Langeslay DJ, Larive CK. Annu. Rev. Anal. Chem. 2011;4:439–465. doi: 10.1146/annurev-anchem-061010-113911. [DOI] [PubMed] [Google Scholar]
- 28.Prien JM, Ashline DJ, Lapadula AJ, Zhang H, Reinhold VN. J. Am. Soc. Mass Spectrom. 2009;20:539–556. doi: 10.1016/j.jasms.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamada S, Sakamoto K, Tsuda H, Yoshida K, Sugiura M, Sugahara K. Biochemistry. 1999;38:838–847. doi: 10.1021/bi981889n. [DOI] [PubMed] [Google Scholar]
- 30.Chai W, Luo J, Lim CK, Lawson AM. Anal. Chem. 1998;70:2060–2066. doi: 10.1021/ac9712761. [DOI] [PubMed] [Google Scholar]
- 31.Chai W, Kogelberg H, Lawson AM. Anal. Biochem. 1996;237:88–102. doi: 10.1006/abio.1996.0205. [DOI] [PubMed] [Google Scholar]
- 32.Venkataraman G, Shriver Z, Raman R, Sasisekharan R. Science (Washington, DC, U. S.) 1999;286:537–542. doi: 10.1126/science.286.5439.537. [DOI] [PubMed] [Google Scholar]
- 33.Mischnick P. Angew. Chem., Int. Ed. 2000;39:1222–1224. doi: 10.1002/(sici)1521-3773(20000403)39:7<1222::aid-anie1222>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 34.Volpi N, Maccari F, Linhardt RJ. Electrophoresis. 2008;29:3095–3106. doi: 10.1002/elps.200800109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zamfir A, Seidler DG, Kresse H, Peter-Katalinić J. Rapid Commun. Mass Spectrom. 2002;16:2015–2024. doi: 10.1002/rcm.820. [DOI] [PubMed] [Google Scholar]
- 36.Staples GO, Bowman MJ, Costello CE, Hitchcock AM, Lau JM, Leymarie N, Miller C, Naimy H, Shi X, Zaia J. Proteomics. 2009;9:686–695. doi: 10.1002/pmic.200701008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bruggink C, Wuhrer M, Koeleman CA, Barreto V, Liu Y, Pohl C, Ingendoh A, Hokke CH, Deelder AM. J. Chromatogr. B. Anal. Technol. Biomed. Life Sci. 2005;829:136–143. doi: 10.1016/j.jchromb.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 38.Costell CE, Contado-Miller JM, Cipollo JF. J. Am. Soc. Mass Spectrom. 2007;18:1799–1812. doi: 10.1016/j.jasms.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thomsson KA, Karlsson NG, Hansson GC. J. Chromatogr. A. 1999;854:131–139. doi: 10.1016/s0021-9673(99)00625-1. [DOI] [PubMed] [Google Scholar]
- 40.Uetrecht C, Rose RJ, van Duijn E, Lorenzen K, Heck AJ. Chem. Soc. Rev. 2010;39:1633–1655. doi: 10.1039/b914002f. [DOI] [PubMed] [Google Scholar]
- 41.Bohrer BC, Merenbloom SI, Koeniger SL, Hilderbrand AE, Clemmer DE. Annu. Rev. Anal. Chem. 2008;1:293–327. doi: 10.1146/annurev.anchem.1.031207.113001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gidden J, Bushnell JE, Bowers MT. J. Am. Chem. Soc. 2001;123:5610–5611. doi: 10.1021/ja015940d. [DOI] [PubMed] [Google Scholar]
- 43.Hofmann J, Hahm HS, Seeberger PH, Pagel K. Nature. 2015;526:241–244. doi: 10.1038/nature15388. [DOI] [PubMed] [Google Scholar]
- 44.Fenn LS, McLean JA. Phys. Chem. Chem. Phys. 2011;13:2196–2205. doi: 10.1039/c0cp01414a. [DOI] [PubMed] [Google Scholar]
- 45.Lee S, Wyttenbach T, Bowers MT. Int. J. Mass Spectrom. Ion Processes. 1997;167:605–614. [Google Scholar]
- 46.Plasencia MD, Isailovic D, Merenbloom SI, Mechref Y, Novotny MV, Clemmer DE. J. Am. Soc. Mass Spectrom. 2008;19:1706–1715. doi: 10.1016/j.jasms.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zucker SM, Lee S, Webber N, Valentine SJ, Reilly JP, Clemmer DE. J. Am. Soc. Mass Spectrom. 2011;22:1477. doi: 10.1007/s13361-011-0179-8. [DOI] [PubMed] [Google Scholar]
- 48.Lee S, Valentine SJ, Reilly JP, Clemmer DE. Int. J. Mass Spectrom. 2012;309:161–167. doi: 10.1016/j.ijms.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Morrison KA, Clowers BH. J. Am. Soc. Mass Spectrom. 2017;28:1–6. doi: 10.1007/s13361-017-1621-3. [DOI] [PubMed] [Google Scholar]
- 50.Campbell JL, Baba T, Liu C, Lane CS, Le Blanc JY, Hager JW. J. Am. Soc. Mass Spectrom. 2017;28:1–8. doi: 10.1007/s13361-017-1663-6. [DOI] [PubMed] [Google Scholar]
- 51.Hernandez O, Isenberg S, Steinmetz V, Glish GL, Maitre P. J. Phys. Chem. A. 2015;119:6057–6064. doi: 10.1021/jp511975f. [DOI] [PubMed] [Google Scholar]
- 52.Seo J, Hoffmann W, Warnke S, Huang X, Gewinner S, Schollkopf W, Bowers MT, von Helden G, Pagel K. Nat. Chem. 2017;9:39–44. doi: 10.1038/nchem.2615. [DOI] [PubMed] [Google Scholar]
- 53.Seo J, Hoffmann W, Warnke S, Bowers MT, Pagel K, von Helden G. Angew. Chem., Int. Ed. 2016;55:14173–14176. doi: 10.1002/anie.201606029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Warnke S, Hoffmann W, Seo J, De Genst E, von Helden G, Pagel K. J. Am. Soc. Mass Spectrom. 2017;28:638. doi: 10.1007/s13361-016-1551-5. [DOI] [PubMed] [Google Scholar]
- 55.Boyarkin OV, Mercier SR, Kamariotis A, Rizzo TR. J. Am. Chem. Soc. 2006;128:2816–2817. doi: 10.1021/ja058383u. [DOI] [PubMed] [Google Scholar]
- 56.Voronina L, Masson A, Kamrath M, Schubert F, Clemmer D, Baldauf C, Rizzo T. J. Am. Chem. Soc. 2016;138:9224–9233. doi: 10.1021/jacs.6b04550. [DOI] [PubMed] [Google Scholar]
- 57.Masson A, Kamrath MZ, Perez MA, Glover MS, Rothlisberger U, Clemmer DE, Rizzo TR. J. Am. Soc. Mass Spectrom. 2015;26:1444–1454. doi: 10.1007/s13361-015-1172-4. [DOI] [PubMed] [Google Scholar]
- 58.Masellis C, Khanal N, Kamrath MZ, Clemmer DE, Rizzo TR. JASMS. 2017 doi: 10.1007/s13361-017-1728-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schindler B, Barnes L, Gray CJ, Chambert S, Flitsch SL, Oomens J, Daniel R, Allouche AR, Compagnon I. J. Phys. Chem. A. 2017;121:2114–2120. doi: 10.1021/acs.jpca.6b11642. [DOI] [PubMed] [Google Scholar]
- 60.Koeniger SL, Merenbloom SI, Valentine SJ, Jarrold MF, Udseth HR, Smith RD, Clemmer DE. Anal. Chem. 2006;78:4161–4174. doi: 10.1021/ac051060w. [DOI] [PubMed] [Google Scholar]
- 61.Mason EA, McDaniel EW. NASA STI/Recon Technical Report A. 1988:89. [Google Scholar]
- 62.Kamrath MZ, Garand E, Jordan PA, Leavitt CM, Wolk AB, Van Stipdonk MJ, Miller SJ, Johnson MA. J. Am. Chem. Soc. 2011;133:6440–6448. doi: 10.1021/ja200849g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Saad OM, Leary JA. Anal. Chem. 2003;75:2985–2995. doi: 10.1021/ac0340455. [DOI] [PubMed] [Google Scholar]
- 64.Shipp EL, Hsieh-Wilson LC. Chem. Biol. 2007;14:195–208. doi: 10.1016/j.chembiol.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 65.Ashikari-Hada S, Habuchi H, Kariya Y, Itoh N, Reddi AH, Kimata K. J. Biol. Chem. 2004;279:12346–12354. doi: 10.1074/jbc.M313523200. [DOI] [PubMed] [Google Scholar]
- 66.Habuchi H, Suzuki S, Saito T, Tamura T, Harada T, Yoshida K, Kimata K. Biochem. J. 1992;285(Pt 3):805–813. doi: 10.1042/bj2850805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maeda N, Ishii M, Nishimura K, Kamimura K. Neurochem. Res. 2011;36:1228–1240. doi: 10.1007/s11064-010-0324-y. [DOI] [PubMed] [Google Scholar]
- 68.Gray C, Thomas B, Upton R, Migas L, Eyers C, Barran P, Flitsch S. Biochim. Biophys. Acta, Gen. Subj. 2016;1860:1688–1709. doi: 10.1016/j.bbagen.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 69.Lalli PM, Corilo YE, Fasciotti M, Riccio MF, de Sa GF, Daroda RJ, Souza GH, McCullagh M, Bartberger MD, Eberlin MN, Campuzano ID. J. Mass Spectrom. 2013;48:989–997. doi: 10.1002/jms.3245. [DOI] [PubMed] [Google Scholar]
- 70.Fasciotti M, Sanvido GB, Santos VG, Lalli PM, McCullagh M, de Sa GF, Daroda RJ, Peter MG, Eberlin MN. J. Mass Spectrom. 2012;47:1643–1647. doi: 10.1002/jms.3089. [DOI] [PubMed] [Google Scholar]
- 71.Huang Y, Dodds ED. Anal. Chem. 2013;85:9728–9735. doi: 10.1021/ac402133f. [DOI] [PubMed] [Google Scholar]
- 72.Pu Y, Ridgeway ME, Glaskin RS, Park MA, Costello CE, Lin C. Anal. Chem. 2016;88:3440–3443. doi: 10.1021/acs.analchem.6b00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gabryelski W, Froese KL. J. Am. Soc. Mass Spectrom. 2003;14:265–277. doi: 10.1016/S1044-0305(03)00002-3. [DOI] [PubMed] [Google Scholar]
- 74.Kailemia MJ, Ruhaak LR, Lebrilla CB, Amster IJ. Anal. Chem. 2014;86:196–212. doi: 10.1021/ac403969n. [DOI] [PMC free article] [PubMed] [Google Scholar]