

Abstract
Antipsychotic drugs remain the standard for schizophrenia treatment. Despite their effectiveness in treating hallucinations and delusions, prolonged exposure to antipsychotic medications leads to cognitive deficits in both schizophrenia patients and animal models. The molecular mechanisms underlying these negative effects on cognition remain to be elucidated. Here we demonstrate that chronic antipsychotic drug exposure increases nuclear translocation of NF-κB in both mouse and human frontal cortex, a trafficking event triggered via 5-HT2A-receptor-dependent downregulation of the NF-κB repressor IκBα. This upregulation of NF-κB activity led to its increased binding at the Hdac2 promoter, thereby augmenting Hdac2 transcription. Deletion of HDAC2 in forebrain pyramidal neurons prevented the negative effects of antipsychotic treatment on synaptic remodeling and cognition. Conversely, virally mediated activation of NF-κB signaling decreased cortical synaptic plasticity via HDAC2. Together, these observations may aid in developing therapeutic strategies to improve the outcome of schizophrenia treatment.
Schizophrenia affects up to 1% of the population, in many cases causing life-long disability1. Antipsychotic drugs, including both typical (e.g., haloperidol) and atypical (e.g., clozapine), were serendipitously discovered in the mid-twentieth century2. Although chronic administration of these agents produces significant reduction or even complete remission of psychotic symptoms such as hallucinations and delusions3,4, the majority of schizophrenia patients have cognitive deficits, which often do not improve upon antipsychotic drug treatment; some patients even deteriorate5-8. This clinical observation is supported by previous reports that long-term antipsychotic medication leads to a decline of cognitive abilities in rodent models9,10. Cognitive impairment in schizophrenia patients is selective and includes impairment in attention, executive function and working memory. Given that these deficits in cognitive processes account for a significant proportion of psychosocial disabilities in schizophrenia patients11,12, it is notable that no study so far has determined the basic molecular mechanism by which chronic antipsychotic drug treatment may lead to a negative effect on cognitive function.
The epigenetic removal of acetyl groups from histone tails by histone deacetylases (HDACs) promotes a compact chromatin structure that represses gene transcription13. Endogenous HDAC2 activity plays a critical role in cognitive performance and appears to generally restrain both structural and functional synaptic plasticity14,15. We have previously showed that chronic treatment with the atypical antipsychotic drug clozapine leads toward a selective augmentation of Hdac2 transcription in mouse frontal cortex16,17, a brain region that is important in cognition and perception and that has been implicated in schizophrenia and antipsychotic drug responses18-21. It remains unknown, however, whether this upregulation of HDAC2 expression is involved in either the therapeutic or negative side effects of atypical antipsychotic medications. Furthermore, the neural substrates and upstream signaling leading to such upregulation of Hdac2 transcription have not been resolved.
In this study, we show that augmentation of HDAC2 expression upon chronic atypical antipsychotic treatment maladaptively affects cortical synaptic remodeling and cognitive processes through a signaling mechanism that involves serotonin 5-HT2A-receptor-dependent upregulation of NF-κB activity.
RESULTS
Chronic atypical antipsychotics upregulate HDAC2 in frontal cortex pyramidal neurons
We had previously shown that chronic treatment with the atypical antipsychotic drug clozapine induces upregulation of Hdac2 transcription in mouse frontal cortex16. We found here similar effects after chronic treatment with the atypical antipsychotic and antipsychotic-like drugs clozapine, risperidone, quetiapine, sulpiride and volinan-serin (M100907) but not with the typical antipsychotic haloperidol (Fig. 1a). Hdac1 and Hdac8 mRNAs were unaffected after treatment with either atypical or typical antipsychotics (Fig. 1b,c). Expression of Hdac1, Hdac2 and Hdac8 mRNA was decreased after chronic treatment with the antidepressant drug fluoxetine (Fig. 1a–c).
Figure 1.
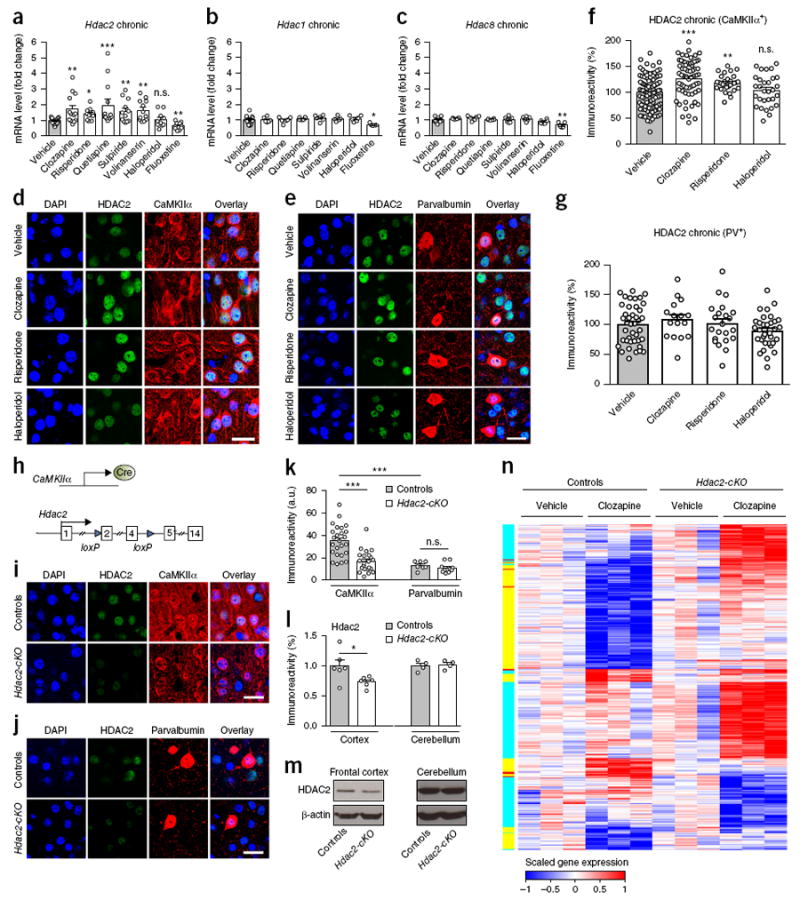
Chronic atypical antipsychotic drug treatment up-regulates HDAC2 in cortical pyramidal neurons. (a–c) Increased expression of Hdac2 mRNA in frontal cortex after chronic atypical antipsychotic treatment. Mice were chronically (21 d) treated (by intraperitoneal injection, i.p.) with clozapine (10 mg/kg), risperidone (4 mg/kg), quetiapine (10 mg/kg), sulpiride (10 mg/kg), volinanserin (1 mg/kg), haloperidol (1 mg/kg), or fluoxetine (20 mg/kg), or vehicle, and killed for analysis 1 d after the last injection. Expression of Hdac1 (n = 5–16 mice per experimental condition), Hdac2 (n = 10–21 mice per experimental condition) and Hdac8 (n = 5–11 mice per experimental condition) was assessed by quantitative real-time PCR (qRT-PCR). (d–g) HDAC2 immunoreactivity levels in CaMKIIα+ or PV+ neurons in mouse frontal cortex after chronic treatment with clozapine, risperidone, haloperidol or vehicle. Representative immunohistochemical images (d,e; see also Supplementary Fig. 1a,b). Quantitative assessment (CaMKIIα+ n = 30–102 cells from 5 or 6 mice per experimental condition; PV+: n = 16–41 cells from 5 or 6 mice per experimental condition; f,g). (h) Strategy for deleting forebrain pyramidal HDAC2 function. (i–k) HDAC2 immunoreactivity levels in frontal cortex CaMKIIα+ or PV+ neurons of Hdac2loxP/loxP:CaMKIIα-Cre mice (Hdac2-cKO) and control littermates. Quantitative assessment (CaMKIIα+, n = 25–27 cells from 5 mice per experimental condition; PV+, n = 7–10 cells from 5 mice per experimental condition; k). Representative immunohistochemical images (I,j). (l,m) Western blots showed decreased HDAC2 protein levels in the frontal cortex, but not cerebellum, of Hdac2-cKO compared to control littermates (n = 4–6 mice per experimental condition). Representative immunoblots are shown (m). (n) Comparative heat maps displaying genes significantly regulated (false discovery rate of 0.05 and fold change (log2) of at least 0.02) by chronic clozapine treatment in either Hdac2-cKO mice or control littermates (n = 3 libraries per treatment per genotype). For data visualization purpose of the heat map, rlog-transformed data from the DESeq2 package were then scaled with a maximum value of 1 and a minimum value of –1 for each gene. Sidebar shows genes significantly regulated by chronic clozapine treatment in Hdac2-cKO (cyan) or control (yellow) mice or significantly regulated by chronic clozapine treatment in both Hdac2-cKO and control mice (red). Mean ± s.e.m. *P < 0.05; **P < 0.01; ***P < 0.001; n.s., not significant. One-way ANOVA with Bonferroni’s post hoc test (a: P = 0.02, F7,48 = 1.22; b: P < 0.001, F7,95 = 1.96; c: P < 0.001, F7,44 = 1.89; f: P < 0.001, F3, 224 = 5.36; g: P > 0.05, F3, 108 = 0.35). Two-way ANOVA with Bonferroni’s post hoc test (k: P = 0.0014, F1,60 = 11.29). Two-tailed unpaired t test (l: cortex: P = 0.036, t10 = 2.41; cerebellum: P = 0.76, t6 = 0.31); nuclei were stained in blue with DAPI (d,e,I,j). Scale bars, 20 μm (d,e,i,j).
Multiple lines of evidence implicate disturbances in cortical pyramidal neurons22-24 and cortical parvalbumin (PV)+ GABAergic interneurons25 as being potentially involved in core psychotic and cognitive symptoms in schizophrenia patients. We used immunohistochemical markers to determine whether either of these neuronal populations showed upregulation of HDAC2 expression after chronic treatment with antipsychotic medications. Consistent with previous studies15, HDAC2 was ubiquitously expressed throughout the brain and was localized primarily within the cell nucleus (Fig. 1d, e and Supplementary Fig. 1a, b). Notably, chronic treatment with clozapine and risperidone, but not with haloperidol, induced upregulation of HDAC2 in CaMKIIα+ cortical pyramidal neurons (Fig. 1d,f and Supplementary Fig. 1a) but not in PV+ cortical GABAergic interneurons (Fig. 1e,g and Supplementary Fig. 1b). This effect of chronic atypical antipsychotic drug treatment did not occur in CaMKIIα+ hippocampal pyramidal neurons (Supplementary Fig. 2a,b) and was absent after subchronic atypical antipsychotic drug treatment (Supplementary Fig. 2c,d).
Upregulation of cortical HDAC2 upon chronic clozapine treatment disrupts synaptic remodeling and cognition
To evaluate the consequences of upregulation of HDAC2 in cortical pyramidal neurons by chronic treatment with atypical antipsychotic drugs in mice, we used a genetic strategy to selectively suppress HDAC2 function in CaMKIIα+ glutamatergic neurons. We crossed loxP-flanked HDAC2 (Hdac2loxP/loxP) mice, which contain loxP sites upstream of exon 2 and downstream from exon 4 in the mouse Hdac2 gene—a mutation that allows us to delete a portion of the oligomerization domain as well as residues in the catalytic domain required for enzymatic activity26—with a second line of mice expressing Cre recombinase under the control of the CaMKIIα promoter (Fig. 1h). We validated the predicted pattern of deletion of HDAC2 in CaMKIIα+ pyramidal neurons but not in PV+ GABAergic interneurons (Fig. 1i–m and Supplementary Fig. 3a–c). We also found that the level of expression of other Hdac tested was unaffected in Hdac2loxP/loxP:CaMKIIα-Cre mice (Hdac2-cKO) as compared to control littermates (Supplementary Fig. 3d), indicating an absence of compensatory events after deletion of HDAC2.
We next sought to determine what influence the lack of forebrain HDAC2 function would have on the potential effects induced by chronic treatment with atypical antipsychotic drugs on cortical pyramidal synaptic remodeling and behavior. Since it was previously shown that HDAC2 negatively regulates synaptic plasticity and cognition15, we hypothesized that upregulation of HDAC2 activity by chronic atypical antipsychotic drug treatment might have a detrimental impact on genes associated with dendritic structural remodeling and behavioral plasticity. To test this possibility, we performed poly(A)+ RNA sequencing in frontal cortex samples from Hdac2-cKO mice and from control littermates treated chronically with either clozapine or vehicle. Broadly, we identified fewer differentially expressed genes in the frontal cortex of control animals that had received chronic clozapine treatment versus vehicle in comparison with the transcriptional profiles obtained in the frontal cortex of Hdac2-cKO mice that had received chronic clozapine treatment versus vehicle (Fig. 1n and Supplementary Table 1). Further gene ontology analysis indicated that substantial repression for several signaling pathways associated with cell morphogenesis, neuron projection and synapse structure occurred in control mice after chronic clozapine treatment (Supplementary Fig. 3e). Notably, the number of differentially expressed genes associated with pathways involved in dendritic growth and neurogenesis was markedly enriched in the frontal cortex of Hdac2-cKO mice treated chronically with clozapine compared to chronic vehicle (Supplementary Fig. 3e). These results support the notion that forebrain HDAC2 function leads to downregulation of synaptic formation and plasticity gene networks upon chronic clozapine treatment.
Dendritic spines are critical structural and functional units with important roles in cognitive processes22. To evaluate the structural consequences of HDAC2-dependent repression of these genes observed after long-lasting exposure to atypical antipsychotic drugs, we measured the effect of chronic clozapine treatment on dendritic spine density in CaMKIIα+ cortical pyramidal neurons in Hdac2-cKO mice and controls. We targeted pyramidal neurons from frontal cortex of adult mice through the injection of adeno-associated virus (AAV8) bearing enhanced yellow fluorescent protein (eYFP) under the control of the CaMKIIα promoter (Supplementary Fig. 4a). In slices prepared 3 weeks after injection, this configuration resulted in robust overexpression of eYFP in frontal cortex CaMKIIα+, but not PV+, neurons (Supplementary Fig. 4b–d), enabling explicit discrimination of single pyramidal cells and their dendritic spines. Notably, chronic treatment with clozapine selectively reduced the density of mature mushroom spines in CaMKIIα+ frontal cortex neurons but not that of stubby or immature thin spines or total spine density (Fig. 2a–e). This synaptic remodeling event required the expression and function of HDAC2 in cortical pyramidal neurons, as the effect of chronic clozapine treatment on spine structure was absent in the frontal cortex of Hdac2-cKO mice (Fig. 2a–e). The pattern of immunoreactivity against synaptophysin, which is specifically localized in presynaptic terminals of active synapses15, was similar in the frontal cortex of mice stereotaxically injected with AAV-eYFP compared to those given mock injections (Supplementary Fig. 4e,f). This indicates the absence of effects of AAV8 infection itself on density of functional synapses. Together, these observations suggest that gain of HDAC2 function upon long-lasting treatment with atypical antipsychotic drugs might impact their antipsychotic-related behavioral profiles.
Figure 2.
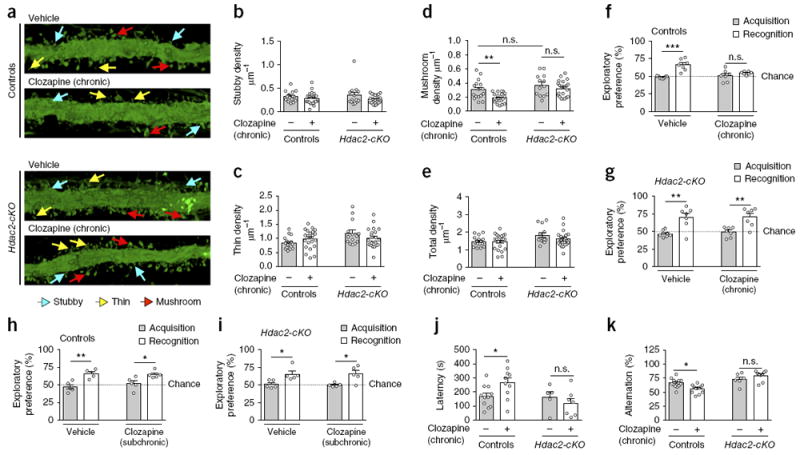
Chronic clozapine treatment negatively regulates synaptic remodeling and cognition via HDAC2. (a–e) Chronic clozapine treatment decreases mature synaptic structural elements in mouse frontal cortex. Hdac2-cKO mice and control littermates were stereotaxically injected with AAV-eYFP. Two days after surgery, mice were treated chronically (21 d) with clozapine (10 mg/kg) or vehicle, and they were killed for analysis one day after the last injection. Representative three-dimensional reconstructions of AAV-injected cortical dendritic segments (a). Stubby (b), thin (c), mushroom (d) and total (e) frontal cortex spine density in Hdac2-cKO mice and control littermates after chronic clozapine treatment (n = 16–22 neurons from 5 or 6 mice per experimental condition). (f–i) Effect of chronic (21 d; n = 7 mice per experimental condition) or subchronic (2 d; n = 5 mice per experimental condition) clozapine treatment on cognitive function in Hdac2-cKO mice and control littermates. Chronic (f,g) but not subchronic (h,i) clozapine treatment impaired novel-object recognition in control mice, an effect that did not occur in Hdac2-cKO littermates. (j) Effect of chronic clozapine treatment on the water-finding task in the test trial (controls, n = 10 or 11 mice per experimental condition; Hdac2-cKO, n = 5–7 mice per experimental condition). (k) Effect of chronic clozapine treatment on short-term spatial recognition memory measured by sequential arm visits (alternation) in the Y-maze test (controls, n = 12–15 mice per experimental condition; Hdac2-cKO littermates, n = 6–9 mice per experimental condition). Mean ± s.e.m. *P < 0.05; **P < 0.01; ***P < 0.001; n.s., not significant. Two-way ANOVA with Bonferroni’s post hoc test (b: P = 0.06, F1,70 = 3.64; c: P = 0.80, F1,70 = 0.06; d: P = 0.001, F1,70 = 11.34; e: P = 0.31, F1,70 = 1.03; f: P < 0.001, F1,24 = 22.19; g: P < 0.001, F1,24 = 28.20; h: P < 0.001, F1,16 = 25.35; i: P < 0.001, F1,16 = 17.89; j: P = 0.03, F1,29 = 4.98; k: P < 0.001, F1,38 = 32.98). The dashed line indicates chance performance (f–i).
To test this possibility, we examined cognitive capacities following chronic atypical antipsychotic drug treatment in Hdac2-cKO mice and controls. We found that a novel-object recognition test as a measure of cognitive performance (Supplementary Fig. 5a) was significantly disrupted upon chronic (Fig. 2f and Supplementary Fig. 5b) but not subchronic (Fig. 2h and Supplementary Fig. 5c) clozapine treatment in control mice. This effect was not observed in Hdac2-cKO littermates (Fig. 2g,i; see also Supplementary Fig. 5d for absence of effect of chronic clozapine treatment on novel-object recognition test in heterozygous Hdac2loxP/+:CaMKIIα-Cre mice compared to controls). Similar findings were observed using water-finding (Fig. 2j) and Y-maze (Fig. 2k) tests as behavior models of latent learning and short-term spatial recognition memory, respectively. This phenomenon prompted us to explore the signaling mechanism through which chronic antipsychotic drug treatment modulates cortical pyramidal HDAC2 expression, as well as direct effects of this pathway on synaptic structural elements and behavioral phenotypes that parallel schizophrenia.
Chronic clozapine treatment augments binding of NF-κB to the Hdac2 promoter
Antipsychotic drugs all present a relatively high affinity for the 5-HT2A receptor3 (Supplementary Fig. 6a), and forebrain 5-HT2A receptors have been involved in both beneficial27,28 and adverse29,30 effects of antipsychotic medications. Our previous findings, obtained after microarray studies followed by high-throughput quantitative real-time PCR assays, showed that specific 5-HT2A-receptor-dependent signaling pathways lead to induction of expression of genes that act as transcription factors or indirectly modulate transcription in numerous tissues, including the CNS18,31. This motivated us to screen the Hdac2 promoter for potential binding sites of transcriptional regulators. Using transcription factor binding databases32, we found a potential recognition element for NF-κB at the promoter region of the Hdac2 gene (Fig. 3a). NF-κB is a family of dimeric transcription factors best studied for its involvement in inflammation and immune responses and that has more recently been shown to regulate synaptic plasticity and brain function33. Previous studies have indicated that p65, one of the prototypical NF-κB subunits, is selectively located at synapses and involved in regulating neuropsychological processes related to cocaine addiction34 and depression35. Based on this knowledge, we used a series of chromatin immunoprecipitation assays with an anti-p65 antibody in the mouse frontal cortex at different regions across the Hdac2 gene (Fig. 3b). p65 binding was enriched at the Hdac2 promoter region located −454 to −299 base pairs upstream of the transcription start site (Fig. 3c), a region that contains the predicted NF-κB binding site (Fig. 3a,b), whereas we detected no binding at other positions along the Hdac2 gene (Fig. 3c). The capacity of NF-κB to interact physically with a DNA fragment that corresponds with a putative NF-κB binding site within the promoter of the mouse Hdac2 gene was validated by fluorescence anisotropy assay (Fig. 3d and Supplementary Fig. 6b,c). We built a homology model of the NF-κB heteromer (p65–p50) bound to the Hdac2 promoter site that was consistent with our experimental results (Fig. 3e).
Figure 3.
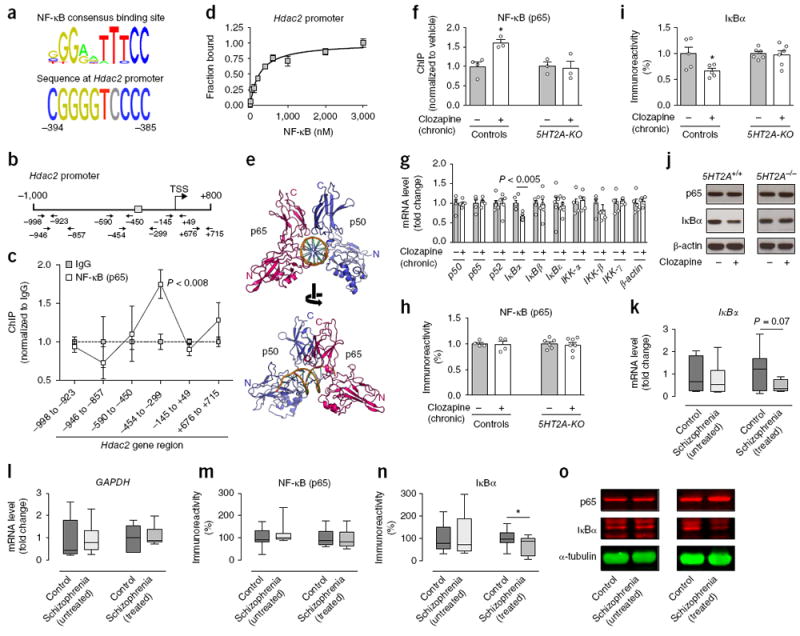
Serotonin 5-HT2A receptor-dependent increased binding of NF-κB to the Hdac2 promoter in mouse and human cortical neurons after chronic atypical antipsychotic drug treatment. (a) Alignment of the vertebrate NF-κB consensus binding site with the promoter of mouse Hdac2 gene. (b) Map of the Hdac2 gene showing position of primers used for qPCR assays. Gray box indicates location of the predicted NF-κB binding site. TSS, transcriptional start site. (c) NF-κB (p65) binds to the promoter region of the Hdac2 gene in mouse frontal cortex (n = 6 mice per experimental condition). (d) Fluorescence anisotropy assay shows physical interaction between NF-κB (p65 and p50) and the Hdac2 promoter (n = 2 experiments performed in triplicate; see also Supplementary Fig. 6b,c). (e) Ribbon representation of NF-κB (p65 and p50) bound to the Hdac2 promoter, viewed down (top) and perpendicular (bottom) from the DNA helical axis. (f) Binding of NF-κB (p65) to the Hdac2 promoter is increased by chronic clozapine treatment in frontal cortex of wild-type mice, but not 5HT2A-KO littermates (n = 3 or 4 mice per experimental condition). (g) Chronic clozapine treatment downregulates the expression of IκBα mRNA in mouse frontal cortex. Expression of p50, p65, p52, IκBα, IκBβ (also known as Nfkbib), IκBε (also known as Nfkbie), IKK-α (also known as Chuk), IKK-β (also known as Ikbkb), IKK-γ (also known as Ikbkg) and β-actin (also known as Actb) was assayed by qRT-PCR (n = 5 or 6 mice per experimental condition). (h–j) Chronic clozapine downregulates protein levels of IκBα (I,j), and not p65 (h,j), in frontal cortex of wild-type mice but not in 5HT2A-KO littermates (n = 4–7 mice per experimental condition). Representative immunoblots are shown (j). (k,l) Expression of IκBα (k) and GAPDH (l) mRNA in postmortem human frontal cortex of schizophrenia subjects and controls, assayed by qRT-PCR (see Supplementary Tables 2 and 3 for demographic information). (m–o) Protein levels of IκBα (n,o), and not p65 (m,o), are decreased in postmortem human frontal cortex of atypical antipsychotic-treated, and not in untreated, schizophrenic subjects. Representative immunoblots are shown (o). Mean ± s.e.m. *P < 0.05; n.s., not significant. Two-tailed unpaired t test (c: –998 to –923: P = 0.55, t10 = 0.61; –946 to –857: P = 0.50, t10 = 0.69; –590 to –450: P = 0.78, t10 = 0.28; –454 to –299: P = 0.005, t10 = 3.54; –145 to +49: P = 0.27, t10 = 1.14; +676 to +715: P = 0.26, t10 = 1.17; g: p50: P = 0.92, t8 = 0.09, p65: P = 0.72, t8 = 0.36, p52: P = 0.95, t10 = 0.06, IκBα: P = 0.0013, t10 = 4.44, IκBβ: P = 0.80, t10 = 0.25, IκBε: P = 0.74, t10 = 0.34, IKK-α: P = 0.68, t8 = 0.41, IKK-β: P = 0.33, t8 = 1.02, IKK-γ: P = 0.64, t8 = 0.47, β-actin: P = 0.66, t8 = 0.44; k: control versus untreated schizophrenics: P = 0.56, t12 = 0.59, control versus treated schizophrenics: P = 0.07, t15 = 1.858; l: control versus untreated schizophrenics: P = 0.95, t15 = 0.06, control versus treated schizophrenics: P = 0.70, t16 = 0.39). Two-tailed paired t test (m: control versus untreated schizophrenics: P = 0.15, t9 = 1.5, control versus treated schizophrenics: P = 0.54, t9 = 0.63; n: control versus untreated schizophrenics: P = 0.54, t9 = 0.62, control versus treated schizophrenics: P = 0.02, t9 = 2.61). Two-way ANOVA with Bonferroni’s post hoc test (f: P = 0.03, F1,9 = 6.24; h: P = 0.94, F1,17 = 0.004; i: P = 0.03, F1,18 = 5.02). The α value was corrected for multiple independent null hypotheses using the Holm’s sequentially Bonferroni method (c,g). Box plots in k–n present, in ascending order, minimum sample value, first quartile, median, third quartile and maximum sample value.
We next evaluated the effect of chronic treatment with clozapine on NF-κB (p65) binding to the Hdac2 promoter in mouse frontal cortex. Notably, chronic clozapine treatment significantly increased NF-κB (p65) binding to the Hdac2 promoter (Fig. 3f). This event did not occur in the frontal cortex of 5-HT2A-null mutant (5HT2A-KO) mice (Fig. 3f; see also Supplementary Fig. 7a,b for absence of effect of chronic atypical antipsychotic drug treatment on HDAC2 immunoreactivity in CaMKIIα+ cortical pyramidal neurons of 5HT2A-KO mice). Together, these results suggest that 5-HT2A-receptor-regulated pathways on cortical neurons modulate transcription of Hdac2 via a signaling mechanism that involves alterations in NF-κB function.
Chronic clozapine treatment augments NF-κB function via serotonin 5-HT2A-receptor-dependent downregulation of IκBα
To gain insight into the ability of chronic treatment with clozapine to enhance NF-κB (p65) binding to the promoter region of the Hdac2 gene, we measured mRNA expression of genes previously shown to function as fundamental modulators of the NF-κB pathway33. We found that chronic clozapine significantly decreased IκBα (also known as Nfkbia) mRNA in mouse frontal cortex (Fig. 3g), whereas this treatment did not affect expression of other genes involved in NF-κB-dependent signaling (Fig. 3g). We next evaluated the effect of chronic clozapine on IκBα protein expression in mouse frontal cortex. Expression of IκBα, but not IκBβ, p65 or p50 (p105), was diminished upon chronic clozapine treatment as compared to vehicle-treated mice, an effect that was not observed in 5HT2A-KO littermates (Fig. 3h–j and Supplementary Fig. 7c; see also Supplementary Fig. 7d for absence of effect of chronic clozapine treatment on IκBα mRNA in the frontal cortex of 5HT2A-KO mice).
To study the relevance of the effects observed in mice, we examined IκBα and p65 protein levels in postmortem frontal cortex of human subjects with schizophrenia untreated or treated with atypical antipsychotics and from individually matched controls (see Supplementary Tables 2 and 3 for demographic information). Equivalent to the lower IκBα expression found in mouse frontal cortex after chronic treatment with clozapine, we observed decreased expression of IκBα but not p65 in the frontal cortex of humans with schizophrenia treated with atypical antipsychotics but not in the untreated group (Fig. 3m–o). The trend for reduced IκBα mRNA levels was also evident in postmortem frontal cortex of medicated schizophrenic subjects (Fig. 3k,l).
To directly interrogate the functional relevance of these findings in mice and humans, and given that NF-κB is retained in the cytoplasm through its interaction with inhibitory IκB proteins, including IκBα33, we tested whether chronic antipsychotic drug treatment affects sub-cellular localization of NF-κB (p65) in frontal cortex pyramidal neurons. Consistent with previous studies34, NF-κB (p65) localization was most intense cytoplasmically in mouse cortical neurons (Fig. 4a). Notably, chronic clozapine treatment selectively increased the nuclear/cytoplasmic ratio of NF-κB (p65) in CaMKIIα+, but not PV+, frontal cortex neurons of wild-type mice (Fig. 4a–e and Supplementary Fig. 8a). This trafficking event after chronic clozapine treatment was not observed in the frontal cortex of 5HT2A-KO mice (Fig. 4a,b).
Figure 4.
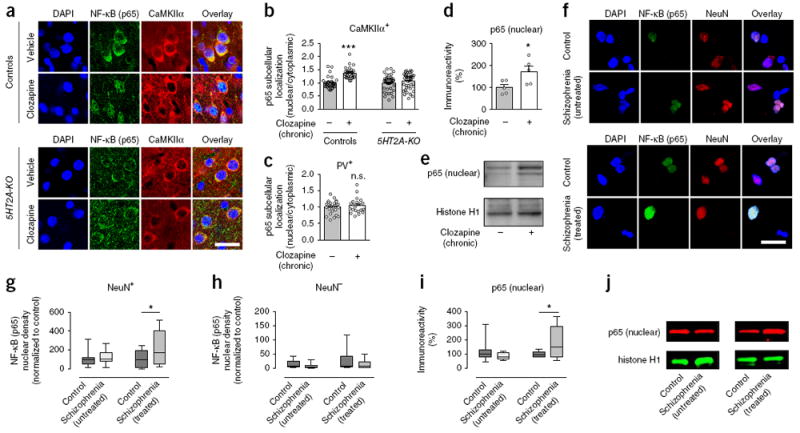
Serotonin 5-HT2A receptor-dependent increased nuclear translocation of NF-κB in mouse and human cortical neurons after chronic atypical antipsychotic drug treatment. (a–d) Chronic clozapine treatment increases nuclear translocation of NF-κB (p65) in frontal cortex pyramidal neurons of wild-type but not 5HT2A-KO mice. Representative immunohistochemical images (a). Quantitative assessment in CaMKIIα+ (n = 30–40 cells from 4 mice per experimental condition, b) and PV+ (n = 20–27 cells from 4 mice per experimental condition, c) cells. (d,e) Western blots showed upregulation of p65 protein in nuclear preparations from the frontal cortex of mice treated chronically with clozapine as compared to vehicle (n = 5 mice per experimental condition). Representative immunoblots are shown (e). (f–h) Increased neuronal nuclear translocation of NF-κB (p65) in the frontal cortex of schizophrenic subjects who were treated with atypical antipsychotic drugs. Postmortem human nuclei were separated by sucrose gradient centrifugation, after which density of NF-κB (p65) was assessed by immunocytochemical staining with antibodies against p65 and the neuronal marker NeuN. Representative immunocytochemical images in frontal cortex nuclei of untreated schizophrenic subjects and individually matched controls (f, top) and in frontal cortex nuclei of atypical antipsychotic-treated schizophrenic subjects and individually matched controls (f, bottom). NF-κB (p65) immunoreactivity levels in neuronal (NeuN+, g) and non-neuronal (NeuN–, h) nuclei. (i,j) Western blots showed upregulation of p65 protein in nuclear preparations from the frontal cortex of atypical antipsychotic-treated schizophrenic subjects as compared to individually matched controls but not in the frontal cortex of untreated schizophrenic subjects as compared to individually matched controls. Representative immunoblots are shown (j). Mean ± s.e.m. *P < 0.05; ***P < 0.001; n.s., not significant. Two-tailed unpaired t test (c: P = 0.47, t45 = 0.72; d: P = 0.03, t8 = 2.45). Two-tailed paired t test (g: control versus untreated schizophrenics: P = 0.44, t9 = 0.80, control versus treated schizophrenics: P = 0.01, t9 = 2.89; h: control versus untreated schizophrenics: P = 0.27, t9 = 1.16, control versus treated schizophrenics: P = 0.24, t9 = 1.25; i: control versus untreated schizophrenics: P = 0.18, t9 = 1.14, control versus treated schizophrenics: P = 0.04, t9 = 2.31). Two-way ANOVA with Bonferroni’s post hoc test (b: P < 0.001, F1, 136 = 11.99). Nuclei were stained in blue with DAPI (a,f). Scale bars, 20 μm (a,f). Box plots present, in ascending order, minimum sample value, first quartile, median, third quartile and maximum sample value (g–i).
To further investigate the effect of chronic atypical antipsychotic drugs on the intracellular translocation of NF-κB, we performed immunocytochemistry assays in nuclear preparations from postmortem frontal cortex of schizophrenic humans and individually matched controls. We used anti-NeuN antibody, whose antigen is a specific marker for neuronal nuclei, to selectively separate neuronal from non-neuronal nuclei in postmortem human brain samples. Notably, consistent with our findings in mice (Fig. 4a–e), we observed a dramatic and selective augmentation in the nuclear accumulation of NF-κB (p65) within neuronal nuclei of frontal cortex samples from treated, but not untreated, humans with schizophrenia (Fig. 4f,g). This functional difference in subcellular distribution of NF-κB (p65) did not occur in non-neuronal frontal cortex nuclei of subjects with schizophrenia treated with atypical antipsychotics (Fig. 4f,h). Similar findings were observed in immunoblot assays with antibodies against p65 in nuclear fractions of frontal cortex from treated, but not from untreated, schizophrenic subjects (Fig. 4i,j and Supplementary Fig. 8b). These findings, along with the absence of changes in p65 expression in the frontal cortex of untreated schizophrenic subjects (Fig. 3m–o), suggest that alterations in nuclear translocation of NF-κB (p65) represent a consequence of continuous exposure to antipsychotic medications rather than a biochemical marker of schizophrenia in postmortem human brain.
5-HT2A-receptor-dependent downregulation of IκBα upon chronic clozapine treatment via MAPK–ERK
We next focused our attention onto the downstream signaling events that mediate downregulation of IκBα expression after chronic clozapine treatment via 5-HT2A receptors. As expected based on previous reports18, we found an induction of IκBα transcription in the frontal cortex of wild-type mice, but not 5HT2A-KO mice, intraperitoneally injected with the 5-HT2A receptor agonist 1-(2,5-dimethoxy-4-iodo-phenyl)-2-aminopropane (DOI; Fig. 5a). Similar effects were observed in mice injected with the serotonin precursor 5-hydroxytryptophan (5-HTP; Fig. 5b). The mitogen-activated protein kinase (MAPK) pathway that leads to the activation of extracellular signal-regulated kinases (ERK1/2) has been linked to neuronal transcriptional events and might also regulate synaptic targets to control plasticity36. Using SL-327, an inhibitor of the MAPK–ERK pathway, we examined the impact of MAPK–ERK cascade signaling on 5-HT2A-receptor modulation of IκBα transcription. Notably, SL-327 prevented 5-HT2A-receptor-dependent activation of IκBα transcription both in mouse frontal cortex (Fig. 5c) and in HEK293 cells (Supplementary Fig. 9a–f), demonstrating that the MAPK–ERK pathway couples activation of the 5-HT2A receptor to induction of IκBα transcription.
Figure 5.
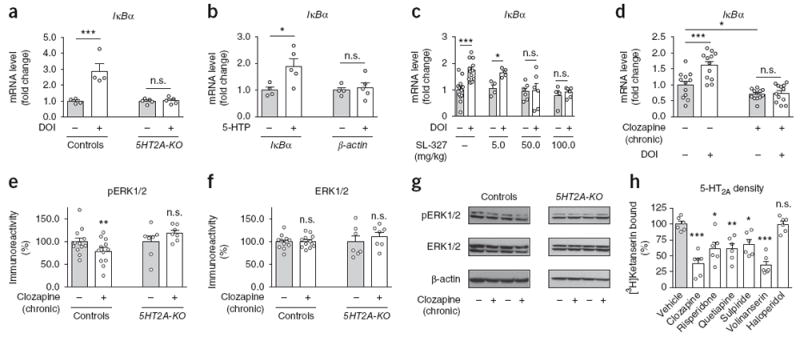
Serotonin 5-HT2A receptor-dependent downregulation of IκBα transcription after chronic clozapine treatment via MAPK–ERK. (a) Changes in expression of IκBα mRNA induced by the 5-HT2A receptor agonist DOI in the frontal cortex of wild-type and 5HT2A-KO mice. Mice were injected intraperitoneally with DOI (2 mg/kg) or vehicle and were killed for analysis after 60 min (n = 4 or 5 mice per group). (b) Changes in expression of IκBα mRNA induced by the 5-HT precursor 5-HTP in mouse frontal cortex. Mice were injected intraperitoneally with 5-HTP (100 mg/kg) or vehicle and were killed for analysis after 60 min (n = 4 or 5 mice per group). (c) The effect of DOI on IκBα mRNA expression is blocked by SL-327 in mouse frontal cortex. Mice were injected with DOI (2 mg/kg) or vehicle after being preinjected with the indicated doses of SL-327 or vehicle (n = 4–12 mice per group). (d) Mice were chronically (21 d) treated with clozapine (10 mg/kg) or vehicle, injected with a single dose of DOI (2 mg/kg) or vehicle 24 h after the last administration of chronic clozapine, and killed for analysis after 60 min (n = 12 mice per group). (e–g) Chronic clozapine treatment downregulates protein levels of ERK1/2 phosphorylation (pERK1/2) in frontal cortex samples of wild-type mice (n = 12 mice per group) but not 5HT2A-KO mice (n = 7 mice per group). Western blots showed no change in ERK1/2 in frontal cortex of wild-type (n = 12 mice per group) or 5HT2A-KO (n = 7 mice per group) animals treated chronically with clozapine as compared to vehicle (e,f). Representative immunoblots are shown (g). (h) Chronic treatment with clozapine (10 mg/kg), risperidone (4 mg/kg), quetiapine (10 mg/kg), sulpiride (10 mg/kg) or volinanserin (1 mg/kg), but not with haloperidol (1 mg/kg), downregulates 5-HT2A receptor density in mouse frontal cortex. Mice were treated chronically (21 d) with the indicated drug, or with vehicle, and killed for analysis 24 h after the last injection. [3H]Ketanserin binding was tested in frontal cortex plasma membrane preparations (n = 6 mice per experimental condition). Mean ± s.e.m. *P < 0.05; **P < 0.01; ***P < 0.001; n.s., not significant. Two-way ANOVA with Bonferroni’s post hoc test (a: P < 0.001, F1,14 = 27.38; c: P < 0.0017, F3,46 = 5.88; e: P = 0.04, F1,34 = 4.47; f: P = 0.43, F1,34 = 0.62). One-way ANOVA with Bonferroni’s post hoc test (d: P < 0.001, F3,44 = 18.97; h: P < 0.001, F6,35 = 14.07). Two-tailed unpaired t test (b: IκBα: P = 0.03, t7 = 2.65, β-actin: P = 0.68, t7 = 0.42).
We next examined whether this pathway is likewise affected by chronic treatment with antipsychotic drugs. As shown (Fig. 3g), chronic clozapine treatment downregulated IκBα mRNA expression in mouse frontal cortex (Fig. 5d). We also found that chronic clozapine treatment eliminated the induction of IκBα transcription upon activation of the 5-HT2A receptor in the mouse frontal cortex (Fig. 5d). We then demonstrated that absence of 5-HT2A-receptor-dependent signaling altered the effect of chronic clozapine treatment on phosphorylated ERK levels in the mouse frontal cortex (Fig. 5e–g). Notably, analysis of [3H]ketanserin binding revealed a marked decrease in frontal cortex 5-HT2A-receptor density only after chronic treatment with those atypical antipsychotic and antipsychotic-like drugs that augment Hdac2 expression but not with haloperidol (Figs. 1a and 5h and Supplementary Fig. 9g). It has been suggested that the p65 subunit of NF-κB interacts with HDAC2 as part of the same protein complex to negatively regulate gene transcription37-39. Our data show that immunoprecipitation with anti-p65 antibodies in mouse frontal cortex did not result in co-immunoprecipitation of anti-HDAC2 immunoreactivity (Supplementary Fig. 10a,b). Together, these data suggest that chronic atypical antipsychotic drug treatment led to elevations in Hdac2 promoter activity within the frontal cortex through a linear signaling pathway that involves 5-HT2A-receptor-dependent downregulation of IκBα expression via hypophosphorylation of ERK1/2.
Inhibition of frontal cortex NF-κB function prevents the effects of chronic clozapine treatment on HDAC2 expression and cognitive deficits
To corroborate the functional significance of changes in frontal cortex NF-κB-dependent transcriptional activity on HDAC2 expression upon chronic clozapine treatment, we next tested the effects of AAV-mediated gene transfer of a dominant-negative IκBα (IκBα-S32A-S36A; dn-IκBα), which is resistant to proteasome degradation and therefore constitutively inhibits NF-κB function40. Using a previously described nine-amino-acid p2A motif for co-expression of two proteins from a single promoter41, we separated Flag-dn-IκBα from eYFP and injected the AAV vector bearing this configuration under the control of the CaMKIIα promoter into mouse frontal cortex (Supplementary Fig. 11a). We verified that the AAV-CaMKIIα∷ Flag-dn-IκBα-p2A-eYFP construct (AAV-dn-IκBα-eYFP) enabled robust and selective overexpression of two proteins from a single open reading frame (Supplementary Fig. 11b,c; see also Supplementary Fig. 11d,e for expression of eYFP in CaMKIIα+ but not PV+ cortical cells and Supplementary Fig. 11f,g for additional validation of the p2A peptide-linked multicistronic vector in vitro).
Injection of AAV-dn-IκBα-eYFP, but not AAV-eYFP, impeded the effects of chronic clozapine treatment on nuclear translocation of NF-κB into the nucleus of eYFP+ cortical neurons (Fig. 6a,c), validating the capability of this virally mediated gene transfer approach to inhibit NF-κB function. Notably, AAV-mediated overexpression of dn-IκBα-eYFP, but not eYFP alone, prevented upregulation of frontal cortex CaMKIIα+ pyramidal HDAC2 expression (Fig. 6b,d) and deficits in the novel-object recognition test (Fig. 6e) after chronic clozapine treatment (see also Supplementary Fig. 11h,i for effects of dn-IκBα on p65-dependent activation of Hdac2 promoter activity in HEK293 cells). These results indicate that manipulation of IκBα function in cortical pyramidal neurons affects the impact of chronic atypical antipsychotic drug treatment on Hdac2 promoter activity and HDAC2-dependent cognitive deficits.
Figure 6.
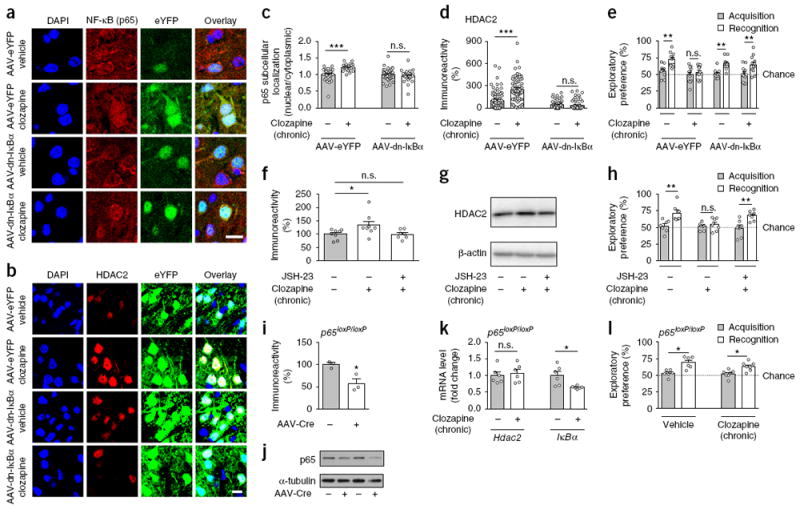
Cortical pyramidal NF-κB is necessary for the effects of chronic atypical antipsychotic drug treatment on HDAC2 expression and cognitive deficits. (a,c) AAV-mediated overexpression of Flag-tagged dominant-negative IκBα (IκBα-S32A-S36A; dn-IκBα) prevents the effect of chronic clozapine treatment on nuclear translocation of NF-κB (p65) in frontal cortex pyramidal neurons. AAV-CaMKIIα∷Flag-dn-IκBα-p2A-eYFP (AAV-dn-IκBα) or AAV-CaMKIIα∷eYFP (AAV-eYFP) were injected into the frontal cortex. Three weeks after surgery, mice were treated chronically (21 d) with clozapine (10 mg/kg) or vehicle and nuclear/cytoplasmic ratios of NF-κB (p65) in eYFP+ neurons were assayed by immunohistochemistry 1 d after the last injection. Representative immunohistochemical images (a). Quantitative assessment (n = 32–42 cells from 4–6 mice per experimental condition, c). (b, d) AAV-mediated overexpression of AAV-Flag-dn-IκBα prevents the effect of chronic clozapine treatment on upregulation of HDAC2 in frontal cortex pyramidal neurons. AAV-Flag-dn-IκBα or AAV-eYFP were injected into the frontal cortex. Representative immunohistochemical images (b). Quantitative assessment (n = 58–75 cells from 4–6 mice per experimental condition, d). (e) AAV-mediated overexpression of AAV-Flag-dn-IκBα prevents the effect of chronic clozapine treatment on deficits in the novel-object recognition test (n = 11 or 12 mice per experimental condition). (f–h) Effects of NF-κB inhibitors on HDAC2 expression and novel-object recognition after chronic clozapine treatment. Mice were infused (i.c.v.) chronically (21 d) with the NF-κB inhibitor JSH-23 and injected (i.p.) with clozapine or vehicle. Experiments were conducted 1 d after the last clozapine administration. Western blots showed that JSH-23 prevents upregulation of frontal cortex HDAC2 after chronic clozapine treatment (n = 7 or 8 mice per experimental condition; f). Representative immunoblots are shown (g). JSH-23 prevents the effect of chronic clozapine treatment on deficits in the novel-object recognition test (n = 7 mice per experimental condition, h). (i–l) Conditional deletion of p65 in the frontal cortex of p65loxP/loxP mice prevents the effects of chronic clozapine treatment on Hdac2 expression and recognition memory. AAV-CaMKIIα∷mCherry-Cre (AAV-Cre) or mock were injected into the frontal cortex, and anti-p65 immunoreactivity was measured by Western blotting 3 weeks after surgery (n = 3 mice per group, i). Representative immunoblots are shown (j). Full-length immunoblots are shown in Supplementary Figure 15. AAV-Cre was injected into the frontal cortex of p65loxP/loxP mice. Three weeks after surgery, mice were treated chronically (21 d) with clozapine (10 mg/kg) or vehicle, and expression of Hdac2 and IκBα was assessed by qRT-PCR (n = 6 mice per experimental condition, k). AAV-mediated deletion of frontal cortex p65 in p65loxP/loxP mice prevents the effects of chronic clozapine treatment on deficits in the novel-object recognition test (n = 6–7 mice per experimental condition, l). Mean ± s.e.m. *P < 0.05; **P < 0.01; ***P < 0.001; n.s., not significant. Two-way ANOVA with Bonferroni’s post hoc test (c: P < 0.001, F1,55 = 13.02; d: P < 0.001, F1, 269 = 26.82; e: P < 0.002, F3,82 = 5.23; h: P < 0.03, F2,36 = 3.56; l: P < 0.001, F1,22 = 26.33). One-way ANOVA with Bonferroni’s post hoc test (f: P < 0.03, F2,20 = 3.84). Two-tailed unpaired t test (i: P = 0.02, t4 = 3.62; k: Hdac2: P = 0.65, t10 = 0.46, IκBα: P = 0.01, t10 = 2.98). The dashed line indicates chance performance (e,h,l). Nuclei were stained in blue with DAPI (a,b). Scale bars, 20 μm (a,b).
We next reasoned that repression of frontal cortex NF-κB function by pharmacological or genetic tools would prevent the effects of chronic clozapine treatment on HDAC2 expression and cognitive capabilities. To test this possibility, we first directly compared the effects of the NF-κB inhibitor JSH-23, administered continuously via intracerebroventricular delivery, with those of chronic clozapine treatment. As above (Fig. 1a,d,f), chronic clozapine treatment induced upregulation of frontal cortex HDAC2 (Fig. 6f,g) and disrupted novel-object recognition (Fig. 6h). Notably, adjunctive JSH-23 treatment prevented these effects induced by chronic clozapine (Fig. 6f-h).
We next aimed to manipulate NF-κB function directly in CaMKIIα+ frontal cortex pyramidal neurons. To do so, we used AAV-mediated transgene expression of Cre tagged with the fluorescent construct mCherry under the control of the CaMKIIα promoter. Virally mediated overexpression of Cre in the frontal cortex of p65loxP/loxP mice induced local and selective knockdown of p65 expression in CaMKIIα+ but not PV+ neurons (Fig. 6i,j and Supplementary Fig. 11j,k). Notably, the effects of chronic clozapine treatment on upregulation of frontal cortex HDAC2 expression (Fig. 6k) and deficits in the novel object recognition test (Fig. 6l) were absent in AAV-Cre-injected mice. However, expression of IκBα remained downregulated after chronic clozapine treatment in the frontal cortex of loxP-flanked p65 mice injected with AAV-Cre (Fig. 6k). This, together with the effect of chronic clozapine treatment on downregulation of IκBα expression in the frontal cortex of Hdac2-cKO mice (Supplementary Fig. 11l,m), confirms that upstream pathways were responsible for the effects of chronic atypical antipsychotic treatment on IκBα transcription.
Virally mediated activation of frontal cortex NF-kB function decreases synaptic plasticity via HDAC2
To directly evaluate the consequences of long-lasting treatment with antipsychotic medications on NF-κB-dependent transcriptional function in cortical pyramidal neurons, we used AAV vectors to overexpress an HA-tagged constitutively active IκB-kinase (IKK-β-S177E-S181E; HA-caIKK-β) that shows dramatically enhanced kinase activity42 and consequently models effects of chronic antipsychotic drug treatment on augmentation of NF-κB function. We verified that the AAV-CaMKIIα∷HA-caIKK-β-p2A-eYFP construct (AAV-caIKK-β-eYFP) enabled robust and selective overexpression of two proteins from a single open reading frame (Supplementary Fig. 12a–c; see also Supplementary Fig. 12d,e for additional validation of the p2A peptide-linked multicistronic vector in vitro).
Using this virally mediated overexpression approach, we next interrogated, via injection of AAV-caIKK-β-eYFP or AAV-eYFP, whether long-lasting changes in transcriptional activity of NF-κB are sufficient to affect HDAC2 expression in the frontal cortex of living mice. After 3 weeks of continuous positive modulation of NF-κB-dependent function, we found a significant increase in the translocation of NF-κB into the nucleus of eYFP+ cortical neurons (Fig. 7a,b), mimicking the effect of chronic antipsychotic drug treatment in mouse models and in the frontal cortex of schizophrenic subjects treated with atypical antipsychotic drugs (Fig. 4). Additionally, AAV-mediated augmentation of NF-κB-dependent function induced selective upregulation of HDAC2 immunoreactivity in eYFP+ frontal cortex neurons (Fig. 7c,d), whereas HDAC1 immunoreactivity was unchanged in AAV-caIKK-β-eYFP neurons in comparison with controls (Supplementary Fig. 12f,g).
Figure 7.
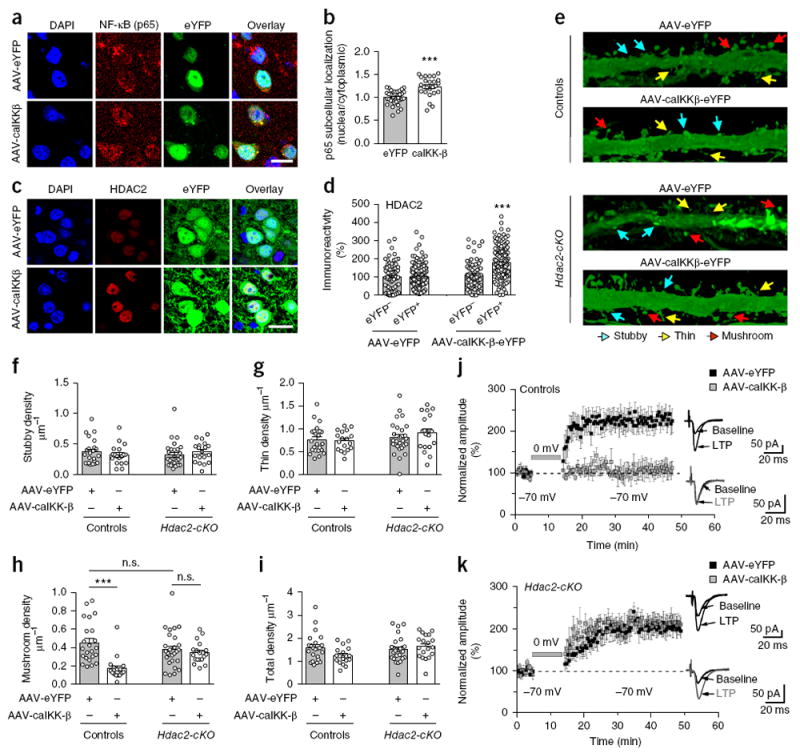
Virally mediated augmentation of NF-κB function in frontal cortex pyramidal neurons impairs synaptic remodeling and synaptic plasticity via HDAC2. (a, b) AAV-mediated over-expression of HA-tagged constitutively active IκB-kinase (IKK-β-S177E-S181E; HA-caIKK-β) increases nuclear translocation of NF-κB (p65) in frontal cortex pyramidal neurons. AAV-CaMKIIα∷HA-caIKK-β-p2A-eYFP (AAV-HA-caIKK-β) or AAV-CaMKIIα∷eYFP (AAV-eYFP) were injected into the frontal cortex, and nuclear/cytoplasmic ratios of NF-κB (p65) in eYFP+ neurons were assayed by immunohistochemistry 3 weeks after surgery. Representative immunohistochemical images (a). Quantitative assessment (n = 25–36 cells from 4 mice per experimental condition, b). (c, d) AAV-mediated over-expression of HA-caIKK-β-eYFP upregulates HDAC2 in eYFP+ frontal cortex neurons. Representative immunohistochemical images (c). Quantitative assessment (n = 80–177 cells from 4 mice per experimental condition, d). (e–i) AAV-caIKK-β decreases mature structural elements via HDAC2 in frontal cortex. Representative three-dimensional reconstructions of AAV-injected cortical dendritic segments (e). Stubby (f), thin (g), mushroom (h) and total (i) frontal cortex spine density in Hdac2-cKO mice and control littermates after injection of either AAV-HA-caIKK-β or AAV-eYFP (n = 18–27 neurons from 5 or 6 mice per experimental condition). (j, k) AAV-mediated over-expression of HA-caIKK-β blocks LTP in frontal cortical layer II/III neurons via HDAC2. Whole-cell patch-clamp recordings from visually identified eYFP+ layer II/III pyramidal neurons were used in conjunction with an LTP-inducing protocol that pairs extracellular stimulation of layer IV with brief depolarization of layer II/III neurons. In control mice (j), LTP was completely blocked in AAV-caIKK-β-eYFP neurons compared to AAV-eYFP neurons (n = 6 neurons from each of 5 mice per experimental condition). In Hdac2-cKO mice (k), LTP in AAV-caIKK-β-eYFP neurons was robust and indistinguishable from that observed in AAV-eYFP neurons (n = 8 neurons from 5 or 6 mice per experimental condition), indicating that the LTP-suppressing effect of AAV-caIKK-β-eYFP required HDAC2. No differences in LTP were observed between AAV-eYFP control (j) and AAV-eYFP or AAV-caIKK-β-eYFP Hdac2-cKO (k) mice (P > 0.05). Mean ± s.e.m. ***P < 0.001; n.s., not significant. Two-tailed unpaired t test (b: P = 0.001, t59 = 5.07). Two-way ANOVA with Bonferroni’s post hoc test (d: P < 0.001, F1, 499 = 40.64; f: P = 0.97, F1,83 = 0.0009; g: P = 0.52, F1,83 = 0.41; h: P < 0.001, F1,83 = 16.46; i: P = 0.34, F1,83 = 0.91; j: P < 0.001, F1,27 = 15.4; k: P = 0.85, F1,27 = 0.60). Nuclei were stained in blue with DAPI (a, c). Scale bars, 20 μm (a, c).
To determine whether such HDAC2 upregulation via NF-κB in frontal cortex regulates synaptic plasticity and behavior, we measured the effect of AAV-caIKK-β-eYFP, or eYFP alone, on cortical synaptic structure, synaptic functional plasticity and animal behavior models of schizophrenia in Hdac2-cKO mice and control littermates. Overexpression of caIKK-β in CaMKIIα+ frontal cortex neurons reduced the density of mature mushroom spines but did not change the density of stubby or immature thin spines or total spine density (Fig. 7e–i). Notably, this synaptic remodeling event required HDAC2 function in cortical pyramidal neurons, as the effect of AAV-caIKK-β-eYFP on spine structure was absent in the frontal cortex of Hdac2-cKO mice (Fig. 7e–i).
Given the effect of NF-κB activity on dendritic spines, we next sought to determine whether NF-κB function also regulates persistent synaptic functional plasticity in frontal neocortical neurons, using a voltage-clamp pairing protocol for inducing long-term potentiation (LTP) of local cortical synapses43. As described above for spine structure analysis, we overexpressed AAV-caIKK-β-eYFP, or eYFP alone, under the control of the CaMKIIα promoter and then prepared acute coronal slices for whole-cell patch-clamp recordings from visually identified eYFP+ layer II/III frontal cortex pyramidal neurons. In control eYFP+ neurons, as expected, pairing layer IV stimulation with brief postsynaptic depolarization induced robust LTP in layer II/III neurons that was sustained for 30–45 min, at which time the experiments were terminated (Fig. 7j). In contrast, no LTP was evident in AAV-caIKK-β-eYFP cortical pyramidal neurons over the same time-course (Fig. 7j), indicating that enhanced NF-κB activity suppressed LTP. To probe whether such regulation of LTP by NF-κB required expression of HDAC2, as was the case for mushroom spine density (Fig. 7e–i), we injected AAV-caIKK-β-eYFP or AAV-eYFP into the frontal cortex of Hdac2-cKO and control littermates and tested for LTP in layer II/III pyramidal neurons. We found robust LTP in caIKK-β-eYFP+Hdac2-cKO neurons, indistinguishable from that in control cells (Fig. 7k), indicating that the LTP-suppressing effects of enhanced NF-κB activity require HDAC2.
To confirm unequivocally the role of HDAC2 in phenotypes resulting from changes in frontal cortex NF-κB activity, we tested whether AAV-caIKK-β-eYFP alters behavior models of psychosis and cognitive deficits in Hdac2-cKO mice and controls. In healthy humans, hallucinogenic drugs, such as lysergic acid diethylamide (LSD) and DOI, and dissociative drugs, such as phencyclidine (PCP) and dizocilpine (MK801), evoke psychotic and cognitive symptoms resembling certain aspects of schizophrenia44-47. We found that Hdac2-cKO mice responded less intensely than their control littermates when the animals were exposed to conditions that model psychosis, such as head-twitch behavior induced by the LSD-like drug DOI (Fig. 8a) and hyperlocomotor activity induced by the phencyclidine-like drug MK801 (Fig. 8b,c, Supplementary Fig. 13a,b and Supplementary Table 4). These results suggest that Hdac2-cKO mice are less predisposed to behavioral phenotypes that model psychosis. Notably, activation of NF-κB function by AAV-caIKK-β-eYFP in frontal cortex CaMKIIα+ pyramidal neurons of control mice exacerbated behavior models of psychosis, such as head-twitch induced by DOI (Fig. 8a) and MK801-dependent locomotor activity (Fig. 8b,c, Supplementary Fig. 13a,b and Supplementary Table 4). It also impaired social interaction tasks (Fig. 8d–f) and led to deficits in cognitive function as defined by the novel-object recognition test (Fig. 8g,h). However, the same AAV-mediated model of prolonged frontal cortex pyramidal NF-κB function augmentation failed to disrupt either behavior models of psychotic symptoms or social and cognitive processes when these behavioral protocols were tested in mice with deleted HDAC2 expression in CaMKIIα+ neurons (Fig. 8a–h and Supplementary Figs. 13 and 14). Notably, exploratory preference in the novel-object recognition task as a measure of rodent cognitive ability was positively correlated with frontal cortex mushroom spine density in control mice (Fig. 8i) but not in Hdac2-cKO littermates (Fig. 8j), which suggests that this phenomenon of cortical pyramidal NF-κB-dependent structural plasticity via HDAC2 dominated behavioral deficits in cognitive-related tasks.
Figure 8.
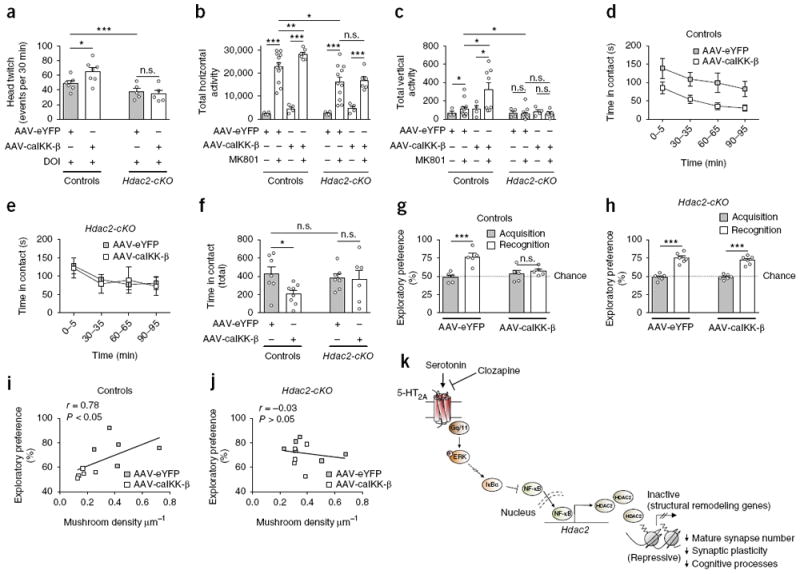
Virally mediated augmentation of NF-κB function in frontal cortex pyramidal neurons exacerbates schizophrenia-related behaviors via HDAC2. (a) AAV-mediated over-expression of HA-caIKK-β increases the head-twitch response induced by the hallucinogen DOI (0.5 mg/kg) in control mice but not in Hdac2-cKO littermates (n = 5 or 6 mice per experimental condition). (b, c) AAV-mediated overexpression of HA-caIKK-β increases hyperlocomotor activity induced by the dissociative drug MK801 (0.3 mg/kg) in control mice (n = 4–12 mice per experimental condition) but not in Hdac2-cKO littermates (n = 4–11 mice per experimental condition). Bar graph summary of the total of MK801-induced horizontal locomotor activity as a summation of horizontal activity from t = 5 to t = 120 min (b). Bar graph summary of the total of MK801-induced vertical activity as a summation of vertical activity from t = 5 to t = 30 min (c). (d–f) AAV-mediated overexpression of HA-caIKK-β promotes social avoidance in control mice but not in Hdac2-cKO littermates. Time-course of social interaction (d, e). Quantitative assessment (n = 6–8 mice per experimental condition, f). (g, h) AAV-HA-caIKK-β, but not AAV-eYFP, impaired novel-object recognition in control mice (n = 5 mice per experimental condition, g), an effect that did not occur in Hdac2-cKO littermates (n = 6 mice per experimental condition, h). (i, j) Novel-object recognition performance correlated with mushroom spine density in control mice (i) but not in Hdac2-cKO (j) littermates. Mean ± s.e.m. *P < 0.05; **P < 0.01; ***P < 0.001; n.s., not significant. Two-way ANOVA with Bonferroni’s post hoc test (a: P < 0.001, F1,19 = 16.45; b: P < 0.001, F3,44 = 41.96; c: P = 0.02, F3,45 = 3.43; f: P = 0.08, F1,24 = 3.12; g: P = 0.0012, F1,16 = 15.34; h: P < 0.001, F1,20 = 123.6). Correlation analysis was conducted using Pearson’s r (i, j). The dashed line indicates chance performance (g, h). (k) Schematic model of unfavorable effects of atypical antipsychotic drug treatment on synaptic plasticity and cognitive behavior. Long-lasting exposure to atypical antipsychotic drugs decreases frontal cortex serotonin 5-HT2A receptor density, which in turn reduces the impact of 5-HT2A receptor-dependent signaling on the MAPK–ERK cascade. This lower 5-HT2A receptor-dependent activation of the MAPK–ERK pathway after chronic clozapine treatment leads to downregulation of IκBα transcription, which consequently augments both nuclear translocation of NF-κB and its binding to the Hdac2 promoter in frontal cortex pyramidal neurons. NF-κB-dependent upregulation of Hdac2 transcription upon chronic atypical antipsychotic treatment results in negative epigenetic regulation of genes involved in structural remodeling and synaptic plasticity. As our data also demonstrate that chronic treatment with atypical antipsychotics reduces the formation of mature spines and impairs cognition through a NF-κB-dependent mechanism involving upregulation of HDAC2, together these observations uncover a previously unsuspected signaling pathway responsible for deleterious effects of these drug agents in terms of synaptic remodeling and cognitive function, and not for their therapeutic actions.
DISCUSSION
Findings in mice suggest that activation of cortical 5-HT2A receptor signaling evokes psychosis-related behavior18,48. Postmortem brain studies demonstrate increased levels of the 5-HT2A receptor in the frontal cortex of antipsychotic-free schizophrenic subjects but not in schizophrenic subjects treated with atypical antipsychotics20,49. Here we showed downregulation of cortical 5-HT2A-receptor density in the frontal cortex of mice chronically treated with atypical antipsychotics but not in mice treated with haloperidol. Together, these findings suggest that a decrease in frontal cortex 5-HT2A-receptor-dependent signaling may contribute to the mechanisms underlying the beneficial effects of chronic antipsychotic treatment on psychotic symptoms. These results, however, do not exclude the possibility of compensatory pathways that may emerge in response to chronic antipsychotic drug exposure and, ultimately, restrain their therapeutic effects50. Our current findings highlight a new mechanism whereby cognitive abilities, including working memory, latent learning and recognition memory, deteriorate upon long-lasting treatment with antipsychotic medications. We demonstrate that chronic treatment with atypical, but not with typical, antipsychotic drugs induces 5-HT2A-receptor-dependent augmentation of HDAC2 epigenetic function via NF-κB, which ultimately leads to detrimental compensatory events along synaptic remodeling and cognitive processes (Fig. 8k). Further clinical work will be necessary to test the extent to which adjunctive treatment with pharmacological inhibitors of this pathway improves brain plasticity and cognition in schizophrenia patients.
We also observed clear differences between transcriptional networks regulated by chronic clozapine treatment in control versus Hdac2-cKO mice. Although our data revealed the basic signaling and neural circuit mechanism responsible for upregulation of Hdac2 transcription upon chronic atypical antipsychotic treatment, it will be interesting to expand this analysis in future studies to characterize the function of these transcriptional networks in synaptic plasticity and memory formation.
Collectively, this study indicates that inhibition of the pathway by which the 5-HT2A receptor augments Hdac2 transcription via NF-κB may serve as a pharmacological approach to improve the currently limited therapeutic range of antipsychotic drug treatment. By extension, our findings emphasize the importance of compensatory pathways as one of the probable mechanisms that counteract positive clinical outcomes of compounds administered chronically.
ONLINE METHODS
Materials and drug administration
1-(2,5-Dimethoxy-4-iodophenyl)-2-aminopropane (DOI), (5R,10S)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine hydrogen maleate (dizocilpine, (+)-MK801), 5-hydroxy-l-tryptophan (5-HTP) and benserazide were purchased from Sigma-Aldrich. 4-Methyl-N1-(3-phenylpropyl)-1,2-benzenediamine (JSH-23) was obtained from Merck Millipore. Clozapine, risperidone, paliperidone quetiapine, norquetiapine, sulpiride, volinanserin (M100907), haloperidol, fluoxetine and α-[amino[(4-aminophenyl)thio]methylene]-2-(trifluoromethyl)benzen eacetonitrile (SL-327) were obtained from R&D Systems. All other chemicals were obtained from standard sources. The injected doses (i.p.) were, unless otherwise indicated: 5-HTP, 100 mg/kg; DOI, 2 mg/kg; clozapine, 10 mg/kg; risperidone, 4 mg/kg; quetiapine, 10 mg/kg; sulpiride, 10 mg/kg; volinanserin, 1 mg/kg; haloperidol, 1 mg/kg; and fluoxetine, 20 mg/kg.
Plasmid construction
All PCR reactions were performed using Pfu Ultra High Fidelity DNA polymerase (Stratagene) in a Mastercycler Ep Gradient Auto thermal cycler (Eppendorf). All the constructs were confirmed by DNA sequencing. The Hdac2 promoter construct (−481 to +141 bp) subcloned into the XhoI and HindIII sites of pGL4.11 [luc2P] plasmid (Promega) has been described previously16. Deletion of the putative NF-κB binding site (−394 to −385) was achieved using the site-directed mutagenesis kit, according to the manufacturer’s protocol (Stratagene). The pcDNA3-RelA-cFlag construct (Flag-p65) was obtained from Addgene (plasmid #20012). For the HA-caIKK-β construct, the pCR-HA-IKK-β vector was obtained from Addgene (plasmid #15470), after which mutations S177E and S181E were inserted using the site-directed mutagenesis kit (Stratagene) according to the manufacturer’s instructions. For the pcDNA3.1-Flag-dn-IκBα construct, mouse IκBα cDNA along with the Flag tag was PCR amplified from mouse frontal cortex cDNA using the following primers: 5′-TTTTaagcttAGCATGGACTACAAGGATGACGATGA CAAAtttcagccagctgggcac-3′ and 5′-TTTTtctagaTTAttataatgtcagacgctggcc-3′. The PCR product was then ligated into the HindIII and XbaI sites of the pcDNA3.1 (+) plasmid, after which mutations S32A and S36A were inserted using the site-directed mutagenesis kit (Stratagene) according to the manufacturer’s instructions. The AAV-CaMKIIα-eYFP-WPRE vector (AAV-CaMKIIα∷ eYFP) was donated by Dr. K. Deisseroth. For the AAV-CaMKIIα∷Flag-dn-IκBα-p2A-eYFP construct, Flag-dn-IκBα (see above) was PCR amplified using the following primers: 5′-TTTTaccggtGCCACCATGGACTACAAGGA TGACGATGACAAATTTCAG-3′ and 5′-TTTTggcgcgccGTCACAGGCCTG CTCCAGGCTACAC-3′. The PCR product was digested and inserted into the AgeI and AscI sites of the AAV-CaMKIIα-hChR2-(T159C)-p2A-eYFP-WPRE construct (donation of Dr. K. Deisseroth). For the AAV-CaMKIIα∷HA-caIKK-β-p2A-eYFP construct, HA-caIKK-β (see above) was PCR amplified using the following primers: 5′-TTTTaccggtGCCACCATGGCTTACCCATACGATG TTCCAGAT-3′ and 5′-TTTTggcgcgccTAATGTCAGACGCTGGCCTCC-3′. The PCR product was digested and inserted into the AgeI and AscI sites of the AAV-CaMKIIα-hChR2-(T159C)-p2A-eYFP-WPRE construct (see above). For the pETDuet-His-TEV-p65-p50 construct, p65 cDNA (residues 19–291; Supplementary Fig. 6b) along with the TEV protease site was PCR amplified from the Flag-p65 primers described above using the primers 5′- TTTgaatt cGAGAACCTTTATTTCCAAGGCCCTTATGTGGAGATCATCGA-3′ and 5′-TTTAAGCTTTCAGTCTGGCAAGTACTGGAACT-3′. The PCR product was then ligated into the EcoRI and HindIII sites of the pETDuet plasmid, after which p50 cDNA (residues 39–350; Supplementary Fig. 6b) was PCR amplified from pcDNA3 p50 cFlag obtained from Addgene (plasmid #20018) using the primers 5′-AAAAAAcatatgGGCCCATACCTTCAAATATTAGAG C-3′ and 5′-TTTTTTctcgagTTATTCAGGGTAGTAGAGAAAGGGTTTC-3′. The PCR product was then ligated into the NdeI and XhoI sites of the pET-Duet plasmid. Generation and subcloning of the human 5-HT2A receptor N-terminally tagged with the c-Myc epitope and C-terminally tagged with enhanced cyan fluorescent protein (eCFP) construct has been described previously20. Introduction of the mutation of I181D into the intracellular loop 2 of c-Myc-5-HT2A-eCFP was performed with the site-directed mutagenesis kit (Stratagene) according to the manufacturer’s instructions. c-Myc-5-HT2A-eCFP and c-Myc-5-HT2A-I181D-eCFP were subcloned into the vector pcDNA5/FRT/TO (Invitrogen) for the subsequent generation of Flp-In-T-REx HEK293 cells.
Transient transfection of HEK293 and Neuro-2a cells
Human embryonic kidney (HEK293) cells (ATCC: CRL-1573) were maintained in Dulbecco’s modified Eagle’s medium supplemented with 10% (v/v) FBS at 37 °C in a 5% CO2 humidified atmosphere. Neuro-2a cells (ATCC: CCL-131) were maintained in Eagle’s Minimum Essential Medium supplemented with 5% (v/v) FBS at 37 °C in a 5% CO2 humidified atmosphere. Transfection was performed using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instructions.
Generation of stable Flp-In-T-REx HEK293 cell lines
To generate Flp-In-T-REx HEK293 cells able to express c-Myc-5-HT2A-eCFP or c-Myc-5-HT2A-I181D-eCFP in an inducible manner, cells were transfected with a mixture containing either c-Myc-5-HT2A-eCFP receptor cDNA in the pcDNA5/FRT/TO vector and pOG44 vector (1:9) or c-Myc-5-HT2A-I181D-eCFP receptor cDNA in the pcDNA5/FRT/TO vector and pOG44 vector (1:9) using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instructions. When co-transfected with the pcDNA5/FRT plasmid into the Flp-In mammalian host cell line, the Flp recombinase expressed from pOG44 mediates integration of the pcDNA5/FRT vector containing the gene of interest into the genome via Flp recombination target (FRT) sites. Cell maintenance and selection were performed as described previously51. Clones resistant to hygromycin were screened for c-Myc-5-HT2A-eCFP and c-Myc-5-HT2A-I181D-eCFP expression by both fluorescence and western blotting. To induce expression of c-Myc-5-HT2A-eCFP or c-Myc-5-HT2A-I181D-eCFP, cells were treated with doxycycline (0.01 μg/mL; 24 h). Dialyzed FBS was used for cell growth to avoid activation of 5-HT2A receptors by 5-HT that is present routinely in serum.
Luciferase reporter assay
Promoter luciferase reporter assays were carried out as previously described, with minor modifications16,17. Briefly, HEK293 cells were plated at a density of 1–5 × 105 in six-well dishes, cultured for 24 h and transfected with the corresponding plasmids. For the effect of Flag-p65 on transcriptional activity of the HDAC2 promoter, cells were transfected with the pcDNA3-RelA-cFlag plasmid (0.0 and 0.1 μg) and with the pGL4.11 [luc2P] plasmid containing the mouse Hdac2 promoter (−481 to +141 bp; 1.0 μg) or with the pGL4.11 [luc2P] plasmid containing the mouse Hdac2Δ (−394 to −385) promoter construct (1.0 μg). For the effect of dominant negative Flag-IκBα-(S32A-S36A), (Flag-dn-IκBα) together with Flag-p65 on HDAC2 promoter activity, cells were transfected with the pGL4.11 [luc2P] plasmid (Promega) containing the mouse Hdac2 promoter (−481 to +141 bp; 1.0 μg), the pcDNA3-RelA-cFlag plasmid (0.0, 0.05 and 0.1 μg) and/or the pcDNA3.1-Flag-dn-IκBα (1.0 μg) or pcDNA3.1. In all assays, cells were transfected with a construct encoding a constitutively active IκB-kinase (IKK-β-S177E-S181E; 0.5 μg). Transfected cells were incubated for 24 h, and the luciferase activity was measured with a luminometer (LMax II 384 Luminescence microplate reader, Molecular Devices) using the Dual-Luciferase reporter assay system (Promega) according to the manufacturer’s instructions. Transfection efficiency was normalized with co-expressed pGL4.75 [hRluc/CMV] (Promega) (0.02 μg).
Expression and purification of NF-κB (p65/p50) heterodimer
DNA sequences coding for Rel homology regions (RHR) of p50 (residues 39–350) and p65 (residues 19–291) were subcloned into the pET-DUET1 expression vector (see above). The plasmid was transformed into Escherichia coli BL21(DE3)pLysS cells and grown at 37 °C. The cells were induced with 0.5 mM IPTG and harvested after 4 h. After sonication, the cell lysate was purified on a Ni-NTA column. The heterodimer was further purified using a MonoS 5/5 column and the final purification step used a Superdex-75 gel filtration column in the buffer 25 mM HEPES (pH 7.5), 100 mM NaCl and 1 mM TCEP.
Fluorescence anisotropy DNA binding assay
Fluorescence anisotropy DNA binding assays were carried out as previously reported, with minor modifications52. Briefly, binding assays were performed using 5 nM fluorescein labeled 19-mer DNA sites. The two sites, PRDII from the IFNB1 promoter with the sequence 5-TTAGTGGGAAATTCCTCT-3′ (see ref. 52) and Hdac2 with the sequence 5′-TTCCTCGGGGTCCCCGCGG-3′ (Fig. 3a), were mixed with the NF-κB p50-p65 heterodimer at different concentrations to a final volume of 300 μL. The reaction buffer was 25 mM HEPES (pH 7.5), 50 mM NaCl and 1 mM TCEP. Fluorescence readings were taken on a PC1 fluorimeter (ISS, Inc.) with excitation and emission filters set at 492 nm and 545 nm, respectively. Before measurement, the DNA/protein mixtures were equilibrated at 20 °C for 20 min.
Anisotropy is calculated as the ratio of the difference between vertical and horizontal emission intensities over the total normalized intensity. Each anisotropy point is the average of 10 measurements. The fraction of DNA bound (B) was calculated using:
where [A]x represents the anisotropy measured at protein concentration X, [A]DNA is the anisotropy of free fluorescent DNA and [A]FINAL is the anisotropy at saturation.
Molecular modeling
Using the Protein Data Bank (PDB) code structure of the p65/p50 NF-κB heterodimer bound to DNA (PDB code 1VKX), the DNA sequence was mutated to the mouse Hdac2 promoter using Coot53. The new heterodimer/DNA complex was submitted to the webserver NPDock54 to complete a simple energy minimization of the heterodimer bound to the mouse Hdac2 promoter DNA site. This modeling was obtained with the assistance of PyMOL (http://www.pymol.org/).
Experimental animals
Experiments were performed on adult (8–20 weeks old) male mice. Animals were housed at 12 h light/dark cycle at 23 °C with food and water ad libitum. The Institutional Animal Use and Care Committee at Virginia Commonwealth University School of Medicine and the Icahn School of Medicine at Mount Sinai approved all experimental procedures.
5HT2A (Htr2a) knockout (KO) mice have been previously described18,31. For experiments involving 5HT2A-KO mice, wild-type littermates on a 129S6/Sv background were used as controls. All subjects were offspring of heterozygote breeding.
Hdac2 knockout (KO) mice were obtained from The Jackson Laboratory (stock number: 022625). Heterozygous Hdac2+/− mice were intercrossed to generate Hdac2−/− mice on a C57BL/6 background. Homozygosity of the Hdac2-null allele resulted in either embryonic lethality or partial lethality during the first few days postnatal as a result of proliferation defects and impaired development (data not shown). These findings are consistent with some26,55 but not all15 of the prior descriptions of global Hdac2 gene deletion. Based on this, we therefore used CaMKIIα-Cre transgenic mice on a C57BL/6 background, in which Cre recombinase is efficiently expressed56 in combination with conditional loss of function of Hdac2 alleles26. In mice, CaMKIIα is expressed postnatally in forebrain glutamatergic pyramidal neurons, beginning 10–14 d after birth55. To delete HDAC2 function specifically in forebrain glutamatergic pyramidal neurons, we bred homozygous Hdac2loxP/loxP mice to the CaMKIIα-Cre transgenic line. In contrast to the global deletion of HDAC2, Hdac2loxP/loxP:CaMKIIα-Cre (Hdac2-cKO) mice were viable and did not display any gross histological or developmental abnormality (data not shown). Hdac2loxP/loxP:CaMKIIα-Cre mice were born at near expected Mendelian ratios (data not shown).
p65loxP/loxP (Relafl) mice were obtained from The Jackson Laboratory (Stock number: 024342). Heterozygous p65loxP/+ mice were intercrossed to generate p65loxP/loxP mice on a C57BL/6 background.
Genomic DNA was isolated from tails for genotyping by PCR analysis. The primer sequences used were as follows:
5HT2A: forward-1 (5′-CTGTGGGATTTTCTTTCTGCTT-3′), forward-2 (5′-GTGTGATGGCTCTTGATTATGC-3′), common reverse (5′-TCTCTTGAT TCCCACTTTGTGGTT-3′);
Hdac2loxP/loxP: Hdac2 wt allele forward (5′-GCACAGGCTACTACTGT GTAGTCC-3′), Hdac2loxP mutant allele: forward (5′-GTCCCTCGA CCTGCAGGAATTC-3′), Hdac2loxP mutant allele reverse (5′-CCACCAC TGACATGTACCCAAC-3′);
CaMKII-Cre, transgene forward (5′-GCGGTCTGGCAGTAAAAACTATC-3′), transgene reverse (5′-GTGAAACAGCATTGCTGTCACTT-3′); internal positive control forward (5′-CTAGGCCACAGAATTGAAAGATCT-3′), internal positive control reverse (5′-GTAGGTGGAAATTCTAGCATCATCC-3′); and
p65loxP/loxP mutant forward (5′- GCTACTTCCATTTGTCACGTCC-3′), p65loxP/loxP wt forward (5′-GGGTACGGGTGAATCTTGACT-3′), p65loxP/loxP common reverse (5′-TGAGGATGGATGGTCCAACT-3′).
Mouse brain samples
The day of the experiment, mice were killed for analysis by cervical dislocation, and bilateral frontal cortex (bregma 1.90 to 1.40 mm) was dissected and either frozen at −80 °C or immediately processed for RNA extraction, chromatin immunoprecipitation and/or biochemical assays. The coordinates were taken according to a published atlas of the C57BL/6 and 129/Sv mouse brains57.
[3H]Ketanserin binding assays in HEK293 cells and mouse frontal cortex
Membrane preparations and [3H]ketanserin binding assays were carried out as previously reported, with minor modifications19. Nonspecific binding was determined in the presence of 10 μM methysergide. Membrane preparations were incubated at 37 °C for 60 min. Free ligand was separated from bound ligand by rapid filtration (MicroBeta filtermat-96, PerkinElmer) under vacuum through GF/C glass fiber filters. The filters were then rinsed twice with 3 mL of ice-cold incubation buffer, air-dried and counted for radioactivity by liquid scintillation spectrometry using a MicroBeta-2 counter (PerkinElmer).
RNA-seq and differential analysis
RNA was purified from bilateral frontal cortex tissue samples (see above) using RNeasy mini kit (Qiagen). Total RNA (5 μg) was used for mRNA library construction following instructions of Illumina RNA sample kit. All libraries were sequenced on an Illumina HiSeq 2500 platform at Genewiz Inc. (South Plainfield, NJ). Samples were quality control analyzed on an Agilient Bioanalyzer. The library was sequenced with 2 × 100 reads, and the reads were aligned with the STAR v2.5.1b58 (mouse genome GRCm38 assembly) and Gencode gene annotation (Release M8, Ensembl 83)59. The matrix counts of gene expression for all samples were computed by featureCounts v1.5.0-p1 (ref. 60). Differentially expressed genes (5% FDR and at least 0.2 log2 fold change) were identified through pairwise comparison using the Bioconductor46 package DESeq2 v1.12.4 (refs. 61,62). Values of scaled gene expression were displayed in the heat map. The RNA-seq data discussed in this paper have been deposited in NCBI’s Gene Expression Omnibus (GEO) under the GEO series accession number GSE93918. The gene enrichment analysis was performed with Homer v4.8.3 (ref. 63). Gene sets were selected from among those that contained at least two genes and gave P values < 0.05 for the enrichment.
Quantitative real-time PCR
Quantitative real-time PCR (qRT-PCR) assays were carried out in quadruplicate as previously described18,20,31,64,65 using a QuantStudio 6 Flex Real-Time PCR System (ThermoFisher Scientific; see Supplementary Table 5 for primer pair sequences). For the effect of DOI administration (i.p.) on induction of IκBα mRNA expression in mouse frontal cortex, experiments were performed as previously reported18,27. For the effect of 5-HTP administration (i.p.) on induction of IκBα mRNA expression in mouse frontal cortex, mice received injections (i.p.) of benserazide (30 mg/kg)66 30 min before 5-HTP or vehicle.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) experiments were performed as previously reported16,17. See Supplementary Table 6 for primer pair sequences.
Postmortem human brain tissue samples
Human brains were obtained at autopsies performed in the Basque Institute of Legal Medicine, Bilbao, Spain. The study was developed in compliance with policies of research and ethical review boards for postmortem brain studies (Basque Institute of Legal Medicine, Spain). We conducted retrospective searches on autopsied humans for previous medical diagnosis and treatment using examiners’ information and records of hospitals and mental health centers. After searching antemortem information, 20 subjects who had met criteria of schizophrenia according to the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV)67 were selected. Toxicological screening for antipsychotics, other drugs and ethanol was performed in blood, urine, liver and gastric contents samples. All subjects who were drug-free before death (as revealed by the absence of prescriptions in medical histories) also gave negative results in the toxicological screening. The toxicological assays were performed at the National Institute of Toxicology, Madrid, Spain, using a variety of standard procedures including radioimmunoassay, enzymatic immunoassay, high-performance liquid chromatography and gas chromatography–mass spectrometry. Controls for the present study were chosen among the collected brains on the basis, whenever possible, of the following cumulative criteria: (i) negative medical information on the presence of neuropsychiatric disorders or drug abuse; (ii) appropriate gender, age, postmortem delay (time between death and autopsy) and freezing storage time to match each subject in the schizophrenia group; (iii) sudden and unexpected death (motor vehicle accidents); and (iv) toxicological screening for psychotropic drugs with negative results except for ethanol. Specimens of prefrontal cortex (Brodmann area 9) were dissected at autopsy (0.5–1.0 g tissue) on an ice-cooled surface and immediately stored at −80 °C until use. The definitive pairs of antipsychotic-untreated schizophrenics and respective matched controls are shown in Supplementary Table 2, and the definitive pairs of atypical antipsychotic-treated schizophrenics and respective matched controls are shown in Supplementary Table 3. Pairs of schizophrenic patients and matched controls were processed simultaneously and under the same experimental conditions. Tissue pH is one of the most widely used tissue quality measurements68-70. Tissue pH values were within a relatively narrow range (control subjects: 6.33 ± 0.05; schizophrenic subjects: 6.23 ± 0.07). An algorithm designated as RIN (RNA integrity number) has been recently developed to analyze the quality of the tissue; it assigns a score from 1 to 10 (10 being the best) based on the ribosomal peaks and the extent of RNA degradation products68,71. All brain samples were assayed for RIN values using the Agilent 2100 Bioanalyzer (Applied Biosystems) as previously reported72 (Supplementary Tables 2 and 3).
Nuclei isolation from mouse frontal cortex
Mouse frontal cortex tissue samples (see above) were homogenized using a Teflon-glass grinder (15–20 up-and-down strokes) at 1,500 rpm in 4.8 mL lysis buffer (0.1% Triton X-100, 0.32 M sucrose, 5 mM CaCl2, 2 mM MgCl2, 1 mM DTT, 10 mM Tris-HCl; pH 7.4), filtered through a cell strainer (40 μm) and centrifuged for 12 min at 4 °C (600g). The pellet was resuspended in 750 μL of PBS and diluted up to 4 mL with 0.2 M sucrose. This was followed by the addition (via slow pipetting) of 4 mL of 1.1 M sucrose to the bottom of the resuspended pellet and centrifugation for 15 min at 4 °C (2,800g). The pellet was then resuspended again in RIPA buffer (100 μL) and sonicated (level 3; 10 s). Samples were centrifuged for 20 min at 4 °C (16,000g), after which supernatants (nuclear fraction) were collected and frozen at −20 °C or immediately processed for western immunoblotting (Supplementary Fig. 8a).
Co-immunoprecipitation in mouse frontal cortex
Mouse frontal cortex tissue samples (see above) were minced into >1-mm-sized pieces using a razor blade. Samples were homogenized in a prechilled Teflon-glass handy homogenizer (10 up-and-down strokes at 1,500 rpm in 5 mL lysis buffer (10 mM KCl, 1.5 mM MgCl2, 0.2% Igepal, 1 mM DTT, 10 mM HEPES; pH 7.9), after which samples were incubated on ice for 15 min and centrifuged for 10 min at 4 °C (900g). Pellets were resuspended in 500 μL resuspension buffer (10 mM EDTA, 1.0 mM EGTA, 0.2% Triton X-100, 0.5 mM DTT, 10 mM HEPES; pH 6.8) and incubated on ice for 15 min, after which samples were centrifuged for 5 min at 4 °C (2,500g). Supernatants (cytosolic fraction) were collected and frozen at −20 °C, whereas pellets were washed once with the resuspension buffer and centrifuged for 5 min at 4 °C (2,500g). The pellet was resuspended in 100 μL nuclear buffer (1.0 mM EDTA, 1.0 mM EGTA, 300 mM NaCl, 10% glycerol, 0.5 mM DTT, 10 mM HEPES; pH 6.8) and centrifuged for 20 min at 4 °C (16,000g), after which supernatants (nuclear fraction) were frozen at −20 °C or immediately processed for co-immunoprecipitation assays. For co-immunoprecipitation assays, samples were resuspended in 1 mL RIPA buffer (150 mM NaCl, 5 mM EDTA, 1% SDS, 1% Triton X-100, 50 mM HEPES; pH 7.4) and rotated at 4 °C for 60 min. After centrifugation for 15 min at 4 °C (16,000g), 900 μL of the supernatant were rotated with 40 μL of protein A/G beads (Santa Cruz Biotechnology, Inc.) at 4 °C for 60 min. After centrifugation for 1 min at 4 °C (16,000g), 400 μL of the supernatant were incubated overnight with 4 μL of anti-p65 antibody (see above) and 40 μL of protein A/G beads at 4 °C. Beads were then washed three times with RIPA buffer. Equal amounts of protein were resolved by SDS-PAGE (see below).
Nuclei isolation from postmortem human brain samples
For multiphoton confocal fluorescence microscope assays, nuclei isolation from postmortem human brain samples was performed as previously reported, with minor modifications73. Briefly, postmortem human frontal cortex tissue samples (see above) were homogenized using a Teflon-glass grinder (10 up-and-down strokes at 1,500 rpm in 8 mL of lysis buffer (0.1% Triton X-100, 0.32 M sucrose, 5 mM CaCl2, 2 mM MgCl2, 1 mM DTT, 10 mM Tris-HCl; pH 7.4), filtered through a cell strainer (40 μm) and centrifuged for 5 min at 4 °C (300g)). The pellet was resuspended in 750 μL of blocking buffer (1% goat serum and 2 mM MgCl2 in TBS buffer) and incubated with gentle rotation for 60–120 min at 4 °C with anti-NeuN (Millipore MAB377, 1:1,000) and DyLight 488-conjugated anti-p65 (Abcam ab139869, 1:500). Samples were transferred to precoated (5% BSA in PBS) 15-mL tubes and diluted with 4 mL of 0.25 M sucrose, after which a layer of 1.1-M sucrose was added. Next, second centrifugation step (15 min, 2,800g), and the resulting pellet was resuspended in 500 μL blocking buffer. Resuspended samples were incubated with gentle rotation for 60 min at 4 °C with Alexa Fluor-568 goat anti-mouse (Invitrogen A-11004, 1:1,000) antibody. Samples were then centrifuged for 6 min at 4 °C (600g), after which pellets were resuspended in 100 μL buffer (2 mM MgCl2 in TBS buffer). Resuspended samples were incubated for 10–15 min at room temperature in the presence of 2% paraformaldehyde and then quenched with the addition of glycine. Finally, 20 μL of sample were added onto glass slides with 4′,6′-diamidino-2-phenylindole dihydrochloride (DAPI)-containing mounting medium. Images were captured with a Carl Zeiss multiphoton confocal fluorescence microscope (CLSM780, Carl Zeiss). An average of 24 nuclei per subject totaling 226–247 nuclei per experimental group were analyzed.
For western immunoblot assays, postmortem frontal cortex tissue samples (1 g) were homogenized in a prechilled Teflon-glass grinder in 20 mL homogenization buffer (0.32 M sucrose, 5 mM Tris-HCl; pH 7.4) and centrifuged for 15 min at 4 °C (1,100g). The supernatants (S1 fraction) were kept on ice until further processing (see below). The pellets (P1 fraction) were resuspended in 5 mL homogenization buffer (see above) and centrifuged for 15 min at 4 °C (1,100g). Pellets were resuspended in 1.2 mL homogenization buffer (nuclear faction) and processed immediately or stored at −80 °C. The S1 fractions (see above) were centrifuged for 10 min at 4 °C (40,000g). The supernatants (S2), which correspond to the cytosolic fraction, were processed immediately or stored at −80 °C. Equal amounts of protein (nuclear and cytoplasmic fractions) were resolved by SDS-PAGE (Supplementary Fig. 8b).
Immunoblot assays
Western blot experiments were performed as previously reported, with minor modifications20,74. Briefly, samples were loaded onto poly-acrylamide gel (10–12%) and submitted to sodium dodecyl sulfate-PAGE. After transfer to nitrocellulose membranes, samples were blocked with 5% nonfat dry milk and 0.5% BSA in TBST buffer (Tris-buffered saline and 0.05 or 1% Tween-20), followed by overnight incubation in primary antibody at 4 °C or 1 h at room temperature (20–25 °C). The following primary antibodies were used: anti-p65 (mouse brain: Santa Cruz sc-372, 1:500; human brain: Santa Cruz, sc-372, 1:500), anti-IκBα (mouse brain: Santa Cruz sc-371, 1:100; human brain: Santa Cruz sc-371, 1:500), anti-IκBβ (mouse brain: Abcam ab7547, 1:1,000), anti-HDAC2 (mouse brain: Abcam ab32117, 1:1,000), anti-p50 (p105; mouse brain: ab32360, 1:1,000), anti-pERK (mouse brain: Cell Signaling 4370, 1:1,000), anti-ERK (mouse brain: Cell Signaling 9102, 1:1,000), anti-histone H1 (mouse brain: Santa Cruz sc-8030, 1:200; human brain: Santa Cruz sc-8030, 1:250), anti-β-actin (mouse brain: Abcam ab8227, 1:3,000 or 1:10,000, Cell Signaling 4697S, 1:5,000; human brain: Sigma-Aldrich, 1:200,000; Neuro-2a cells: Abcam ab8227, 1:3,000, Cell Signaling 4697S, 1:5,000), anti-α-tubulin (mouse brain: Abcam ab7291, 1:3,000, Abcam ab6160, 1:5,000; human brain: Abcam ab7291, 1:5,000), anti-hemagglutinin (Neuro-2a cells: Cell Signaling 2367S, 1:1,000; mouse brain: Cell Signaling 2367S, 1:1,000) and anti-Flag (mouse brain: Wako 018-22381, 1:2,500; Neuro-2a cells: Abcam ab8227, 1:3,000). Incubation with the secondary antibody (1:5,000–20,000) coupled to peroxidase (Amersham Biosciences) was performed at room temperature for 90 min, followed by repeated washing with TBST. Immunoreactive proteins were visualized with enhanced chemiluminescence (Thermo Scientific) according to the manufacturer’s instructions. In each case, the blots were stripped and reprobed for a control protein to control loading amounts. All immunoblots were quantified by densitometry using NIH Image 1.62 software. Western blot experiments in postmortem human brain samples were analyzed with the Odyssey infrared imaging system (LI-COR Biosciences). In those experiments, the nitrocellulose membrane was incubated with the primary antibody in blocking solution (see above). After washing, the membranes were incubated with secondary antibodies (anti-mouse IgG DyLight 800 (Cell Signaling 5257S, 1:15,000) and anti-rabbit IgG DyLight 680 (Thermo Scientific SA5-10042, 1:5,000)). The membranes were then scanned on an Odyssey infrared imaging system, and images were acquired and analyzed according to the manufacturer’s instructions.
Virally mediated gene transfer
Adeno-associated virus (AAV) serotype 8 was used for CaMKIIα∷eYFP, CaMKIIα∷HA-caIKK-β-p2A-eYFP, CaMKIIα∷Flag-dn-IκBα-p2A-eYFP and CaMKIIα∷mCherry-Cre, all produced by the University of North Carolina at Chapel Hill Vector Core75. AAV-eYFP, AAV-caIKK-β-eYFP, AAV-dn-IκBα-eYFP and AAV-Cre were injected into the frontal cortex by stereotaxic surgery according to standard methods. Mice were anesthetized with a combination of ketamine (100 mg/kg) and xylazine (10 mg/kg) during the surgery. The virus was delivered bilaterally with a Hamilton syringe at a rate of 0.1 μL/min for a total volume of 1.0 μL on each side. The following coordinates were used: +1.6 mm rostrocaudal, −2.4 mm dorsoventral, +2.6 mm mediolateral from bregma (relative to dura) with a 10° lateral angle. The coordinates were taken according to a published atlas of the C57BL/6 mouse strain57. Nissl staining of the coronal brain slice was taken from the mouse brain atlas with author’s permission (Supplementary Fig. 4b)57. All experiments were performed at least 3 weeks after surgery, when transgene expression is maximal75.
Immunohistochemistry
Immunohistochemistry assays were performed as previously reported with minor changes76. The animals were deeply anesthetized with pentobarbital (10 mg/kg). Transcardial perfusion was performed with 10 mL PBS, followed by 30 mL of freshly prepared 4% paraformaldehyde (PFA) in PBS at room temperature. Brains were removed, immersion-fixed in 4% PFA in PBS at 4 °C (overnight) and stored at 30% sucrose in PBS at 4 °C for at least 48 h. Screening of immunoreactive cells used a series of 30-μm-thick coronal sections from frontal cortex prepared on a sliding cryostat (Leica CM3050-S) equipped with a freezing stage. The free-floating sections were transferred to 24-well dishes containing PBS. Coronal brain sections were washed with PBS and incubated in 10% normal goat serum with 0.1% Triton X-100 in PBS for 60 min at 4 °C. The sections were then incubated overnight in the same solution containing the following antibodies: anti-HDAC2 (Abcam ab32117, 1:500), anti-synaptophysin (Sigma-Aldrich S5768, 1:1,000), anti-CaMKIIα (Thermo Scientific MA1-048, 1:200), anti-parvalbumin (Sigma-Aldrich P3088, 1:500), anti-eYFP (Invitrogen A-11122, 1:1,000), anti-p65 (Thermo Scientific RB-1638-P0, 1:100), anti-hemagglutinin (Cell Signaling 2367S, 1:500), anti-Flag (Sigma-Aldrich F1804, 1:500) or anti-HDAC1 (Abcam ab7028, 1:500). The sections were rinsed five times in PBS for 10 min each time and incubated for 1 h with Alexa Fluor-488 or -568 dye-conjugated goat anti-rabbit antibody (1:3,000) and Alexa Fluor-488 or -568 dye-conjugated goat anti-mouse antibody (1:3,000). Following incubation, the sections were washed three times with PBS, and the immunostained sections were examined by confocal fluorescence microscopy (CLSM780, Carl Zeiss). Counterstaining with DAPI allowed us to distinguish cortical areas and laminar borders. Images were taken of frontal cortex pyramidal neurons in cortical layers II/III because previous findings suggest that alterations in dendritic structure and signaling responses of layer II/III cortical pyramidal neurons may contribute to cognitive impairments in schizophrenia patients22,24,77,78. Similar findings were observed in layer V fontal cortex pyramidal neurons (data not shown).
Dendritic spine analysis
For spine analysis, apical dendritic segments 50–150 μm away from the soma were randomly chosen from AAV-infected cells that express eYFP. Note that eYFP signal was detected in layers II/III and layer V (Supplementary Fig. 4b), which correlates with expression of eYFP under the CaMKIIα promoter in frontal cortex pyramidal neurons. Images were taken of pyramidal neurons in frontal cortical layers II/III (see above for discussion). Similar findings were observed in frontal cortex layer V (data not shown). Images were acquired from a 4% paraformaldehyde-fixed 100-μm coronal slice, using a confocal fluorescence microscope (CLSM780, Carl Zeiss). CaMKIIα+ pyramidal neurons in layers III-V expressing eYFP were confirmed by their characteristic triangular somal shape. To qualify for spine analysis, dendritic segments had to satisfy the following requirements: (i) the segment had to be completely filled (all endings were excluded), (ii) the segment must have been at least 50 μm from the soma and (iii) the segment could not overlap with other dendritic branches. Dendritic segments were imaged using a 100× lens (numerical aperture 1.46; Carl Zeiss) and a zoom of 2.5. Pixel size was 0.03 μm in the xy plane and 0.01 μm in the z plane. Images were taken with a resolution of 1,024 × 300 (the y dimension was adjusted to the particular dendritic segment to expedite imaging), the pixel dwell time was 1.27 μm/s and the line average was set to 4. We analyzed an average of 4–6 dendrites per neuron on 4–5 neurons per mouse, totaling 1,207–2,217 dendritic spines per experimental group. For quantitative analysis of spine size and shape, NeuronStudio was used with the rayburst algorithm described previously. NeuronStudio classifies spines as stubby, thin or mushroom on the basis of the following values: (i) aspect ratio, (ii) head-to-neck ratio and (iii) head diameter. Spines with a neck can be classified as either thin or mushroom, and those without a neck are classified as stubby. Spines with a neck are labeled as thin or mushroom on the basis of head diameter. These parameters have been verified by comparison with trained human operators fully blinded across groups.
Whole-cell patch-clamp recordings and LTP induction
Acute frontal cortex slices were prepared from 3–4-month-old Hdac2 cKO mice and control litter-mates that had been previously injected with AAV-caIKK-β-eYFP, or with eYFP alone, into frontal cortex as described above. Mice were deeply anesthetized with isoflurane and decapitated. The brain was rapidly removed in ice-cold (~4 °C) sucrose-artificial cerebrospinal fluid (sucrose-aCSF), bubbled with (95% O2–5% CO2) and consisting of 233.7 mM sucrose, 26 mM NaHCO3, 3 mM KCl, 8 mM MgCl2, 0.5 mM CaCl2, 20 mM glucose and 0.4 mM ascorbic acid. Coronal slices (350 μm) were cut using a vibrating microtome (Leica VT1000S) and allowed to equilibrate in aCSF at room temperature for ~1 h before transferring to the recording chamber. The recording aCSF was composed of 117 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 2.5 mM CaCl2, 1.2 mM NaH2PO4, 24.9 mM NaHCO3 and 11.5 mM glucose. During recording, slices were maintained at 31 °C and perfused (1.5 mL/min) with oxygenated aCSF in an immersion chamber containing the GABAA receptor antagonist gabazine (10 μM). Whole-cell recordings were performed with glass micropipettes pulled from borosilicate glass capillaries using a P-87 micropipette puller (Sutter Instruments, Novato, CA). The pipette resistance was 3–4 M∷. Electrodes were filled with an intracellular solution containing 124 mM potassium-gluconate, 10 mM HEPES, 10 mM phosphocreatine di(Tris), 0.2 mM EGTA, 4 mM Mg2ATP and 0.3 mM Na2GTP at pH 7.3 and 280–290 mOsm. Cortical neurons from layers II/III expressing eYFP, with pyramidal shaped somata, were chosen for recording. Fluorescent neurons were visualized using an upright fluorescent microscope (BX50WI, Olympus) with 40× water immersion lens and an IR-1000 infrared CCD monochrome video camera (DAGE MTI). Gigaseal and further access to the intracellular neuronal compartment were achieved in voltage-clamp mode, with the holding potential set at −70 mV. Soon after rupturing the membrane, the intra-cellular neuronal fluid reached equilibrium with the pipette solution without significant changes in either series resistance or membrane capacitance values. All recordings were made in voltage-clamp mode using a Multiclamp 200B amplifier (Molecular Devices, Sunnyvale, CA). Analog signals were low-pass filtered at 2 kHz and digitized at 5 kHz with the use of a Digidata 1440A interface and pClamp10 software (Molecular Devices, Sunnyvale, CA). The pairing protocol used to induce long-term potentiation (LTP) has been described previously43 and consisted of pairing presynaptic stimulation with a brief period of postsynaptic depolarization. A tungsten concentric bipolar electrode (FHC Inc, Bowdoin, ME) was placed at the base of layer IV, and whole cell recordings were made from eYFP-identified layer II/III pyramidal neurons in the same column in prefrontal cortex. Excitatory postsynaptic currents (EPSCs) were evoked at a constant rate of 0.05 Hz throughout the entire experiment. EPSCs evoked by L4 stimulation were recorded at −70 mV during a baseline period of ~5–10 min, after which the cell was briefly depolarized to 0 mV for 10 min without changing stimulation rate. After this period of pairing, the holding potential was then returned to −70 mV and recordings continued for an additional 35–40 min at the same rate of stimulation. Series resistance was measured at the beginning and at the end of each recording. If series resistance changed >30% at the end of the recording, that cell was discarded. Offline analysis was performed using Clampfit (Molecular Devices) and Mini Analysis software (Synaptosoft Inc, Fort Lee, NJ). Mean EPSC amplitudes were measure across four different experimental groups and then normalized to their respective averaged baseline values. Data are represented in percent change from average baseline values.
Osmotic minipump surgery and infusion
Mice were anesthetized as detailed above. For intracerebroventricular (i.c.v.) infusion, mice were surgically implanted into either left or right (randomly assigned) lateral ventricle with a subcutaneous Alzet minipump (model 1004; Durect-Alzet) and lateral guide cannulae (Brain Infusion Kit 2; Alzet). The following coordinates were used: −0.5 mm anteroposterior, ±1.0 mm mediolateral, −2.0 mm dorsoventral from bregma. One day before surgery, cannulae (28-gauge stainless steel) were filled with JSH-23 (1.0 μM) or vehicle (40% hydroxypropyl β-cyclodextrin/PBS; Wako), and the pedestal within the assembly was affixed by vinyl tubing to a minipump loaded with JSH-23 or vehicle. The minipump was activated to initiate a continued delivery at 0.11 μL/h over 21 d. The surgical procedure began with an incision over the skull, and the skin was spread apart under the scapulae to create an area for positioning the minipump on the back. Cannulae were permanently fixed to the skull with Loctite skull adhesive (Henkel). Cannulae, tubing and minipumps were all secured under the skin using Vetbond tissue adhesive (3M) and two staples.
Head-twitch behavioral response
Behavioral testing took place between 9:00 a.m. and 6:00 p.m. Head-twitch behavioral assay was performed, as previously described16.
Locomotor activity
Motor function (locomotor activity) was assessed using a computerized three-dimensional activity monitoring system, as previously reported16.
Social interaction test
Mice were individually housed in a homecage for 2 d before the trial. In the first 5-min trial, an age-matched intruder mouse was introduced into the resident’s home cage under bright light conditions. Behavior was recorded on video for blind scoring. The duration of social interactions (close following, inspection, anogenital sniffing and other social body contacts, except aggressive behavior), aggression (attacking/biting and tail rattling) and escape behaviors were scored. Four trials, with an intertrial interval of 30 min, were used to analyze social behavior, using the same intruder mouse. Intruder mice had been selected based on their lack of aggressive behavior.
Novel-object recognition (NOR) test
Mice were habituated for 10 min to the NOR arena for three consecutive days before the first NOR test. On the days of testing, mice were given a 10-min acquisition trial and a 5-min recognition trial, separated by a 24 h intertrial return to their home cage. During the acquisition trial, the animals were allowed to explore two different objects (A and B). During the recognition trial, the animals explored a familiar object (A) from the acquisition trial and a novel object (C). Behavior was recorded on video for blind scoring of object exploration. Object exploration was defined as the animal licking, sniffing or touching the object with the forepaws while sniffing. The exploration time of each object (in s) was recorded manually by the use of two stopwatches. The exploratory preference (100 × (time spent exploring the novel object/total exploration time)) was then calculated for retention trials. If the exploration time in the acquisition or retention trials to either object was < 5 s, the data were excluded from analysis. This rarely occurred and did not affect our ability to complete the analysis using the data from the remaining animals of that group.
Spontaneous alternation behavior in a Y-maze test
Spontaneous alternation behavior comprised the tendency for mice to alternate their (conventionally) nonreinforced choices of Y-maze arms on successive opportunities79. Each arm or the Y-maze was 40 cm long, 12 cm high, 3 cm wide at the bottom and 10 cm wide at the top. The arms converge in an equilateral triangular central area that is 4 cm at its longest axis. Each mouse was placed individually at the center of the apparatus and allowed to move freely through the maze during an 8-min session. The number of arm entries was recorded visually. Alternation was defined as successive entries into the three arms on overlapping triplet sets. The alternation was calculated as the ratio of actual to possible alternations (defined as the total number of arm entries minus 2) multiplied by 100. Spontaneous alternation (%), defined as successive entries into the three arms on overlapping triplet sets, is associated with spatial short-term memory.
Latent learning in the water-finding test
The apparatus consisted of an open field (30 × 50 × 15 cm high) with an alcove (10 × 10 × 10 cm) in the middle of one of the long walls of the enclosure. The floor of the open field was divided into 15 identical squares for measuring locomotor activity. A drinking tube, identical to those used in the home cages, was inserted into the center of the alcove ceiling with its tip 6.5 cm (in the training trial) or 7.5 cm (in the test trial) above the floor. The test consisted of two trials: a training trial (the first day) and test trial (the second day). In the training trial, mice not deprived of water were placed individually into one corner of the open field of the apparatus. Each mouse was allowed 3 min to explore the environment; the 3 min was counted from the time the mouse started to explore. During this time, ambulation was measured by counting the number of times the animals crossed from one square to another in the open field. The frequency of touching, sniffing or licking of the water tube in the alcove (number of approaches) was also recorded. Animals that did not start exploring after 3 min had elapsed or that did not find the drinking tube during the 3 min exploratory period were omitted from the test trial. The mice were immediately returned to their home cages after the training trial. The mice were deprived of water for 24 h before the test trial. In the test trial, mice were again individually placed on the test apparatus 24 h after the single test trial. The time until the mouse moved out of the corner was measured as the starting latency. In addition, the time taken to enter the alcove (entering latency) and the time between entering the alcove and drinking the water (finding latency) were scored. Thus, the drinking latency consisted of the sum of the entering and finding latencies.
Statistical analysis
Statistical analyses were performed with GraphPad Prism software version 6. For all ChIP and mRNA data, fold changes relative to controls were determined using the corrected Ct method16,31. In immunoblot assays in postmortem human brain, the theoretical amount of protein in each sample was obtained by interpolation of the integrated optical density in the standard curve and compared with the real amount of protein loaded into the gel well74,80. Immunohistochemical images were acquires using confocal fluorescence microscope (see above) at identical settings for each of the conditions. Images were quantified using NIH Image 1.62 by an experimenter blind to treatment, viral injection and/or genotype groups. For quantifying HDAC1 or HDAC2 immunoreactivity in mouse tissue sections, the mean signal intensities of HDAC1 or HDAC2 were measured in CaMKIIα+, PV+ or eYFP+ cells. For quantifying synaptophysin immunoreactivity after chronic clozapine treatment, the mean signal intensity of synaptophysin immunoreactivity in the different layers of the frontal cortex and regions of the hippocampus (DAPI) was measured in both hemispheres of treated mice and controls. For the quantification of synaptophysin immunoreactivity in stereotaxically injected mice, the mean signal intensity of synaptophysin immunoreactivity was measured in the different layers of the frontal cortex (DAPI) in the stereotaxically injected site (eYFP+ neuronal bodies) and the contralateral (mock-injected) hemisphere. For calculating p65 subcellular localization (nuclear/cytoplasmic), the mean signal intensity of nuclear p65 (i.e., p65 signal overlapping the DAPI-stained area) was divided by the mean signal intensity of cytoplasmic p65 (i.e., CaMKIIα+ or eYFP+ area not overlapping with DAPI-stained area). For quantifying neuronal nuclear p65 in postmortem human brain samples, the mean intensity signal of p65 in NeuN+ or NeuN– nuclei was quantified (see also above). Microscope images were taken by an observer who was blind to the experimental conditions. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported in our previous publications16. Data distribution was assumed to be normal, but this was not formally tested. Animals were randomly allocated into the different experimental groups. Data points were excluded based on previously established criterion and were set to ± 2 s.d. from the group mean. The statistical significance of experiments involving three or more independent variables on a continuous dependent variable was assessed by three-way ANOVA. The statistical significance of experiments involving three or more groups and two or more treatments was assessed by two-way ANOVA followed by Bonferroni’s post hoc test. Statistical significance of experiments involving three or more groups was assessed by one-way ANOVA followed by Bonferroni’s post hoc test. Statistical significance of experiments involving two groups was assessed by Student’s t test. The effects on mRNA expression of multiple genes or ChIP assays at different regions of a particular gene were assessed by correcting the α value for multiple independent null hypotheses. This was performed by using the Holm’s sequentially rejective Bonferroni method as follows: α′ = 1 − (1 − α)(1/N), where α = 0.05 and N = the number of independent null hypothesis (for example, number of modulated genes)16,31,81. Correlation analysis was conducted using the Pearson’s r. The level of significance was set at P = 0.05. Al values included in the figure legends represent mean ± s.e.m. A Life Sciences Reporting Summary is available.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supplementary Material
Acknowledgments
The authors thank M. Fribourg, P. Roussos (Icahn School of Medicine at Mount Sinai), P. Bos, S. Bowers, A. Ellaithy (Virginia Commonwealth University School of Medicine), A. Gallitano (The University of Arizona), T. Nabeshima (Fujita Health University), M. Hiramatsu (Meijo University) and K. Yamada (Nagoya University) for their critical review of the manuscript; F. Isoda and C. Mobbs for their help in promoter assays; A. Meredith for his help in evaluating immunohistological assays; S. Morgello and the Manhattan HIV Brain Bank for providing control brain cortex; H. Morishita (Icahn School of Medicine at Mount Sinai) for the donation of CaMKIIα-Cre mice; D. Benson (Icahn School of Medicine at Mount Sinai) for the generous gift of Neuro2A cells; K. Deisseroth (Stanford University) for providing the p2A construct; J. Gingrich (Columbia University) for the donation of 5-HT2A knockout mice; E. Olson (University of Texas Southwestern Medical Center), R. Bassel-Duby (University of Texas Southwestern Medical Center) and E. Nestler (Icahn School of Medicine at Mount Sinai) for their gift of loxP-flanked Hdac2 mice; K. Hideshima and A. Hojati for assistance with biochemical assays; and the staff members of the Basque Institute of Legal Medicine for their cooperation in the study. NIH R01 MH084894 (J.G.M.), NIH R01 MH111940 (J.G.M.), Dainippon Sumitomo Pharma (J.G.M.), NARSAD (J.G.M.), the Japan Society for the Promotion of Science (JSPS) 15H06719 and 16K19786 (D.I.), NIH R01 MH104491 (G.W.H.), NIH R01 MH086509 (S.A.), NIH P50 MH096890 (S.A.), MINECO/ERDF SAF2009-08460 (J.J.M. and L.F.C.), SAF2013-45084R (J.J.M. and L.F.C.), Basque Government IT616-13 (J.J.M.), NIH R21 MH103877 (S.D.) and NIH R01 MH090264 (S.J.R.) participated in the funding of this study. RNA-seq analysis was supported in part through the computational resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai and the NIH infrastructure grant S10OD018522. C.M. and A.G.B. were recipients of a postdoctoral and a predoctoral fellowship from the Basque Government, respectively. D.I. was a recipient of postdoctoral fellowships from JSPS (Young Scientists JSPS 23-3454) and the Uehara Memorial Foundation.
Footnotes
AUTHOR CONTRIBUTIONS
D.I., M.d.l.F.R. and J.G.-M. designed experiments, analyzed data and wrote the manuscript. D.I. and M.d.l.F.R. performed experiments. J.G.-M. supervised the research. S.A.G., J.M.S., M.I., T.H., J.L.M., A.G.-B., M.K., G.E.M. and J.F.L.-G. assisted with experiments. N.K., supervised by G.W.H., performed electrophysiological studies. C.M., supervised by L.F.C. and J.J.M., performed assays in postmortem brain samples. L.F.C. and J.J.M. obtained and classified postmortem human brain samples. V.S., supervised by C.R.E., performed fluorescence anisotropy assays. A.K., supervised by S.D., helped with nuclei separation assays. Y.J., supervised by S.A., helped with experiments in postmortem human brain. J.S. and Y.G. performed biostatistical analyzes. D.J.C., supervised by S.J.R., helped with synaptic structure assays. All authors discussed the results and commented on the manuscript.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
Note: Any Supplementary Information and Source Data files are available in theonline version of the paper.
References
- 1.van Os J, Kapur S. Schizophrenia. Lancet. 2009;374:635–645. doi: 10.1016/S0140-6736(09)60995-8. [DOI] [PubMed] [Google Scholar]
- 2.Crilly J. The history of clozapine and its emergence in the US market: a review and analysis. Hist Psychiatry. 2007;18:39–60. doi: 10.1177/0957154X07070335. [DOI] [PubMed] [Google Scholar]
- 3.Meltzer HY. Update on typical and atypical antipsychotic drugs. Annu Rev Med. 2013;64:393–406. doi: 10.1146/annurev-med-050911-161504. [DOI] [PubMed] [Google Scholar]
- 4.Miyamoto S, Miyake N, Jarskog LF, Fleischhacker WW, Lieberman JA. Pharmacological treatment of schizophrenia: a critical review of the pharmacology and clinical effects of current and future therapeutic agents. Mol Psychiatry. 2012;17:1206–1227. doi: 10.1038/mp.2012.47. [DOI] [PubMed] [Google Scholar]
- 5.Husa AP, et al. Lifetime use of antipsychotic medication and its relation to change of verbal learning and memory in midlife schizophrenia - an observational 9-year follow-up study. Schizophr Res. 2014;158:134–141. doi: 10.1016/j.schres.2014.06.035. [DOI] [PubMed] [Google Scholar]
- 6.Fervaha G, et al. Antipsychotics and amotivation. Neuropsychopharmacology. 2015;40:1539–1548. doi: 10.1038/npp.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goldberg TE, et al. The effect of clozapine on cognition and psychiatric symptoms in patients with schizophrenia. Br J Psychiatry. 1993;162:43–48. doi: 10.1192/bjp.162.1.43. [DOI] [PubMed] [Google Scholar]
- 8.Nielsen RE, et al. Second-generation antipsychotic effect on cognition in patients with schizophrenia--a meta-analysis of randomized clinical trials. Acta Psychiatr Scand. 2015;131:185–196. doi: 10.1111/acps.12374. [DOI] [PubMed] [Google Scholar]
- 9.Schröder N, de Lima MN, Quevedo J, Dal Pizzol F, Roesler R. Impairing effects of chronic haloperidol and clozapine treatment on recognition memory: possible relation to oxidative stress. Schizophr Res. 2005;73:377–378. doi: 10.1016/j.schres.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 10.Rosengarten H, Quartermain D. The effect of chronic treatment with typical and atypical antipsychotics on working memory and jaw movements in three- and eighteen-month-old rats. Prog Neuropsychopharmacol Biol Psychiatry. 2002;26:1047–1054. doi: 10.1016/s0278-5846(02)00221-x. [DOI] [PubMed] [Google Scholar]
- 11.Kellendonk C, Simpson EH, Kandel ER. Modeling cognitive endophenotypes of schizophrenia in mice. Trends Neurosci. 2009;32:347–358. doi: 10.1016/j.tins.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Millan MJ, et al. Cognitive dysfunction in psychiatric disorders: characteristics, causes and the quest for improved therapy. Nat Rev Drug Discov. 2012;11:141–168. doi: 10.1038/nrd3628. [DOI] [PubMed] [Google Scholar]
- 13.Gräff J, Tsai LH. The potential of HDAC inhibitors as cognitive enhancers. Annu Rev Pharmacol Toxicol. 2013;53:311–330. doi: 10.1146/annurev-pharmtox-011112-140216. [DOI] [PubMed] [Google Scholar]
- 14.Gräff J, et al. Epigenetic priming of memory updating during reconsolidation to attenuate remote fear memories. Cell. 2014;156:261–276. doi: 10.1016/j.cell.2013.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guan JS, et al. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kurita M, et al. HDAC2 regulates atypical antipsychotic responses through the modulation of mGlu2 promoter activity. Nat Neurosci. 2012;15:1245–1254. doi: 10.1038/nn.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurita M, et al. Repressive epigenetic changes at the mGlu2 promoter in frontal cortex of 5-HT2A knockout mice. Mol Pharmacol. 2013;83:1166–1175. doi: 10.1124/mol.112.084582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.González-Maeso J, et al. Hallucinogens recruit specific cortical 5-HT(2A) receptor-mediated signaling pathways to affect behavior. Neuron. 2007;53:439–452. doi: 10.1016/j.neuron.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 19.Moreno JL, et al. Allosteric signaling through an mGlu2 and 5-HT2A heteromeric receptor complex and its potential contribution to schizophrenia. Sci Signal. 2016;9:ra5. doi: 10.1126/scisignal.aab0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.González-Maeso J, et al. Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature. 2008;452:93–97. doi: 10.1038/nature06612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moreno JL, et al. Identification of three residues essential for 5-hydroxytryptamine 2A-metabotropic glutamate 2 (5-HT2A·mGlu2) receptor heteromerization and its psychoactive behavioral function. J Biol Chem. 2012;287:44301–44319. doi: 10.1074/jbc.M112.413161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 2000;57:65–73. doi: 10.1001/archpsyc.57.1.65. [DOI] [PubMed] [Google Scholar]
- 23.Black JE, et al. Pathology of layer V pyramidal neurons in the prefrontal cortex of patients with schizophrenia. Am J Psychiatry. 2004;161:742–744. doi: 10.1176/appi.ajp.161.4.742. [DOI] [PubMed] [Google Scholar]
- 24.Hill JJ, Hashimoto T, Lewis DA. Molecular mechanisms contributing to dendritic spine alterations in the prefrontal cortex of subjects with schizophrenia. Mol Psychiatry. 2006;11:557–566. doi: 10.1038/sj.mp.4001792. [DOI] [PubMed] [Google Scholar]
- 25.Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012;35:57–67. doi: 10.1016/j.tins.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Montgomery RL, et al. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 2007;21:1790–1802. doi: 10.1101/gad.1563807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moreno JL, et al. Persistent effects of chronic clozapine on the cellular and behavioral responses to LSD in mice. Psychopharmacology (Berl) 2013;225:217–226. doi: 10.1007/s00213-012-2809-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yadav PN, Kroeze WK, Farrell MS, Roth BL. Antagonist functional selectivity: 5-HT2A serotonin receptor antagonists differentially regulate 5-HT2A receptor protein level in vivo. J Pharmacol Exp Ther. 2011;339:99–105. doi: 10.1124/jpet.111.183780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams AA, et al. Reduced levels of serotonin 2A receptors underlie resistance of Egr3-deficient mice to locomotor suppression by clozapine. Neuropsychopharmacology. 2012;37:2285–2298. doi: 10.1038/npp.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McOmish CE, Lira A, Hanks JB, Gingrich JA. Clozapine-induced locomotor suppression is mediated by 5-HT2A receptors in the forebrain. Neuropsychopharmacology. 2012;37:2747–2755. doi: 10.1038/npp.2012.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.González-Maeso J, et al. Transcriptome fingerprints distinguish hallucinogenic and nonhallucinogenic 5-hydroxytryptamine 2A receptor agonist effects in mouse somatosensory cortex. J Neurosci. 2003;23:8836–8843. doi: 10.1523/JNEUROSCI.23-26-08836.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sandelin A, Alkema W, Engström P, Wasserman WW, Lenhard B. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32:D91–D94. doi: 10.1093/nar/gkh012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gutierrez H, Davies AM. Regulation of neural process growth, elaboration and structural plasticity by NF-κB. Trends Neurosci. 2011;34:316–325. doi: 10.1016/j.tins.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Russo SJ, Mazei-Robison MS, Ables JL, Nestler EJ. Neurotrophic factors and structural plasticity in addiction. Neuropharmacology. 2009;56(Suppl 1):73–82. doi: 10.1016/j.neuropharm.2008.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Koo JW, Russo SJ, Ferguson D, Nestler EJ, Duman RS. Nuclear factor-kappaB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc Natl Acad Sci USA. 2010;107:2669–2674. doi: 10.1073/pnas.0910658107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thomas GM, Huganir RL. MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci. 2004;5:173–183. doi: 10.1038/nrn1346. [DOI] [PubMed] [Google Scholar]
- 37.Ashburner BP, Westerheide SD, Baldwin AS., Jr The p65 (RelA) subunit of NF-kappaB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol. 2001;21:7065–7077. doi: 10.1128/MCB.21.20.7065-7077.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ito K, et al. Histone deacetylase 2-mediated deacetylation of the glucocorticoid receptor enables NF-kappaB suppression. J Exp Med. 2006;203:7–13. doi: 10.1084/jem.20050466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Y, et al. HDAC-mediated deacetylation of NF-κB is critical for Schwann cell myelination. Nat Neurosci. 2011;14:437–441. doi: 10.1038/nn.2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang G, et al. Hypothalamic programming of systemic ageing involving IKK-β, NF-κB and GnRH. Nature. 2013;497:211–216. doi: 10.1038/nature12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Szymczak-Workman AL, Vignali KM, Vignali DA. Design and construction of 2A peptide-linked multicistronic vectors. Cold Spring Harb Protoc. 2012;2012:199–204. doi: 10.1101/pdb.ip067876. [DOI] [PubMed] [Google Scholar]
- 42.Huynh QK, et al. Characterization of the recombinant IKK1/IKK2 heterodimer Mechanisms regulating kinase activity. J Biol Chem. 2000;275:25883–25891. doi: 10.1074/jbc.M000296200. [DOI] [PubMed] [Google Scholar]
- 43.Feldman DE. Timing-based LTP and LTD at vertical inputs to layer II/III pyramidal cells in rat barrel cortex. Neuron. 2000;27:45–56. doi: 10.1016/s0896-6273(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 44.Vollenweider FX, Vollenweider-Scherpenhuyzen MF, Bäbler A, Vogel H, Hell D. Psilocybin induces schizophrenia-like psychosis in humans via a serotonin-2 agonist action. Neuroreport. 1998;9:3897–3902. doi: 10.1097/00001756-199812010-00024. [DOI] [PubMed] [Google Scholar]
- 45.Schmid Y, et al. Acute effects of lysergic acid diethylamide in healthy subjects. Biol Psychiatry. 2015;78:544–553. doi: 10.1016/j.biopsych.2014.11.015. [DOI] [PubMed] [Google Scholar]
- 46.Morris BJ, Cochran SM, Pratt JA. PCP: from pharmacology to modelling schizophrenia. Curr Opin Pharmacol. 2005;5:101–106. doi: 10.1016/j.coph.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 47.Kristiansen LV, Huerta I, Beneyto M, Meador-Woodruff JH. NMDA receptors and schizophrenia. Curr Opin Pharmacol. 2007;7:48–55. doi: 10.1016/j.coph.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 48.Nichols DE. Psychedelics. Pharmacol Rev. 2016;68:264–355. doi: 10.1124/pr.115.011478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Muguruza C, et al. Dysregulated 5-HT(2A) receptor binding in postmortem frontal cortex of schizophrenic subjects. Eur Neuropsychopharmacol. 2013;23:852–864. doi: 10.1016/j.euroneuro.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marder E, Goaillard JM. Variability, compensation and homeostasis in neuron and network function. Nat Rev Neurosci. 2006;7:563–574. doi: 10.1038/nrn1949. [DOI] [PubMed] [Google Scholar]
- 51.Lopez-Gimenez JF, Vilaró MT, Milligan G. Morphine desensitization, internalization, and down-regulation of the mu opioid receptor is facilitated by serotonin 5-hydroxytryptamine2A receptor coactivation. Mol Pharmacol. 2008;74:1278–1291. doi: 10.1124/mol.108.048272. [DOI] [PubMed] [Google Scholar]
- 52.Escalante CR, Shen L, Thanos D, Aggarwal AK. Structure of NF-kappaB p50/p65 heterodimer bound to the PRDII DNA element from the interferon-beta promoter. Structure. 2002;10:383–391. doi: 10.1016/s0969-2126(02)00723-2. [DOI] [PubMed] [Google Scholar]
- 53.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tuszynska I, Magnus M, Jonak K, Dawson W, Bujnicki JM. NPDock: a web server for protein-nucleic acid docking. Nucleic Acids Res. 2015;43:W425–W430. doi: 10.1093/nar/gkv493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morris MJ, Mahgoub M, Na ES, Pranav H, Monteggia LM. Loss of histone deacetylase 2 improves working memory and accelerates extinction learning. J Neurosci. 2013;33:6401–6411. doi: 10.1523/JNEUROSCI.1001-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cabungcal JH, et al. Perineuronal nets protect fast-spiking interneurons against oxidative stress. Proc Natl Acad Sci USA. 2013;110:9130–9135. doi: 10.1073/pnas.1300454110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hof PR, et al. Comparative Cytoarchitectonic Atlas of the C57BL/6 and 129/Sv Mouse Brains. Elsevier; Amsterdam: 2000. [Google Scholar]
- 58.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Harrow J, et al. GENCODE: producing a reference annotation for ENCODE. Genome Biol. 2006;7(Suppl. 1):1–9. doi: 10.1186/gb-2006-7-s1-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 61.Gentleman RC, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Renthal W, et al. Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron. 2007;56:517–529. doi: 10.1016/j.neuron.2007.09.032. [DOI] [PubMed] [Google Scholar]
- 65.Covington HE, III, et al. Antidepressant actions of histone deacetylase inhibitors. J Neurosci. 2009;29:11451–11460. doi: 10.1523/JNEUROSCI.1758-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Baumann MH, Williams Z, Zolkowska D, Rothman RB. Serotonin (5-HT) precursor loading with 5-hydroxy-l-tryptophan (5-HTP) reduces locomotor activation produced by (+)-amphetamine in the rat. Drug Alcohol Depend. 2011;114:147–152. doi: 10.1016/j.drugalcdep.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders: DSM-IV. 4. Washington, DC: 1994. [Google Scholar]
- 68.Stan AD, et al. Human postmortem tissue: what quality markers matter? Brain Res. 2006;1123:1–11. doi: 10.1016/j.brainres.2006.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Preece P, Cairns NJ. Quantifying mRNA in postmortem human brain: influence of gender, age at death, postmortem interval, brain pH, agonal state and inter-lobe mRNA variance. Brain Res Mol Brain Res. 2003;118:60–71. doi: 10.1016/s0169-328x(03)00337-1. [DOI] [PubMed] [Google Scholar]
- 70.Li JZ, et al. Systematic changes in gene expression in postmortem human brains associated with tissue pH and terminal medical conditions. Hum Mol Genet. 2004;13:609–616. doi: 10.1093/hmg/ddh065. [DOI] [PubMed] [Google Scholar]
- 71.Imbeaud S, et al. Towards standardization of RNA quality assessment using user-independent classifiers of microcapillary electrophoresis traces. Nucleic Acids Res. 2005;33:e56. doi: 10.1093/nar/gni054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.García-Sevilla JA, et al. Reduced platelet G protein-coupled receptor kinase 2 in major depressive disorder: antidepressant treatment-induced upregulation of GRK2 protein discriminates between responder and non-responder patients. Eur Neuropsychopharmacol. 2010;20:721–730. doi: 10.1016/j.euroneuro.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 73.Kozlenkov A, et al. Differences in DNA methylation between human neuronal and glial cells are concentrated in enhancers and non-CpG sites. Nucleic Acids Res. 2014;42:109–127. doi: 10.1093/nar/gkt838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.González-Maeso J, Rodríguez-Puertas R, Meana JJ, García-Sevilla JA, Guimón J. Neurotransmitter receptor-mediated activation of G-proteins in brains of suicide victims with mood disorders: selective supersensitivity of alpha(2A)-adrenoceptors. Mol Psychiatry. 2002;7:755–767. doi: 10.1038/sj.mp.4001067. [DOI] [PubMed] [Google Scholar]
- 75.Aschauer DF, Kreuz S, Rumpel S. Analysis of transduction efficiency, tropism and axonal transport of AAV serotypes 1, 2, 5, 6, 8 and 9 in the mouse brain. PLoS One. 2013;8:e76310. doi: 10.1371/journal.pone.0076310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chan P, et al. Epsilon-sarcoglycan immunoreactivity and mRNA expression in mouse brain. J Comp Neurol. 2005;482:50–73. doi: 10.1002/cne.20377. [DOI] [PubMed] [Google Scholar]
- 77.Sweet RA, Henteleff RA, Zhang W, Sampson AR, Lewis DA. Reduced dendritic spine density in auditory cortex of subjects with schizophrenia. Neuropsychopharmacology. 2009;34:374–389. doi: 10.1038/npp.2008.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jaaro-Peled H, Ayhan Y, Pletnikov MV, Sawa A. Review of pathological hallmarks of schizophrenia: comparison of genetic models with patients and nongenetic models. Schizophr Bull. 2010;36:301–313. doi: 10.1093/schbul/sbp133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hughes RN. The value of spontaneous alternation behavior (SAB) as a test of retention in pharmacological investigations of memory. Neurosci Biobehav Rev. 2004;28:497–505. doi: 10.1016/j.neubiorev.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 80.González-Maeso J, et al. Effects of age, postmortem delay and storage time on receptor-mediated activation of G-proteins in human brain. Neuropsychopharmacology. 2002;26:468–478. doi: 10.1016/S0893-133X(01)00342-6. [DOI] [PubMed] [Google Scholar]
- 81.Shaffer JP. Multiple hypothesis testing. Annu Rev Psychol. 1995;46:561–584. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.