

Graphical abstract
1. INTRODUCTION
The importance of nickel enzymes, where nickel serves as an essential cofactor, in Archaea, bacteria, plants, and primitive eukaryotes, is well documented.1 Despite the fact that no enzyme utilizing Ni has been found in mammalian species, the impact of Ni biochemistry on human health is also significant.2 Indeed, nickel is known to cause cancer by an epigenetic mechanism,3 which appears to involve substitution of Ni(II) for Fe(II) in non-heme iron dioxygenases that are involved in DNA and histone demethylation.4 Furthermore, exposure to nickel compounds can also elicit an immune reaction: nickel contact dermatitis is one of the most common allergies in the modern world,5 and the molecular basis for the immune reaction is now beginning to emerge.6 Exposure to high levels of nickel has also been shown to impair the normal homeostasis of essential metal ions.7 Nonetheless, nickel is among the metals included in a group of “possibly essential elements” for animals and humans.8
Experiments using animal models have shown that nickel may be beneficial for bone composition and strength, for optimal reproduction, for energy metabolism, and for sensory function.8 The molecular basis for these functions is, however, not known. It is not surprising that human nickel deficiency has never been reported, probably because normal nickel intake greatly exceeds the estimated 25–35 μg/day metabolic requirement.8
In this Review, the current knowledge of the biochemistries, structures, and reaction mechanisms of enzymes whose active sites require nickel, and utilize it in a nonredox role, are discussed. In addition to excluding redox-active nickel sites (see the pertinent Reviews in this Thematic Issue), proteins isolated in proteomics facilities using nickel affinity columns that contain nickel, but whose native active site does not contain nickel, are also excluded. The focus is on the enzymes, and data from synthetic model systems is included only when it enhances knowledge of the reaction mechanism.
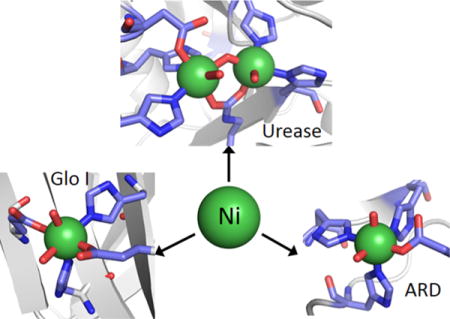
The selection of nickel as a catalytic center for biological reactions is related to its flexible coordination geometry,9 which makes this metal a very versatile element for biological applications. To date, eight microbial nickel-containing enzymes have been well-characterized, including urease, hydrogenase, CO-dehydrogenase, acetyl-CoA synthase, methyl-CoM reductase, Ni-superoxide dismutase, acireductone dioxygenase, and glyoxalase I, while a few other possible nickel-dependent enzymes are emerging.10 The biological roles of nickel enzymes are conveniently divided into redox and nonredox roles.11 Unlike the more abundant biological redox metals iron and copper, aquated Ni(II) ions have no biologically relevant redox chemistry, as water will oxidize and reduce at potentials less extreme than those of the metal ion. Thus, the ligand environment is critical in adjusting the redox potential of Ni(II) into a biologically accessible range. In terms of nickel enzymes, this is usually achieved via coordination of anionic S-donors such as sulfide (as in carbon monoxide dehydrogenase) or, more commonly, thiolate sulfur in the form of cysteine ligands, which stabilizes the Ni(II/III) redox couple, or by coupling processes to sulfur redox chemistry (e.g., in methyl coenzyme M reductase).11 Thus, S-donor ligands are strongly associated with Ni redox enzymes, which include a novel superoxide dismutase, and several other enzymes including hydrogenase and CO-dehydrogenase/acetyl coenzyme A synthase that use Ni(II/III) redox chemistry to catalyze reactions that are involved in biological C1 chemistry. These enzymes have been extensively reviewed recently,12 and are covered by Reviews elsewhere in this Thematic Issue (see Valentine et al.,13 Lubitz,14 and Ragsdale15). In addition to adjusting redox potentials, the S-donor rich ligand environments often favor coordinatively unsaturated complexes with low-spin electronic structures.9
Nickel is also employed in enzymes as a Lewis acid catalyst, and in contrast to the redox enzymes, the coordination environments of these enzymes are composed exclusively of O/N-donor ligands. The coordination chemistry typically favors six-coordinate Ni(II) complexes that invariably have high-spin electronic structures.9 Three enzymes that utilize Ni(II) as a Lewis acid, urease, acireductone dehydrogenase, and glyoxalase I, are discussed here, with the goal of updating other comprehensive reviews through mid-2013.16
2. UREASE
2.1. Biological Significance
Urease (urea aminohydrolase EC 3.5.1.5) is an important enzyme in the global nitrogen cycle, and is found in plants, algae, fungi, and several microorganisms.17 It catalyzes the rapid hydrolytic decomposition of urea.18
Two important records characterize its history: in 1926, urease from Canavalia ensiformis (jack bean urease, JBU) was the first enzyme to be crystallized, providing clear evidence that enzymes are proteins,19 while in 1975 the requirement for nickel in JBU catalysis was established, providing the first example for the biological essentiality of this metal as an enzyme cofactor.20 Since then, significant steps toward a complete understanding of the chemistry and biochemistry of nickel in the urease system have been achieved.21
Urea is produced in large amounts by vertebrates as the catabolic product of nitrogen-containing compounds; each human produces ca. 10 kg of urea per year.22 Considering the long half-life for the nonenzymatic decomposition of urea in water that occurs through an elimination step to produce ammonia and cyanic acid (t1/2 ∼ 40 years)23 and the even longer half-life for the nonenzymatic urea hydrolysis, never observed experimentally24 but estimated as being ca. 520 years,25 severe environmental problems could arise in the absence of an efficient enzymatic degradation of urea. On the other hand, the hydrolysis of urea catalyzed by urease18 has a half-time of a few microseconds, with a kcat/Km that is 3 × 1015 times higher than the rate of the uncatalyzed reaction, making urease the most efficient hydrolase known, an efficiency ascribed to the presence of two Ni(II) ions in the active site of the enzyme.25
The overall hydrolysis of the products generated by the urease catalyzed reaction causes a pH increase that has important consequences in medical and agricultural settings. Urease is the main virulence factor in a large variety of lethal human pathogens such as Mycobacterium tuberculosis,26 Yersinia enterocolitica,27 and Cryptococcus neoformans.28 Infections of the urinary and gastrointestinal tracts in human and animals by ureolytic bacteria such as Proteus mirabilis29 can cause kidney stone formation, catheter encrustation, pyelonephritis, ammonia encephalopathy, and hepatic coma.17b,30 A further example of a ureolytic pathogen is represented by Helicobacter pylori, a bacterium able to survive in the acidic environment of the stomach by exploiting the pH increase caused by the urease activity, and acting as the major cause of pathologies, including cancer induced by gastroduodenal infections.31 In the agricultural context, bacterial urease activity is widespread in the environment and especially in soils fertilized with urea, a nitrogen fertilizer used worldwide.32 Although necessary for crop nitrogen uptake, the efficiency of soil nitrogen fertilization with urea is severely decreased by the urease activity itself, because the soil pH increase associated with urea hydrolysis causes the release of large amounts of ammonia nitrogen into the atmosphere. The damage to plants from ammonia toxicity, as well as seedling damage, causes significant environmental and economic problems.33 In plants, the primary role of urease is to salvage urea nitrogen during germination, for nutrition purposes,34 although insecticidal activity as a plant defense system has also been reported.35 In particular, plant ureases from Glycine max (soybean) and C. ensiformis display insecticidal activity that is unrelated to the enzyme activity.36 One of the urease isoforms found in jack bean, termed canatoxin, appears to generate an entomotoxic peptide, pepcanatox, in insects possessing cysteine and aspartate protease activity in their digestive systems. A recombinant analogue of this peptide, jaburetox, also features insecticidal activity.37 The crystal structure of urease from jack bean has revealed that this peptide is found in a region connecting domains that in bacterial ureases constitute separate subunits.38
2.2. Enzymology
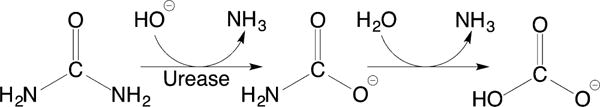
The process of enzymatic urea decomposition occurs in two steps: the first is the hydrolysis of urea to give ammonia and carbamate, which then spontaneously reacts in water to yield a second molecule of ammonia and bicarbonate (Scheme 1).18a
Scheme 1.
Ureases exhibit typical Michaelis–Menten behavior.21e The observed values of Km fall in the 0.2–32 mM range, with typical values around 1–4 mM, and are largely independent of pH.21e On the other hand, kcat and kcat/Km strongly depend on pH, and bell-shaped profiles are observed, with one activity maximum in the 7.0–7.5 pH range and another maximum in the 6.0–6.5 pH range.39 These curves imply the presence of general acidic groups with pKa’s of ca. 5 and ca. 6.9 and general basic groups with pKa’s of ca. 6.3 and ca. 9. Although enzyme inactivation at pH values lower than ca. 5 is observed due to irreversible denaturation and loss of the essential nickel ions, a distinct group of ureases possess an optimum pH in the range 2.0–4.5.21e The amino acid sequence for these acidic enzymes does not differ from that of the neutral ureases as far as the active site residues are concerned, and the factors that determine this difference in the pH-dependent activity profiles are still unknown.
For ureases, weak substrate and product inhibition are observed. In particular, JBU shows uncompetitive inhibition by urea (Ki ca. 1–6 M) and noncompetitive inhibition by ammonium ions (Ki = 2–118 mM).21e Furthermore, urea is not the only substrate for the enzyme, and hydrolytic activity is also observed for urea analogues such as formamide, acetamide, N-methylurea, N-hydroxyurea, N,N′-dihydroxyurea, semicarbazide, and thiourea, as well as different kinds of phosphoric acid amides and esters.40 The values of kcat for these alternative substrates are ca. 2–3 orders of magnitude lower than that observed for urea, and the kinetics are poorly understood because they act simultaneously as enzyme substrates and inhibitors.21e
2.3. Spectroscopy
The presence of six-coordinate octahedral Ni(II) in JBU was first revealed by optical absorption spectroscopic studies,20,41 and later confirmed by X-ray absorption spectroscopic studies on the same protein, which further suggested the presence of Ni(II) ions coordinated to three histidine N atoms at 2.04 Å, two O atoms at 2.07 Å, and one O atom at 2.25 Å.42 Magnetic susceptibility experiments on JBU indicated the presence of weakly antiferromagnetically coupled (J = −6.3 cm−1) high-spin (S = 1) closely spaced octahedral Ni(II) ions.43 This conclusion received further support from later extended X-ray absorption fine structure (EXAFS) studies, which indicated the appearance of a new peak in the Fourier transform upon addition of β-mercaptoethanol (BME) to JBU that could be fit using a model that involved the presence of two Ni(II) ions separated by 3.26 Å and bridged by the thiolate group of BME.44 In the case of bacterial urease, X-ray absorption spectroscopy studies carried out on urease from Klebsiella aerogenes (KAU)44 and Bacillus pasteurii (BPU)45 provided a picture of the active site essentially identical to that of JBU.
2.4. Structure
The first report of the crystal structure of urease dates back to 1995 in the case of KAU,46 and now the total number of full structures of ureases in the Protein Data Bank amounts to 43 (Table 1). Of these, 38 are bacterial ureases, four are of JBU (C. ensiformis) and one is from pigeon pea (Cajanus cajan).
Table 1.
Selected Structural Parameters for Urease Structures Currently Available in the PDB
| PDB code | enzyme source | enzyme form | pHa | Tb (K) | metal, ligandsc | release date | flap status | resolution (Å) | ref |
|---|---|---|---|---|---|---|---|---|---|
| 2KAU | K. aerogenes | wild type | 7.5 | 298 | 2Ni | July 1995 | closed | 2.20 | 46 |
| 1KRA | K. aerogenes | wild type | 7.5 | 298 | – | October 1995 | closed | 2.30 | 47 |
| 1FWJ | K. aerogenes | wild type | 7.5 | 298 | 2Ni, water | October 1997 | closed | 2.20 | 48 |
| 1EJX | K. aerogenes | wild type | 7.5 | 100 | 2Ni, water | July 2003 | disordered | 1.60 | unpublished |
| 1EJW | K. aerogenes | wild type | 7.5 | 298 | 2Ni, water | November 2003 | closed | 1.90 | unpublished |
| 1A5K | K. aerogenes | αLys217Glu | 7.5 | 298 | – | May 1998 | closed | 2.20 | 49 |
| 1A5M | K. aerogenes | αLys217Ala | 7.5 | 298 | – | May 1998 | closed | 2.00 | 49 |
| 1A5N | K. aerogenes | αLys217Ala | 7.2 | 298 | 2Ni, formate | May 1998 | disordered | 2.40 | 49 |
| 1A5L | K. aerogenes |
αLys217Cys αCys219Ala |
7.5 | 298 | – | May 1998 | disordered | 2.20 | 49 |
| 1A5O | K. aerogenes |
αLys217Cys αCys219Ala |
7.2 | 298 | 2Ni, formate | May 1998 | closed | 2.50 | 49 |
| 1FWA | K. aerogenes | αCys319Ala | 7.5 | 298 | 2Ni, water | October 1997 | closed | 2.00 | 48 |
| 1FWB | K. aerogenes | αCys319Ala | 6.5 | 298 | 2Ni, water | October 1997 | closed | 2.00 | 48 |
| 1FWC | K. aerogenes | αCys319Ala | 8.5 | 298 | 2Ni, water | October 1997 | closed | 2.00 | 48 |
| 1FWD | K. aerogenes | αCys319Ala | 9.4 | 298 | 2Ni, water | October 1997 | closed | 2.00 | 48 |
| 1FWF | K. aerogenes | αCys319Asp | 7.5 | 298 | 2Ni, water | October 1997 | disordered | 2.00 | 48 |
| 1FWG | K. aerogenes | αCys319Ser | 7.5 | 298 | 2Ni, water | October 1997 | closed | 2.00 | 48 |
| 1FWH | K. aerogenes | αCys319Tyr | 7.5 | 298 | 2Ni, water | October 1997 | open | 2.00 | 48 |
| 1EJS | K. aerogenes | αHis219Asn | 7.5 | 298 | 2Ni, water | September 2000 | closed | 2.00 | 50 |
| 1EJT | K. aerogenes | αHis219Gln | 7.5 | 298 | 2Ni, water | September 2000 | closed | 2.00 | 50 |
| 1KRB | K. aerogenes | αHis219Ala | 7.5 | 298 | 2Ni, water | October 1995 | closed | 2.50 | 47 |
| 1KRC | K. aerogenes | αHis320Ala | 7.5 | 298 | 2Ni, water | October 1995 | closed | 2.50 | 47 |
| 1EJU | K. aerogenes | αHis320Asn | 7.5 | 298 | 2Ni, water | September 2000 | disordered | 2.40 | 50 |
| 1EJV | K. aerogenes | αHis320Gln | 7.5 | 298 | 2Ni, water | September 2000 | disordered | 2.00 | 50 |
| 1FWI | K. aerogenes | αHis134Ala | 7.5 | 298 | 1Ni, water | October 1997 | disordered | 2.50 | 51 |
| 1EJR | K. aerogenes | αAsp221Ala | 7.5 | 298 | 2Ni, water | September 2000 | disordered | 2.00 | 50 |
| 1EF2 | K. aerogenes | wild type | 7.5 | 298 | 2Mn, water | March 2000 | closed | 2.00 | 52 |
| 2UBP | B. pasteurii | native | 6.8 | 100 | 2Ni, water | November 1999 | open | 2.00 | 53 |
| 1UBP | B. pasteurii | native | 6.8 | 100 | 2Ni, BME | March 1999 | open | 1.65 | 54 |
| 3UBP | B. pasteurii | native | 6.8 | 100 | 2Ni, DAP | December 1999 | closed | 2.00 | 53 |
| 4UBP | B. pasteurii | native | 6.8 | 100 | 2Ni, AHA | March 2000 | open | 1.55 | 55 |
| 1IE7 | B. pasteurii | native | 6.8 | 100 | 2Ni, PHO | April 2001 | open | 1.85 | 56 |
| 1S3T | B. pasteurii | native | 6.8 | 100 | 2Ni, H3BO3 | April 2004 | open | 2.10 | 57 |
| 4AC7 | B. pasteurii | native | 6.8 | 100 | 2Ni, citrate | January 2013 | open | 1.50 | 58 |
| 4CEU | B. pasteurii | native | 6.5 | 100 | 2Ni, water | November 2013 | open | 1.58 | unpublished |
| 4CEX | B. pasteurii | native | 6.5 | 100 | 2Ni, fluoride | November 2013 | open | 1.59 | unpublished |
| 1E9Z | H. pylori | native | 6.5 | 100 | 2Ni, water | November 2001 | closed | 3.00 | 59 |
| 1E9Y | H. pylori | native | 6.5 | 100 | 2Ni, AHA | November 2001 | open | 3.00 | 59 |
| 3QGA | H. mustelae | native | 7.4 | 100 | 2Fe | August 2011 | disordered | 3.00 | 60 |
| 3LA4 | C. ensiformis | native | 8.8 | 100 | 2Ni, PHO | July 2010 | open | 2.05 | 38 |
| 4H9M | C. ensiformis | native | n.a.d | n.a. | 2Ni, AHA | October 2012 | open | 1.52 | unpublished |
| 4GY7 | C. ensiformis | native | n.a. | n.a. | 2Ni, PHO | November 2012 | open | 1.49 | unpublished |
| 4GOA | C. ensiformis | native | n.a. | n.a. | 2Ni, fluoride | August 2013 | open | 2.20 | unpublished |
| 4G7E | C. cajan | native | n.a. | n.a. | 2Ni, water | June 2013 | open | 2.20 | 61 |
pH of crystallization.
Temperature for data collection.
AHA, acetohydroxamic acid; BME, β-mercaptoethanol; DAP, diamidophosphate; PHO, phosphate.
Not available.
Four structures of KAU refer to the resting state of the recombinant wild type enzyme, at different levels of refinement or temperature of diffraction data collection, while one structure involves the apo form and another one the manganese-substituted enzyme. In addition, 20 structures of KAU mutants are available. In the case of BPU, two structures involve the resting state of the enzyme, while seven additional structures report on complexes with inhibitors. Two structures of H. pylori urease (HPU) are available, with significantly lower resolution as compared to the structures of KAU and BPU: one structure refers to the resting state and the other to the acetohydroxamic acid (AHA) complex. Five structures are available for plant urease, four of which are from JBU, complexed with either AHA or phosphate (PHO) and one from pigeon pea (PPU) in the native state, while the only available structure of the iron-containing alternative urease from H. mustelae does not show any modeled electron density in the active site in addition to the metal ions and ligated residues.
Figure 1 shows the protein architecture for KAU, BPU, HPU, and JBU. KAU and BPU are representative of most bacterial ureases, with a quaternary structure composed of a trimer of trimers of the type (αβγ)3, with α, β, and γ being three different subunits.46,53 The active site is found in the α subunit, giving rise to three active sites per biological unit. In other bacterial ureases, the quaternary structure is made of only two subunits to form (αβ)3 trimers: the α subunit is highly homologous to that found in KAU and BPU, and the β subunit is a fusion peptide made of the β and γ subunits found in the KAU and BPU urease. In the case of HPU, the (αβ)3 trimer further tetramerizes, producing the spherically shaped tetramer of trimers [(αβ)3]4, containing 12 active sites59 that correspond to the shape of the enzyme estimated using electron microscopy.62 Finally, plant ureases are generally made up of a dimer of homotrimers (α3)2, where the α subunit is derived from the fusion of the corresponding αβγ subunits found in bacteria.38
Figure 1.
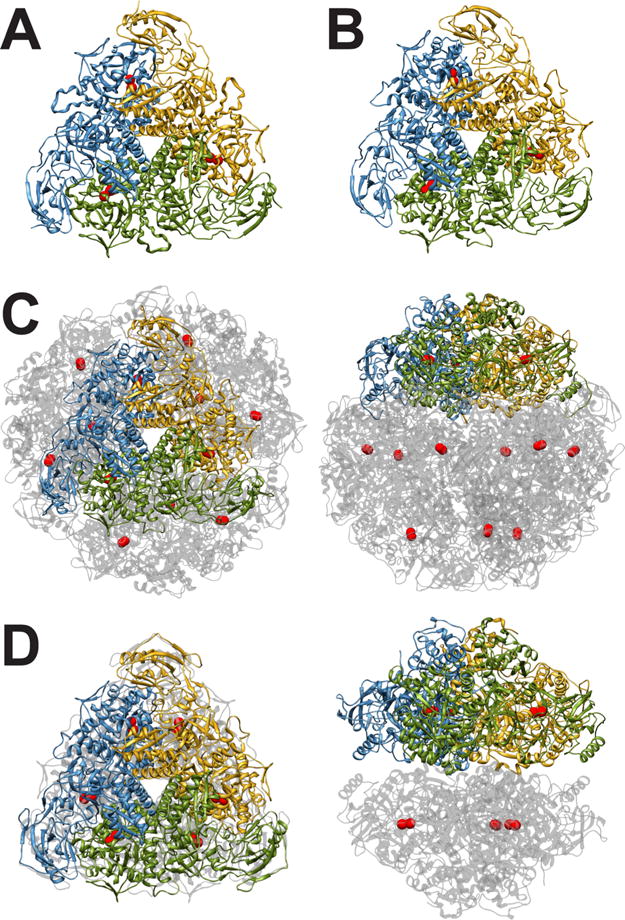
(A) Ribbon scheme of the functional oligomer (αβγ)3 of B. pasteurii urease. (B) Ribbon scheme of the functional oligomer (αβγ)3 of K. aerogenes urease. (C) Ribbon scheme of the functional oligomer [(αβ)3]4 of H. pylori urease seen through the ternary axis (left panel) and rotated by 90° along the horizontal axis (right panel). (D) Ribbon scheme of the functional oligomer [(α)3]2 of C. ensiformis urease seen through the ternary axis (left panel) and rotated by 90° along the horizontal axis (right panel).
In all cases, the secondary and tertiary structures of ureases are very similar. The α subunits consist of a TIM barrel domain and a β-sheet domain, the β subunits are located on the external surface of the trimer and are mainly composed of β-sheet, and the γ subunits consist of domains containing both α-helices and β-sheet. An important feature of the structures of ureases is the conformation of a helix-turn-helix motif in the α-subunit, flanking the active site cleft. This motif is found in an open or closed conformation, and is thought to be important in modulating the afflux of substrate and the efflux of products to and from the active site during catalysis, respectively, as well as moving the catalytically essential and conserved αHis323 residue (BPU numbering) by about 5 Å to and from the active site metal center (Table 1, Figure 2).
Figure 2.
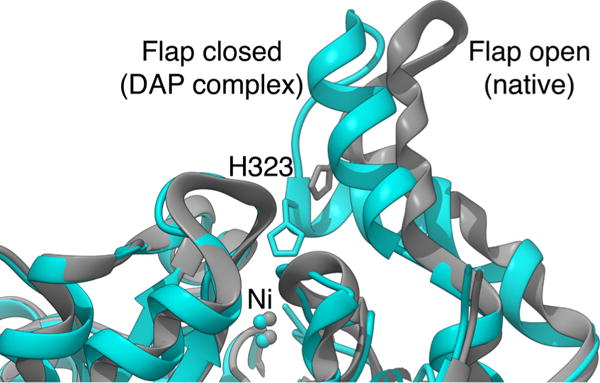
Ribbon scheme of the active site flap of B. pasteurii urease, highlighting the open and closed conformations observed in the native and the DAP-inhibited structures, respectively.
A consensus has been reached on the structure of the coordination environment of the Ni(II) ions in the active site of urease. Indeed all structures so far determined indicate the presence of a dinuclear active site, with the two Ni(II) ions separated by 3.5–3.7 Å, bridged by the oxygen atoms of a carbamylated lysine residue, and bound to two histidines (Figure 3). One Ni(II) ion is additionally bound to an aspartate carboxylate oxygen. The coordination geometry of each Ni(II) ion is completed by a terminally bound water molecule (W1 and W2) and by a nickel-bridging hydroxide ion (WB). The assignment of the protonation state for these solvent-derived nickel ligands was suggested53 by the value of the first dissociation constant for Ni(H2O)62+ (pKa = 10.6),63 which supports the hypothesis that the terminally bound solvent molecules are neutral waters. Moreover, in water-bridged bimetallic complexes, the first pKa for the bridging water decreases significantly to very acidic values, while the pKa for hydroxide deprotonation is slightly lower than the pKa of the first ionization of a water bound to a single Ni ion.63 Therefore, the estimated pKa for the deprotonation of the Ni bridging hydroxide (ca. 9–10) suggests that, at pH 8.0, the nickel-bridging solvent molecule is a hydroxide ion. Overall, in native urease two different types of ligands bridge the binuclear Ni cluster, the carboxylate group of the carbamylated lysine and the hydroxide ion, accounting for the observation of weak antiferromagnetic coupling.43
Figure 3.
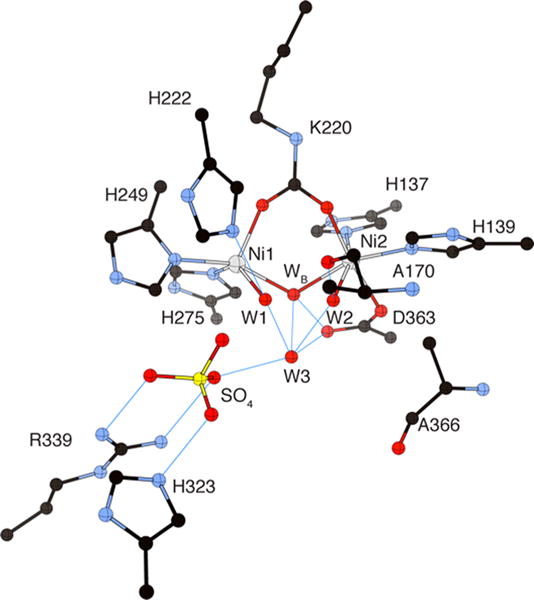
CrystalMaker drawing of the crystallographic structural models for the active site obtained for B. pasteurii urease (PDB code 2UBP) in the native state. The nickel ions are represented in gray, while CPK coloring is used for all other atoms. Hydrogen bonds are shown as thin blue lines. The BPU residue-numbering scheme (all residues belonging to the α subunit) is used. The residue indicated with the letter “K” is the carbamylated lysine.
An extended network of hydrogen bonds stabilizes the nickel-bound solvent molecules in the active site: W1 is 2.9 Å from αHis222 Nε (the following discussion follows the BPU residue-numbering scheme), which is protonated and acts as a hydrogen-bonding donor, as deduced from the interaction of αHis222 Nδ with the peptide NH group of αAsp224 (at 2.9 Å). In contrast, W2 forms a strong hydrogen bond with αAla170 O (at 2.9 Å), which acts as hydrogen bond acceptor. This ligand arrangement yields one pentacoordinated Ni(II) ion (Ni1) with a distorted square-pyramidal geometry and one Ni(II) ion (Ni2) hexacoordinated with a distorted octahedral geometry. An additional water molecule (W3) is part of a hydrogen-bonding network completing a tetrahedral cluster of four water/hydroxide molecules in close proximity to the Ni(II) ions, hinting at the existence of an active site cavity designed to stabilize a tetrahedral transition state and/or intermediate. In the structure of apo-KAU (PDB code 1KRA, Table 1), where the Ni(II) ions were chemically removed by lowering the pH in the presence of chelating agents, a treatment that decarbamylates αLys217, the positions of the remaining ligands are the same as in the holoenzyme, indicating a preorganized metal binding site structure.
The positions of conserved amino acid residues not involved in Ni binding but thought to be important in the catalytic mechanism (αAla170, αHis222, αGly280, αHis323, αArg339, and αAla366) are largely invariant in BPU and KAU, except for αHis323, due to the different conformation of the active site flap (see Figure 2). Some differences are observed in the case of native HPU: in particular, the carbamylated lysine appears to bridge the two nickel ions using only one of the two terminal oxygen atoms, a consequence of an apparent rotation of the terminal −NH−CO2− moiety along the N–C bond by about 90° with respect to the position found in BPU and KAU. In addition, some distances between the nickel ions and the coordinating residues are very large (ranging between 2.6 and 2.7 Å) as compared to those found in KAU and BPU (2.0–2.1 Å), with an unusual orientation of the histidine imidazole rings around the metal ions, a very short (2.1 Å) Ni–Ni distance, and no bridging hydroxide. These differences are most likely ascribed to the much lower resolution of this structure as compared to those of KAU and BPU (see Table 1).
All structural investigations on urease mutants have been carried out using KAU, and the residue-numbering scheme for this urease will be used to describe the attempts to relate their structures to the enzymatic mechanism. The structure of the αHis219Ala mutant (PDB code 1KRB), which features a much lower affinity for the substrate than the wild type enzyme (Km = 1100 mM vs 2.3 mM) and a ca. 30-fold decrease of kcat,64 and thus indicates a role for αHis219 (αHis222 in B. pasteurii residue numbering) in substrate binding, shows structural identity with native KAU. The structure of the αHis320Ala mutant, (PDB code 1KRC)47 shows a root-mean-square deviation for Cα atoms of only 0.1 Å from wild type KAU, and displays only a small change in Km but a ca. 30 000-fold decrease in kcat with respect to the wild type enzyme,64 while not showing the pKa = 6.5 observed for wild type urease.64,65 This indicates a role of αHis320 (αHis323 in B. pasteurii residue numbering) in the catalytic mechanism. The removal of one nickel histidine ligand in the catalytically inactive mutant αHis134Ala64 resulted in an enzyme featuring only one Ni(II) ion in the active site (PDB code 1FWI),51 with the position of all remaining active site residues, including the carbamylated lysine, invariant with respect to the wild type enzyme structure. This indicates the importance of αHis134 (αHis137 in B. pasteurii residue numbering) in stabilizing a dinuclear nickel center for catalysis. Chemical modification of αCys319, located on the flexible flap covering the active site of KAU, blocks enzyme activity,66 while the αCys319Ala mutant is still ca. 50% as active as the wild type urease.67 Structures of the αCys319Ala mutant were determined atpH6.5 (PDB code 1FWB),pH7.5 (PDB code 1FWA),pH 8.5 (PDB code 1FWC), and pH 9.4 (PDB code 1FWD),48 with no significant structural differences observed between the structure of the wild type and mutant enzymes except for a much reduced mobility of the flexible flap covering the active site in the mutant. The structures of αCys319Asp (PDB code 1FWF), αCys319Ser (PDB code 1FWG), and αCys319Tyr (PDB code 1FWH), which feature, respectively, 0.03, 4.5, and 0% of the activity observed for the wild type, also indicate the same active site environment but with a much higher mobility of the flap.48 This evidence indicates that αCys319 (αCys322 in B. pasteurii residue numbering), largely conserved in ureases, is somehow involved in catalysis, possibly having a role in positioning other key residues that are located on the mobile flap or in the active site, in a conformation best suited for catalysis. The structures of the αLys217Glu (PDB code 1A5K), αLys217Ala (PDB code 1A5M), and αLys217Cys/αCys319Ala (PDB code 1A5L) mutants of KAU, which were structurally characterized in order to establish the importance of the carbamylated lysine ligand for urease activity,49 reveal the complete absence of bound Ni ions, indicating the need for the longer side chain of the carbamylated αLys217 (αLys220 in B. pasteurii residue numbering) in order to bind the Ni(II) ions in the active site defined by the four His ligands. The addition of formate to these mutants allowed for a chemical rescue of the enzyme, with the structures of the αLys217Ala–formate–Ni complex (PDB code 1A5N) and αLys217Cys–formate–Ni complex (PDB code 1A5O) revealing the presence of a dinuclear Ni center bridged by formate instead of the carbamate group of αLys217 as in wild type KAU.49
Several classes of molecules (diphenols, quinones, hydroxamic acids, phosphoramides, and thiols) have been tested as urease inhibitors in medicine and agriculture.17b,21e,30,68 However, the efficiency of the presently available inhibitors is low, and negative side effects have been reported.17b,30,69 A structure-based molecular design of new and efficient urease inhibitors requires the knowledge of the structures of urease complexed with competitive inhibitors. The structure of BPU crystallized in the presence of BME (PDB code 1UBP)54,70 revealed the presence of the inhibitor bound to the Ni(II) ions in the active site through the thiolate group, in agreement with the presence of chargetransfer transitions observed in the near-UV41b and magnetic circular dichroism71 spectra of JBU in the presence of thiolate competitive inhibitors (Figure 4).
Figure 4.
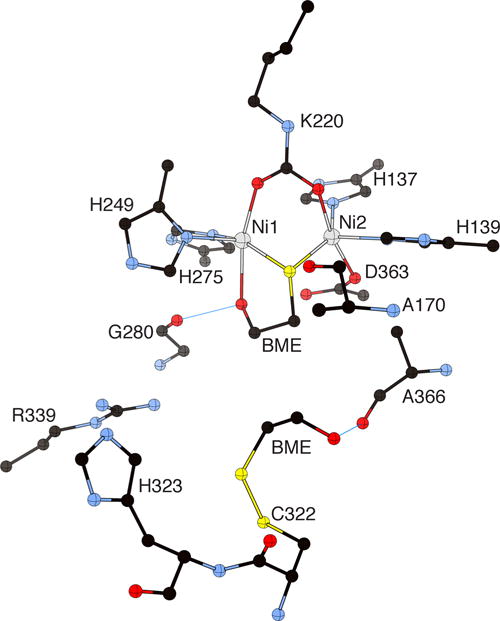
CrystalMaker drawing of the crystallographic structural model for the active site obtained for B. pasteurii urease complexed with β-mercaptoethanol (BME) (PDB code 1UBP). The nickel ions are represented in gray, while CPK coloring is used for all other atoms. Hydrogen bonds are shown as thin blue lines. The BPU residue-numbering scheme (all residues belonging to the α subunit) is used. The residue indicated with the letter “K” is the carbamylated lysine.
The structure further revealed that the thiolate group of BME bridges the two Ni(II) ions, consistent with magnetic and spectroscopic evidence.42c,43,44,71 The crystal structure of BME-inhibited BPU further revealed the chelating mode of the inhibitor to the metal center, with the alcoholic moiety interacting with Ni1 assisted by a hydrogen bond with the carbonyl oxygen of the conserved αGly280 (the BPU residue-numbering scheme is used here for urease-inhibitor complex structures). Finally, a second molecule of BME is found to form a disulfide bond with αCys322, a residue located on the mobile flap, with a hydrogen bond that arises between its α-hydroxyl group and the carbonyl oxygen atom of αAla366, positioned on a neighboring loop. This interaction decreases the flexibility of the flap, sealing the entrance to the active site by steric hindrance, an inhibition mode that could be inferred for all cases in which a thiol acts as a urease inhibitor.41,66b,72
Acetohydroxamic acid (AHA) belongs to a class of compounds known for their urease inhibitory properties.72,73 AHA is a slow-binding inhibitor for plant, bacterial, and fungal ureases, and has also been used for medical treatment against pathogenic bacteria.74 Structures of urease complexed with AHA are available for BPU (PDB code 4UBP),55 HPU (PDB code 1E9Y),59 and a mutant of KAU (PDB code 1FWE).48 In all cases, the hydroxamate oxygen atom bridges the two Ni(II) ions, while the carbonyl oxygen of AHA chelates Ni 1 in a similar fashion as observed for BME (Figure 5).
Figure 5.
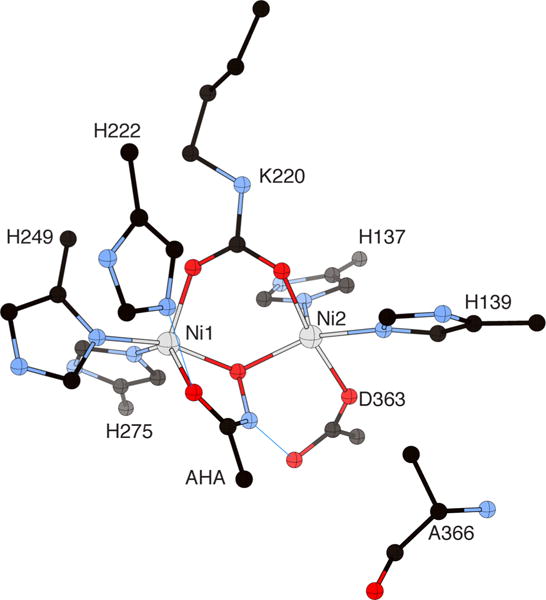
CrystalMaker drawing of the crystallographic structural model for the active site obtained for urease from B. pasteurii complexed with acetohydroxamic acid (AHA) (PDB code 4UBP). The nickel ions are represented in gray, while CPK coloring is used for all other atoms. Hydrogen bonds are shown as thin blue lines. The BPU residue-numbering scheme (all residues belonging to the α subunit) is used. The residue indicated with the letter “K” is the carbamylated lysine.
This binding mode is assisted by two H-bonds between the carbonyl AHA O atom and αHis222 NεH, and between the AHA-NH group and the Oδ2 atom of the nickel-bound αAsp363 residue. In the case of BPU, the latter interaction induces a 35° rotation around the Cβ–Cγ bond of the side chain of αAsp363, revealing the viability of such a conformation change during enzyme catalysis.
Phosphate (PHO) is an additional competitive inhibitor for urease,75 and the inhibition is pH-dependent, with values of Ki increasing in the pH range 5–8.72,76 At pH higher than 7.5–8.0 there is no competitive inhibition. The analysis of this pH dependence suggested that phosphate inhibition of urease involves at least two protonation sites, with pKa’s of ca. 7.2 and 6.5, associated with the inhibitor or with active site residues. The structure of BPU crystallized in the presence of phosphate (PDB code 1IE7)56 revealed the unprecedented presence of a tetrahedral phosphate molecule bound to the binuclear nickel center through three atoms, a phosphate oxygen that bridges the two Ni(II) ions and two oxygen atoms that are bound terminally to each Ni center, with the fourth phosphate oxygen pointing away from the metal center and toward the cavity opening (Figure 6).
Figure 6.
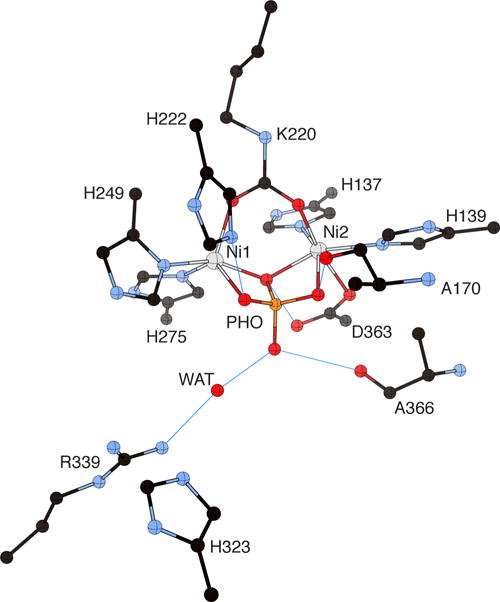
CrystalMaker drawing of the crystallographic structural model for the active site obtained for B. pasteurii urease complexed with phosphate (PHO) (PDB code 1IE7). The nickel ions are represented in gray and phosphorus is in orange, while CPK coloring is used for all other atoms. WAT = solvent molecule. Hydrogen bonds are shown as thin blue lines. The BPU residue-numbering scheme (all residues belonging to the α subunit) is used. The residue indicated with the letter “K” is the carbamylated lysine.
The H-bonding network established between the inhibitor and the active site residues suggests that the inhibitor is formally bound as the neutral phosphoric acid H3PO4 in the pH range 5–6.5, with the pKa observed at ca. 6.5 assigned to the production of the H2PO4− species that exists in the pH range 6.5–7.2. Above this pH, loss of the second phosphate proton to form HPO42− further destabilizes the interaction of the inhibitor with the enzyme.56
Boric acid, B(OH)3, is a competitive inhibitor of urease with maximum activity in the pH range 6–9.72,77 The structure of BPU crystallized in the presence of B(OH)3 (PDB code 1S3T) has been determined (Figure 7).57 In the structure, a molecule of the inhibitor bridges the Ni(II) ions with two oxygen atoms, displacing three water molecules and leaving in place the bridging hydroxide. The third inhibitor oxygen points away from the metal center, toward the cavity opening. The geometry and coordination number of the Ni(II) ions are largely invariant as compared to the resting state of the enzyme. The detailed analysis of the H-bonding network surrounding the inhibitor established that the protonation state corresponds to the neutral B(OH)3 molecule.
Figure 7.
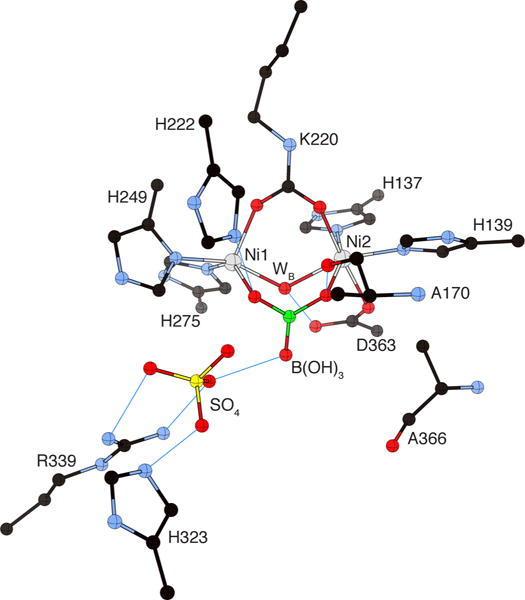
CrystalMaker drawing of the crystallographic structural model for the active site obtained for B. pasteurii urease complexed with boric acid B(OH)3 (PDB code 1S3T). The nickel ions are represented in gray and boron is in green, while CPK coloring is used for all other atoms. WB = nickel-bridging hydroxide. Hydrogen bonds are shown as thin blue lines. The BPU residue-numbering scheme (all residues belonging to the α subunit) is used. The residue indicated with the letter “K” is the carbamylated lysine.
The most effective inhibitors of urease so far discovered are amide and ester derivatives of phosphoric or thiophosphoric acid.21e In the latter case, the inhibitory activity is effective only upon conversion to the oxygen analogue.78 Regardless of the nature of the derivative, the actual inhibitor is diamidophosphate (DAP), obtained by the hydrolysis of the precursor.79 The structure of BPU crystallized in the presence of phenyl-phosphorodiamidate (PPD) (PDB code 3UBP)53 indeed revealed the presence of a molecule of DAP, produced in situ by the hydrolysis of PPD and production of phenol (Figure 8). A specific hydrogen-bonding pattern stabilizes the inhibitor and directs its orientation in the cavity, allowing the assignment of the Ni-bound DAP atoms to either oxygen or nitrogen. DAP is bound to Ni1 and to Ni2 through one oxygen atom and one nitrogen atom, respectively. The second DAP oxygen symmetrically bridges the two nickel ions, while the second DAP nitrogen atom points away toward the cavity opening. The Ni1-bound oxygen atom receives a hydrogen bond from αHis222 NεH, while the Ni2-bound DAP NH2 group forms two hydrogen bonds with the carbonyl oxygen atoms of αAla170 and αAla366. The Ni-bridging DAP oxygen atom is at hydrogen bonding distance from the αAsp363 Oδ2 atom. The DAP molecule thus replaces the tetrahedral cluster of four water/hydroxide molecules found in native BPU. In DAP-inhibited BPU, the flap is in the closed conformation, which allows the formation of two additional hydrogen bonds between the distal DAP NH2 group and the αAla366 O and αHis323 Nε atoms.
Figure 8.
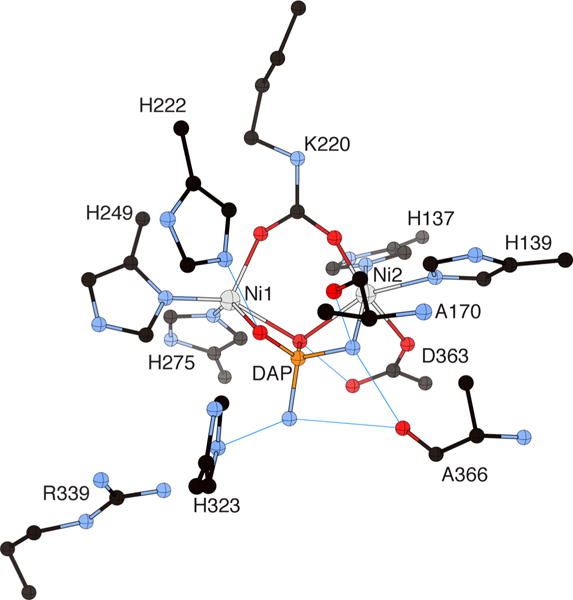
CrystalMaker drawing of the crystallographic structural model for the active site obtained for B. pasteurii urease complexed with diaminophosphate (DAP) (PDB code 3UBP). The nickel ions are represented in gray and phosphorus is in orange, while CPK coloring is used for all other atoms. Hydrogen bonds are shown as thin blue lines. The BPU residue-numbering scheme (all residues belonging to the α subunit) is used. The residue indicated with the letter “K” is the carbamylated lysine.
Citrate is a poor urease inhibitor at concentrations higher than 800 mM, while at lower concentrations (100–600 mM) the inhibition is masked by an activating effect.58 The structure of a complex between citrate and BPU (Figure 9) shows the presence of a carboxylate group that binds the nickel ions in the active site, with several hydrogen bonds involving the polar groups of the inhibitor with the surrounding residues.58
Figure 9.
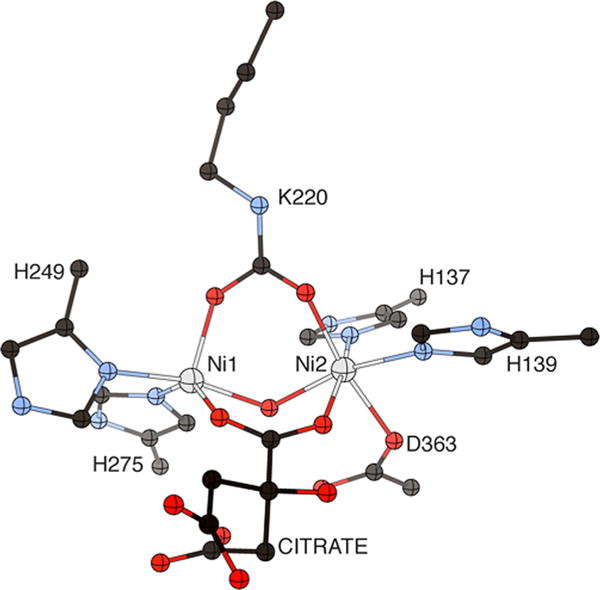
CrystalMaker drawing of the crystallograhic structural model for the active site obtained for B. pasteurii urease complexed with citrate (PDB code 4AC7). The nickel ions are represented in gray, while CPK coloring is used for all other atoms. The BPU residue-numbering scheme (all residues belonging to the alpha subunit) is used. The residue indicated with the letter “K” is the carbamylated lysine.
Fluoride has been reported to act as mixed competitive/ uncompetitive,41a competitive,80 pseudo-uncompetitive slowbinding81 or competitive slow-binding82 inhibitor in a series of inconsistent kinetic studies. Two structures of fluoride-inhibited ureases have been deposited in the PDB (PDB codes 4GOA for JBU and 4CEX for BPU, at 2.20 Å and 1.59 Å resolution, respectively), but are not yet released.
2.5. Mechanism
In the absence of structural data regarding reaction intermediates that have short lifetimes, the available structures of urease in the resting hydrated state, bound to boric acid (an analogue of the substrate), and with diamidophosphate (an analogue of the intermediate or transition state of the hydrolysis reaction) provided a basis for the proposal of a reaction mechanism for enzymatic urea hydrolysis (Figure 10).16a
Figure 10.
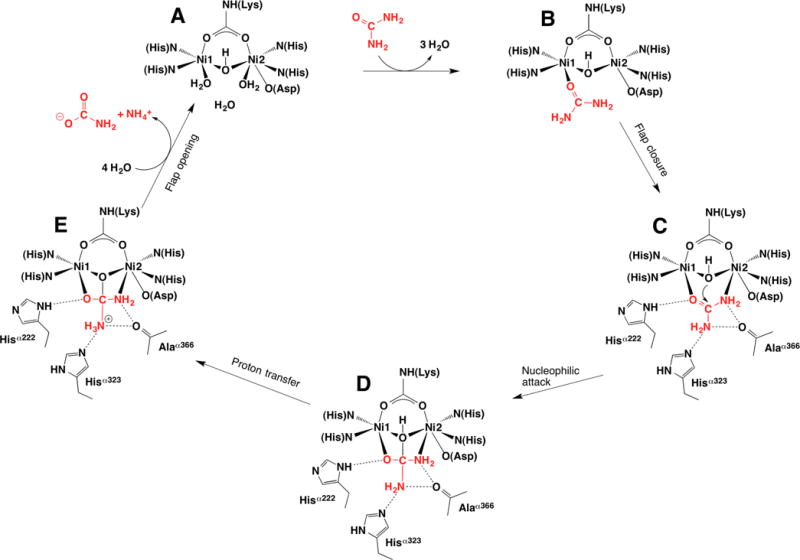
Structure-based urease catalytic mechanism of the enzymatic hydrolysis of urea. The BPU residue-numbering scheme is used.
In the following discussion, the BPU residue-numbering scheme will be used. The presence of the tetrahedral water/ hydroxide cluster in the proximity of the dinickel center in the native enzyme, together with the binding mode of DAP, suggests an orientation-specific mode of substrate binding in the enzyme active site that is designed to stabilize a tetrahedral transition state. The mechanism requires that, upon urea entrance into the active site channel with the flap in the open conformation, the structurally characterized hydrated active site of the resting enzyme (Figure 10A) evolves toward an initial substrate-bound intermediate where urea replaces the three water molecules bound to the Ni(II) ions (Figure 10B). In particular, urea initially binds the more electrophilic and coordinatively unsaturated Ni1 using the urea carbonyl oxygen, an event that must be concomitant with the displacement of water molecules from the active site due to steric hindrance. The Ni1-bound urea O atom is stabilized in this position through the formation of an H-bond donated by the NεH group of αHis222, analogous to what is observed in the structures of native BPU, as well as in BPU complexed with boric acid, diaminophosphate, phosphate, and acetohydroxamic acid. This step is supported by docking and density functional quantum chemistry calculations,83 which also suggest that flap closure facilitates urea coordination to Ni2 via its −NH2 group. This interaction is stabilized by a specific H-bonding network (H-bond donors on the urea carbonyl oxygen side and H-bond acceptors on the urea amide group side) that locates and steers the substrate to a precise orientation thus allowing hydrolysis to occur (Figure 10C). Urea is a poor chelating ligand because of the low Lewis base character of its NH2 groups. However, the formation of strong H-bonds with the nearby carbonyl oxygen atoms could enhance the basicity of the NH2 group and facilitate the interaction of the amide nitrogen with Ni2. The viability of a bidentate urea coordination mode is supported by the crystal structure of BPU in complex with citrate58 and B(OH)3;57 boric acid has a triangular shape and dimensions similar to those of urea, is isoelectronic with it, and has the same neutral charge, so it can be considered an inert structural analogue of the substrate. In the structure of urease complexed with B(OH)3, two of the borate hydroxide moieties replace the water molecules terminally bound to Ni1 and Ni2, while the third borate hydroxide replaces the distal solvent molecule. This substrate-binding mode involves a direct role of both Ni ions in binding and activating the substrate, therefore providing a rationale for the presence of a bimetallic active site in urease and explaining the low reactivity of urease containing a single Ni ion.64 The presence of the Ni-bridging hydroxide in the complex of urease with B(OH)3, placed at 2.1 Å from the B atom, in a direction almost perpendicular to the plane of the molecule, suggests its role as the nucleophile attacking the carbon atom of urea, to yield a tetrahedral transition state/intermediate (Figure 10D).
The bridging urea binding mode is the most efficient method to render the central carbon atom of urea electron poor and therefore prone to nucleophilic attack by the bridging hydroxide. The kinetic inertia of a doubly coordinated nucleophile could be overcome by the weakening of the Ni–OH bonds upon substrate binding, as suggested by kinetic data on the inhibition of KAU with fluoride,81 by the structure and reactivity of inorganic models,84 and by theoretical studies.83 The high reactivity of the nickel-bridging hydroxide is also supported by the ability of the enzyme to hydrolyze PPD79a and perhaps phosphate, where the enzyme would simply induce an oxygen atom (or hydroxide) exchange on the phosphate moiety. In this framework, only molecules able to react with the bridging hydroxide may bind the enzyme in a tridentate mode, as observed for DAP and PHO. This would explain why DAP strongly inhibits urease when DAP is formed by enzymatic hydrolysis of PPD, whereas it is a weak inhibitor if externally added to the native enzyme.79a The nucleophilic attack by the Ni-bridging hydroxide onto the sp2 carbon atom of urea yields a tetrahedral transition state containing an sp3 carbon. The formation of a tetrahedral intermediate located between the two Ni(II) ions is supported by the structure of urease containing DAP in the active site (formed in situ from PPD) that replaces the cluster of four water/ hydroxide molecules.53 The tetrahedral DAP bound to the dinickel center represents an ideal transition state, or intermediate, analogue of the enzymatic reaction, while the structure of the PHO-inhibited urease supports the idea that the enzyme active site cavity is made to stabilize tetrahedral moieties. Closure of the flap would also be responsible for the stabilization of the catalytic transition state through the formation of multiple H-bonds with active site residues. The nucleophilic attack onto the Ni-bridging urea molecule profoundly modifies the electronic structure of the substrate and, in particular, increases the pKa of the distal urea N atom not involved in Ni-binding, as supported by density functional theoretical calculations.83 Furthermore, after the formation of the tetrahedral intermediate, the nickel-bridging −OH group, now part of a diamino-(hydroxy)methanolate moiety (Figure 10D), must now have a very low pKa, and can therefore transfer the proton to the distal urea NH2 group, forming a C–NH3+ bond.53 This proton transfer step could be mediated by αAsp363 Oδ2 through a dihedral rotation along the Cα–Cβ bond of the aspartate bound to Ni2, a movement observed to occur in the case of the AHA–BPU complex,55 to bring this carboxylic oxygen atom, shown to be deprotonated,85 close to the bridging hydroxide or, alternatively, to the distal −NH2 urea group. The higher pKa observed in the pH vs urease activity profile (ca. 9.5) corresponds to the estimated pKa for the bridging hydroxide, suggesting that indeed the bridging hydroxide moiety must carry a proton for the enzyme to work properly.
By moving closer to the active site upon closure of the flap, the neutral imidazole side chain of the conserved αHis323 stabilizes the nascent C–NH3+ group (Figure 10D, E).53 Therefore, in this structure-based mechanism the bridging hydroxide acts as both the nucleophile and the general acid, while αHis323 acts as a general base, not by deprotonating the hydrolytic water, but by stabilizing the positive charge that develops on the transition state. This scheme is supported by the structure of DAP-inhibited BPU, in which the active site flap is in the closed conformation, allowing αHis323 to approach the Ni environment in the active site, and by the 103-fold reduction in kcat upon mutation of αHi320 in KAU (corresponding to αHis323 in BPU).50 The structure of the PHO–BPU complex also reveals that another active site residue, αAla366, is important for the modulation of the protonation state of the distal urea −NH2 group by adopting two different conformations, acting as a molecular switch able to provide stabilization for the protonation of the distal urea NH2 group. The formation of the C–NH3+ moiety after proton transfer, and its stabilization by the catalytic histidine, causes the breakage of the distal C–N bond, with the subsequent release of ammonia.83 The resulting carbamate spontaneously decomposes into another molecule of ammonia and hydrogen carbonate. The opening of the active site flexible flap facilitates the release of products thus allowing bulk water to rehydrate the active site, regenerating the resting state of the enzyme (Figure 10A). These steps could possibly occur in a concerted manner. This structure-based mechanism is in agreement with all available kinetics data, in particular with the pH dependence of the enzyme activity and with the uncompetitive inhibition by fluoride, suggested to replace the bridging hydroxide81 and therefore strengthening the hypothesis of the bridging hydroxide acting as nucleophile.
An alternative reaction mechanism has been proposed that features different roles for the two Ni(II) ions, with Ni1 binding and activating urea, and Ni2 binding and activating the nucleophilic water by turning it into a hydroxide ion.18b,21a,40,46 This mechanism entails a reaction intermediate that would bridge the two Ni ions in a bidentate mode via two O atoms. It has been recognized, however, that this mechanism raises two problems:21a first, the general base with a pKa = 9.018b,86 required for deprotonation of the Ni2-bound water molecule (pKa = 10.663) at the optimum pH for enzyme activity (pH 8), which is not found anywhere in the vicinity of the active site; second, the role of αHis323 as general acid, which must be protonated at pH 8.0 even though it has a pKa of 6.5.64 An attempt to resolve these problems involved the introduction of the so-called reverse protonation hypothesis,21a which would result in only 0.3% of all urease molecules being in the optimal protonation state for catalysis at pH 8.0, resulting in a very low enzymatic efficiency.
The presence of nickel as an enzyme metal cofactor in urease has intrigued the bioinorganic chemistry community since its discovery.20 The structure-based mechanism discussed above could help to explain the requirement for Ni(II) ions instead of the more common and less toxic d10 closed-shell Zn(II) commonly observed in hydrolytic enzymes. The latter ion has large positive charge density, which renders it able to act as a Lewis acid by polarizing substrates and preparing them for nucleophilic attack by hydroxide, and is resilient toward deleterious redox state changes. However, these properties are also applicable to Ni(II), which additionally features an open-shell d8 electronic configuration that induces stereoelectronic restraints not available in the case of Zn(II). This property could be exploited by Ni(II) to drive the two substrates, urea and water, into the optimal spatial topology necessary for catalysis. Furthermore, Ni(II) has a higher affinity toward nitrogenous-based ligands than Zn(II),87 thereby better stabilizing the binding of a urea NH2 group. Finally, Ni(II) ions possess multiple available binding sites due to their preference for octahedral coordination sphere (as opposed to Zn(II), mostly tetrahedral88), thus facilitating both the bridging binding mode of urea and the stabilization of the metal ion binding to the protein through multiple sites for amino acid ligands.
Attempts to substitute the essential Ni(II) with other ions such as Zn(II), Co(II), and Mn(II) have been carried out to check the viability of a urease-based catalysis with nonnative metals. Removal of both Ni(II) ions by treating JBU with EDTA at low pH causes irreversible denaturation of the protein.89 Removal of a single Ni(II) ion from JBU was obtained by dialysis in the presence of citrate, and substitution of the more labile metal ion with Zn(II) or Co(II) produced catalytically inert urease.90 This inactivation was also observed for KAU reconstituted with Zn(II), Co(II), and Cu(II), while in the case of the Mn derivative ca. 0.3% of the activity was retained.91 The crystal structure of the Mn derivative of KAU was determined (PDB code 1EF2, Table 1), with active site features essentially identical to those of the native Ni-bound form.52 Some differences were observed concerning a significantly higher disorder of the Mn-bound aspartate carboxylate group. Recently, an alternative urease in Helicobacter mustelae (Hm) has been reported, which is characterized by the absence of Ni(II), by inactivation in the presence of oxygen, and by Fe(II)-induced expression, observations that suggested the presence of Fe(II) ions in its active site.92 This hypothesis was later confirmed, and the reported crystal structure showed little or no changes as compared to the Ni(II) enzyme, while the catalytic efficiency appeared to be much smaller.60 All together, these observations suggest that, in addition to the identity of the metal ions, the precise position and mobility of the metal ligands, as well as those of protein residues in the active site not involved in metal binding, are important in achieving optimal urease activity.
2.6. Conclusions
The health and environmental effects of urease activity are very significant. The structural and functional studies on urease that have been carried out in past years have paved the way toward the structure-based design and development of new and efficient inhibitors. These could lead to improved therapeutic strategies for the treatment of infections by ureolytic bacterial pathogens as well as to modern agricultural applications by enhancing the efficiency of nitrogen soil fertilization.68b
3. GLYOXALASE I
3.1. Biological Significance and Enzymology
Glyoxalase I ((R)-S-lactoylglutathione methylglyoxal lyase (isomerizing), EC 4.4.1.5, Glo I) is a widely distributed enzyme found in bacteria,93 fungi,94 protoctista,95 plants,96 and animals94 that catalyzes the conversion of a hemithioacetal to a 2-hydroxythioester (Scheme 2).97 It is not ubiquitous, as the gene for Glo I is not found in the protozoans Entamoeba histolytica, Giardia lamblia, and Trypanosoma brucei.98
Scheme 2.
The hemithioacetal is derived from the spontaneous reaction of an α-ketoaldehyde and a thiol. The most important α-ketoaldehyde is methylglyoxal, which is a naturally occurring toxic metabolite that derives from both enzymatic and nonenzymatic processes.97 Methylglyoxal accumulates when Glo I is suppressed or when glutathione (γ-glutamyl-L-cysteinylglycine, GSH), the most common, though not exclusive,98,99 thiol cofactor involved is depleted, leading to glycation products.97 Methylglyoxal is capable of forming adducts with nucleic acids and amino acids leading to protein, DNA, and RNA modifications and a number of cytotoxic effects.97 In these ways, Glo I has been linked to oxidative stress and to the aging process.100 Thus, inhibitors of Glo I have been found to be potent antimalarial agents for Plasmodium falciparum.101 An antibiotic strategy targeting bacterial Glo I enzymes has also been suggested.102 In addition, increased expression of Glo I supports the high glycolytic rates found in tumors, and the overexpression of Glo I in multidrug resistant tumors points to the possible role of methyglyoxal toxicity in chemotherapy.103
Glo I is the first of two enzymes in the glyoxalase system. The second enzyme in this pathway, Glo II, hydrolyzes the thioester produced by Glo I to produce the corresponding α-hydroxy acid (Scheme 2). Thus, the glyoxalase system has been accepted as an important pathway for detoxifying and controlling the level of methylglyoxal in cells by converting it to D-lactate, as opposed to L-lactate arising from glycolysis.104 However, this role has been challenged based in part on organisms or mutants that lack a functional glyoxalase system, but have no observable effect.98,105
When the enzyme system was discovered in 1913, it was thought to be a single enzyme that could convert α-ketoaldehydes directly to α-hydroxy acids.106 Thirty-seven years later, the two activities now associated with Glo I and Glo II were identified.107 In fact, such an enzyme has recently been discovered in E. coli and converts methylglyoxal to lactate without employing a thiol or going through a thioester.108 This “Glo III” has several homologues, including human DJ-1,109 which is linked to early onset Parkinson’s disease. However, Glo III apparently does not require a metal ion,108b although its activity is enhanced by Fe(II) and inhibited by Zn(II) and Cu (II)108b
Early work on the bioinorganic chemistry of Glo I enzymes using metal activation assays of apoenzyme showed that the enzymes from mammals, yeast, and Pseudomonas putida94,110 were activated by Zn(II) as well as by other divalent metals.111 Based on these precedents, all Glo I enzymes were assumed to be Zn metalloenzymes until the discovery that E. coli Glo I was not active with Zn(II) bound, but was maximally activated by Ni(II) ions, and to a lesser extent by Co(II), Mn(II), and Cd(II) (kcat = 338 s−1; Km = 27 ± 0.4 μM; kcat/Km = 12.4 × 106 M−1 s−1 using methylglyoxal and GSH; % E. coli Glo I activity: 100 Ni(II); 30.9 Co(II); 8.1 Mn(II); 6.8 Cd(II)).112 This report was followed by others resulting from studies in Gram-negative bacteria,111b,113 Gram-positive bacteria,114 and the trypanosome protozoans Leishmania major99a,115 and Trypanosoma cruzi116 that found metal activation characteristics similar to that of the E. coli enzyme. Thus, two classes of Glo I have emerged based on metal activation: class I, which can be activated by Zn(II), and class II, which cannot be activated by Zn(II) and is maximally activated by Ni(II) ions.16b
3.2. Structure
The origin of the active site metal preferences in the two classes of enzyme derives from subtle structural differences between the dimers formed by both classes of the enzyme. The primary structure of Glo I enzymes (Figure 11) also separates into two classes that correlate with the metal preferences for activation.93
Figure 11.

Sequence alignments of selected class I and class II glyoxalase I enzymes created using Clustal W2. Amino acids are colored by property (hydrophobic (red), acidic (blue), basic (purple), other (green)). Metal binding residues are highlighted in yellow. Residues marked with an asterisk (∗) are invariant; those marked by other symbols represent low (:) and moderate (.) variability. The N-terminal extension and additional loops found in class I enzymes are highlighted in blue. The S. cerevisiae sequence was truncated after 226 of 326 residues.
Class I (Zn(II) activated) is associated with a protein that has a longer length (∼180 amino acids vs ∼130 amino acids) and features an N-terminal extension as well as additional loops that are absent in the Ni(II)-activated class II enzymes. It is now recognized that most eukaryotic Glo I enzymes fall into class I, while most bacterial Glo I enzymes fall into class II.97 The correlation between amino acid sequence and metal preference that distinguishes class II from class I enzymes is supported by the apparent exceptions in this distribution. Exceptions include the P. putida enzyme,110 which is a bacterial class I enzyme and resembles the eukaryotic enzymes in terms of metal activity and amino acid sequence; one of three enzymes from Pseudomonas aeruginosa,113a GloA1, GloA2, and GloA3, the first two of which belong to class II and the latter to class I and feature the corresponding polypeptide lengths and metal preferences; and the Glo I from L. major,99a,115 which is a class II enzyme from a eukaryote and features the shorter amino acid sequence and Ni(II) activation.
Crystal structures of class I enzymes (mouse, PDB code 2ZA0;117 and human,118 with S-(N-hydroxy-N-p-iodophenylcarbamoyl)glutathione, 1QIN, with S-p-nitrobenzy-loxycarbonylglutathione, 1QIP, with benzylglutathione, 1FRO, with an N-hydroxypyridone inhibitor, 3VW9, and a Q33E/ E172Q double mutant, 1BH5) and class II enzymes (L. major, 2C21;99a L. infantum;119 E. coli,120 apo, 1FA8; with Ni(II) bound, 1F9Z; with Co(II) bound, 1FA6; with Zn(II) bound, 1FA5; and with Cd(II) bound, 1FA7; C. acetobutylicum,114 with Zn(II) bound, 2QH0, or Ni(II) bound, 3HDP) are available. Comparisons of the structures show that both classes of Glo I are members of the βαβββ protein structure superfamily and are homodimers, or in some cases fused dimers (e.g., S. cerevisiae121 and P. falciparum122), composed of monomer units that have two structurally equivalent domains that provide one Glu and one His involved in binding the metal at the dimer interface (Figure 12).
Figure 12.
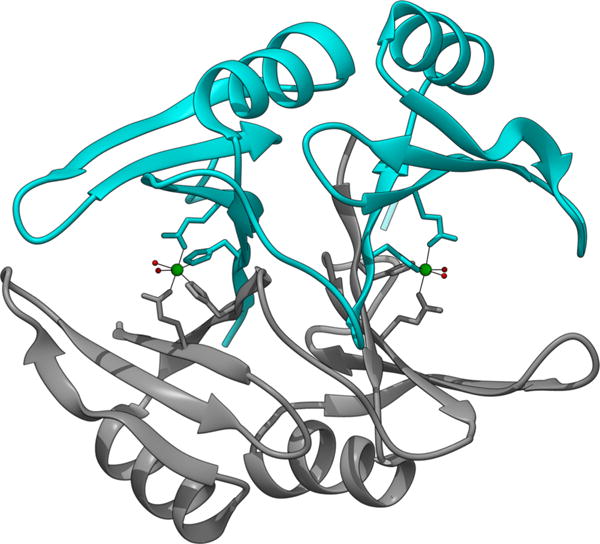
Ribbon diagram of the crystal structure of E. coli Glo I, (PDB code 1F9Z) showing the two subunits of the homo dimer in cyan and gray and the location of the two Ni sites (green spheres) at subunit interfaces.
The N-terminal extension in the class I enzymes wraps around the second subunit, and the longer of the extended loops forms three turns of a helix that sterically restricts the metal site in the class I enzyme.118a Despite the structural similarity of the two metal sites in the homodimeric class II enzyme, the sites are nonequivalent in terms of metalation. Metal binding studies suggest that the enzyme saturates at one Ni(II) ion,112b and NMR has been used both to confirm the stoichiometry and to demonstrate asymmetry in the two metal binding sites not detected by crystallography.123
Both classes of enzyme feature conserved His-Glu-His-Glu residues (His5, Glu56, His74, and Glu122 in the E. coli sequence) that form the protein ligands to the metal sites (Figure 11). The human enzyme and several other enzymes from multicellular eukaryotes feature substitution of Gln for the first His residue. However, the presence of the His residue, rather than Gln, in the amino acid sequence from several class I enzymes (Figure 11) argues against this substitution being a determinant of the enzyme class. Mutation of His5 to Gln in the E. coli enzyme resulted in lower affinity for all divalent metal ions, but did confer some low level activity on the Zn(II) complex.112b
The available Glo I crystal structures provide strong evidence that activity correlates with the geometry of the metal site, with active divalent metals having octahedral geometry (Figure 13).118b,120,124
Figure 13.
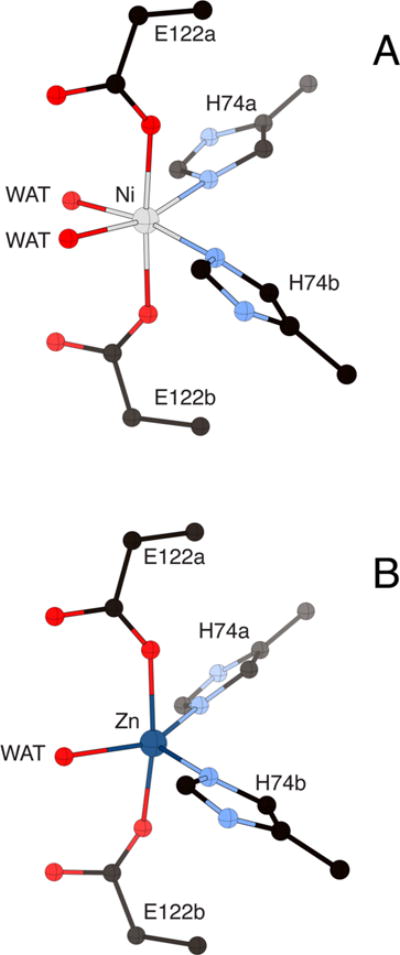
Comparison of the metal site structure of the Ni(II) complex (panel A, PDB code 1F9Z) and the Zn(II) complex (panel B, 1FA5) of E. coli Glo I showing the change in coordination number and geometry for the two metals. The nickel and zinc ions are represented in gray and dark blue, respectively, while CPK coloring is used for all other atoms. WAT = solvent molecules. Protein residues are distinguished by letters indicating the two different subunits of the enzyme.
In the Ni(II) complex of E. coli Glo I, the Ni is six-coordinate with octahedral geometry and features two cis-His ligands and two monodentate trans-Glu ligands, where pairs of His and Glu residues are provided by each protein subunit, and thus the active site forms at the subunit interface.120 The remaining two coordination positions are occupied by water molecules.120 The lack of a Cys residue in the Ni complexes is consistent with the nonredox role of the Ni-containing active sites in Glo I enzymes.11 The same overall structure is also observed for the metal sites in the less active Co(II) complex and the weakly active Cd(II) complex.120 However, the Zn complex has no activity and forms a five-coordinate site with trigonal-bipyramidal geometry that is missing one aqua ligand (Figure 13).120 These crystallographic results are consistent with the structural information regarding the metal sites obtained in solution by X-ray absorption spectroscopy (XAS).125 The results from the E. coli enzyme contrast with the Zn(II) site in human Glo I, where the enzyme can support a six-coordinate Zn complex using Gln33, Glu99, His126, Glu172, and two aqua ligands,118b similar to the Ni complex of E. coli Glo I. The relationship between coordination geometry and activity is further supported by the structural data from Glo I isolated from C. acetobutylicum that has a different quaternary structure such that the ligands forming the active sites are derived from the same polypeptide chain.114 Nonetheless, it has the shorter amino acid sequence typical of the bacterial enzymes and is activated by Ni(II), but not by Zn(II) ions, and therefore belongs to class II. The structure of the Ni(II) complex reveals that it is six-coordinate with a structure similar to that of the E. coli enzyme, and a five-coordinate Zn(II) complex with one aqua ligand.114
3.3. Mechanism
The metal preference of the active site, Zn(II) vs Ni(II), does not appear to be a factor in optimizing catalysis, as the kcat/Km values are similar (∼106−107 M−1 s−1) for class I and II enzymes.112b Kinetic studies of metal-substituted E. coli Glo I (Ni(II), Co(II), Cd(II), Mn(II), and Fe(II)) reveal little effect on Km for the hemithioacetal of methylglyoxal and GSH, indicating that substrate binding is not affected.112b A roughly 5-fold difference is observed in kcat/Km from Cd(II) to Ni(II).112b
Solvent isotope exchange studies of class I enzymes using deuterated α-ketoaldehydes indicate that proton abstraction by a catalytic base to form an enediol(ate) is a feature of the reaction mechanism for Glo I (Figure 14).126
Figure 14.
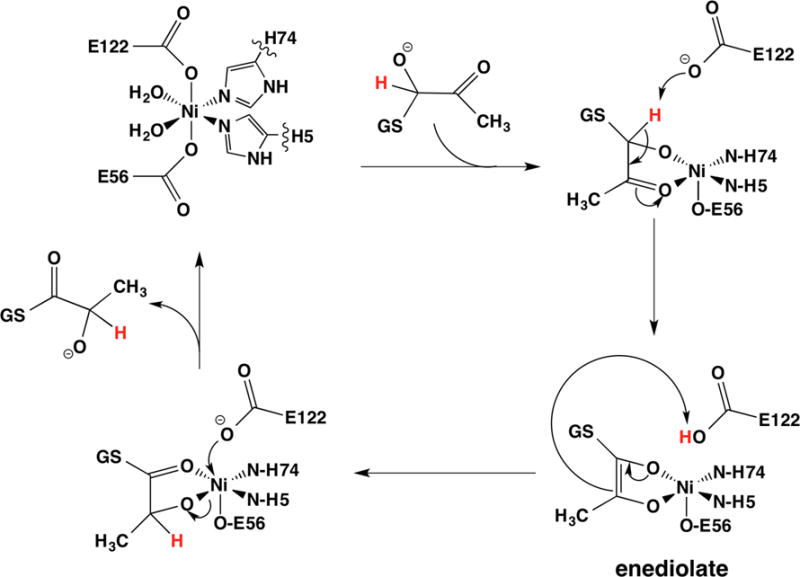
Putative reaction mechanism for the isomerization catalyzed by Glo I that involves coordination of the substrate.
Whether the reaction proceeds by the pre-equilibrium shown in Scheme 2, involving one substrate (hemithioacetal), as long assumed, or a two substrate ordered mechanism involving coordination of the thiol followed by reaction of the α-ketoaldehyde in the active site to form the hemithioacetal, or both, is controversial.127 At least in the case of the class I Glo I from yeast, kinetic studies designed to maximize the difference between these two mechanisms point to the two substrate ordered mechanism.128 When the class II enzyme from E. coli was examined using α-deuterated phenylglyoxal substrate and GSH, no kinetic isotope effect (KIE) was observed for the Ni(II) enzyme.129 In contrast, when the Cd(II)-bound E. coli enzyme kinetics were examined, the observed kcat(H)/kcat(D) of 2.3 indicated that proton transfer was partially rate-limiting, and the results were consistent with a mechanism where proton abstraction as well as other steps contributed to the mechanism of product formation.129 One possible explanation for the difference between the Ni(II)- and Cd(II)-bound enzyme kinetics is that differences in the extent of polarization of the substrate result from different metals in the active site, with Cd(II) being less polarizing and therefore less efficient at lowering the energy barrier for proton abstraction. Differences in aqua ligand exchange rates may also play a role,129 particularly for the Ni(II) enzyme where a slower on-rate for the substrate might suppress the observation of the KIE.
Once formed, the enediol(ate) can be protonated at the alternate C atom to complete the isomerization. Based in part on crystal structures of inhibitor complexes (vide supra), mechanisms involving coordination of the enediolate have been proposed (Figure 14),118b as have mechanisms that involve activating metal-bound water molecules to serve as catalytic bases without binding the substrate to the metal.130 The crystal structures reveal the apparent importance of two cis-aqua ligands in the active site, which could indicate the need for two cis-coordination positions, or a mechanism that employs proton transfers involving both aqua ligands. Coordination of the substrate with loss of a carboxylate ligand has the attractive feature that the Ni(II) site is always charge neutral, and consistent with model chemistry.131 Efforts to distinguish these mechanisms have employed kinetics and XAS studies of the E. coli Ni(II) enzyme in solution in the presence of various inhibitors, mutations, etc.,132 and the crystal structure of human Glo I obtained in the presence of the transition state analogue, S-(N-hydroxy-N-p-iodophenylcarbamoyl)glutathione.118b The latter structure reveals a five-coordinate Zn center in which both water molecules and Glu172 are displaced upon binding the inhibitor, which binds in a bidentate fashion. Glu172 was proposed to play a role as the catalytic base in the deprotonation and reprotonation of the substrate, a role that is consistent with computational models.133 Both R- and S-enantiomers of substrates are bound and stereospecifically reprotonated. The presence of two chemically equivalent Glu ligands suggests that one (Glu172) might be the catalytic base for the S-enantiomer, while the other (Glu99) serves that role for the R-enantiomer.133c The corresponding residues in the E. coli enzyme are Glu122 and Glu56, and XAS studies of a hydroxamate inhibitor (L-γ-glutamyl-N-hydroxy-N-methyl-L-glutaminylglycine) complex are consistent with substitution of both aqua ligands, although evidence for the release of a carboxylate was not specifically observed.132 Although changes in metal ligands greatly affect metal binding affinities, mutation of Glu56 in E56A-E. coli Glo I resulted in the reduction of enzymatic activity to <4% of wild type under conditions of metal saturation,129 consistent with similar mutations of class I enzymes118c and the putative role for the carboxylate as a catalytic base.
3.4. Conclusions
Despite 100 years of research on glyoxalase I enzymes, fundamental questions remain concerning the physiological role of these enzymes and the reaction mechanism. The relatively recent discovery that the class II glyoxalase I enzymes are maximally activated by Ni(II) and not by Zn(II) raises additional questions about the advantages conferred by Ni(II) activation. In any event, the characterization of the Ni(II) Glo I enzymes has provided an excellent example of a nonredox biological role for nickel.
4. ACIREDUCTONE DIOXYGENASE
4.1. Biological Significance
Acireductone (1,2-dihydroxy-5-(methylthio)pent-1-en-3-one) dioxygenase holds a special place in nickel bioinorganic chemistry: the enzyme from Klebsiella oxytoca is the only metalloenzyme that uses the same protein, but different metal ions (Fe(II) and Ni(II)), to catalyze distinct chemical reactions (Figure 15).9,16c,134 Furthermore, it is also the only example of a nickel-containing dioxygenase.
Figure 15.
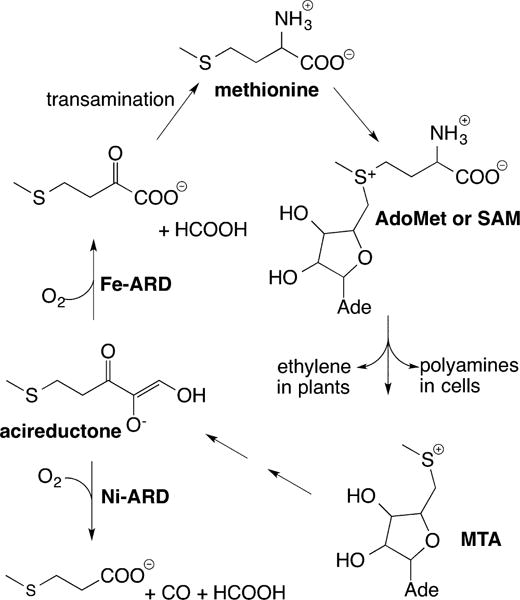
Simplified diagram of the methionine salvage pathway, illustrating key products, intermediates, and catalysis by Ni-ARD vs Fe-ARD.
The Fe(II)-dependent enzyme, Fe-ARD (EC 1.13.11.54; E2′ or ARD′), catalyzes a key step in the methionine salvage pathway (Figure 15),16c that is, the conversion of acireductone to 4-(methylthio)-2-oxobutanoate, the α-keto acid precursor of methionine, and formate. The Ni(II)-dependent enzyme, Ni-ARD (EC 1.13.11.53; E2 or ARD), catalyzes the conversion of acireductone to 3-(methylthio)propionate, formate, and carbon monoxide.16c Ni-ARD is not found in eukaryotes,135 and the precise function of Ni-ARD in K. oxytoca and other bacteria is not clear.136 The Ni-ARD pathway is generally regarded as providing a shunt in the methionine salvage pathway that aids in the regulation of methionine.16c,136b However, the CO product is a known neurotransmitter in mammals,137 an enzyme substrate for carbon monoxide dehydrogenase (see Review by Ragsdale15 in this Thematic Issue), an important structural element in the active site of Ni,Fe hydrogenase (see Review by Lubitz14 in this Thematic Issue), and a signaling molecule for the transcriptional regulator CooA138 in bacteria.139
The methionine salvage pathway (Figure 15) plays a critical role in regulating metabolism in both prokaryotes and eukaryotes.16c,140 Reaction of methionine with ATP produces S-adenosylmethionine (SAM), which is used by a number of methyl transferases and in the biosynthesis of polyamines and in ethylene production in plants.16c Both are essential biological products, as polyamines are involved in cell growth and division,141 and ethylene is required for plant development and fruit and vegetable ripening.142 Methylthioadenosine (MTA) is a byproduct of these two processes that has a strong regulatory effect on the reactions involving SAM.143 As such, the intracellular concentration of MTA is tightly controlled by the methionine salvage pathway.144
4.2. Enzymology
The CO-producing enzyme that was later shown to contain Ni(II) was initially purified and characterized from K. oxytoca as a monomeric protein.136a Both Fe-ARD and Ni-ARD were obtained upon overexpression of a single open-reading frame in E. coli that produced identical 20.2 kDa proteins that could be separated by chromatography.134 The only difference between them was found to be the metal ion present: Ni(II) for the CO-producing enzyme and Fe(II) for the enzyme on the methionine salvage pathway.134 The protein has micromolar affinity for metals and a slightly higher affinity for Ni(II) than for Fe(II) (Kd ≤ 0.1 μM (Ni); Kd ≤ 0.4 μM (Fe)).16c,145 Further, the two enzymes could be interconverted by removing the metal present and reconstituting the apoenzyme with the alternative metal ion.134 It was also observed that reconstitution with other metal ions could confer the same type of activity as Ni-ARD (e.g., Mn(II) and particularly Co(II)) and that Mg(II) incorporation led to Fe-ARD activity.16c,134 In fact, examination of the composition of native enzyme from K. oxytoca showed that 20% contained Co(II) in place of Ni(II).135 The ability of Co(II) to substitute for Ni(II) and the general metal ion promiscuity are also features of the nonredox Ni(II) enzyme glyoxalase I (vide supra).
Despite the fact that ARD catalyzes an oxidation, the metal ions involved are not redox active. Neither Ni-ARD nor Fe-ARD exhibits an EPR signal under either anaerobic or aerobic conditions and in the presence or absence of acireductone, consistent with both metals being divalent and a lack of redox activity.135 Neither enzyme exhibits electronic absorptions below 300 nm at millimolar concentrations,135 and both give well-defined hyperfine-shifted NMR spectra,145,146 observations that are consistent with high-spin divalent metal centers in both cases. These results were confirmed by X-ray absorption spectroscopy (XAS) analysis of the enzyme and enzyme–substrate (ES) complexes,145,146 which are consistent with six-coordinate high-spin metal sites that do not exhibit an edge energy shift in the ES complex that is large enough to support a change in the metal ion oxidation state. This is perhaps not surprising given that acireductone reacts readily with O2 under basic conditions to give the products that are produced by Fe-ARD catalysis.135 Thus, Ni-ARD appears to alter the oxidative chemistry of acireductone.
Analysis of enzyme kinetics is consistent with an ordered mechanism that requires binding of both acireductone and O2 in order to produce products.135 Both enzymes have similar kinetic constants (Table 2), and this, plus the fact that many metals will substitute for Ni(II) or Fe(II), indicates similar mechanisms for the two enzymes that do not involve metal-based redox chemistry (vide infra).
Table 2.
Kinetic Constants for K. oxytoca ARDs
| Ni-ARD | Fe-ARD | |
|---|---|---|
| Km(O2) | 110 μM | 47 μM |
| Km(R) | 50 μM | 52 μM |
| Ks(R) | 25 nM | 53 nM |
| kcat | 5.0 × 10−2 s−1 | 2.6 × 10−2 s−1 |
4.3. Structure
Beginning with the identification of invariant potential metal ligands in the amino acid sequences of ARD proteins (Figure 16), early structural work on the Ni-ARD from K. oxytoca using NMR and XAS played an important role in identifying the Ni(II) ligands and in establishing the interaction between the metal site and acireductone.146
Figure 16.
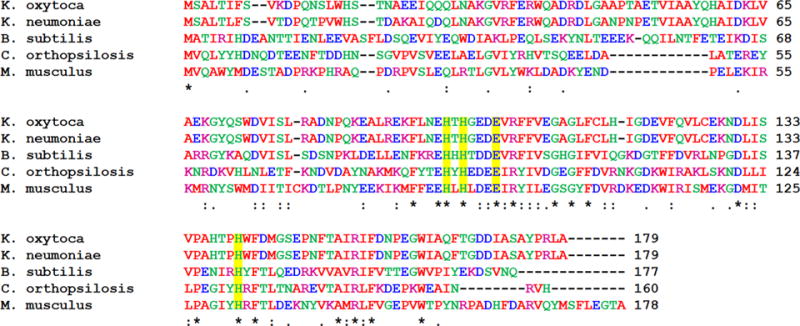
Sequence alignments of selected ARD enzymes created using Clustal W2. The sequences are numbered from Met0, since this residue is cleaved in the mature enzyme. Amino acids are colored by property (hydrophobic (red), acidic (blue), basic (purple), other (green)). Metal binding residues are highlighted in yellow. Residues marked with an asterisk (∗) are invariant; those marked by other symbols represent low (:) and moderate (.) variability.
The presence of paramagnetic Ni(II) led to missing or broadened resonances for residues 95–103, 135–144, and 162–166 in the assignment of sequential amino acid residues. These regions include putative metal ligands His96, His98, Glu102, His137, and His140.147 Three of these His residues are invariant among known ARD sequences, though His137 is not conserved (Figure 16). From the hyperfine-shifted NMR spectrum obtained on the Ni(II) complex, three His ligands were observed, two coordinated via the Nε and one through the Nδ.146 The hyperfine-shifted NMR of the ES complex obtained upon anaerobic addition of substrate gave a complex pattern of hyperfine-shifted resonances that indicated direct coordination of the substrate.146 This pattern is similar to that obtained from a model Ni(II) hydroxamate complex.148 His → Ala mutations of the four His residues identified by NMR showed that the mutation of His137 had no effect on enzyme activity,16c,145 while mutation of the invariant His residues inactivated the enzyme and resulted in decreased solubility.145 Similarly, mutation of Glu102 to Ala resulted in reduced solubility and metal affinity that indicated it was also a ligand.145 Replacement of His98 with Ser led to a variant that was soluble and well folded, and displayed reduced metal binding capacity,145 but was nonetheless able to produce CO from oxidation of acireductone in the presence of high concentrations of Ni(II). XAS data on the wild-type Ni-ARD showed that Ni(II) was six-coordinate with O/N-donor ligands, of which three to four were His imidazole ligands.146 A similar structure was obtained upon addition of substrate, except that the best model involved only two to three His imidazole ligands, suggesting that a His ligand was lost upon substrate coordination.146 NMR and XAS data obtained on Fe-ARD gave very similar results,145 indicating that Fe(II) is bound in the same site as Ni(II). The difference between the two proteins that leads to distinct chemical and physical properties has been attributed to a structural entropy switch,149 and not to differences in metal site structure. This entropy switch was shown to interconvert the protein tertiary structure between conformations that featured a closed, solvent inaccessible (Ni-ARD), or an open, solvent accessible (Fe-ARD), active site.149
The first molecular structure of ARD was obtained using NMR spectral data on the K. oxytoca enzyme,150 and this was subsequently refined using residual dipolar couplings (PDB 1ZRR, Figure 17).151
Figure 17.
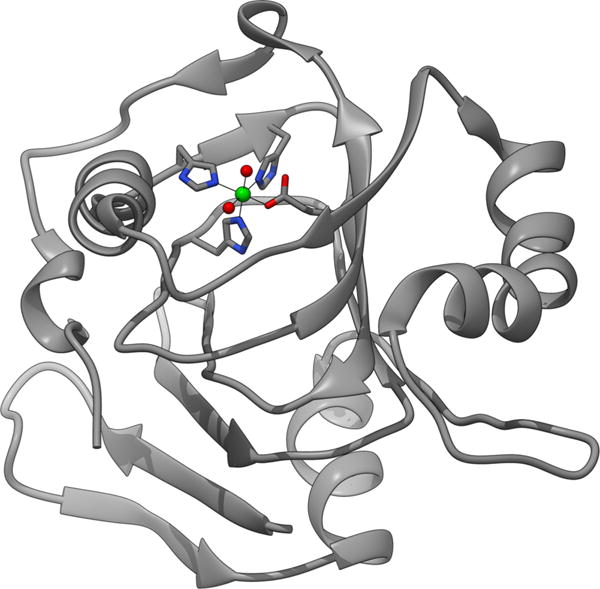
Ribbon diagram of the NMR structure of K. oxytoca Ni-ARD (PDB code 1ZRR) showing the cupin fold and the location of the metal ion (green sphere) with the ligand environment shown as sticks.
The structure showed that ARD belongs to the cupin structural superfamily. The structure that emerged provided a model of the active site (Figure 18) that was later shown to be consistent with the metal site structure in the crystal structure of mouse ARD (PDB 1VR3) with an unknown metal (assumed to be Ni(II)) bound.152
Figure 18.
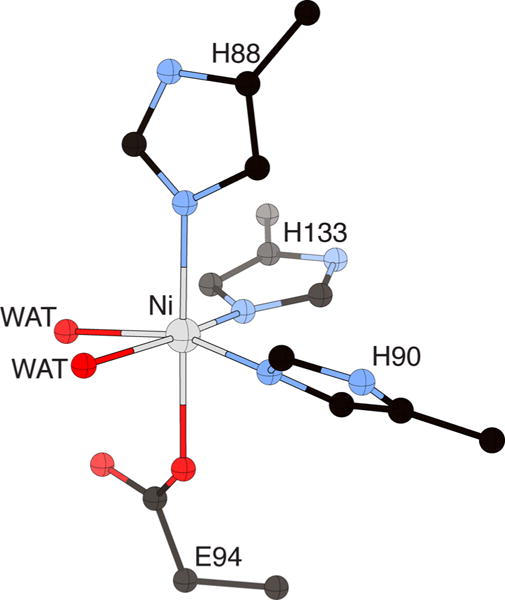
Metal site structure of K. oxytoca Ni-ARD (PDB code 1ZRR), showing the His3Glu coordination of the metal site and the two cis-aqua ligands in the positions used in binding substrate. The nickel ion is represented in gray, while CPK coloring is used for all other atoms. WAT = solvent molecules.
The bacterial and mouse enzymes show similar protein structures, despite the relatively low level of sequence homology between the two proteins (23% identity; 41% similarity, Figure 16). The metal sites are shown to involve ligation by the four protein ligands identified by spectroscopy and mutagenesis (His96, His98, Glu102, and His140 in the K. oxytoca structure), leaving two cis sites available for coordination of substrate (Figure 18).
4.4. Mechanism
The analysis of the enzyme kinetics is consistent with a sequential, ordered mechanism, in which both acireductone and O2 bind before product is released.16c,135 Ni-ARD does not bind O2 in the absence of acireductone, but a red shift in the electronic absorption spectrum of the substrate from 305 to 345 nm indicates that acireductone binds to the enzyme in the absence of O2.135 XAS, model studies, and computational chemistry are consistent with a bidentate binding mode for acireductone (vide supra).146,148,153
The reaction with O2 is proposed to proceed via formation of a C-1 organic peroxide.135 Given the lack of evidence for metal-centered redox chemistry in either the resting enzyme or ES complex and the reactivity of acireductone in the absence of ARDs (vide supra), this peroxide is derived from O2 attack on the coordinated ligand, rather than a mechanism involving O2 activation at the metal. Dioxygenases exhibit two limiting mechanisms, both of which are exemplified by mononuclear, non-heme, Fe-containing dioxygenases.154 In one mechanism, a reduced, redox active metal (e.g., Fe(II)) is used to activate dioxygen for oxidation of the substrate. In the alternate mechanism, the metal serves to activate the substrate toward reaction with O2 by acting as a Lewis acid. This appears to be the case for dioxygenases that require Fe(III). The first mechanism requires a redox active metal capable of reducing O2, while the latter mechanism does not require a change in the redox state of the metal. The use of Ni(II) in a dioxygenase to activate O2 is unexpected from the standpoint that Ni(II) in an O/N-rich ligand environment, such as that found for Ni-ARD, does not typically have biologically accessible redox chemistry (see the Introduction). The structural characterizations of Ni-ARD (vide supra) show no evidence of the cysteine thiolate ligation that might give rise to redox chemistry, and no evidence of redox state change upon substrate binding, and are therefore consistent with a nonredox role for the Ni center. The absence of Cys ligation in the active site of Ni-ARD is also not unexpected for a dioxygenase, since nickel thiolates are sensitive to oxidation by dioxygen155 and no example of a dioxygenase featuring Cys ligation of the metal center is known.154 However, NiSOD, with two Cys ligands, clearly defies this rationalization (see Review by Valentine et al.13 in this Thematic Issue).
Formation of the C-1 peroxide could proceed by a one-electron radical mechanism leading to the formation of superoxide, or via a two-electron mechanism that forms peroxide. Evidence supporting a radical mechanism (or a mechanism involving radical side reactions), as opposed to a carbanion, was obtained using cyclopropyl-substituted acireductone as a suicide inhibitor.135 The course of the reaction of the C-1 peroxide catalyzed by Ni-ARD and Fe-ARD was studied using a 14C- and 2H-labeled desthio analogue of acireductone and 18O.136a,140b These studies established that formate was derived from C-1 and that the O atoms from O2 were incorporated into formate and the carboxylic acid product for both enzymes, and not into CO for Ni-ARD. Conversely, when 2-14C-labeled substrate was used, all of the label was found in the CO molecule produced by Ni-ARD catalysis, establishing C-2 as the source of CO.136a These results were interpreted in terms of a mechanism involving the formation of an organic cyclic peroxy intermediate (Figure 18).16c,135
The difference between Ni-ARD and Fe-ARD was proposed to lie in the nature of the cyclic peroxide formed: a five-membered ring produced by attack of the C-1 peroxide on C-3 and a four-membered cyclic peroxide produced by attack at the C-2 position (Figure 19).135 These reactions were proposed to be supported by different coordination modes of acireductone to Ni and to Fe (the “chelate hypothesis”) involving coordination of the C-1 oxygen in both cases, but coordination of the C-2 oxygen atom gave a five-membered chelate ring with Fe(II) and coordination of the C-3 oxygen atom produced a six-membered chelate ring with Ni(II).135
Figure 19.
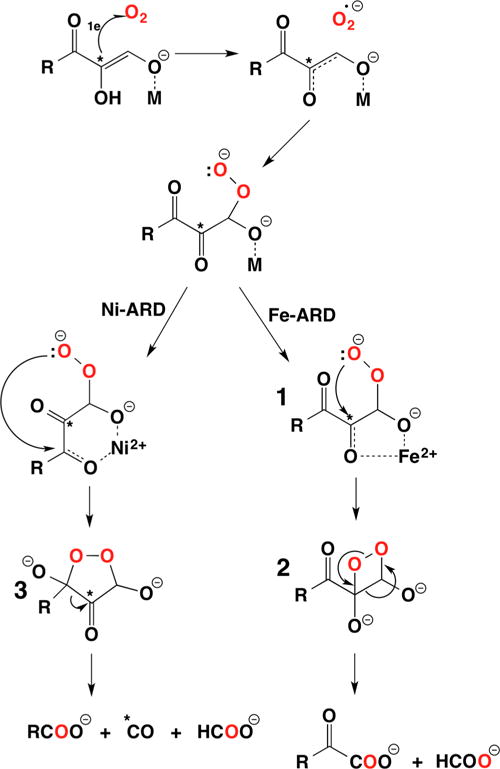
Proposed reaction mechanism illustrating the chelate hypothesis to explain the regioselectivity of the reactions catalyzed by Ni-ARD vs Fe-ARD. The results of incorporation of 18O and 14C labeling studies are indicated by the red O atoms and the asterisks (∗).
(Adapted with permission from ref 1. Copyright 2007 John Wiley & Sons, Ltd.)
Recent model studies support the proposed mechanism for Ni-ARD, but challenge the view that the coordination mode of the organic substrate determines the regiospecificity of the oxidation.156 A Ni(II) complex with a bulky acireductone derivative, [(6-Ph2TPA)Ni(PhC(O)C(OH)C(O)Ph)]ClO4, where 6-Ph2TPA = N,N-bis((6-phenyl-2-pyridyl)methyl)-N-(2-pyridylmethyl)amine), was studied and found to catalyze the oxidation of the substrate with the regiospecificity of Ni-ARD (Figure 20).156b NMR showed that the substrate binds to Ni(II) to form a six-membered chelate ring like that shown in Figures 19 and 20.156b Mechanistic studies of the model are consistent with an initial two-electron oxidation of the substrate to form 1,3-diphenylpropantrione and HO2−,157 which then form a five-membered cyclic peroxy intermediate like that shown in Figure 19, resulting in the formation of the expected products, including CO.157 However, when the corresponding Fe(II) complex was studied, it was shown to bind the acireductone derivative in the same manner, forming a six-membered chelate ring, and gave the products expected for Ni-ARD catalysis upon reaction with O in dry solvent.156a When O2 was added to the reaction, the products expected for Fe-ARD catalysis were obtained.156a Thus, a change of regiospecificity was demonstrated without a change in the structure of the chelate. This change in regiospecificity was attributed to hydration of the triketone reaction intermediate, which in turn leads to the formation of a cyclic peroxy intermediate (Figure 20).156a This chemistry can be related to the tertiary structure of the enzyme, which features a closed site protected by a hydrophobic tryptophan loop in Ni-ARD, but has a more open and solvent exposed site in Fe-ARD.149
Figure 20.
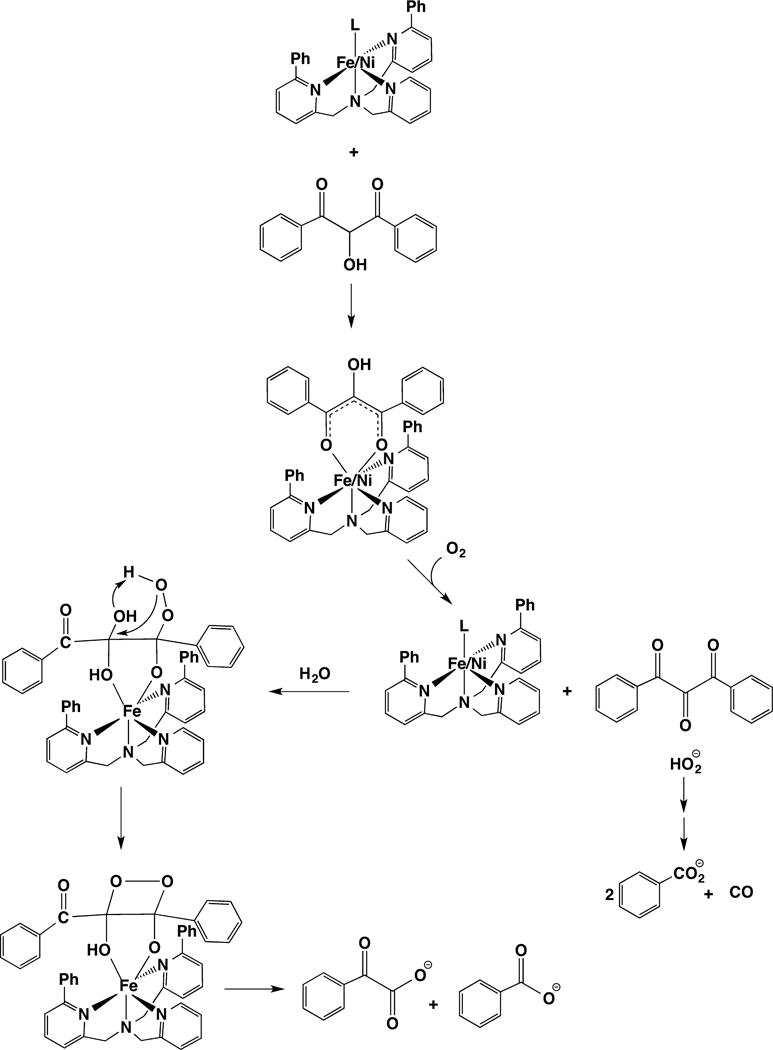
Model chemistry illustrating the role of substrate hydration in determining the regioselectivity of the reactions catalyzed by Ni-ARD vs Fe-ARD.
The “chelate hypothesis” for the different regioselectivities of the oxidations catalyzed by Ni-ARD and Fe-ARD has also been challenged based on computational chemistry.153 The results of mixed quantum-classical molecular dynamics simulations coupled with density functional theoretical calculations agree with the model studies that acireductone binds forming a six-membered chelate ring to both Ni(II) and Fe(II).153 Following electrophilic attack of O on C-1 of the substrate, the five-membered cyclic peroxo intermediate is formed.153 However, regioselectivity is proposed to be determined by the electronic structure of the metal center, which affects the O–O cleavage step; Ni-ARD immediately produces the products, while Fe-ARD forms an additional intermediate that proceeds through an epoxy-like transition state to yield products (Figure 21).153
Figure 21.
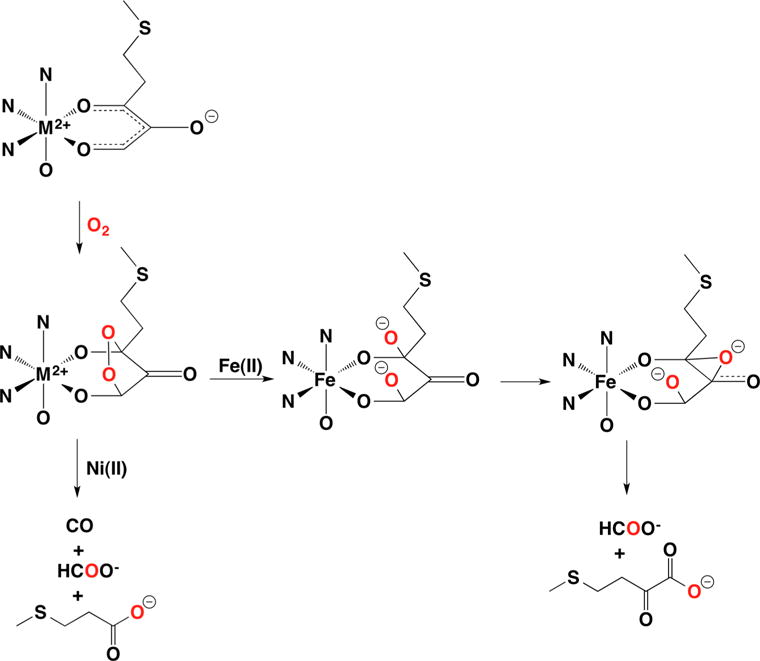
Mechanisms for Ni-ARD vs Fe-ARD catalysis from computational modeling indicate that the electronic structure of the metal ions leads to additional intermediates in the Fe-ARD reaction pathway.
The additional intermediates are partially stabilized by charge transfer mediated by Fe(II) that does not occur for Ni(II).153 Thus, the proposed Fe-ARD mechanism relies on transient redox chemistry, while Ni(II) is a simple Lewis acid. In this regard, it is worth noting that the additional intermediates suggested for Fe-ARD would not be available to the nonenzymatically catalyzed reaction, nor would they be stabilized by transient redox chemistry for the Mg-substituted ARD, both of which give the Fe-ARD products.
4.5. Conclusions
Bacterial ARDs are an interesting case of one protein that produces two enzymes. Depending on the metal ion bound, Fe(II) or Ni(II), the enzymes provide either on-pathway catalysis (Fe-ARD) or an off-pathway shunt that also produces carbon monoxide (Ni-ARD). Ni-ARD is also the only known Ni(II) dioxygenase and, since it lacks any redox activity, is an example of a dioxygenase that activates the substrate (acireductone) by interaction with a Lewis acid, which also alters the course of the oxidation relative to the nonenzymatic oxidation. Mechanistically, it is clear from model and computational chemistry that a difference in the coordination of the substrate is not required in order to obtain the different products observed for Ni-ARD and Fe-ARD. However, whether this difference arises in the enzyme from reaction with water, as suggested by the chemical models, or from different reaction pathways being favored by the electronic structures of the two metals, as suggested by theory, remains an open question.
Biographies
Michael J. Maroney was born and raised in Ames, Iowa. He received a B.S. degree in chemistry from Iowa State University and a Ph.D. from the University of Washington (with Norman J. Rose). After a brief stint as a research chemist at Chevron Research Company, he did postdoctoral research in organometallic chemistry at Northwestern University (with William C. Trogler) and in bioinorganic chemistry at the University of Minnesota (with Lawrence Que, Jr.), where he worked on the structure and function of dinuclear non-heme iron proteins. He joined the chemistry faculty at the University of Massachusetts—Amherst in 1985. His research interests include spectroscopic, theoretical, and model studies of metal sites in proteins aimed at elucidating structure–function relationships, with an emphasis on nickel-containing enzymes and proteins involved in bacterial nickel homeostasis.

Stefano Ciurli was born in 1960 in Rosignano Marittimo (Livorno, Italy). He received his laurea degree in chemistry from the University of Pisa (Italy) in 1986, with a thesis on the synthesis and structure of macrocyclic coordination compounds of low-valent transition metals, carried out in the laboratory of Prof. Carlo Floriani at the Department of Chemistry of Columbia University (NY). He then received his Ph.D. degree in inorganic chemistry from Harvard University (Cambridge, MA) in 1990 under the guidance of Prof. Richard H. Holm, with a thesis on the synthetic, structural, and spectroscopic characterization of vanadium/rhenium/nickel heterometal iron–sulfur clusters. Subsequently he completed two years of postdoctoral studies with Prof. Ivano Bertini and Prof. Claudio Luchinat at the University of Bologna (Italy) working on NMR studies of iron–sulfur proteins. In 1992 he started his independent career at the University of Bologna, studying structure–function relationships in copper- and iron-containing redox metalloproteins, as well as initiating his early work on urease. He became Professor of General and Inorganic Chemistry in 2001. Since then, in addition to pursuing structural investigations on urease, he became interested in the molecular basis of nickel trafficking, approaching, in the most recent years, the metal-mediated protein–DNA interactions involved in nickel-sensing and nickel-dependent gene regulation.

Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Sigel A, Sigel H, Sigel RKO, editors. Nickel and Its Surprising Impact on Nature. John Wiley & Sons, Ltd.; Chichester, England: 2007. [Google Scholar]
- 2.Zambelli B, Ciurli S. Nickel and human health. In: Sigel A, Sigel H, Sigel RKO, editors. Interrelations between Essential Metal Ions and Human Diseases. Vol. 13 Springer Science and Business Media B.V.; Dordrecht, Germany: 2014. [Google Scholar]
- 3.Chen H, Giri NC, Zhang R, Yamane K, Zhang Y, Maroney M, Costa M. J Biol Chem. 2010;285:7374. doi: 10.1074/jbc.M109.058503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Giri NC, Passantino L, Sun H, Zoroddu MA, Costa M, Maroney MJ. Biochemistry. 2013;52:4168. doi: 10.1021/bi400274v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Giri NC, Sun H, Chen H, Costa M, Maroney MJ. Biochemistry. 2011;50:5067. doi: 10.1021/bi101668x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Torres F, das Gracas M, Melo M, Tosti A. Clin, Cosmet Invest Dermatol. 2009;2:39. doi: 10.2147/ccid.s3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmidt M, Raghavan B, Müller V, Vogl T, Fejer G, Tchaptchet S, Keck S, Kalis C, Nielsen PJ, Galanos C, Roth J, Skerra A, Martin SF, Freudenberg MA, Goebeler M. Nature. 2010;11:814. doi: 10.1038/ni.1919. [DOI] [PubMed] [Google Scholar]
- 7.Anke M, Trüpschuch A, Gunstheimer G. In: Trace Elements in Man and Animals 10. Roussel AM, Anderson RA, Favrier AE, editors. Springer; New York: 2002. [Google Scholar]
- 8.Nielsen FH. J Trace Elem Exp Med. 1998;11:251. [Google Scholar]
- 9.Martin-Diaconescu V, Maroney MJ. In: Comprehensive Inorganic Chemistry II. Reedijk J, Poppelmeier KR, editors. Elsevier; Oxford: 2013. [Google Scholar]
- 10.Li Y, Zamble DB. Chem Rev. 2009;109:4617. doi: 10.1021/cr900010n. [DOI] [PubMed] [Google Scholar]
- 11.Maroney MJ. Curr Opin Chem Biol. 1999;3:188. doi: 10.1016/S1367-5931(99)80032-5. [DOI] [PubMed] [Google Scholar]
- 12.Appel AM, Bercaw JE, Bocarsly AB, Dobbek H, DuBois DL, Dupuis M, Ferry JG, Fujita E, Hille R, Kenis PJA, Kerfeld CA, Morris RH, Peden CHF, Portis AR, Ragsdale SW, Rauchfuss TB, Reek JNH, Seefeldt LC, Thauer RK, Waldrop GL. Chem Rev. 2013;113:6621. doi: 10.1021/cr300463y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sheng Y, Abreu IA, Cabelli DE, Maroney MJ, Miller AF, Teixeira M, Valentine JS. Chem Rev. 2014 doi: 10.1021/cr4005296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lubitz W, Ogata H, Rudiger O, Reijerse E. Chem Rev. 2014 doi: 10.1021/cr4005814. [DOI] [PubMed] [Google Scholar]
- 15.Can M, Armstrong FA, Ragsdale SW. Chem Rev. 2014 doi: 10.1021/cr400461p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Ciurli S. Urease: recent insights in the role of nickel. In: Sigel A, Sigel H, Sigel RKO, editors. Nickel and Its Surprising Impact in Nature. Vol. 10 John Wiley & Sons, Ltd.; Chichester, U.K.: 2007. [Google Scholar]; (b) Sukdeo N, Daub E, Honek JF. Nickel and Its Surprising Impact in Nature. Vol. 2 John Wiley & Sons, Ltd.; Chichester, England: 2007. Biochemistry of the nickel-dependent Glyoxalase I enzymes. [Google Scholar]; (c) Pochapsky TC, Ju T, Dang M. Nickel in acireductone dioxygenase. In: Sigel A, Sigel H, Sigel RKO, editors. Nickel and Its Surprising Impact in Nature. Vol. 2 John Wiley & Sons, Ltd.; Chichester: 2007. [Google Scholar]
- 17.(a) Hausinger RP. Microbiol Rev. 1987;51:22. doi: 10.1128/mr.51.1.22-42.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mobley HLT, Hausinger RP. Microbiol Rev. 1989;53:85. doi: 10.1128/mr.53.1.85-108.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Blakeley RL, Hinds JA, Kunze HE, Webb EC, Zerner B. Biochemistry. 1969;8:1991. doi: 10.1021/bi00833a032. [DOI] [PubMed] [Google Scholar]; (b) Dixon NE, Riddles PW, Gazzola C, Blakeley RL, Zerner B. Can J Biochem. 1980;58:1335. doi: 10.1139/o80-181. [DOI] [PubMed] [Google Scholar]
- 19.Sumner JB. J Biol Chem. 1926;69:435. [Google Scholar]
- 20.Dixon NE, Gazzola C, Blakeley R, Zerner B. J Am Chem Soc. 1975;97:4131. doi: 10.1021/ja00847a045. [DOI] [PubMed] [Google Scholar]
- 21.(a) Karplus PA, Pearson MA, Hausinger RP. Acc Chem Res. 1997;30:330. [Google Scholar]; (b) Ciurli S, Benini S, Rypniewski WR, Wilson KS, Miletti S, Mangani S. Coord Chem Rev. 1999;190–192:331. [Google Scholar]; (c) Hausinger RP, Karplus PA. Urease. In: Messerschmidt A, Huber R, Poulos T, Wieghardt K, editors. Handbook of Metalloproteins. John Wiley & Sons, Ltd.; Chichester, U.K.: 2001. [Google Scholar]; (d) Ciurli S, Mangani S. Nickel containing enzymes. In: Bertini I, Sigel A, Sigel H, editors. Handbook on Metalloproteins. Marcel Dekker; New York: 2001. [Google Scholar]; (e) Krajewska B. J Mol Catal B: Enzym. 2009;59:9. [Google Scholar]; (f) Zambelli B, Musiani F, Benini S, Ciurli S. Acc Chem Res. 2011;44:520. doi: 10.1021/ar200041k. [DOI] [PubMed] [Google Scholar]
- 22.Krebs HA, Henseleit K. Hoppe-Seyler’s Z Physiol Chem. 1932;210:33. [Google Scholar]
- 23.Shaw WHR, Bordeaux JJ. J Am Chem Soc. 1955;77:4729. [Google Scholar]
- 24.Blakeley RL, Treston A, Andrews RK, Zerner B. J Am Chem Soc. 1982;104:612. [Google Scholar]
- 25.Callahan BP, Yuan Y, Wolfenden R. J Am Chem Soc. 2005;127:10828. doi: 10.1021/ja0525399. [DOI] [PubMed] [Google Scholar]
- 26.Lin W, Mathys V, Ang ELY, Koh VHQ, Martínez Gömez JM, Ang MLT, Zainul Rahim SZ, Tan MP, Pethe K, Alonso S. Infect Immun. 2012;80:2771. doi: 10.1128/IAI.06195-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Young GM, Amid D, Miller VL. J Bacteriol. 1996;178:6487. doi: 10.1128/jb.178.22.6487-6495.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cox GM, Mukherjee J, Cole GT, Casadevall A, Perfect JR. Infect Immun. 2000;68:443. doi: 10.1128/iai.68.2.443-448.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jones BD, Lockatell CV, Johnson DE, Warren JW, Mobley HLT. Infect Immun. 1990;58:1120. doi: 10.1128/iai.58.4.1120-1123.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mobley HLT, Island MD, Hausinger RP. Microbiol Rev. 1995;59:451. doi: 10.1128/mr.59.3.451-480.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Montecucco C, Rappuoli R. Nat Rev Mol Cell Biol. 2001;2:457. doi: 10.1038/35073084. [DOI] [PubMed] [Google Scholar]
- 32.Stangel PJ. In: Nitrogen in Crop Production. Hauck RD, editor. American Society of Agronomy; Madison, WI: p. 1984. [Google Scholar]
- 33.Bremner JM, Mulvaney RL. In: Soil Enzymes. Burns RG, editor. Academic Press; New York: 1978. [Google Scholar]
- 34.Zonia LE, Stebbins NE, Polacco JC. Plant Physiol. 1995;107:1097. doi: 10.1104/pp.107.4.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Follmer C, Real-Guerra R, Wasserman GE, Olivera-Severo D, Carlini CR. Eur J Biochem. 2004;271:1357. doi: 10.1111/j.1432-1033.2004.04046.x. [DOI] [PubMed] [Google Scholar]
- 36.Carlini CR, Grossi-de-Sa MF. Toxicon. 2002;40:1515. doi: 10.1016/s0041-0101(02)00240-4. [DOI] [PubMed] [Google Scholar]
- 37.Mulinari F, Staniscuaski F, Bertholdo-Vargas LR, Postal M, Oliveira-Neto OB, Rigden DJ, Grossi-de-Sa MF, Carlini CR. Peptides. 2007;28:2042. doi: 10.1016/j.peptides.2007.08.009. [DOI] [PubMed] [Google Scholar]
- 38.Balasubramanian A, Ponnuraj K. J Mol Biol. 2010;400:274. doi: 10.1016/j.jmb.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 39.(a) Mulrooney SB, Zakharian T, Schaller RA, Hausinger RP. Arch Biochem Biophys. 2001;394:280. doi: 10.1006/abbi.2001.2536. [DOI] [PubMed] [Google Scholar]; (b) Krajewska B, Ciurli S. Plant Physiol Biochem. 2005;43:651. doi: 10.1016/j.plaphy.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 40.Zerner B. Bioorg Chem. 1991;19:116. [Google Scholar]
- 41.(a) Dixon NE, Blakeley RL, Zerner B. Can J Biochem. 1980;58:481. doi: 10.1139/o80-064. [DOI] [PubMed] [Google Scholar]; (b) Blakeley RL, Dixon NE, Zerner B. Biochim Biophys Acta. 1983;744:219. [Google Scholar]
- 42.(a) Hasnain SS, Piggot B. Biochem Biophys Res Commun. 1983;112:279. doi: 10.1016/0006-291x(83)91827-2. [DOI] [PubMed] [Google Scholar]; (b) Alagna L, Hasnain SS, Piggot B, Williams DJ. Biochem J. 1984;220:591. doi: 10.1042/bj2200591. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Clark PA, Wilcox DE, Scott RA. Inorg Chem. 1990;29:579. [Google Scholar]
- 43.Clark PA, Wilcox DE. Inorg Chem. 1989;28:1326. [Google Scholar]
- 44.Wang S, Lee MH, Hausinger RP, Clark PA, Wilcox DE, Scott RA. Inorg Chem. 1994;33:1589. [Google Scholar]
- 45.Benini S, Ciurli S, Nolting HF, Mangani S. Eur J Biochem. 1996;239:61. doi: 10.1111/j.1432-1033.1996.0061u.x. [DOI] [PubMed] [Google Scholar]
- 46.Jabri E, Carr MB, Hausinger RP, Karplus PA. Science. 1995;268:998. [PubMed] [Google Scholar]
- 47.Jabri E, Karplus PA. Biochemistry. 1996;35:10616. doi: 10.1021/bi960424z. [DOI] [PubMed] [Google Scholar]
- 48.Pearson MA, Overbye Michel L, Hausinger RP, Karplus PA. Biochemistry. 1997;36:8164. doi: 10.1021/bi970514j. [DOI] [PubMed] [Google Scholar]
- 49.Pearson MA, Schaller RA, Michel LO, Karplus PA, Hausinger RP. Biochemistry. 1998;37:6214. doi: 10.1021/bi980021u. [DOI] [PubMed] [Google Scholar]
- 50.Pearson MA, Park IS, Schaller RA, Michel LO, Karplus PA, Hausinger RP. Biochemistry. 2000;39:8575. doi: 10.1021/bi000613o. [DOI] [PubMed] [Google Scholar]
- 51.Park IS, Michel LO, Pearson MA, Jabri E, Karplus PA, Wang S, Dong J, Scott RA, Koehler BP, Johnson MK, Hausinger RP. J Biol Chem. 1996;271:18632. doi: 10.1074/jbc.271.31.18632. [DOI] [PubMed] [Google Scholar]
- 52.Yamaguchi K, Cosper NJ, Stalhandske C, Scott RA, Pearson MA, Karplus PA, Hausinger RP. J Biol Inorg Chem. 1999;4:468. doi: 10.1007/s007750050333. [DOI] [PubMed] [Google Scholar]
- 53.Benini S, Rypniewski WR, Wilson KS, Miletti S, Ciurli S, Mangani S. Structure. 1999;7:205. doi: 10.1016/S0969-2126(99)80026-4. [DOI] [PubMed] [Google Scholar]
- 54.Benini S, Rypniewski WR, Wilson KS, Ciurli S, Mangani S. J Biol Inorg Chem. 1998;3:268. doi: 10.1007/s007750100254. [DOI] [PubMed] [Google Scholar]
- 55.Benini S, Rypniewski WR, Wilson KS, Miletti S, Ciurli S, Mangani S. J Biol Inorg Chem. 2000;5:110. doi: 10.1007/s007750050014. [DOI] [PubMed] [Google Scholar]
- 56.Benini S, Rypniewski WR, Wilson KS, Ciurli S, Mangani S. J Biol Inorg Chem. 2001;6:778. doi: 10.1007/s007750100254. [DOI] [PubMed] [Google Scholar]
- 57.Benini S, Rypniewski WR, Wilson KS, Mangani S, Ciurli S. J Am Chem Soc. 2004;126:3714. doi: 10.1021/ja049618p. [DOI] [PubMed] [Google Scholar]
- 58.Benini S, Kosikowska P, Cianci M, Mazzei L, Vara AG, Berlicki L, Ciurli S. J Biol Inorg Chem. 2013;18:391. doi: 10.1007/s00775-013-0983-7. [DOI] [PubMed] [Google Scholar]
- 59.Ha NC, Oh ST, Sung JY, Cha KA, Lee MH, Oh BH. Nat Struct Biol. 2001;8:505. doi: 10.1038/88563. [DOI] [PubMed] [Google Scholar]
- 60.Carter EL, Tronrud DE, Taber SR, Karplus PA, Hausinger RP. Proc Natl Acad Sci USA. 2011;108:13095. doi: 10.1073/pnas.1106915108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Balasubramanian A, Durairajpandian V, Elumalai S, Mathivanan N, Munirajan AK, Ponnuraj K. Int J Biol Macromol. 2013;58:301. doi: 10.1016/j.ijbiomac.2013.04.055. [DOI] [PubMed] [Google Scholar]
- 62.Austin JW, Doig P, Stewart M, Trust TJ. J Bacteriol. 1992;174:7470. doi: 10.1128/jb.174.22.7470-7473.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Basolo F, Pearson RG. Mechanisms of Inorganic Reactions. 2nd. John Wiley & Sons, Ltd.; New York: p. 1967. [Google Scholar]
- 64.Park IS, Hausinger RP. Protein Sci. 1993;2:1034. doi: 10.1002/pro.5560020616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Park IS, Hausinger RP. J Protein Chem. 1993;12:51. doi: 10.1007/BF01024914. [DOI] [PubMed] [Google Scholar]
- 66.(a) Todd MJ, Hausinger RP. J Biol Chem. 1991;266:24327. [PubMed] [Google Scholar]; (b) Todd MJ, Hausinger RP. J Biol Chem. 1991;266:10260. [PubMed] [Google Scholar]
- 67.Martin PR, Hausinger RP. J Biol Chem. 1992;267:20024. [PubMed] [Google Scholar]
- 68.(a) Amtul Z, Rahman AU, Siddiqui RA, Choudhary MI. Curr Med Chem. 2002;9:1323. doi: 10.2174/0929867023369853. [DOI] [PubMed] [Google Scholar]; (b) Kosikowska P, Berlicki Ł. Expert Opin Ther Pat. 2011;21:945. doi: 10.1517/13543776.2011.574615. [DOI] [PubMed] [Google Scholar]
- 69.Krogmeier MJ, McCarty GW, Bremner JM. Proc Natl Acad Sci USA. 1989;86:1110. doi: 10.1073/pnas.86.4.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Benini S, Ciurli S, Rypniewski WR, Wilson KS, Mangani S. Acta Crystallogr, Sect D: Biol Crystallogr. 1998;54:409. doi: 10.1107/s0907444997013085. [DOI] [PubMed] [Google Scholar]
- 71.Finnegan MG, Kowal AT, Werth MT, Clark PA, Wilcox DE, Johnson MK. J Am Chem Soc. 1991;113:4030. [Google Scholar]
- 72.Todd MJ, Hausinger RP. J Biol Chem. 1989;264:15835. [PubMed] [Google Scholar]
- 73.(a) Kobashi K, Hase JI, Uehara K. Biochim Biophys Acta. 1962;62:380. doi: 10.1016/0006-3002(62)91067-3. [DOI] [PubMed] [Google Scholar]; (b) Dixon NE, Gazzola C, Watters JJ, Blakeley R, Zerner B. J Am Chem Soc. 1975;97:4130. doi: 10.1021/ja00847a044. [DOI] [PubMed] [Google Scholar]
- 74.Burne RA, Chen YYM. Microbes Infect. 2000;2:533. doi: 10.1016/s1286-4579(00)00312-9. [DOI] [PubMed] [Google Scholar]
- 75.Howell SP, Sumner JB. J Biol Chem. 1934;104:619. [Google Scholar]
- 76.Krajewska B, Zaborska W. J Mol Catal. 1999;6B:75. [Google Scholar]
- 77.(a) Krajewska B, Zaborska W, Leszko M, Brzozka Z. Pol J Chem. 1999;73:359. [Google Scholar]; (b) Breitenbach JM, Hausinger RH. Biochem J. 1988;250:917. doi: 10.1042/bj2500917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.(a) Christianson CB, Byrnes BH, Carmona G. Fert Res. 1990;26:21. [Google Scholar]; (b) Creason GL, Schmidt MR, Douglass EA, Hendrickson LL. Soil Biol Biochem. 1990;22:209. [Google Scholar]
- 79.(a) Andrews RK, Dexter A, Blakeley RL, Zerner B. J Am Chem Soc. 1986;108:7124. [Google Scholar]; (b) McCarthy GW, Bremner JM, Lee JS. Plant Soil. 1990;127:269. [Google Scholar]; (c) Faraci WS, Yang BV, O’Rourke D, Spencer RW. Bioorg Med Chem. 1995;3:605. doi: 10.1016/0968-0896(95)00043-g. [DOI] [PubMed] [Google Scholar]
- 80.Saboury AA, Moosavi-Movahedi AA. J Enz Inhib. 1997;12:273–279. doi: 10.3109/14756369709035819. [DOI] [PubMed] [Google Scholar]
- 81.Todd MJ, Hausinger RP. Biochemistry. 2000;39:5389. doi: 10.1021/bi992287m. [DOI] [PubMed] [Google Scholar]
- 82.Krajewska B, Zaborska W, Leszko M. J Mol Catal B: Enzym. 2001;14:101. [Google Scholar]
- 83.Musiani F, Arnofi E, Casadio R, Ciurli S. J Biol Inorg Chem. 2001;6:300. doi: 10.1007/s007750000204. [DOI] [PubMed] [Google Scholar]
- 84.Barrios AM, Lippard SJ. J Am Chem Soc. 1999;121:11751. [Google Scholar]
- 85.Suarez D, Diaz N, Merz KM. J Am Chem Soc. 2003;125:15324. doi: 10.1021/ja030145g. [DOI] [PubMed] [Google Scholar]
- 86.Todd MJ, Hausinger RP. J Biol Chem. 1987;262:5963. [PubMed] [Google Scholar]
- 87.Martell AE, Smith RM. Critical Stability Constants. Plenum Press; New York: 1974. [Google Scholar]
- 88.Laitaoja M, Valjakka J, Janis J. Inorg Chem. 2013;52:10983. doi: 10.1021/ic401072d. [DOI] [PubMed] [Google Scholar]
- 89.Dixon NE, Gazzola C, Asher CJ, Lee DW, Blakeley RL, Zerner B. Can J Biochem. 1980;58:474. doi: 10.1139/o80-063. [DOI] [PubMed] [Google Scholar]
- 90.King GJ, Zerner B. Inorg Chim Acta. 1997;255:381. [Google Scholar]
- 91.Park IS, Hausinger RP. Biochemistry. 1996;35:5345. doi: 10.1021/bi952894j. [DOI] [PubMed] [Google Scholar]
- 92.Stoof J, Breijer S, Pot RG, van der Neut D, Kuipers EJ, Kusters JG, van Vliet AH. Environ Microbiol. 2008;10:2586. doi: 10.1111/j.1462-2920.2008.01681.x. [DOI] [PubMed] [Google Scholar]
- 93.Suttisansanee U, Honek JF. Semin Cell Dev Biol. 2011;22:285. doi: 10.1016/j.semcdb.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 94.Aronsson AC, Marmstal E, Mannervik B. Biochem Biophys Res Commun. 1978;81:1235. doi: 10.1016/0006-291x(78)91268-8. [DOI] [PubMed] [Google Scholar]
- 95.Wyllie S, Fairlamb AH. Semin Cell Dev Biol. 2011;22:271. doi: 10.1016/j.semcdb.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Deswal R, Sopory SK. FEBS Lett. 1991;282:277. doi: 10.1016/0014-5793(91)80494-n. [DOI] [PubMed] [Google Scholar]
- 97.Sousa Silva M, Gomes RA, Ferreira AEN, Ponces Freire A, Cordeiro C. Biochem J. 2013;453:1. doi: 10.1042/BJ20121743. [DOI] [PubMed] [Google Scholar]
- 98.Sousa Silva M, Ferreira AEN, Gomes RA, Sousa Silva M, Vicente Miranda H, Ferreira AE, Cordeiro CA. Int J Med Microbiol. 2012;302:225. doi: 10.1016/j.ijmm.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 99.(a) Ariza A, Vickers TJ, Greig N, Armour KA, Dixon MJ, Eggleston IM, Fairlamb AH, Bond CS. Mol Microbiol. 2006;59:1239. doi: 10.1111/j.1365-2958.2006.05022.x. [DOI] [PubMed] [Google Scholar]; (b) Newton GL, Arnold K, Price MS, Sherrill C, Delcardayre SB, Aharonowitz Y, Cohen G, Davies J, Fahey RC, Davis C. J Bacteriol. 1996;178:1990. doi: 10.1128/jb.178.7.1990-1995.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Smith K, Borges A, Ariyanayagam MR, Fairlamb AH. Biochem J. 1995;312:465. doi: 10.1042/bj3120465. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Tabor H, Tabor CW. J Biol Chem. 1975;250:2648. [PubMed] [Google Scholar]
- 100.(a) Penninckx M. Enzyme Microb Technol. 2000;26:737. doi: 10.1016/s0141-0229(00)00165-4. [DOI] [PubMed] [Google Scholar]; (b) Xue M, Rabbani N, Thornalley PJ. Semin Cell Dev Biol. 2011;22:293. doi: 10.1016/j.semcdb.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 101.Urscher M, Alisch R, Deponte M. Semin Cell Dev Biol. 2011;22:262. doi: 10.1016/j.semcdb.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 102.Clugston SL, Honek JF. J Mol Evol. 2000;50:491. doi: 10.1007/s002390010052. [DOI] [PubMed] [Google Scholar]
- 103.Thornalley PJ, Rabbani N. Semin Cell Dev Biol. 2011;22:318. doi: 10.1016/j.semcdb.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 104.Ekwall K, Mannervik B. Biochim Biophys Acta. 1973;297:297. doi: 10.1016/0304-4165(73)90076-7. [DOI] [PubMed] [Google Scholar]
- 105.(a) Gomes RA, Sousa Silva M, Vicente Miranda H, Ferreira AE, Cordeiro CA, Freire AP. FEBS J. 2005;272:4521. doi: 10.1111/j.1742-4658.2005.04872.x. [DOI] [PubMed] [Google Scholar]; (b) Bito A, Haider M, Hadler I, Breitenbach M. J Biol Chem. 1997;272:21509. doi: 10.1074/jbc.272.34.21509. [DOI] [PubMed] [Google Scholar]
- 106.(a) Dakin HD, Dudley HW. J Biol Chem. 1913;14:423. [Google Scholar]; (b) Neuberg C. Biochem Z. 1913;49:502. [Google Scholar]
- 107.(a) Racker E. J Biol Chem. 1951;190:685. [PubMed] [Google Scholar]; (b) Crook EM, Law K. Biochem J. 1952;52:492. doi: 10.1042/bj0520492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.(a) Misra K, Banerjee AB, Ray S, Ray M. Biochem J. 1995;305:999. doi: 10.1042/bj3050999. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Subedi KP, Choi D, Kim I, Min B, Park C. Mol Microbiol. 2011;81:926. doi: 10.1111/j.1365-2958.2011.07736.x. [DOI] [PubMed] [Google Scholar]
- 109.Lee JY, Song J, Kwon K, Jang S, Kim C, Baek K, Kim J, Park C. Hum Mol Genet. 2012;21:3215. doi: 10.1093/hmg/dds155. [DOI] [PubMed] [Google Scholar]
- 110.Saint-Jean AP, Phillips KR, Creighton DJ, Stone MJ. Biochemistry. 1998;37:10345. doi: 10.1021/bi980868q. [DOI] [PubMed] [Google Scholar]
- 111.(a) Davis KA, Williams GR. Biochim Biophys Acta. 1966;113:393. doi: 10.1016/s0926-6593(66)80078-4. [DOI] [PubMed] [Google Scholar]; (b) Sukdeo N, Clugston SL, Daub E, Honek JF. Biochem J. 2004;384:111. doi: 10.1042/BJ20041006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.(a) Clugston SL, Barnard J, Kinach R, Miedema D. Biochemistry. 1998;37:8754. doi: 10.1021/bi972791w. [DOI] [PubMed] [Google Scholar]; (b) Clugston SL, Yajima R, Honek JF. Biochem J. 2004;377:309. doi: 10.1042/BJ20030271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.(a) Sukdeo N, Honek JF. Biochim Biophys Acta, Proteins Proteomics. 2007;1774:756. doi: 10.1016/j.bbapap.2007.04.005. [DOI] [PubMed] [Google Scholar]; (b) Kizil G, Wilks K, Wells D, Ala’Aldeen DA. J Med Microbiol. 2000;49:669. doi: 10.1099/0022-1317-49-7-669. [DOI] [PubMed] [Google Scholar]
- 114.Suttisansanee U, Lau K, Lagishetty S, Rao KN, Swaminathan S, Sauder JM, Burley SK, Honek JF. J Biol Chem. 2011;286:38367. doi: 10.1074/jbc.M111.251603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Vickers TJ, Greig N, Fairlamb AH. Proc Natl Acad Sci USA. 2004;101:13186. doi: 10.1073/pnas.0402918101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Greig N, Wyllie S, Vickers TJ, Fairlamb AH. Biochem J. 2006;400:217. doi: 10.1042/BJ20060882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kawatani M, Okumura H, Honda K, Kanoh N, Muroi M, Dohmae N, Takami M, Kitagawa M, Futamura Y, Imoto M, Osada H. Proc Natl Acad Sci USA. 2008;105:11691. doi: 10.1073/pnas.0712239105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.(a) Cameron AD, Olin B, Ridderstrom M, Mannervik B, Jones TA. EMBO J. 1997;16:3386. doi: 10.1093/emboj/16.12.3386. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Cameron AD, Ridderstrom M, Olin B, Kavarana MJ, Creighton DJ, Mannervik B. Biochemistry. 1999;38:13480. doi: 10.1021/bi990696c. [DOI] [PubMed] [Google Scholar]; (c) Ridderstrom M, Cameron AD, Jones TA, Mannervik B. J Biol Chem. 1998;273:21623. doi: 10.1074/jbc.273.34.21623. [DOI] [PubMed] [Google Scholar]
- 119.Barata L, Sousa Silva M, Schuldt L, da Costa G, Tomas AM, Ferreira AE, Weiss MS, Ponces Freire A, Cordeiro C. Acta Crystallogr, Sect F: Struct Biol Cryst Commun. 2010;66:571. doi: 10.1107/S1744309110010754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.He MM, Clugston SL, Honek JF, Matthews BW. Biochemistry. 2000;39:8719. doi: 10.1021/bi000856g. [DOI] [PubMed] [Google Scholar]
- 121.Marmstal E, Aronsson AC, Mannervik B. Biochem J. 1979;183:23. doi: 10.1042/bj1830023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Iozef R, Rahlfs S, Chang T, Schirmer H, Becker K. FEBS Lett. 2003;554:284. doi: 10.1016/s0014-5793(03)01146-3. [DOI] [PubMed] [Google Scholar]
- 123.Su Z, Sukdeo N, Honek JF. Biochemistry. 2008;47:13232. doi: 10.1021/bi8013278. [DOI] [PubMed] [Google Scholar]
- 124.Holm L, Kääriäinen S, Rosenström P, Schenkel A. Bioinformatics. 2008;24:2780. doi: 10.1093/bioinformatics/btn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Davidson G, Clugston SL, Honek JF, Maroney MJ. Inorg Chem. 2000;39:2962. doi: 10.1021/ic0001208. [DOI] [PubMed] [Google Scholar]
- 126.(a) Creighton DJ, Hamilton DS. Arch Biochem Biophys. 2001;387:1. doi: 10.1006/abbi.2000.2253. [DOI] [PubMed] [Google Scholar]; (b) Vander Jagt DL, Han LP. Biochemistry. 1973;12:5161. doi: 10.1021/bi00749a022. [DOI] [PubMed] [Google Scholar]; (c) Douglas KT, Quilter AJ, Shinkai S, Ueda K. Biochim Biophys Acta. 1985;829:119. doi: 10.1016/0167-4838(85)90075-5. [DOI] [PubMed] [Google Scholar]; (d) Hall SS, Doweyko AM, Jordan F. J Am Chem Soc. 1976;98:7460. doi: 10.1021/ja00439a077. [DOI] [PubMed] [Google Scholar]; (e) Chari RV, Kozarich JW. J Biol Chem. 1981;256:9785. [PubMed] [Google Scholar]
- 127.(a) Mannervik B, Bartfai T, Górna-Hall B. J Biol Chem. 1974;249:901. [PubMed] [Google Scholar]; (b) Mannervik B, Górna-Hall B, Bartfai T. Eur J Biochem. 1973;37:270. doi: 10.1111/j.1432-1033.1973.tb02985.x. [DOI] [PubMed] [Google Scholar]
- 128.Lages NF, Cordeiro C, Sousa Silva M, Ponces Freire A, Ferreira AEN. PLoS One. 2012;7:e32749. doi: 10.1371/journal.pone.0032749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mullings KY, Sukdeo N, Suttisansanee U, Ran Y, Honek JF. J Inorg Biochem. 2012;108:133. doi: 10.1016/j.jinorgbio.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 130.(a) Sellin S, Eriksson LE, Mannervik B. Biochemistry. 1982;21:4850. doi: 10.1021/bi00263a004. [DOI] [PubMed] [Google Scholar]; (b) Sellin S, Rosevear PR, Mannervik B, Mildvan AS. J Biol Chem. 1982;257:10023. [PubMed] [Google Scholar]
- 131.Rudzka K, Arif AM, Berreau LM. J Am Chem Soc. 2006;128:17018. doi: 10.1021/ja0601336. [DOI] [PubMed] [Google Scholar]
- 132.Davidson G, Clugston SL, Honek JF, Maroney MJ. Biochemistry. 2001;40:4569. doi: 10.1021/bi0018537. [DOI] [PubMed] [Google Scholar]
- 133.(a) Feierberg I, Cameron AD, Aqvist J. FEBS Lett. 1999;453:90. doi: 10.1016/s0014-5793(99)00703-6. [DOI] [PubMed] [Google Scholar]; (b) Feierberg I, Luzhkov V, Aqvist J. J Biol Chem. 2000;275:22657. doi: 10.1074/jbc.M000726200. [DOI] [PubMed] [Google Scholar]; (c) Himo F, Siegbahn PE. J Am Chem Soc. 2001;123:10280. doi: 10.1021/ja010715h. [DOI] [PubMed] [Google Scholar]
- 134.Dai Y, Wensink PC, Abeles RH. J Biol Chem. 1999;274:1193. doi: 10.1074/jbc.274.3.1193. [DOI] [PubMed] [Google Scholar]
- 135.Dai Y, Pochapsky TC, Abeles RH. Biochemistry. 2001;40:6379. doi: 10.1021/bi010110y. [DOI] [PubMed] [Google Scholar]
- 136.(a) Wray JW, Abeles RH. J Biol Chem. 1993;268:21466. [PubMed] [Google Scholar]; (b) Sekowska A, Dénervaud V, Ashida H, Michoud K, Haas D, Yokota A, Danchin A. BMC Microbiol. 2004;4:9. doi: 10.1186/1471-2180-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Johnson RA, Johnson FK. Curr Opin Neurol. 2000;13:709. doi: 10.1097/00019052-200012000-00016. [DOI] [PubMed] [Google Scholar]
- 138.Aono S. Acc Chem Res. 2003;36:825. doi: 10.1021/ar020097p. [DOI] [PubMed] [Google Scholar]
- 139.Gullotta F, di Masi A, Coletta M, Ascenzi P. Biofactors. 2012;38:1. doi: 10.1002/biof.192. [DOI] [PubMed] [Google Scholar]
- 140.(a) Abeles R, Frey P, Jencks W. Biochemistry. Jones and Bartlett; Boston, MA: p. 1992. [Google Scholar]; (b) Wray JW, Abeles RH. J Biol Chem. 1995;270:3147. doi: 10.1074/jbc.270.7.3147. [DOI] [PubMed] [Google Scholar]
- 141.Wallace HM. Essays Biochem. 2009;46:1. doi: 10.1042/bse0460001. [DOI] [PubMed] [Google Scholar]
- 142.(a) Taiz L. Plant Physiology. Benjamin/Cummings Publishing Co.; Redwood City, CA: p. 1991. [Google Scholar]; (b) Schaller GE. BMC Biol. 2012;10 doi: 10.1186/1741-7007-10-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Schlenk F. Adv Enzymol Relat Areas Mol Biol. 1983;54:195. doi: 10.1002/9780470122990.ch4. [DOI] [PubMed] [Google Scholar]
- 144.Shapiro SK, Barrett A. Biochem Biophys Res Commun. 1981;102:302. doi: 10.1016/0006-291x(81)91521-7. [DOI] [PubMed] [Google Scholar]
- 145.Chai SC, Ju T, Dang M, Goldsmith RB, Maroney MJ, Pochapsky TC. Biochemistry. 2008;47:2428. doi: 10.1021/bi7004152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Al-Mjeni F, Ju T, Pochapsky TC, Maroney MJ. Biochemistry. 2002;41:6761. doi: 10.1021/bi012209a. [DOI] [PubMed] [Google Scholar]
- 147.Mo HP, Dai Y, Pochapsky SS, Pochapsky TC. J Biomol NMR. 1999;14:287. doi: 10.1023/a:1008396624784. [DOI] [PubMed] [Google Scholar]
- 148.Szajna E, Dobrowolski P, Fuller AL, Arif AM, Berreau LM. Inorg Chem. 2004;43:3988. doi: 10.1021/ic040002a. [DOI] [PubMed] [Google Scholar]
- 149.Ju T, Goldsmith RB, Chai SC, Maroney MJ, Pochapsky SS, Pochapsky TC. J Mol Biol. 2006;363:823. doi: 10.1016/j.jmb.2006.08.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Pochapsky TC, Pochapsky SS, Ju T, Mo H, Al-Mjeni F, Maroney MJ. Nat Struct Biol. 2002;9:966. doi: 10.1038/nsb863. [DOI] [PubMed] [Google Scholar]
- 151.Pochapsky SS, Sunshine JC, Pochapsky TC. J Am Chem Soc. 2008;130:2156. doi: 10.1021/ja710187x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Xu Q, Schwarzenbacher R, Krishna SS, et al. Proteins: Struct, Funct, Bioinf. 2006;64:808. doi: 10.1002/prot.20947. [DOI] [PubMed] [Google Scholar]
- 153.Sparta M, Valdez CE, Alexandrova AN. J Mol Biol. 2013;425:3007. doi: 10.1016/j.jmb.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 154.Que L, Ho RYN. Chem Rev. 1996;96:2607. doi: 10.1021/cr960039f. [DOI] [PubMed] [Google Scholar]
- 155.(a) Mirza SA, Pressler MA, Kumar M, Day RO, Maroney MJ. Inorg Chem. 1993;32:977. [Google Scholar]; (b) Grapperhaus CA, Darensbourg MY. Acc Chem Res. 1998;31:451. [Google Scholar]
- 156.(a) Allpress CJ, Grubel K, Szajna-Fuller E, Arif AM, Berreau LM. J Am Chem Soc. 2013;135:659. doi: 10.1021/ja3038189. [DOI] [PubMed] [Google Scholar]; (b) Szajna E, Arif AM, Berreau LM. J Am Chem Soc. 2005;127:17186. doi: 10.1021/ja056346x. [DOI] [PubMed] [Google Scholar]
- 157.Berreau LM, Borowski T, Grubel K, Allpress CJ, Wikstrom JP, Germain ME, Rybak-Akimova EV, Tierney DL. Inorg Chem. 2011;50:1047. doi: 10.1021/ic1017888. [DOI] [PubMed] [Google Scholar]