

Abstract
Membrane potential imaging using voltage-sensitive dyes can be combined with other optical techniques for a variety of applications. Combining voltage imaging with Ca2+ imaging allows correlating membrane potential changes with intracellular Ca2+ signals or with Ca2+ currents. Combining voltage imaging with uncaging techniques allows analyzing electrical signals elicited by photorelease of a particular molecule. This approach is also a useful tool to calibrate the change in fluorescence intensity in terms of membrane potential changes from different sites permitting spatial mapping of electrical activity. Finally, combining voltage imaging with optogenetics, in particular with channelrhodopsin stimulation, opens the gate to novel investigations of brain circuitries by allowing measurements of synaptic signals mediated by specific sets of neurons. Here we describe in detail the methods of membrane potential imaging in combination with other optical techniques and discus some important applications.
Keywords: Calcium imaging, Calcium channels, Photolysis, Signal calibration, Channelrhodopsin
1 Introduction
The advantages of the optical measurements of membrane potential (Vm), over electrode recordings, are the possibility to record voltage changes at high spatial resolution and from a very large number of sites and the ability to probe structures too small to tolerate electrodes. Combining this measurement with another optical measurement or with an optical stimulation at the same spatial resolution and scale allows investigating and understanding the voltage-dependence of a variety of physiological phenomena or the changes of Vm produced by activation of a particular signaling pathway as well as by a well-defined stimulation protocol. In this chapter we describe in detail the main optical techniques that have been so far successfully combined with Vm imaging: calcium (Ca2+) imaging, photorelease of caged compounds and optogenetic photostimulation. At the end of this chapter we discuss the case of Vm imaging combined with channelrhodopsin (ChR) stimulation, which is becoming a fundamental experimental approach in neurophysiology.
Combined optical measurements have been widely used in biology for a long time. In molecular biology, for instance, the concomitant localization of two or more proteins is achieved by using fluorescent antibodies with different absorption and emission spectra. Combined imaging of two different physiological parameters requiring sequences of signal acquisitions was more difficult to achieve until 20 years ago. Thus, before the introduction of more sophisticated imaging systems, combined optical recordings at relatively slow rate and using two indicators have been utilized to measure intracellular pH and Ca2+ (Martinez-Zaguilan et al. 1991) as well as slow Vm changes and Ca2+ signals (Kremer et al. 1992). Combined functional imaging is therefore an experimental approach that evolved in the last two decades in parallel with advancements in imaging technology. The correlation of a Vm recording with a measurement of a Ca2+ signal is perhaps the most important case of dual optical measurement of a functional signal (Canepari et al. 2008). Indeed, crucial information on Ca2+ influx from the extracellular space comes from the fact that Ca2+ signals are directly correlated with Vm when they originate from Ca2+ channels in the plasma membrane. This category includes voltage-gated Ca2+ channels (VGCCs), glutamate receptors (in particular NMDA receptors) and other Ca2+ permeable pores. In all these cases, the charge influx associated with Ca2+ contributes, to a different extent, to the current underlying depolarization. In addition, the biophysical properties of several Ca2+ signals in the plasma membrane also depend on membrane depolarization. For instance, Ca2+ influx via VGCCs is voltage-dependent since depolarization is necessary for channel opening. Similarly, Ca2+ influx via NMDA receptors is voltage-dependent since depolarization is necessary to unblock the channel from Mg2+. In contrast, Ca2+ signals are not directly correlated with Vm when they are due to Ca2+ flux through internal membranes. For example Ca2+ release from internal store is not directly correlated with Vm. In this case, Ca2+ flux does not contribute to the current underlying the Vm change.
Among photostimulations combined to Vm imaging, chemical stimulation delivered by photorelease of a molecule from a caged compound is a powerful tool to explore many physiological phenomena. Notably, uncaging of principal neurotransmitter such as glutamate or GABA, allows investigating how local activation of synaptic receptors produces or modulates Vm in different sub-cellular sites (Vogt et al. 2011b). Vm imaging can be also combined with ChR stimulation expressed in sub-sets of well identified neurons. This application may provide new information on the function of local neuronal circuits (Tsuda et al. 2013).
The techniques of Vm imaging described in this chapter are achieved by recording signals from organic voltage-sensitive dyes. Unless specified otherwise, this type of signal is expressed as a fractional change in fluorescence intensity ΔF/F0 = (F(t) − F(t0))/F(t0), where F(t) is the recorded fluorescence intensity at a given time and F(t0) is the fluorescence intensity at the resting membrane potential (resting fluorescence). Finally, most of the examples in this chapter are recordings from individually-stained single-neurons described in the previous chapter.
2 Principles of Combining Two Optical Techniques
The combination of two optical techniques is typically achieved by using two photosensitive molecules. These molecules can be indicators, for instance fluorescence dyes, or molecules that produce a stimulation of the cell upon light activation. If one of the two molecules is used for photostimulation, for instance a caged compound or a photo-sensitive protein, achieving simultaneous imaging requires that this molecule is not excited by the same wavelength used for excitation of the imaging indicator. This requirement is straightforward to achieve for many caged compounds that are activated exclusively by UV light and that can be combined with indicators excited in the visible range. In contrast, combining imaging techniques with ChR stimulation is more difficult since ChR is activated by light over a broad visible spectrum. If the two molecules are both fluorescent indicators, for instance a voltage-sensitive dye and a Ca2+ indicator, combined imaging can be achieved in three different ways depending on the optical properties of the two indicators. First, the two indicators may have separate absorption spectra but a largely overlapping region in the emission spectra. Second, the two indicators may have a large overlapping region in their absorption spectra but well separated emission spectra. Third, the two indicators may have a separation both in the absorption and in the emission spectra.
Figure 4.1 shows three examples of spectral representations for the different cases and the optical arrangements to combine the recording of the two signals. In the first case (Fig. 4.1a), the fluorescence from the two dyes can be excited using two separate wavelengths and the emitted light from the two indicators is recorded using a single detector. In this case, it is never possible to achieve a simultaneous measurement since the imaging experiment requires alternating between the two different excitation wavelengths. In the second case (Fig. 4.1b), because the excitation spectra are overlapping, fluorescence from both indicators can be excited using the same excitation wavelength. Emitted light can be recorded by two different detectors after splitting the emitted light with a secondary dichroic mirror. Thus, by synchronizing data acquisition of the two detection systems, it is possible to achieve simultaneous imaging of the fluorescence signals from the two indicators. Finally, in the third case (Fig. 4.1c), the combined excitation and detection of the two fluorescence systems can be done using two excitation wavelengths and two detection systems simultaneously.
Fig. 4.1.
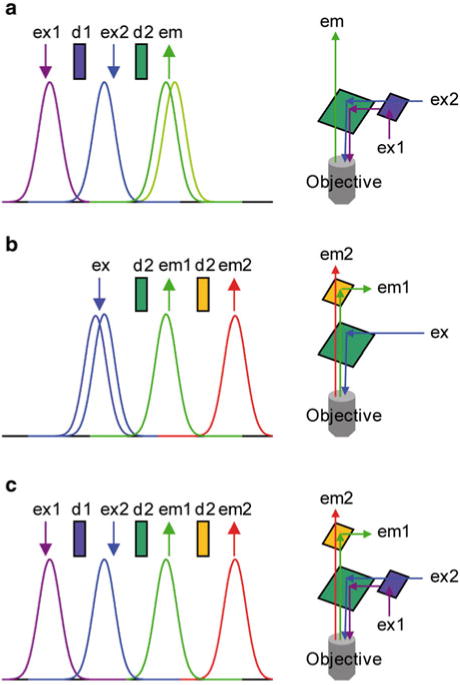
Alternatives for combined imaging using two-indicators. (a) ( Left) Ideal absorption and emission spectra of two indicators with separate absorption spectra and overlapping emission spectra. Arrows indicated by “ex” and “em” are positioned over ideal excitation and emission. Rectangles indicated by “d” are in position of ideal dichroic mirrors. ( Right) Optical arrangements for excitation and recording of fluorescence using either one or two light sources. (b) ( Left) Same as (a) but for two indicators with overlapping absorption spectra and separate emission spectra. ( Right) Optical arrangements for excitation and recording of fluorescence using either one or two light detection systems. (c) ( Left) Same as (a) and (b) but for two indicators with separate absorption and emission spectra. ( Right) Optical arrangements for excitation and recording of fluorescence using either one or two light sources and light detection systems
The optimization of recording signals from two indicators depends on the availability of probes with narrow excitation and emission bands. This is not the case for the available fast voltage sensitive dyes. Most of the styryl voltage dyes are characterized by a broad excitation spectrum in the visible range and a broad emission spectrum in the red-IR region (Fluhler et al. 1985). Figure 4.2 shows the absorption and emission spectra of the hydrophobic voltage-sensitive dye di-8-ANEPPS which are similar to those of di-4-ANEPPS and of the water soluble indicators JPW-1114 (di-2-ANEPEQ) and JPW-3028 (di-1-ANEPEQ) all based on the same chromophore. Similar spectral properties are also shared by many of the commonly used “RH” dyes such as RH-795, RH-237, RH-421 and RH-414. With these dyes, well separated spectra are difficult to obtain using commercially available Ca2+ indicators excited in the visible spectral range. Nevertheless, the combined recordings are still possible due to a large Stokes shift of charge-shift voltage probes (~150 nm). In addition, more efficient combined recordings can be achieved using Ca2+ indicators excited in the UV spectral range.
Fig. 4.2.
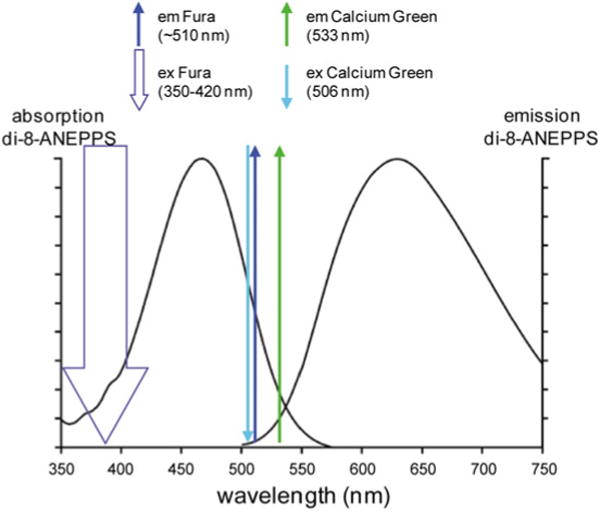
Absorption and emission spectra of di-8 ANEPPS in combination with the ideal absorption and emission of Calcium Green and Fura calcium indicators. Left and right curves are the absorption and emission spectra as reported by Invitrogen—Molecular Probes. The thick downward arrow represents the excitation of the ratiometric indicator Fura. The thin downward arrows represent the excitation peak of Calcium Green. The upward arrows represent the emission peaks of the two indicators
Both voltage dyes and Ca2+ indicators can be loaded into cells either by extracellular bath application of the indicator or by intracellular injection using either a patch-electrode or a sharp microelectrode. For extracellular loading, Ca2+ dyes are commercially available in membrane-permeant AM-ester form. Once in the cell, the acetyl-ester is hydrolyzed by the endogenous esterases and the released free indicator remains in the cytoplasm (Yuste 2000). For intracellular loading, Ca2+ indicators are available as water-soluble potassium salts. When injected into cells from a patch electrode, the dye can quickly equilibrate in the cytosol at a given concentration permitting quantitative estimate of Ca2+ signals (Eilers and Konnerth 2000). In general, intracellular loading of the Ca2+ indicator is done in conjunction with the intracellular loading of the voltage sensitive dye. It is also possible to load the cell with the voltage sensitive dye first and include the Ca2+ indicator in the pipette used for re-patching the labeled cell. In this protocol, Vm imaging is carried out first, immediately after re-patching, during which time the cell is loaded with the Ca2+ indicator from the recording pipette.
3 Combining Vm and Ca2+ Imaging
3.1 Combining Vm and Ca2+ Imaging in Sequential Way
Commonly used voltage sensitive dyes such as di-4-ANEPPS, di-8-ANEPPS and Di-2-ANEPEQ (JPW-1114) have wide excitation spectrum in the blue/green region and wide emission spectrum in the red/IR region (Fig. 4.2). If combined with Fluo, Calcium Green or Oregon Green Ca2+ indicators, both dyes can be excited by a blue wavelength (which, however, is not optimal for the voltage-sensitive dye) and green (Ca2+) and red (Vm) fluorescence can be recorded separately. Voltage sensitive dye excitation is optimal at 532 nm (Canepari et al. 2010). Thus, if combined with UV-excitable Fura indicators, the voltage-sensitive dye can be excited at its optimal visible wavelength and, as with the previous combination, the two emission signals can be recorded separately.
Although both types of combinations allow simultaneous measurement of Vm and Ca2+ fluorescence, many studies were initially performed through sequential recordings of the two optical signals. Combined Vm and Ca2+ imaging from individual neurons using blue-excitable/green-emitting Ca2+ indicators has been also used in several other studies. In the barrel cortex, the high-affinity indicator Oregon Green BAPTA-1 was combined with the voltage sensitive dye JPW-1114 in recordings from individual neurons and with the voltage indicator RH-1691 in in vitro and in vivo network recordings (Berger et al. 2007). In prefrontal cortical neurons, dendritic recordings were obtained combining JPW-1114 either with the high-affinity Ca2+ indicator Calcium Green-1 or with the low-affinity Ca2+ indicator Fluo-5F (Milojkovic et al. 2007).
The use of UV-excitable Fura indicators minimizes the spectral overlap with the voltage-sensitive dye. Additionally, the voltage-sensitive dye is not excited during Ca2+-imaging preventing a substantial photodynamic damage that can occur during relatively long recording periods (Canepari et al. 2008). UV-excitable Fura indicators with different equilibrium constants (Kd) are commercially available (see Table 4.1). This permits the choice of the most suitable indicator for the measurement of either the Ca2+ influx (high-affinity indicator) or the change in intracellular free Ca2+ concentration (low-affinity indicator).
Table 4.1.
Dissociation constants and buffering capacities of Fura dyes
| Dye | Kd (μM) | Kdye(300 μM) | Kdye(1 mM) |
|---|---|---|---|
| Fura-2 | 0.224a | ~1,300 | ~4,500 |
| Fura-5F | 0.4a | ~750 | ~2,500 |
| Bis-fura-2 | 0.525a | ~570 | ~1,900 |
| Fura-4F | 0.77a | ~390 | ~1,300 |
| Fura-6F | 5.3a | ~57 | ~190 |
| Fura-FF | 10b | ~30 | ~100 |
| Mag-fura-2 | 25c–40d | ~7.5–12 | ~25–40 |
A list of commercially available Fura dyes with dissociation constant (Kd) and buffering capacities (Kdye) at the two concentrations of 300 μM and 1 mM
Invitrogen—Molecular Probes handbook (for fura-2 and bis-fura-2 at 1 mM Mg2+)
In contrast to the indicators excited in the visible range, Fura indicators allow ratiometric measurements. When excited above their isosbestic wavelength (~360 nm), the changes of fluorescence associated with an increase in Ca2+ concentration are negative. Imaging above the isosbestic wavelength has several advantages. First, the resting fluorescence, which corresponds to nominally 0 Ca2+, is significantly higher than with dyes that increase their fluorescence with Ca2+. Second, the achievable imaging contrast is high because healthy cells are characterized by very low resting Ca2+, and the dye in adjacent structures, exposed to millimolar Ca2+ concentrations, is essentially not fluorescent. Third, since the dynamic range of the fluorescence is ~1 after subtraction of fluorescence at saturating Ca2+, these measurements allow a better quantitative estimation of the Ca2+ signals. An example of Vm and Ca2+ optical signals recorded sequentially from the two indicators in three different regions on the dendritic tree of a Purkinje neuron stained by JPW-1114 and Fura-FF is illustrated in Fig. 4.3. Using an excitation band of 387 ± 5 nm for Fura-FF (Fig. 4.3a), the Ca2+ increase associated with a climbing fibre excitatory postsynaptic potential (EPSP) corresponds to a negative ΔF/F0 (Fig. 4.3b). Sequential Vm and Ca2+ recording using Fura Ca2+ indicators has been done in CA1 hippocampal pyramidal neurons (Canepari et al. 2007), in prefrontal cortex layer-5 pyramidal neurons (Milojkovic et al. 2007) and in cerebellar Purkinje neurons (Canepari and Vogt 2008). Sequential recording (as well as signal averaging) is meaningful only if repeated application of the same stimulation protocol results in the same response. This requirement must be confirmed experimentally by comparing individual recordings as shown in Fig. 4.3c. In this example, the Vm and the Ca2+ optical signals were recorded from a dendritic location on a cerebellar Purkinje neuron in response to four repetitions of climbing fibre activation separated by 1 min. The results showed that signals were practically identical in four individual trials (gray traces) permitting the correlation of Vm and Ca2+ optical signals recorded sequentially in response to the same stimulus.
Fig. 4.3.
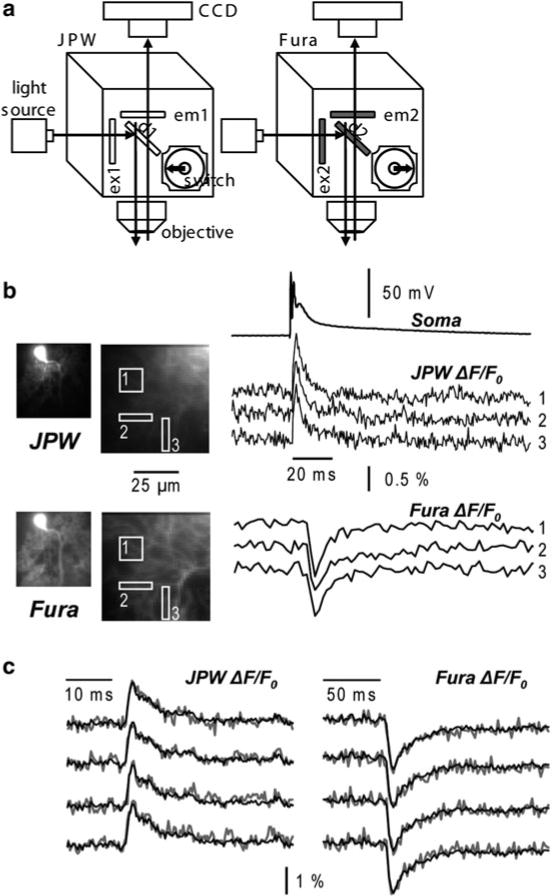
Combining Vm and Ca2+ imaging using JPW-1114 and a UV excitable Ca2+ indicator. (a) Schematic of the imaging apparatus for sequential Vm and Ca2+ imaging using a Fura indicator. Filters for voltage-sensitive dye (JPW): ex1 = 525 ± 25 nm, d1 > 570 nm and em1 > 610 nm. Filters for Ca2+ indicator (Fura): ex2 = 387 ± 6 nm, d1 > 470 nm and em1 = 510 ± 42 nm. (b) Vm and Ca2+ fractional changes of fluorescence from cerebellar Purkinje neuron dendrites related to a climbing fibre EPSP recorded from the locations 1–3 reported in the fluorescence images on the left. Somatic recording of the climbing fibre EPSP is the upper trace. (c) Individual Vm ( left) and Ca2+ ( right) fractional changes of fluorescence ( gray traces) related to a climbing fibre EPSP. Superimposed black traces are the averages of the four trials
An important aspect of combined Vm and Ca2+ imaging is the interpretation of Ca2+ optical signals, which depends on how much the Ca2+ indicator perturbs the physiological Ca2+ homeostasis. The buffering capacity of a Ca2+ indicator, Kdye, defined as the ratio between the dye-bound Ca2+ and the free Ca2+ in the presence of the indicator depends on the dissociation constant and on the concentration of the indicator (Table 4.1). The perturbation of the physiological Ca2+ introduced by the Ca2+ indicator can be evaluated by comparing the parameter Kdye with the endogenous buffering capacity of the cell (Kcell). The interpretation of Ca2+ optical signals is simplified when Kdye is either much larger or much smaller than Kcell, i.e. when most of Ca2+ binds to the indicator or when the fraction of Ca2+ bound to the indicator is negligible compared to the total Ca2+ (Canepari et al. 2008). Without addressing in detail the issue of calibration of Ca2+ optical signals (Neher 2000) in this volume focused on Vm imaging, we will note that, in the first case, the ΔF/F0 signal associated with Ca2+ increase is approximately linear with the total intracellular Ca2+ signal. In the second case, the time course of the ratio (F − Fmin)/(Fmax − F), where Fmin and Fmax are the fluorescence intensities at 0 and saturating Ca2+ respectively, is linear with the physiological intracellular free Ca2+ concentration change (Δ[Ca2+]i). In many instances, Ca2+ signals are due to Ca2+ influx through a Ca2+ channel in the plasma membrane. The contribution to the change in membrane potential due to the Ca2+ influx depends on the Ca2+ permeability of the channel relative to its permeability to other ions, in particular to Na+ and K+. This is different for VGCCs, AMPA receptors, NMDA receptors and other Ca2+ permeable pores such as transient receptor potential (TRP) channels. Vice-versa, because opening or unblocking of Ca2+ channels often depends, in a non-linear manner, on the local membrane potential, the Ca2+ influx is often, but not always, larger when the voltage related depolarizing optical signal is larger (Canepari et al. 2007). Because this bi-directional relationship is complex, the analysis of Vm and Ca2+ signals from multiple sites on individual neurons is not straightforward and requires careful interpretation.
A notable application of combined Vm and Ca2+ imaging is the study of synaptic plasticity where the supra-linear Ca2+ signal plays an important role. The supra-linear Ca2+ signal occurs when two stimulation protocols are combined to evoke the coincident activity (pairing protocol) which results in a Ca2+ signal that is larger than the sum of the two Ca2+ signals associated with the application of individual stimulation protocols. A supra-linear Ca2+ signal is always caused by a supra-linear Δ[Ca2+]i, but not necessarily by a supra-linear Ca2+ influx through the plasma membrane. Indeed, the supra-linear Ca2+ signal might be caused by Ca2+ release from internal stores or by the saturation of the endogenous Ca2+ buffer. However, in the latter case, if the buffering capacity of the dye dominates over the endogenous buffering capacity of the cell and the dye is not saturated, the presence of the indicator will cancel the supra-linear Δ[Ca2+]i and no Ca2+ dependent ΔF/F0 will be observed. Thus, in these conditions, detection of supra-linear Ca2+ dependent ΔF/F0 signals which are not due to Ca2+ release from internal stores always corresponds to a supra-linear Ca2+ influx and it must always correlate with an increase in the depolarizing Vm signal. In contrast, if the buffering of the dye is negligible compared to the endogenous buffering of the cell, a supra-linear Δ[Ca2+]i signal that is due to the saturation of the endogenous Ca2+ buffer can be measured reliably. The two different types of supra-linear Ca2+ signals not involving Ca2+ release from stores are illustrated by the two following examples.
In measurements from CA1 hippocampal pyramidal neurons (Canepari et al. 2007), cells were loaded with the voltage-sensitive dye JPW-3028 and 300 μM Bis-Fura-2. The purpose of these experiments was to analyze, over large regions of the dendritic tree, the Vm and Ca2+ optical signals associated with back-propagating action potentials, with excitatory postsynaptic potentials (EPSPs) and with pairing of these two membrane potential transients using an LTP induction protocol. Pyramidal neurons in the CA1 region are characterized by relatively low endogenous buffering capacity in the dendrites (~100) and even lower buffering capacity (~20) in the spines (Sabatini et al. 2002). Thus, the buffering capacity of 300 μM Bis-Fura-2 (Kd ~500 in the presence of Mg2+, see Table 4.1) is ~6 times larger than the buffering capacity of the dendrite and ~30 times larger than the buffering capacity of the spine. Ca2+ signals associated either with back-propagating action potentials or with EPSPs were mediated by Ca2+ influx through voltage-gated Ca2+ channels and/or through NMDA receptors. As shown in Fig. 4.4a, the pairing of the two stimulating protocols elicited a supra-linear Ca2+ signal. In the presence of the NMDA receptor blocker AP-5, since the measurements were done using a non-saturating concentration of a high-affinity indicator, the supra-linear Ca2+ signal must have been caused by supra-linear Ca2+ influx mediated by recruitment of additional VGCCs. In agreement with this expectation, at each site where a supra-linear Ca2+ signal was observed, the Vm related optical signal during the pairing protocol had a larger peak depolarization compared to the signals associated with unpaired stimulations (Fig. 4.4a).
Fig. 4.4.
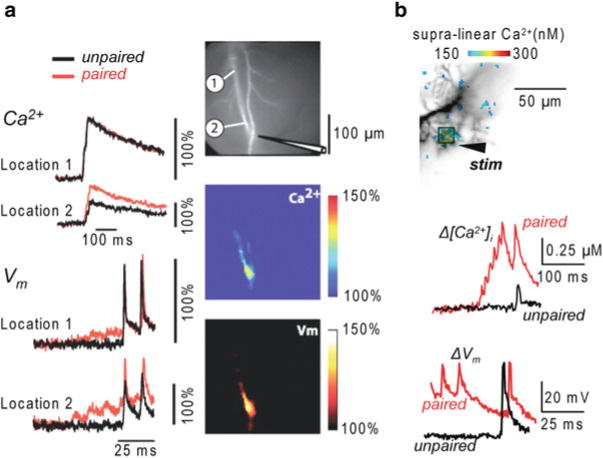
Combining Vm and Ca2+ imaging using high- and low-affinity Fura indicators. (a) ( Left) Ca2+ and Vm fractional changes of fluorescence corresponding to back-propagating action potentials (bAPs) and paired EPSP–bAP signals from two locations on the dendritic tree of a CA1 hippocampal pyramidal neuron. Signals are superimposed to reveal the region-specific increase in peak depolarization during paired activity. ( Right) Fluorescence image of the dendritic arbor of three hippocampal pyramidal neurons. The stimulating electrode is indicated schematically. ( Middle and bottom panels) Color-coded spatial maps of the Ca2+ signal and of the Vm signal during paired activity respectively; signals were scaled using the signals in unpaired conditions. Experiments are in the presence of the NMDA receptor blocker AP-5. Supra-linear Ca2+ correlated with larger depolarization. Reproduced from Canepari et al. 2007 with the permission of Wiley-Blackwell. (b) ( Top) Recorded dendrites of a cerebellar Purkinje neuron; supra-linear Δ[Ca2+]i from the difference between Δ[Ca2+]i associated with the pairing protocol and Δ[Ca2+]i associated with the unpaired PF-EPSPs and CF-EPSP in color-coded scale. ( Bottom) Δ[Ca2+]i and ΔVm from the region square region depicted above associated with one unpaired ( black traces) and one paired CF-EPSP. Supra-linear Ca2+ is not associated with larger depolarization. Modified from Canepari and Vogt 2008
In measurements from cerebellar Purkinje neurons (Canepari and Vogt 2008), cells were loaded with 1 mM of the Ca2+ indicator Fura-FF and with the voltage sensitive dye JPW-1114. Purkinje neurons have an exceptionally high dendritic Kcell estimated at ~2,000 (Fierro and Llano 1996). Therefore, the addition of a low affinity Ca2+ indicator such as Fura-FF (Kd ~10), even at millimolar concentrations, does not significantly alter the physiological homeostasis and it is possible to record changes in free intracellular Ca2+ concentration (Δ[Ca2+]i signals Canepari et al. 2008). In this particular preparation, it was possible to calibrate Vm signals in terms of membrane potential. As shown in Fig. 4.4b, pairing the stimulation of the large climbing fibre synaptic potential with local parallel fibres stimulation generated a local dendritic supra-linear Δ[Ca2+]i signal which was independent of Ca2+ release from stores (Brenowitz and Regehr 2005). Although the Δ[Ca2+]i signal originated from Ca2+ influx through VGCCs, in Purkinje neurons, in contrast to CA1 pyramidal neurons, the Vm signal during the pairing protocol did not have larger peak depolarization compared to the Vm signals associated with unpaired stimulations (Fig. 4.4b). In this case the Ca2+ influx associated with the climbing fibre synaptic potential was the same in paired and unpaired conditions. The corresponding Δ[Ca2+]i signal, however, was larger following paired stimulation because the Ca2+ influx associated with the parallel fibre stimulation locally and transiently saturated the endogenous Ca2+ buffer (Canepari and Vogt 2008). This supra-linear Ca2+ signal could be correctly interpreted only when recorded using a low-affinity indicator. When the cell was injected with 10 mM Bis-Fura-2, Kdye became ~10 times larger than Kcell and supra-linear Ca2+ signals were abolished (Canepari and Vogt 2008).
3.2 Simultaneous Vm and Ca2+ Imaging
Sequential Vm and Ca2+ imaging produce valid results if identical responses are evoked in repetitive measurements. Many physiological processes, however, are stochastic, showing large trial-to-trial variability. In these cases, the study of the time variability contains important biological information and simultaneous Vm and Ca2+ optical measurement is required. Simultaneous Vm and Ca2+ imaging was initially achieved using the Ca2+ indicator Calcium Orange, which has excitation peak at ~550 nm and emission peak at ~580 nm (Sinha et al. 1995; Sinha and Saggau 1999), or with blue-excitable indicators (Bullen et al. 1997; Bullen and Saggau 1998). These possibilities may have, however, some inherent problems. First, the small overlap between the emission spectra of the two dyes may produce a “bleeding” of the Vm signal into the Ca2+ recording channel that contaminates the signal. In some situations, such emission cross talk can be mathematically corrected (Sinha et al. 1995). Second, while Vm recordings of synaptic or action potentials can be often achieved within short time, the complete time-course of the corresponding Ca2+ signal may require longer acquisition intervals (Canepari and Vogt 2008). In this case, the voltage sensitive dye is exposed to high intensity light for a longer time than what would be required for the Vm measurement alone, increasing both bleaching of the dye and phototoxicity. Simultaneous Vm and Ca2+ imaging was also performed using the combination of an absorption voltage sensitive dye with a fluorescent Ca2+ indicator. Using bath application of the absorption dye RH-482 and a low-affinity Ca2+ indicator Magnesium Green, Sabatini and Regehr could correlate the Ca2+ signal underlying synaptic release with the excitation of pre-synaptic terminals in cerebellar synapses (Sabatini and Regehr 1996; Sabatini and Regehr 1997). In these experiments, simultaneous Ca2+ and Vm measurements were done by using excitation light wavelength of ~500 nm and recording simultaneously fluorescence from the Ca2+ indicator Magnesium Green and the transmitted light from voltage-sensitive dye RH-482 at 710 nm from a single location with two photodiodes. The important limitation of this approach is that the absorption measurements from multiple sites on individual neurons are substantially less sensitive compared to fluorescence measurements (Antic and Zecevic 1995). In the above example, extensive temporal and spatial signal averaging over a large region of the cerebellar slice, which could be obtained with absorption signals, permitted signal detection from presynaptic terminals (Sabatini and Regehr 1997).
The above mentioned problems of simultaneous Vm and Ca2+ imaging using the same excitation wavelength can be overcome by using a Fura indicator. The realization of this measurement, however, required the design of a dedicated apparatus (Vogt et al. 2011a) to optimize the signal-to-noise ratio (S/N). In principle, di-8-ANEPPS and JPW-1114 could be excited by blue light in the configuration shown in Fig. 4.1c. However, narrow-band laser excitation at 532 nm or at 543 nm is associated with much larger sensitivity (Canepari et al. 2010) permitting single-trial recordings. These excitation wavelengths are longer than the emission peak of Fura indicators. To allow transmission of Vm and Ca2+ emission wavelengths, while reflecting intermediate excitation wavelength (543 nm), the apparatus included a dual-band pass dichroic mirror as shown in the schematic in Fig. 4.5a. Multiple wavelengths dichroic mirrors are commercially available from several companies (e. g. Semrock). The mirror that was used in this case reflected 543 nm and transmitted lower green and red wavelengths corresponding to the emissions of Fura and JPW1114 indicators respectively. This optimal configuration allowed resolving dendritic excitation and associated Ca2+ signals from relatively small regions in single trial measurements. In the example shown in Fig. 4.5b, simultaneous ΔVm and Δ[Ca2+]i signals, calibrated as described in Canepari and Vogt (2008), associated with one climbing fibre-EPSP and a train of five parallel fibre EPSPs at 100 Hz were compared in two small dendritic regions. ΔVm and Δ[Ca2+]i signals associated with large depolarization could be observed in both regions for the climbing fibre EPSP but only near the parallel fibre stimulating electrode, for the parallel fibre EPSPs train. These measurements, where signals of ~1 % change of fluorescence could be resolved from regions of few microns, represent the current state-of-the-art in sensitivity of simultaneous Vm and Ca2+ imaging from single cells.
Fig. 4.5.
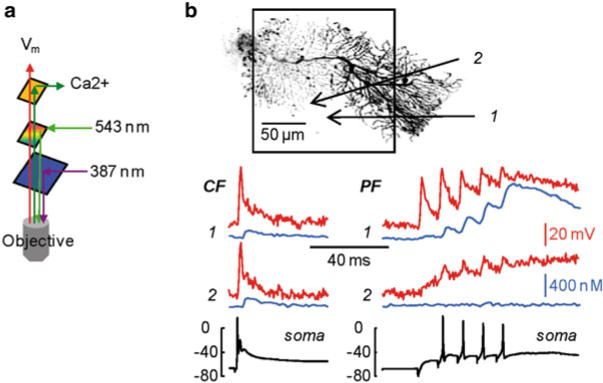
Simultaneous Vm and Ca2+ imaging. (a) Schematic of the optical configuration to achieve simultaneous Vm and Ca2+ imaging; 543 and 387 nm are used for voltage sensitive dye and Fura indicator excitations respectively; low green and red emissions of the two indicators are separated and detected with two cameras. (b) ( Top) Reconstruction of a cerebellar Purkinje neuron with the dendritic area in recording position outlined; two sites of interest ( 1 and 2) are indicated by the arrows. ( Bottom) ΔVm ( red traces) and Δ[Ca2+]i ( blue traces) signals associated with a climbing EPSP ( left) or with a train of five parallel fibre EPSPs at 100 Hz ( right) from the two sites of interest; somatic electrical recordings shown below. Modified from Vogt et al. 2011a
3.3 Simultaneous Optical Measurement of Vm and Ca2+ Currents
Simultaneous Vm and Ca2+ imaging can be also used for the study of native fast Ca2+ currents based on derivation of Ca-currents from optical signals., We have already stated that the ΔF/F0 Ca2+ signal is proportional to the change in concentration of Ca2+ ions bound to the indicator. If, in addition, the Ca2+ bound to the indicator is also proportional to the total free Ca2+ flux entering the cell through the plasma membrane, then the Ca2+ current can be estimated by the derivative of ΔF/F0 (Sabatini and Regehr 1998). This estimate is possible with low-affinity indicators (Kao and Tsien 1988) where the equilibration of the dye-Ca2+ binding reaction is faster than the Ca2+ influx. It was recently demonstrated that the reaction between Ca2+ and the low-affinity indicator Oregon Green 488 BAPTA-5N is faster than a typical VGCC-mediated current, i.e. the ΔF/F0 signal from this indicator matches the kinetics of the Ca2+ current integral (Jaafari et al. 2014). It follows that the kinetics of the ΔF/F0 derivative is proportional to the kinetics of the Ca2+ current and can be calibrated in terms of “Ca2+ current volume density”. The simultaneous Vm optical measurement allows studying the voltage dependence of the current, i.e. the membrane potential of activation and deactivation of the channel. Technically, simultaneous Vm and Ca2+ imaging using Oregon Green 488 BAPTA-5N requires blue illumination for both indicators (see previous paragraph) and high-speed acquisitions (20 kHz frame rate) to investigate the time-course of the Ca2+ current at high temporal resolution. Figure 4.6a shows an example of a CA1 hippocampal pyramidal neuron filled with 1 mM of Oregon Green 488 BAPTA-5N and the voltage sensitive dye JPW1114. Figure 4.6b shows the Ca2+ current volume density (ICa/V) associated with an action potential monitored optically in the proximal apical dendritic segment of a CA1 hippocampal pyramidal neuron. The ICa/V and Vm signals were measured under two conditions: for an action potential starting at Vm = −60mV and for anaction potential starting at Vm = −80mV (Fig. 4.6b). When the action potential started at Vm = −80 mV, ICa/V was larger and started at a more hyperpolarized Vm (Fig. 4.6c), indicating that at Vm = −80 mV the Ca2+ current contained a component mediated by low-voltage activated (T-type) Ca2+ channels (Cueni et al. 2009). The example shown in Fig. 4.6 illustrates how simultaneous optical measurement of Vm and Ca2+ currents can drastically improve our understanding of the physiological function of Ca2+ channels by permitting the exploration of local biophysics of native channels during physiological activity.
Fig. 4.6.
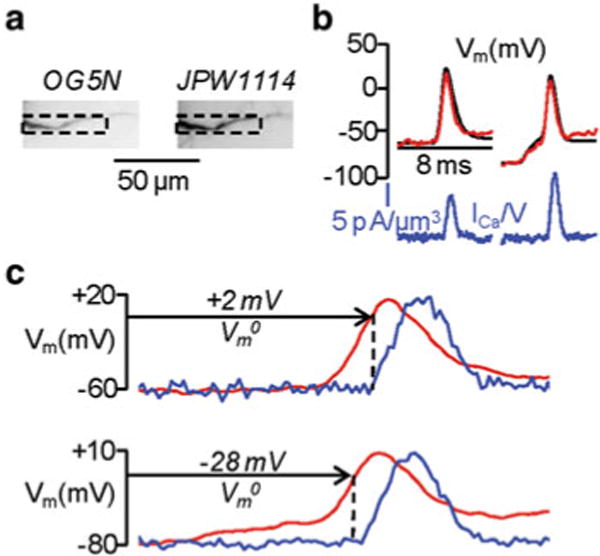
Dendritic Ca2+ currents associated with an action potential at different starting Vm· (a) Fluorescence images of a CA1 hippocampal pyramidal neuron filled with Oregon Green 488 BAPTA-5 N ( left image) and the voltage sensitive dye JPW1114 ( right image); the region of interest from where fluorescence was averaged is outlined. (b) Ca2+ currents ( blue) associated with one action potential starting either at Vm = −60 or −80 mV; Vm recorded optically ( red) and with the electrode ( black) superimposed. (c) Ca2+ currents normalized and superimposed to the action potential in the two cases; Vm at the current onset (Vm0) indicated. Modified from Jaafari et al. 2014
4 Combining Vm Imaging with Photostimulation
4.1 Combining Vm Imaging with Glutamate or GABA Photorelease
Membrane potential imaging can be combined with uncaging techniques to investigate how the photorelease of biologically active molecules changes the membrane potential in different regions of a cell. In particular, the caged compounds 4-Methoxy-7-nitroindolinyl-caged-L-glutamate (MNI-glutamate, Canepari et al. 2001) and 1-(4-Aminobutanoyl)-4-[1,3-bis(dihydroxyphosphoryloxy)propan-2-yloxy]-7-nitroindoline (DPNI-GABA, Trigo et al. 2009) can be used to obtain fast photorelease of either L-glutamate or GABA in sub-cellular regions of variable size and to investigate the regulation of membrane potential. An example of this methodological approach is the study of shunting inhibition (Mann and Paulsen 2007). Propagation of action potentials in the axon or back-propagation of action potentials in the dendrites can be strongly regulated by local changes of membrane conductance due to opening of GABAA receptors. Simultaneous Vm imaging and GABA photorelease is an ideal approach to investigate shunting inhibition. In the CA1 hippocampal pyramidal neuron shown in Fig. 4.7a, the effect of local GABA photorelease on the back-propagating action potential in the apical dendrite was explored. DPNI-GABA was applied using a patch pipette positioned close to the distal part of the dendrite at ~200 μm from the soma. The fluorescence change associated with a back-propagating action potential under control conditions (Fig. 4.7b, left traces) was compared with a back-propagating action potential occurring 15 ms after an episode of GABA photorelease (Fig. 4.7b, right traces) from four regions of interest. The amplitude of the somatic action potential was the same in both cases, but local GABA uncaging decreased the amplitude of the fluorescence change associated with the back-propagating action potential progressively with the distance from the soma. Although shunting inhibition induced by GABA uncaging is produced by non-physiological co-activation of synaptic and extra-synaptic GABA receptors, this experiment represents a useful model of this phenomenon since it permits a clear separation of presynaptic and postsynaptic effects, a clear localization of the activated GABA receptors and the possibility to perform pharmacological studies that cannot be done using presynaptic stimulation. Thus, combining membrane potential imaging with GABA photorelease shows the spatial restriction of the shunting effect and the resulting rapid loss in action potential backpropagation.
Fig. 4.7.
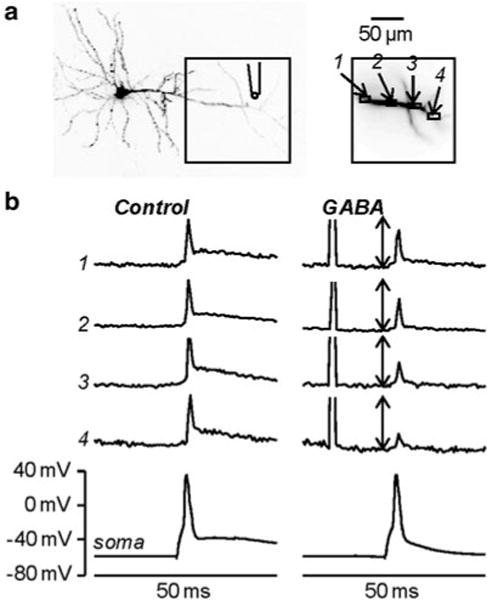
Investigating shunting inhibition using combined Vm imaging and local GABA photorelease. (a) Image of a CA1 hippocampal pyramidal neuron; the dendritic area in recording position is outlined and four regions of interest are shown on the right; the position of the pipette used for caged-glutamate application is illustrated. (b) Vm optical signals from the regions 1–4 and somatic recordings associated with one back-propagating action potential elicited by somatic current injection in control condition ( left) and 15 ms after an episode of 1 ms GABA photorelease ( right). The optical signals are normalized to the peaks of the spike in control condition; the length of the double-arrow corresponds to the peak. Modified from Vogt et al. 2011b
4.2 Using Glutamate Uncaging to Calibrate Voltage-Sensitive Dyes Signals
The calibration of Vm optical signals on an absolute scale (in mV) provides direct measurements of Vm signal amplitudes from multiple locations. This information can be used to construct and analyze the dynamic spatial maps of signal amplitudes throughout neuronal processes. The temporally and spatially well resolved maps of activity are one of the key elements in understanding the rules governing dendritic signal processing and plasticity. The absolute calibration of optical Vm signals, however, is not straightforward and often the exact calibration is not possible. Voltage-sensitive dye recording of membrane potential transients belongs to a class of indirect measurements. The quantity that is being measured directly, by photodetectors, is light intensity, and the quantity that needs to be monitored is derived from a known relationship between the light intensity and Vm. It is convenient that the relationship between light intensity and Vm is strictly linear over the entire physiological range for many voltage sensitive dyes (e.g. Gupta et al. 1981; Loew and Simpson 1981; Wu and Cohen 1993). In a multisite recording, the fractional change in light intensity is proportional to Vm, but also, to a different extent at different sites, to an additional factor: the ratio of inactive dye to active dye. The inactive dye is bound to connective tissue or any other membranes that do not change potential while the active dye is bound to the excitable membrane being monitored. The inactive dye contributes to the resting fluorescence only, and the light from active dye contributes to the resting fluorescence and also carries the signal. In experiments utilizing intracellular application of the dye, inactive dye would be bound to intracellular membranes and organelles. It is a general rule that the ratio of active dye to inactive dye is different for different regions of the object and is also unknown. It follows that the sensitivity of recording from different regions of the object will be different and the calibration of all detectors cannot be done by calibrating the optical signal from any single site. In this situation, the calibration is absolute and straightforward if a calibrating electrical signal that has known amplitude at all locations is available. An all-or-none action potential signal is ideal for this purpose and can be used to create a sensitivity profile of the measuring system. This type of calibration was used to scale the amplitudes of sub-threshold signals in mitral cells of the olfactory bulb (Djurisic et al. 2004). If action potentials are not available, another type of calibrating electrical signal can be a slow electrical change spreading with minimal attenuation over relatively long distances. Such condition occurs, for instance, in cerebellar Purkinje neurons (Roth and Häusser 2001; Canepari et al. 2008).
The possibility to achieve uniform depolarization over the entire dendritic tree using L-glutamate photolysis may extend the possibility to calibrate fluorescence signals to other cell types. This calibration procedure is based on the principle that if the ionotropic glutamate receptor becomes the dominant conductance in a particular neuronal compartment, its reversal potential will determine the membrane potential of the compartment. Thus, if dominance of glutamate receptor conductance is obtained by L-glutamate uncaging, the resulting Vm change in different compartments of the neuron will be the same and can be used to calibrate voltage-sensitive dye imaging data. The idea of a calibration protocol based on L-glutamate uncaging relies on saturating depolarization in the visualized area (Vogt et al. 2011b). The fractional change of fluorescence will then correspond to the resting membrane potential in each visualized site where photolysis occurs.
In the CA1 hippocampal pyramidal neuron of Fig. 4.8a, one action potential was evoked by somatic current injection. The fractional changes of fluorescence associated with the back-propagating action potential from four ROIs on the apical dendrites are shown in Fig. 4.8b (left traces). The amplitude of the signal is variable and could not be directly correlated with the absolute change of membrane potential. After addition of 1 μM TTX, to block action potentials, and of 50 μM CTZ, to prolong depolarization by blocking AMPA receptor desensitization, L-glutamate was photoreleased over the whole field of view. The recording was performed starting from the resting Vm of the cell. The size of the fluorescence change at each site and the absolute somatic depolarization elicited by uncaging increased with the amount of released L-glutamate, i.e. with the exposure time of the UV light, eventually reaching saturation. The saturating Vm corresponded to the reversal potential of glutamate receptors. Using this information, the amplitude of the back-propagating action potential signals was converted into the change in membrane potential. This method of calibration of voltage-sensitive dyes signals, based on L-glutamate photorelease, can only be applied to cell types characterized by dominant glutamate conductance.
Fig. 4.8.
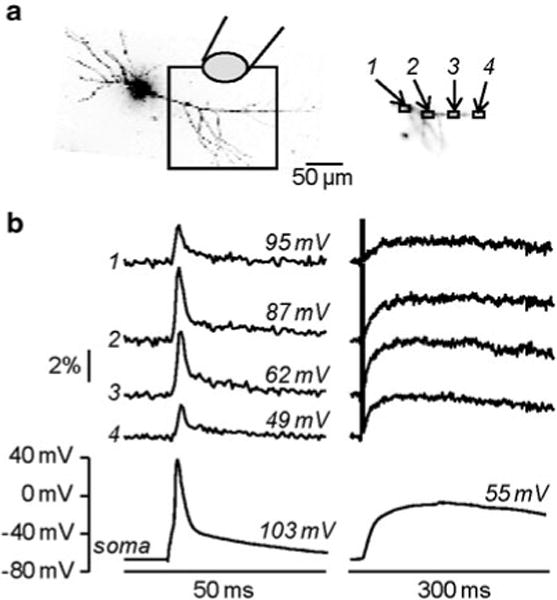
Calibration of fractional changes of fluorescence in terms of membrane potential using L-glutamate (a) Image of a CA1 hippocampal pyramidal neuron; the dendritic area in recording position is outlined and four regions of interest are shown on the right; caged-glutamate is applied over the visualized area of the dendrite and wide-field illumination was used for photolysis. (b) ( Left) Vm optical signals from the regions of interest 1–4 and somatic recordings associated with one back-propagating action potential. ( Right) After addition of 1 μM TTX and 50 μM CTZ, Vm optical signals from the same regions and somatic recordings associated with 1 ms L-glutamate photo-release. The peaks of the somatic signal are indicated over their respective traces; the sizes of the estimated peak spikes in the regions of interest are indicated over their respective traces. Modified from Vogt et al. 2011b
4.3 Combining Vm Imaging with ChR Stimulation
For control of neuronal activity, optogenetic approaches based on light-sensitive channels and other photo-reactive molecules enable highly selective control of genetically-defined populations of neurons (Mancuso et al. 2011; Yizhar et al. 2011). Such techniques offer high time resolution and high spatial precision, allowing stimulation of either individual neurons or large neuronal populations. A commonly used protein for this application is Channelrhodopsin2 (ChR2, Zhang et al. 2006). Combination of optical stimulation using ChR2, with membrane potential imaging is in principle a potent approach to investigate electrical activity both at cellular and at network level. The accomplishment of this type of experiment, however, requires voltage-sensitive dyes that are excitable by red wavelengths since activation of ChR2 occurs at visible wavelengths < 600 nm. Indicators with these characteristics have been developed by Rina Hildensheim and Amiram Grinvald to investigate electrical activity from the entire cortex in vivo without optical interference of hemoglobin absorption (Grinvald and Hildensheim 2004). More recently, red or near-IR excitable or fluorescent indicators have been developed by Joseph Wuskell, Ping Yan and Leslie Loew to monitor electrical activity in the heart (Matiukas et al. 2005) and in the brain (Wuskell et al. 2006) or to be injected in individual cells (Zhou et al. 2007; Yan et al. 2012). Among these new voltage sensitive dyes, five have been specifically tested to be used in combination with ChR2 stimulation (Kee et al. 2008). In the example shown in Fig. 4.9a (modified from Tsuda et al. 2013), ChR2 was selectively expressed in interneurons located in the molecular layer, providing inhibitory post-synaptic potentials (IPSPs) to cerebellar Purkinje neurons when the light-sensitive channel was excited. In this experiment, the cerebellar slice was stained with the voltage sensitive dye Di-2-ANBDQPQ (Zhou et al. 2007) and excited at 658 nm, beyond the excitation spectrum of ChR2 (Kee et al. 2008). Transient photo-stimulation of the molecular layer with blue light produced an IPSP in the nearby Purkinje neurons. As shown in Fig. 4.9a, the IPSP elicited by photo-stimulation of the molecular layer could be measured optically. Notably, the kinetics of the optical signal was identical to that of the IPSP measured with a patch electrode. In another study, ChR2 was selectively expressed in GABAergic interneurons of the hippocampal stratum lacunosum moleculare and the regulation of electrical activity in different areas was characterized by staining the slice with the indicator DI-4-ANBDQPQ (Kee et al. 2008) and by performing Vm imaging (Leão et al. 2012). Thus, using this strategy, it was possible to determine that interneurons from the stratum lacunosum molecular differentially modulate CA3 and entorhinal inputs to hippocampal CA1 neurons.
Fig. 4.9.
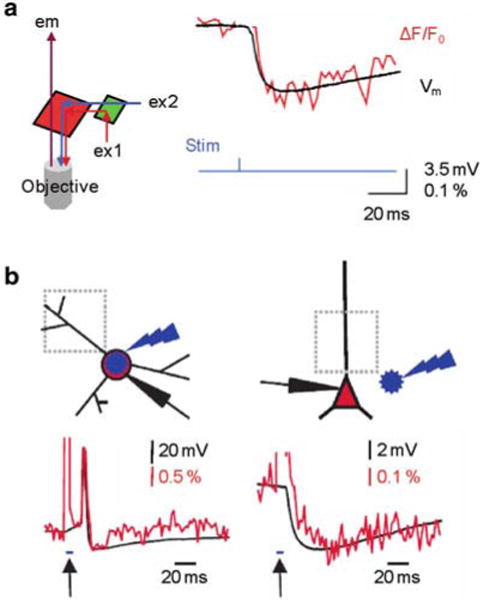
Combining Vm imaging with ChR2 stimulation. (a) ( Left) Schematic of the optical configuration to achieve this combination; red and blue illumination are used for voltage sensitive dye and ChR2 excitations respectively; IR fluorescence emission is detected. ( Right) IPSP recorded electrically (with a patch electrode) and optically from a the soma of a Purkinje neuron following ChR2 excitation at interneurons located in the molecular layer; modified from Tsuda et al. 2013 (b) Imaging of light evoked action potential in a ChR2 expressing interneuron ( left) and of IPSP in CA1 pyramidal neuron induced by ChR stimulation at interneurons ( right). ( Top) Schematic illustrations of the experiment on the top with site of blue flash and imaging region. ( Bottom) Simultaneous optical dendritic and electrical somatic recordings; arrows indicate blue light application (5 ms); modified from Willadt et al. 2014
Using water soluble indicators excitable with red light (Zhou et al. 2007; Yan et al. 2012), electrical activity can be imaged in combination with ChR2 photostimulation from individually stained cells (Willadt et al. 2014). The clear advantage of this approach is that the higher sensitivity of Vm imaging in individually stained neurons permits optical measurements of smaller membrane potential changes from well identified sub-cellular compartments. In the examples shown in Fig. 4.9b, ChR2 was expressed by all GABAergic interneurons and individual hippocampal cells were filled with the indicator Di-2-ANBDQPTEA. When electrical and optical recordings were performed from a GABAergic interneuron, ChR2 photo-stimulation of the recorded cell generated an action potential that could be monitored both electrically and optically from proximal dendritic segment (Fig. 4.9b, left). In contrast, when electrical and optical recordings were performed from a CA1 pyramidal neuron, ChR2 photo-stimulation of pre-synaptic interneurons generated an IPSP that was recorded both electrically and optically from the proximal part of the apical dendrite (Fig. 4.9b, right). In conclusion, these initial studies indicate that combining membrane potential imaging with optogenetic photostimulation is likely to become a standard approach in the near future.
Contributor Information
Nadia Jaafari, Inserm U836, Grenoble Institute of Neuroscience, Team 3, Grenoble Cedex 09, France; Laboratoire Interdisciplinare de Physique (CNRS UMR 5588), Université Joseph Fourier, Bat. E45, 140 avenue de la physique, Domaine univ., 38400 St Martin dἨères, B.P. 87, 38402 St Martin dἨères cedex, France.
Kaspar E. Vogt, International Institute for Integrative Sleep Medicine, University of Tsukuba, Tsukuba, Ibaraki, Japan
Peter Saggau, Allen Institute for Brain Science, 551N 34th Street, Seattle, WA 98103, USA.
Loew M. Leslie, Department of Cell Biology, R. D. Berlin Center for Cell Analysis and Modeling, University of Connecticut Health Center, Farmington, CT 06030-1507, USA
Dejan Zecevic, Department of Cellular and Molecular Physiology, Yale University School of Medicine, New Haven, CT 06520, USA.
Marco Canepari, Inserm U836, Grenoble Institute of Neuroscience, Team 3, Grenoble Cedex 09, France; Laboratoire Interdisciplinare de Physique (CNRS UMR 5588), Université Joseph Fourier, Bat. E45, 140 avenue de la physique, Domaine univ., 38400 St Martin dἨères, B.P. 87, 38402 St Martin dἨères cedex, France.
References
- Antic S, Zecevic D. Optical signals from neurons with internally applied voltage-sensitive dyes. J Neurosci. 1995;15:1392–1405. doi: 10.1523/JNEUROSCI.15-02-01392.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger T, Borgdorff A, Crochet S, Neubauer FB, Lefort S, Fauvet B, Ferezou I, Carleton A, Lüscher HR, Petersen CC. Combined voltage and calcium epifluorescence imaging in vitro and in vivo reveals subthreshold and suprathreshold dynamics of mouse barrel cortex. J Neurophysiol. 2007;97:3751–3762. doi: 10.1152/jn.01178.2006. [DOI] [PubMed] [Google Scholar]
- Brenowitz SD, Regehr WG. Associative short-term synaptic plasticity mediated by endocannabinoids. Neuron. 2005;45:419–431. doi: 10.1016/j.neuron.2004.12.045. [DOI] [PubMed] [Google Scholar]
- Bullen A, Patel SS, Saggau P. High-speed, random-access fluorescence microscopy: I. High-resolution optical recording with voltage-sensitive dyes and ion indicators. Biophys J. 1997;73:477–491. doi: 10.1016/S0006-3495(97)78086-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullen A, Saggau P. Indicators and optical configuration for simultaneous high-resolution recording of membrane potential and intracellular calcium using laser scanning microscopy. Pflugers Arch. 1998;436:788–796. doi: 10.1007/s004240050703. [DOI] [PubMed] [Google Scholar]
- Canepari M, Djurisic M, Zecevic D. Dendritic signals from rat hippocampal CA1 pyramidal neurons during coincident pre- and post-synaptic activity: a combined voltage- and calcium imaging study. J Physiol. 2007;580:463–484. doi: 10.1113/jphysiol.2006.125005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canepari M, Nelson L, Papageorgiou G, Corrie JE, Ogden D. Photochemical and pharmacological evaluation of 7-nitroindolinyl-and 4-methoxy-7-nitroindolinyl-amino acids as novel, fast caged neurotransmitters. J Neurosci Meth. 2001;112:29–42. doi: 10.1016/s0165-0270(01)00451-4. [DOI] [PubMed] [Google Scholar]
- Canepari M, Vogt KE. Dendritic spike saturation of endogenous calcium buffer and induction of postsynaptic cerebellar LTP. PLoS One. 2008;3:e4011. doi: 10.1371/journal.pone.0004011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canepari M, Vogt K, Zecevic D. Combining voltage and calcium imaging from neuronal dendrites. Cell Mol Neurobiol. 2008;58:1079–1093. doi: 10.1007/s10571-008-9285-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canepari M, Willadt S, Zecevic D, Vogt KE. Imaging inhibitory synaptic potentials using voltage sensitive dyes. Biophys J. 2010;98:2032–2040. doi: 10.1016/j.bpj.2010.01.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cueni L, Canepari M, Adelman JP, Lüthi A. Ca2+ signaling by T-type Ca2+ channels in neurons. Pflugers Arch. 2009;457:1161–1172. doi: 10.1007/s00424-008-0582-6. [DOI] [PubMed] [Google Scholar]
- Djurisic M, Antic S, Chen WR, Zecevic D. Voltage imaging from dendrites of mitral cells: EPSP attenuation and spike trigger zones. J Neurosci. 2004;24:6703–6714. doi: 10.1523/JNEUROSCI.0307-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eilers J, Konnerth A. Dye loading with patch pipettes. In: Yuste R, Lanni F, Konnerth A, editors. Imaging neurons a laboratory manual. Cold Spring Harbour Laboratory Press; New York, NY: 2000. [Google Scholar]
- Fierro L, Llano I. High endogenous calcium buffering in Purkinje cells from rat cerebellar slices. J Physiol. 1996;496:617–625. doi: 10.1113/jphysiol.1996.sp021713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluhler E, Burnham VG, Loew LM. Spectra, membrane binding, and potentiometric responses of new charge shift probes. Biochemistry. 1985;24:5749–5755. doi: 10.1021/bi00342a010. [DOI] [PubMed] [Google Scholar]
- Grinvald A, Hildesheim R. VSDI: a new era in functional imaging of cortical dynamics. Nat Rev Neurosci. 2004;5:874–885. doi: 10.1038/nrn1536. [DOI] [PubMed] [Google Scholar]
- Gupta RK, Salzberg BM, Grinvald A, Cohen LB, Kamino K, Lesher S, Boyle MB, Waggoner AS, Wang CH. Improvements in optical methods for measuring rapid changes in membrane potential. J Mem Biol. 1981;58:123–137. doi: 10.1007/BF01870975. [DOI] [PubMed] [Google Scholar]
- Hyrc KL, Bownik JM, Goldberg MP. Ionic selectivity of low-affinity ratiometric calcium indicators: mag-Fura-2, Fura-2FF and BTC. Cell Calcium. 2000;27:75–86. doi: 10.1054/ceca.1999.0092. [DOI] [PubMed] [Google Scholar]
- Jaafari N, De Waard M, Canepari M. Imaging fast calcium currents beyond the limitations of electrode techniques. Biophys J. 2014;107:1280–1288. doi: 10.1016/j.bpj.2014.07.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao JP, Tsien RY. Ca2+ binding kinetics of fura-2 and azo-1 from temperature-jump relaxation measurements. Biophys J. 1988;53:635–639. doi: 10.1016/S0006-3495(88)83142-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kee MZ, Wuskell JP, Loew LM, Augustine GJ, Sekino Y. Imaging activity of neuronal populations with new long-wavelength voltage-sensitive dyes. Brain Cell Biol. 2008;36:157–172. doi: 10.1007/s11068-009-9039-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer SG, Zeng W, Skorecki KL. Simultaneous fluorescence measurement of calcium and membrane potential responses to endothelin. Am J Physiol. 1992;263:1302–1309. doi: 10.1152/ajpcell.1992.263.6.C1302. [DOI] [PubMed] [Google Scholar]
- Leão RN, Mikulovic S, Leão KE, Munguba H, Gezelius H, Enjin A, Patra K, Eriksson A, Loew LM, Tort OLM interneurons differentially modulate CA3 and entorhinal inputs to hippocampal CA1 neurons. Nat Neurosci. 2012;15:1524–1530. doi: 10.1038/nn.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loew LM, Simpson LL. Charge-shift probes of membrane potential: a probable electrochromic mechanism for p-aminostyrylpyridinium probes on a hemispherical lipid bilayer. Biophys J. 1981;34:353–365. doi: 10.1016/S0006-3495(81)84854-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso JJ, Kim J, Lee S, Tsuda S, Chow NB, Augustine GJ. Optogenetic probing of functional brain circuitry. Exp Physiol. 2011;96:26–33. doi: 10.1113/expphysiol.2010.055731. [DOI] [PubMed] [Google Scholar]
- Mann EO, Paulsen O. Role of GABAergic inhibition in hippocampal network oscillations. Trends Neurosci. 2007;30:343–349. doi: 10.1016/j.tins.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Martinez-Zaguilan R, Martinez GM, Lattanzio F, Gillies RJ. Simultaneous measurement of intracellular pH and Ca2+ using the fluorescence of SNARF-1 and fura-2. Am J Physiol. 1991;260:297–307. doi: 10.1152/ajpcell.1991.260.2.C297. [DOI] [PubMed] [Google Scholar]
- Matiukas A, Mitrea BG, Pertsov AM, Wuskell JP, Wei MD, Watras J, Millard AC, Loew LM. New near-infrared optical probes of cardiac electrical activity. Am J Physiol Heart Circ Physiol. 2005;290:H2633–H2643. doi: 10.1152/ajpheart.00884.2005. [DOI] [PubMed] [Google Scholar]
- Milojkovic BA, Zhou WL, Antic SD. Voltage and calcium transients in basal dendrites of the rat prefrontal cortex. J Physiol. 2007;585:447–468. doi: 10.1113/jphysiol.2007.142315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naraghi M. T-jump study of calcium binding kinetics of calcium chelators. Cell Calcium. 1997;22:255–268. doi: 10.1016/s0143-4160(97)90064-6. [DOI] [PubMed] [Google Scholar]
- Neher E. Some quantitative aspects of calcium fluorimetry. In: Yuste R, Lanni F, Konnerth A, editors. Imaging neurons a laboratory manual. Cold Spring Harbour Laboratory Press; New York, NY: 2000. [Google Scholar]
- Roth A, Häusser M. Compartmental models of rat cerebellar Purkinje cells based on simultaneous somatic and dendritic patch-clamp recordings. J Physiol. 2001;535:445–472. doi: 10.1111/j.1469-7793.2001.00445.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BS, Oertner TG, Svoboda K. The life cycle of Ca2+ ions in dendritic spines. Neuron. 2002;33:439–452. doi: 10.1016/s0896-6273(02)00573-1. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG. Timing of neurotransmission at fast synapses in the mammalian brain. Nature. 1996;384:170–172. doi: 10.1038/384170a0. [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG. Control of neurotransmitter release by presynaptic waveform at the granule cell to purkinje cell synapse. J Neurosci. 1997;17:3425–3435. doi: 10.1523/JNEUROSCI.17-10-03425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG. Optical measurement of presynaptic calcium currents. Biophys J. 1998;74:1549–1563. doi: 10.1016/S0006-3495(98)77867-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron. 1999;23:399–409. doi: 10.1016/s0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Sinha SR, Patel SS, Saggau P. Simultaneous optical recording of evoked and spontaneous transients of membrane potential and intracellular calcium concentration with high spatio-temporal resolution. J Neurosci Meth. 1995;60:46–49. doi: 10.1016/0165-0270(94)00219-7. [DOI] [PubMed] [Google Scholar]
- Sinha SR, Saggau P. Simultaneous optical recording of membrane potential and intracellular calcium from brain slices. Methods. 1999;18:204–214. doi: 10.1006/meth.1999.0773. [DOI] [PubMed] [Google Scholar]
- Trigo FF, Papageorgiou G, Corrie JE, Ogden D. Laser photolysis of DPNI-GABA, a tool for investigating the properties and distribution of GABA receptors and for silencing neurons in situ. J Neurosci Meth. 2009;181:159–169. doi: 10.1016/j.jneumeth.2009.04.022. [DOI] [PubMed] [Google Scholar]
- Tsuda S, Kee MZ, Cunha C, Kim J, Yan P, Loew LM, Augustine GJ. Probing the function of neuronal populations: combining micromirror-based optogenetic photostimulation with voltage-sensitive dye imaging. Neurosci Res. 2013;75:76–81. doi: 10.1016/j.neures.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt KE, Gerharz S, Graham J, Canepari M. High-resolution simultaneous voltage and Ca2+ imaging. J Physiol. 2011a;589:489–494. doi: 10.1113/jphysiol.2010.200220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt KE, Gerharz S, Graham J, Canepari M. Combining membrane potential imaging with L-glutamate or GABA photorelease. PLoS One. 2011b;6:e24911. doi: 10.1371/journal.pone.0024911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willadt S, Canepari M, Yan P, Loew LM, Vogt KE. Combined optogenetics and voltage sensitive dye imaging at single cell resolution. Front Cell Neurosci. 2014;8:311. doi: 10.3389/fncel.2014.00311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JY, Cohen LB. Fast multisite optical measurement of membrane potential. In: Mason WT, editor. Biological techniques: fluorescent and luminescent probes for biological activity. Academic; New York, NY: 1993. [Google Scholar]
- Wuskell JP, Boudreau D, Wei MD, Jin L, Engl R, Chebolu R, Bullen A, Hoffacker KD, Kerimo J, Cohen LB, Zochowski MR, Loew LM. Synthesis, spectra, delivery and potentiometric responses of new styryl dyes with extended spectral ranges. J Neurosci Methods. 2006;151:200–215. doi: 10.1016/j.jneumeth.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Yan P, Acker CD, Zhou WL, Lee P, Bollensdorff C, Negrean A, Lotti J, Sacconi L, Antic SD, Kohl P, Mansvelder HD, Pavone FS, Loew LM. Palette of fluorinated voltage-sensitive hemicyanine dyes. Proc Natl Acad Sci U S A. 2012;109:20443–20448. doi: 10.1073/pnas.1214850109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhar O, Fenno LE, Davidson TJ, Mogri M, Deisseroth K. Optogenetics in neural systems. Neuron. 2011;71:9–34. doi: 10.1016/j.neuron.2011.06.004. [DOI] [PubMed] [Google Scholar]
- Yuste R. Loading brain slices with AM Esters of calcium indicators. In: Yuste R, Lanni F, Konnerth A, editors. Imaging neurons a laboratory manual. Cold Spring Harbour Laboratory Press; New York, NY: 2000. [Google Scholar]
- Zhang F, Wang LP, Boyden ES, Deisseroth K. Channelrhodopsin-2 and optical control of excitable cells. Nat Methods. 2006;3:785–792. doi: 10.1038/nmeth936. [DOI] [PubMed] [Google Scholar]
- Zhou W-L, Yan P, Wuskell JP, Loew LM, Antic SD. Intracellular long-wavelength voltage-sensitive dyes for studying the dynamics of action potentials in axons and thin dendrites. J Neurosci Meth. 2007;164:225–239. doi: 10.1016/j.jneumeth.2007.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]