

Abstract
Allelic variation at the Zea mays (maize) pericarp color1 (p1) gene has been attributed to epigenetic gene regulation. A p1 distal enhancer, 5.2 kb upstream of the transcriptional start site, has demonstrated variation in DNA methylation in different p1 alleles/epialleles. In addition, DNA methylation of sequences within the 3’ end of intron 2 also plays a role in tissue-specific expression of p1 alleles. We show here a direct evidence for small RNAs’ involvement in regulating p1 that has not been demonstrated previously. The role of mediator of paramutation1 (mop1) was tested in the maintenance of somatic silencing at distinct p1 alleles: the non-paramutagenic P1-wr allele and paramutagenic P1-rr’ epiallele. The mop1-1 mutation gradually relieves the silenced phenotype after multiple generations of exposure; P1-wr;mop1-1 plants display a loss of 24-nt small RNAs and DNA methylation in the 3’ end of the intron 2, a region close to a Stowaway transposon. In addition, a MULE sequence within the proximal promoter of P1-wr shows depletion of 24nt siRNAs in mop1-1 plants. Release of silencing was not correlated with small RNAs at the distal enhancer region of the P1-wr allele. We found that the somatic silencing of the paramutagenic P1-rr’ is correlated with significantly reduced H3K9me2 in the distal enhancer of P1-rr’; mop1-1 plants, while symmetric DNA methylation is not significantly different. This study highlights that the epigenetic regulation of p1 alleles is controlled both via RdDM as well as non-RdDM mechanisms.
Introduction
Epigenetic gene regulation affects genome integrity by maintaining, over generations, silencing in repetitive sequences and transposons [1]. In plants, epigenetic regulation plays a key role in phenomena such as genomic imprinting [2] and paramutation [3–7] mainly, by enforcing a silenced chromatin state through DNA methylation and/or chromatin modifications, leading to gene silencing. The silenced states are then maintained transgenerationally by epigenetic machinery [8]. Several components involved in the maintenance of transcriptional gene silencing have been identified in plants, the detailed mechanisms that may be distinct among different plant species are not fully understood.
Paramutation has remained an intriguing epigenetic process; it is defined as an interaction between two alleles, in which a silenced (somatic repression) allele suppresses a homologous active allele in trans (establishment) and leads to a heritable change in expression [6,9]. The newly repressed allele is meiotically heritable and is capable of suppressing another homologous naïve allele. Paramutation was first observed in the maize r1 (red1) gene [10], and later found in b1 (booster1) [11], pl1 (purple plant1) [12], p1 (pericarp color1) [13], and lpa1-241 (low phytic acid1) [14]. Paramutation-like phenomena have also been reported in other plants, fungi, and animals (For review see [9]). The mechanism of paramutation in maize has been extensively characterized at the b1 locus, but many details remain elusive. Seven direct tandem repeats located 100 kb upstream of the b1 coding region are critical and sufficient to mediate paramutation [15,16]. These tandem repeats demonstrate differential DNA methylation and chromatin states between paramutable (B-I) and paramutagenic (B’) alleles [15,17]. Several genes involved in maize paramutation have been identified and these turned out to be homologs of components involved in RNA-directed DNA methylation (RdDM) in Arabidopsis. These genes are required for various aspects of paramutation and siRNA biogenesis, including mediator of paramutation1 (Mop1) (encodes RDR2, an RNA-dependent RNA polymerase) [18], required to maintain repression6 (Rmr6)/Mop3 (encodes NRPD1, the largest Pol IV subunit) [19,20], Mop2/Rmr7 (encodes NRPD2/E2, the second largest Pol IV and Pol V subunits) [21,22], Rmr1 (encodes a Rad54-like ATPase) [23], and rmr2 (encodes a novel plant specific protein) [24]. Mutations in these genes disrupt paramutation and drastically reduce the abundance of 24-nt heterochromatic siRNAs (“siRNAs” hereafter). This implies that a siRNA-mediated mechanism is involved in paramutation although siRNAs alone are not sufficient for paramutation in certain cases [16,25].
The maize Mop1 gene is required for the establishment of paramutation at r1, b1, pl1, and p1 loci [18,26]. However, Mop1 differentially maintains somatic repression at these loci; the mop1-1 mutation disrupts the silencing at B’ and Pl’, but has a gradual effect at P1-rr’ [18,26]. Additionally, Mop1 is involved in the maintenance of transcriptional silencing at Mutator (Mu) transposons, transgenes, and the non-paramutagenic P1-wr allele [18,27–29]. Notably, it takes multiple generations in the presence of mop1-1 to disrupt the silencing of MuDR and p1 alleles [18,30]. Another component encoded by Mop2/Rmr7 (NRPD2/E2) also shows different requirements for establishment and somatic repression of various paramutagenic alleles. The Mop2-1 mutation contained an amino acid change in a highly conserved motif essential for polymerase activity in the second largest subunit of Pol IV and Pol V [21]. Paramutation of B-I to B’ is abolished by Mop2-1 in a dominant fashion, while somatic repression is released at B’ in a recessive manner. However, Mop2-1 acts recessive for establishment of paramutation at P1-rr’ and does not release somatic repression of P1-rr’ even after three generations of exposure [21]. By contrast, maize rmr1 (required to maintain repression1) encodes a SNF2 protein and is required for accumulation of a majority of 24-nt siRNAs, plays a role in maintaining the repressed chromatin states at Pl’ and transgenes, but it is not required for establishment of the Pl’ paramutation [23,28].
The above-mentioned genetic interactions exemplify the complexity of the mechanisms underlying the establishment of paramutation and maintenance of somatic repression, demonstrating that components of silencing machinery are not always shared between the two processes. In maize, another well-characterized gene system, p1, offers an opportunity to dissect the mechanism required for the establishment and maintenance of paramutation. The p1 gene encodes an R2R3 MYB transcription factor which regulates the 3-deoxyflavonoid biosynthetic pathway leading to the accumulation of red phlobaphene pigment in floral tissues [31,32]. Multiple alleles of p1 can be identified by their distinct expression patterns in pericarp and cob glumes. For instance, P1-wr specifies white pericarp and red cob and P1-rr conditions red pericarp and red cob tissues [33]. Paramutation at P1-rr has arisen independently twice; once spontaneously as P1-pr [34,35] and once through transgene induction of P1-rr’ [13]. The sequences required for paramutation were identified as being part of a transcriptional enhancer called the P1.2/distal enhancer (DE) [13,36]. The P1.2 contains a MULE fragment and p1 repeats and is present ~5 kb upstream of the TSS and repeated twice at the 3’ end of the gene. Paramutation of P1-rr is not fully penetrant and can lead to a range of pericarp pigmentation phenotypes. The amount of reduction of pigmentation in P1-rr’ is inversely correlated with both increased paramutagenicity and increased DNA methylation of the DE [13,35].
The silent pericarp phenotype of P1-wr is extremely stable and correlates with hypermethylation of the distal enhancer (DE) region, which is located between 5135 and 4637 bp upstream of the TSS [37] and overlaps with the P1.2 promoter element of P1-rr identified as an enhancer in transgenic experiments [36]. In addition to P1-wr, other silenced p1 epialleles such as P1-pr [38], P1-rr’ and P1-prTP[39], p1-ww:DP [40], and P1-wr* [41] are epigenetically regulated. A wealth of information related to epigenetic regulation of p1 has come from previous studies involving the dominant Unstable factor for orange1 (Ufo1) mutation which is involved in maintaining somatic repression of silenced alleles. These studies of P1-Ufo1-1 interactions have established different regions of p1 associated with changes of DNA methylation and histone modifications in different p1 alleles and epialleles. For example, hypomethylation of the DE correlates with increased transcription of p1 in pericarp and thus leading to enhanced pigmentation and hypomethylation of sequences in the intron 2 region lead to a gain of cob pigmentation [38–43].
The current study was designed to understand the role of epigenetics in maintenance of allelic expression patterns observed in different alleles/epialleles of p1 in maize. The specific questions addressed are: (1) Are there separate mechanisms for the regulation for p1 alleles/epialleles that have been derived via paramutation or non-paramutation types of gene silencing mechanisms? (2) Are p1 alleles regulated via the RdDM pathway? (3) Why do the requirements of somatic repression of P1-rr’ differ from other paramutagenic loci? To address these questions, changes in epigenetic states were compared at the paramutagenic P1-rr’ and non-paramutagenic P1-wr alleles in the presence or absence of mop1-1. This study provides evidence for the regulation of p1 alleles by RdDM and chromatin modifications.
Results
mop1-1 affects DNA methylation of an intron 2 region of P1-wr
The stably inherited P1-wr colorless pericarp phenotype can be altered in the presence of the mop1 mutation after multiple generations of exposure [18]. The gain of the pericarp pigment phenotype observed in P1-wr; mop1-1 was attributed to RNA-based mechanisms that may participate in tissue-specific silencing, however no molecular mechanism has been demonstrated for P1-wr; mop1-1 reactivation. To determine the underlying cause of mop1-1-induced reactivation of P1-wr pericarp expression, we performed genomic bisulfite sequencing for DNA methylation analysis on two regulatory regions, the DE and the 3’ end of intron 2 (F8C) of p1 (Fig 1). The DE and F8C regions are known to contain cis-acting transcriptional enhancer elements for p1 expression in pericarp [36,43] and cob glume pigmentation [41], respectively. The epigenetic state of these regulatory regions can also be affected by Ufo1-1, which induces transcriptional reactivation associated with hypomethylation [41,43]. We found that the P1-wr DE region remained hypermethylated in mop1-1 as compared to Mop1 plants (Fig 1B and S1 Fig). However, the F8C region of intron 2 showed a reduction of CHG and CHH methylation in mop1-1 (Fig 1B). The F8C region was divided into two consecutive sub-regions, Int2-1 and Int2-2, to further dissect the DNA methylation affected by mop1-1. The 196 bp Int2-1 was previously implicated as a cob-specific regulatory region, whereas no regulatory function has been associated with the 291 bp Int2-2 region [41]. In the presence of mop1-1, DNA methylation in the Int2-2 region is reduced at CHG (71.9% to 41.2%)(P = 0.0065) and CHH (46.1% to 36.8%)(P = 0.0508)contexts, whereas in Int2-1 the change is not significant (Fig 1C and 1D). When the methylation level of individual cytosines was plotted for the F8C region, the reduction of CG, CHG, and CHH methylation in P1-wr;mop1-1 was more pronounced in the first 78 bp of the Int2-2 region (Fig 1D). These results identified the Int2-2 region as a target of mop1-1 mediated DNA methylation.
Fig 1. Cytosine methylation level of two regulatory regions of P1-wr.
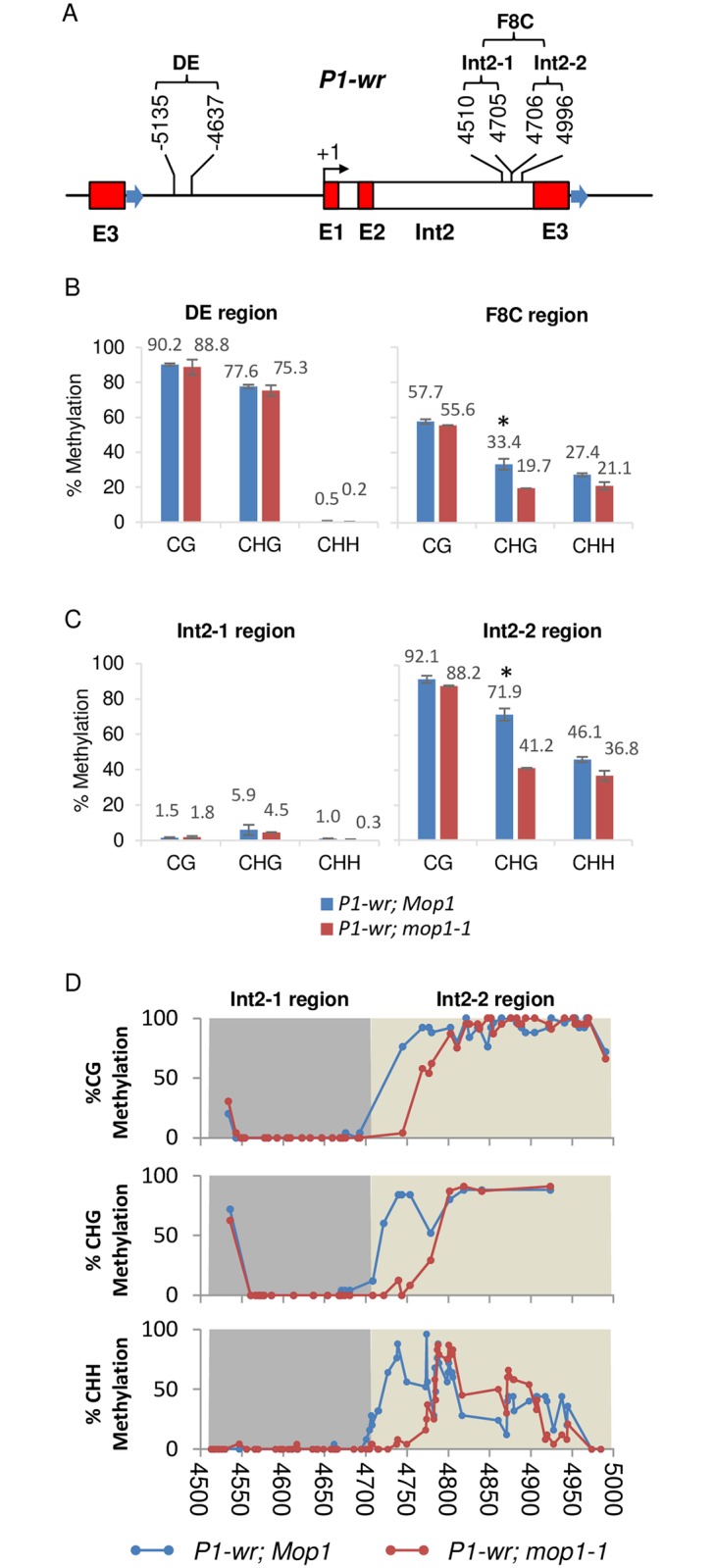
(A) Line diagram representing P1-wr gene structure. Block arrows represent sequences of tandem repeat.. The transcription start site is shown as a bent arrow marked +1. Red boxes labeled E1, E2 and E3 represent exon 1, 2 and 3, respectively. Introns are open boxes. Sequence coordinates of DE and F8C regions analyzed by bisulfite sequencing are shown above the gene structure (accession no. EF165349). (B) Overall DNA methylation level at CG, CHG and CHH (H is A, C, or T) contexts for DE and F8C regions in mop1-1 (P1-wr/P1-wr; mop1-1/mop1-1) and Mop1 (P1-wr/P1-wr; Mop1/mop1-1) plants. Average methylation is calculated as the mean of at least two biological replicates with error bars indicating the SE of the mean. * represents means are significantly different (P ≤ 0.05) as determined by Student’s t test. (C) Comparison of DNA methylation changes at P1-wr sequences within Int2-1 and Int2-2 regions of F8C in the presence of mop1-1. Percent methylation is shown on the y-axis. (D) DNA methylation of individual cytosines (circles) of the Int2-1 and Int2-2 regions.
Small RNA sequencing identifies regions of P1-wr targeted by RdDM
To identify whether the methylation of P1-wr was directed by siRNAs, small RNA profiling was done on young cob samples from P1-wr;Mop1 and P1-wr;mop1-1 plants. As expected from the function of Mop1 and previous work [44], 24-nt siRNAs accumulation was reduced globally in mop1-1 (S3 Fig). Small RNAs were aligned against a single copy of P1-wr and represented as an aggregate of sRNAs targeting this multi-copy gene. The Int2-2 region showed a complete loss of 24-nt siRNA abundance in P1-wr; mop1-1 plants (Fig 2A and 2B, and Table 1), which correlates with the reduction of DNA methylation level. As shown in Fig 1D the DNA methylation in all three contexts was significantly lower in P1-wr; mop1-1 plants (position 4700–4778). The lost siRNAs are downstream but adjacent to the loss of DNA methylation suggesting that the siRNAs help to recruit the machinery needed for the DNA methylation that spreads further than the siRNA targets. In addition, seven other regions of P1-wr had a loss of 24-nt siRNAs in mop1-1 plants; however, these regions were not assayed for DNA methylation (See Table 1). All of the affected regions have sequences with homology to DNA transposons or are nearby such transposon sequences. One region (-325 to -155) that showed substantial reduction of siRNA accumulation contains several inverted repeat sequences of a MULE transposon (Fig 2B). This region is part of the proximal enhancer, which contains additional partial fragments of Tourist, MULE, and a captured intron of GRMZM2G341379. Although the distal enhancer DE has a regulatory role on p1 expression [36], it did not show any small RNA accumulation in Mop1 or mop1-1, indicating that the maintenance of DNA methylation of DE is not Mop1-dependent. This is consistent with the DNA methylation results for DE, which showed very little CHH methylation and the level was not affected in the absence of Mop1 (Fig 1B). See S4 Fig for small RNA abundances for 20–23 nt classes which had very low abundance across P1-wr.
Fig 2. 24-nt siRNA abundance at the P1-wr gene.

(A) Line diagram of P1-wr showing gene structure and different regulatory elements (see Fig 1 legend for details). Abundance of 24-nt siRNA was shown on sense and antisense strands from P1-wr;Mop1/mop1-1 (3 plants) and P1-wr;mop1-1/mop1-1 (2 plants) samples as reads per million (RPM) and normalized to the 22-nt size class (see Methods). Green, grey, and red shaded areas indicate regulatory regions, TEs, and exons, respectively. Proximal enhancer is shown as region between black dashed lines. (B) Repetitive elements and 24-nt siRNA abundance in the proximal enhancer region. (C) 24-nt siRNA abundance in the Int2-1, Int2-2, and downstream regions. siRNA abundances from P1-wr;Mop1 samples (blue) and from P1-wr;mop1-1 (red) are shown. Coordinates for each graph are shown below. Black arrows indicate inverted repeats.
Table 1. Targets of 24-nt siRNAs accumulation at P1-wr.
| Feature | Start* | End | P1-wr; Mop1 (RPM**) | P1-wr; mop1-1 (RPM) |
|---|---|---|---|---|
| Tourist | -1726 | -1676 | 74 | 1 |
| Proximal enhancer MULE | -576 | -526 | 5 | 0 |
| Proximal enhancer MULE | -376 | -126 | 233 | 6 |
| Harbinger | 2274 | 2324 | 7 | 0 |
| hAT | 3424 | 3524 | 23 | 0 |
| Int2-2 | 4824 | 4874 | 5 | 0 |
| Stowaway | 5024 | 5124 | 83 | 7 |
| exon 3 | 5324 | 5374 | 7 | 0 |
*Coordinates relative to TSS, accession no. EF165349
**RPM = Reads Per Million
mop1-1-induced reactivation of paramutagenic P1-rr’ is associated with a minor reduction of CHH methylation of the distal enhancer
To compare the regulation of a paramutagenic epiallele of p1, P1-rr’ was introgressed with mop1-1 and Mop1. A previous study showed that the functional Mop1 is required for establishing P1-rr’ paramutation [18]. In the mop1-1 background, P1-rr’ expression was reactivated and the pericarp and cob glume pigmentation was observed. The P1.2/DE region (containing the transcriptional enhancer) is sufficient to establish transcriptional silencing associated with p1 paramutation. Since the P1-rr’ silenced state is associated with hypermethylation at the DE region of P1-rr (Fig 3A), we investigated whether the DNA methylation would change at the P1-rr’ DE region in mop1-1. DNA bisulfite sequencing of P1-rr DE region showed a minor reduction in the overall symmetric and asymmetric methylation in P1-rr’; mop1-1 as compared to P1-rr’; Mop1 plants (Fig 3B). Notably, in the mop1-1 homozygous plants, CG and CHH methylation were reduced from 94.0 ± 0.8% to 84.6 ± 4.0% and 7.1 ± 1.7% to 3.8 ± 1.1%, respectively. However, comparing the methylation at individual sites, most of the CHH sites which were methylated (>0%) in Mop1 plants showed reduced methylation in mop1-1 (Fig 4A). In addition, bisulfite sequencing of the F8C region in intron 2 revealed that Int2-1 showed low levels of DNA methylation in this region, whereas Int2-2 had statistically non-significant reductions of methylation in all contexts for Mop1 and mop1-1 (Figs 3C and 4B).
Fig 3. DNA methylation level of native P1-rr and paramutagenic P1-rr’ allele in Mop1 and mop1-1 background.
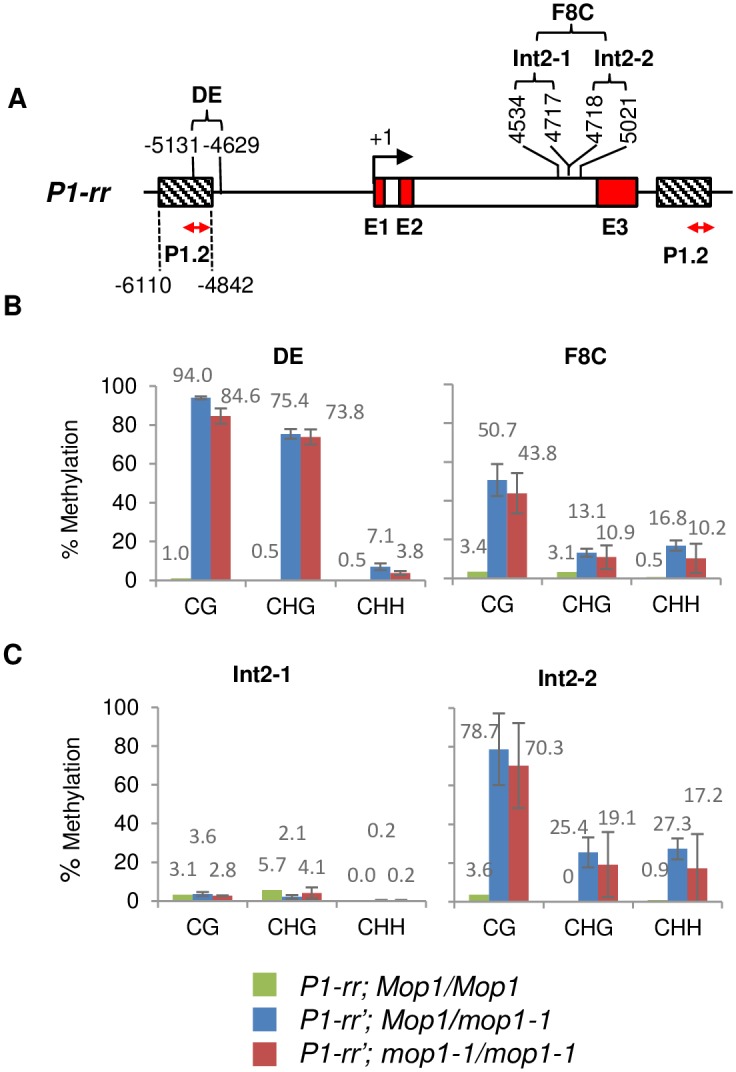
(A) Line diagram depicting P1-rr gene structure (Accession: AF427146). The exons and introns are shown as red filled and open boxes, respectively. The transcription start site is indicated by a bent arrow. The DNA methylation of the 503 bp DE region shown above the gene structure was analyzed by bisulfite sequencing. Double-headed arrow represents the region analyzed by ChIP-qPCR (Fig 5). Hatched boxes indicate the P1.2 region. (B) Average DNA methylation in CG, CHG and CHH contexts at the DE and F8C regions of P1-rr and P1-rr’ in the Mop1 or mop1-1 plants. (C) Average DNA methylation of the Int2-1 and Int2-2 sub-regions of F8C. The percentage methylation is shown on the y-axis. Average methylation is calculated as the mean of two biological replicates with error bars indicating the SE of the mean. Comparisons of Mop1 and mop1-1 plants were not significant using the Student’s t test at a P of ≤ 0.05. A single P1-rr sample was analyzed so there is no error bar for P1-rr (the data is consistent with a previous study from Sekhon et al, 2012 [39]).
Fig 4. DNA methylation of individual cytosine sites of native and paramutagenic P1-rralleles in Mop1 and mop1-1 background.

Bisulfite sequencing results for individual cytosine sites in the (A) DE region and (B) Int2-1 (dark grey) and Int2-2 (light grey) regions. Each site is indicated by a circle. P1-rr; Mop1/Mop1, P1-rr’; Mop1/mop1-1, and P1-rr’; mop1-1/mop1-1 are green, blue, and red, respectively. Data are the mean of two biological replicates (except P1-rr which is one plant).
mop1-1 mediated reactivation of P1-rr’ is associated with reduced H3K9me2
In the absence of significant DNA methylation differences we tested if somatic silencing is associated with another chromatin modification. ChIP-qPCR was performed to compare repressive H3K9me2 mark levels with the p1 expression using pericarps from P1-rr, P1-rr’, P1-rr’;Mop1, and P1-rr’;mop1-1. H3K9me2 was enriched three-fold within the DE in paramutagenic P1-rr’ compared to naïve P1-rr (Fig 5A). The increased methylation at both DNA and H3K9 sites in P1-rr’ implies that this region is under tight epigenetic suppression. The results also showed that the absence of Mop1 caused a ~30% reduction of H3K9me2 levels within the DE region (Fig 5B). The p1 transcripts, measured by qRT-PCR, were higher in the mop1-1 background (Fig 5C), suggesting that reduction of H3K9me2 may be sufficient to release some level of P1-rr’ silencing, although several generations are required for this release.
Fig 5. H3K9me2 and transcript analysis at P1-rr’ in Mop1 and mop1-1.
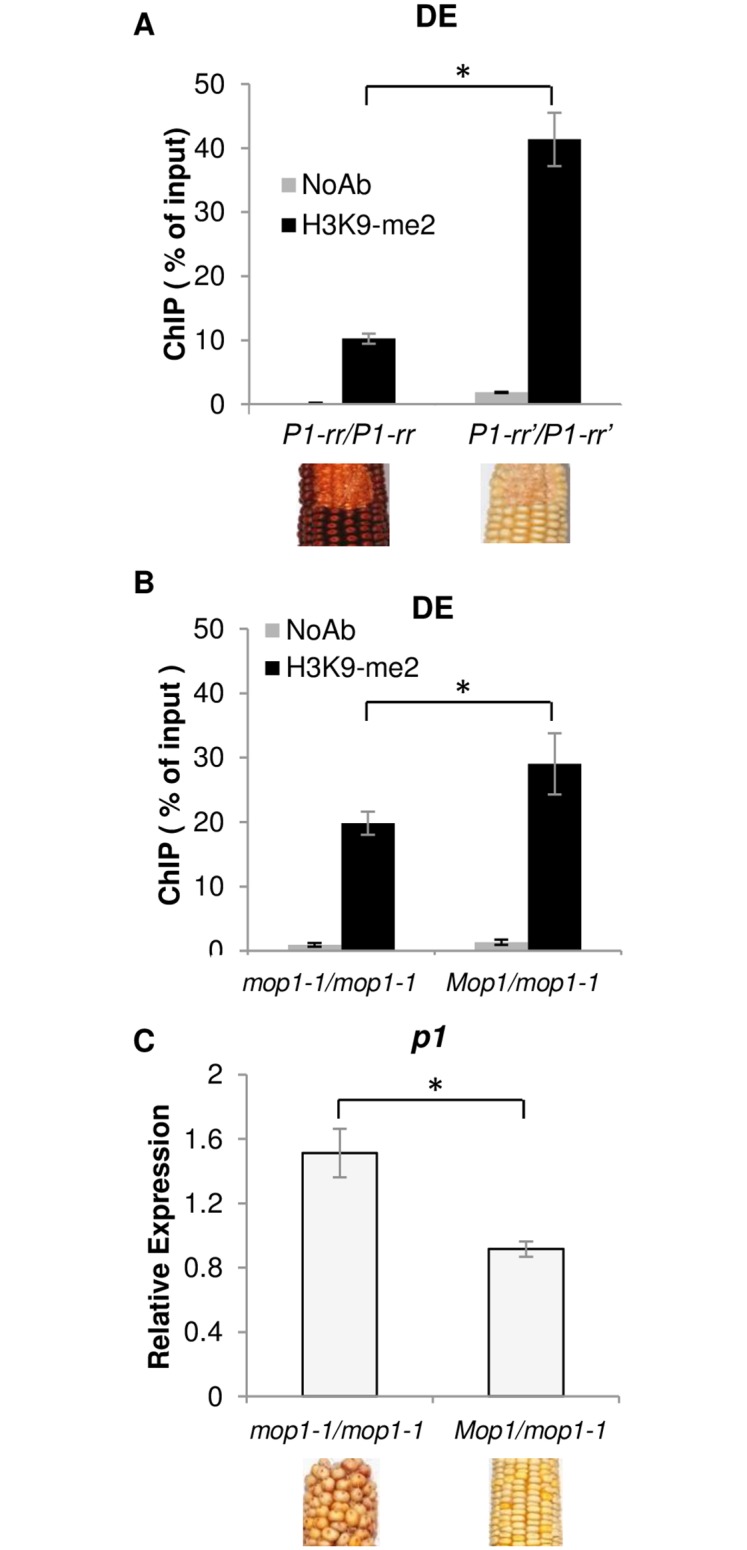
(A) ChIP-qPCR of H3K9me2 of the DE region of P1-rr and P1-rr’ plants. (B) ChIP-qPCR of H3K9me2 of the DE region of P1-rr’ plants in Mop1 or mop1-1 plants and (C) their corresponding p1 expression from qRT-PCR. Ears are shown for the corresponding phenotypes in which the pericarp samples were used for ChIP experiments. Data are presented as the mean of three biological replicates with error bars indicating SE of the mean. * P<0.05 using an unpaired t-test.
Discussion
The maize p1 gene is regulated via epigenetic mechanisms that generate tissue-specific expression patterns. In addition to tissue-specific gene silencing, certain p1 alleles/epialleles are affected by paramutation. Mop1 is required for establishment of paramutation via biogenesis of siRNA from repetitive components of the genome. In this study two alleles of p1 (P1-wr and P1-rr’) that differ in their paramutagenicity were used to characterize their interaction with mop1-1. Our results show that the tissue-specific somatic derepression of these two p1 alleles by mop1-1 involve different regions; this is discussed below.
Silencing of P1-wr in pericarp can be attributed to two distinct regulatory regions
In a previous study, promoter::GUS transgenes of the p1 promoter were used and two enhancers of p1 expression were identified, the P1.2 (containing the DE region) and proximal enhancer, which may act together to give the strongest expression of p1 [36]. In the current study, we have further characterized the key regulatory elements of p1 that are subjects of epigenetic regulation and hence influence p1 activity. While the DE of P1-wr is not a target of 24-nt siRNAs, the proximal enhancer is targeted by Mop1-dependent 24-nt siRNAs and likely subjected to RdDM (Fig 2). The proximal enhancer sequence contains the remnants of a MULE element with a captured intron and these transposon sequences are frequently the targets of RdDM. For example, in the case of FWA silencing, DNA methylation of the tandemly repeated promoter elements derived from ancestral transposon sequences has been well characterized [45]. Our results show these sRNAs originate from the MULE fragments present within this proximal enhancer of P1-wr. The proximal enhancer sequence similarity between of P1-wr and P1-rr alleles is over 98%. Although, we did not characterize the proximal enhancer at P1-wr, high levels of DNA methylation have been observed in the proximal enhancer and surrounding regions in P1-rr and its epiallele P1-pr [38]. The proximal enhancer region in these p1 alleles resembles a Mop1-dependent mCHH island, regions near genes with high levels of CHH DNA methylation which mark the boundary between genic chromatin and flanking heterochromatin [46,47]. Li et al. (2015) identified that loss of mCHH islands in mop1-1 can lead to reduction of CG and CHG methylation upstream of the mCHH island and can cause increased gene expression of the proximal gene. The modest up-regulation of P1-wr in mop1-1 after multiple generations is possibly due to the gradual loss of repressive epigenetic marks in the proximal enhancer, although further tests of DNA methylation and/or other chromatin modifications are needed to confirm this speculation. The DE, on the other hand, may be regulated via a non-RdDM mechanism, as it is hypermethylated at symmetric sites, hypomethylated at asymmetric sites (this study, see Fig 1; Sekhon and Chopra 2009), and lacks siRNA production (this study, see Fig 2), which are some the hallmarks of non-RdDM heterochromatin [48]. This suggests that the DE is not involved in mop1-1 activation of P1-wr, in contrast to its interaction with Ufo1-1 which reduces the CG and CHG methylation of the DE [43]. This implies that P1-wr regulation may be attributed to a non-RdDM mechanism through the DE and a possible RdDM mediated mechanism through the proximal enhancer.
In this study, sRNA changes were also observed in another sequence, Int2-2, present within intron 2 of P1-wr (Fig 2, Table 1). The loss of RdDM corresponds to the reduction of DNA methylation at Int2-2 in the presence of mop1-1 (Fig 1C and 1D). This region is nearby a Stowaway element from which DNA methylation may spread. This Stowaway element also shows loss of 24-nt sRNAs within this int2-2 region (Fig 2C). Presence of MITES in rice gene introns has been predicted to be transcribed into double stranded RNAs with a role in gene regulation [49,50]. We observed the presence of DNA methylation in the region adjoining to Int2-2 in the absence of siRNAs corresponding to that region. It has been demonstrated in maize that heterochromatin spreads from retrotransposons to the unique sequences [51]. Interestingly, there is a sharp boundary of CHH methylation between Int2-1 and Int2-2 in Mop1 and this boundary shifts towards the Stowaway element in the presence of mop1-1 (Fig 1D). Our previous study of a silenced epiallele, P1-wr*, found that the Int2-1 region conferred cob-specific p1 gene expression [41]. P1-wr is a tandemly repeated multi-copy allele, so these intronic cis regulatory elements may act as long distance enhancers of the proximal gene copy. Combining our results, these cis regulatory elements in adjoining intronic sequences may thus play important tissue-specific roles in p1 regulation.
P1-rr’ silencing is maintained through H3K9me2 of the distal enhancer sequence
The P1.2 region of P1-rr contains P1-wr DE homologous sequences and has been shown to be necessary for P1-rr’ paramutation in addition to its role as a positive regulator of p1 expression [13,36]. This region is repeated three times throughout the P1-rr gene and contains a MULE sequence, which makes it a likely target of RdDM. Compared to P1-rr, the P1.2 region of P1-rr’ is highly methylated and contains >7% CHH methylation, a hallmark of RdDM (Fig 3B). Indeed, the absence of Mop1 results in about a 50% reduction of CHH methylation, but the changes of CG and CHG methylation levels are minimal (3–10% reduction see Fig 3B). In addition, our results and a previous study [39] have found that the P1.2 is also a target for epigenetic regulation via histone modifications and highly enriched for H3K9me2 in P1-rr’ (Fig 5A). The absence of Mop1 leads to a reduction of the H3K9me2 inversely proportional to the up-regulation of p1 expression (Fig 5B and 5C). A positive correlation exists between H3K9me2 levels and DNA methylation in plants as well as for several other systems [52,53]. Previous studies have shown that chromatin structure, rather than DNA methylation, is the primary effector that is responsible for silencing of an allele of the maize anthocyanin regulatory gene Pl-Blotched as compared with the uniformly expressed Pl-Rhodes allele [54]. Additionally, a chromatin-mediated mechanism was postulated when the expression of transposon mudrA is progressively reactivated in mop1-1 and loss of DNA methylation precedes reactivation [27,30]. While RdDM is needed for the establishment of paramutation at P1-rr’, its contribution to maintenance of silencing is modest, as the silenced state of the P1.2 enhancer in P1-rr’ is only slightly alleviated in mop1-1 (Figs 3–5). This indicates that after paramutation has been established, other mechanisms affecting the chromatin epigenetic state, such as histone modifications, are involved in maintaining the transcriptional silencing of P1-rr’. This epiallele appears to be under regulation of both RdDM and non-RdDM maintenance of its chromatin state, thus behaving like a majority of RdDM loci which only show small reductions in CG and CHG methylation in mop1-1 [48]. It has been shown that small RNAs can be involved in maintaining high levels of H3K9me2 independent of CHG methylation in Arabidopsis thaliana [55]. This can explain the reduction of H3K9me2 in the P1-rr’ DE (Fig 5B) while CHG DNA methylation remains high in the presence of mop1-1 (Fig 3B). Another possibility is that other chromatin-related genes down regulated in mop1-1 [56] are causing a reduction of H3K9me2, although this seems less likely as a global reduction of H3K9me2 has not been reported for mop1-1. It also cannot be ruled out whether the proximal enhancer plays any role in mop1-1 activation of P1-rr’; since this region is identical among P1-wr, P1-rr, and P1-rr’, they are likely to behave similarly.
Sequence variation in transcriptional enhancers determine p1 allele participation in paramutation
The P1.2 region of P1-rr’ is capable of transgene-induced transcriptional silencing and participates in paramutation [13,36]. It has also been demonstrated that a P1.2::GUS transgene only suppresses P1-rr and not P1-wr [13]. Our results show that P1-rr’ has higher CHH methylation within the DE (Fig 3B), whereas CHH sites are barely methylated at the DE of P1-wr (Fig 1B). In another study, a paramutagenic P1-rr epiallele, P1-pr, had slightly lower levels of CHH methylation at DE (4.5%) [35]. Because the CHH methylation is reduced by mop1-1 it seems likely that the DE is a valid RdDM target, although at low levels. The effect of mop1-1 at distinct regions of P1-wr and P1-rr’ may be attributed to the different small RNAs derived from structurally-modified regulatory elements of these alleles. A MULE, repetitive element, and a fragment of a hAT element are present near the DE region of the P1.2 repeat of P1-rr, while the DE of P1-wr contains only the 3’ end of the P1.2 region [37] which does not have any transposon sequences. Another allele, p1-ww, similarly lacks the transposon sequences in the P1.2 region and does not gain DNA methylation when heterozygous with the paramutagenic P1-pr [35]. A doppia element likewise has been implicated in paramutation of r1 [57]. Similar to P1.2, the repeats that determine b1 paramutation (b1TR) contain a 413 bp sequence that acts as a transcriptional enhancer and is required for transgene-induced paramutation via RNA mediated silencing [16].
Differential requirements of somatic repression of paramutagenic alleles may be due to the balance of RdDM and maintenance methylation
The maintenance of a paramutagenic state and somatic repression in different genes has distinct requirements because these two states are variably impacted in the presence of different mutants. For example, rmr1 is needed for somatic repression of pl’, but it is not needed for somatic repression of B’ or establishment of paramutation of pl’, B’, or R-r’ [23]. The presence of mop1-1 immediately releases transcriptional silencing of B’ [26]. Unlike B’, P1-rr’ silencing is not immediately released by mop1-1 [18]. The only other RdDM mutant that has been tested with P1-rr’ is Mop2-1, which is needed for establishment of paramutation but not for somatic repression [21]. The uncloned Ufo1-1 mutant has also been shown to be required for somatic repression of P1-rr’ and B’, however its role in the establishment of paramutation at either loci has not been reported [39]. Our study has demonstrated that symmetric DNA methylation of the important paramutagenic region of p1 remains high in the presence of mop1-1 even after multiple generations of exposure. Thus the DE of P1-rr’ appears to be the target of Mop1-mediated RdDM for maintenance of paramutagenicity and Mop1-independent maintenance of CG and CHG methylation, needed to maintain somatic repression. This could explain the different responses of B’ and P1-rr’ to mop1-1, with B’ somatic repression depending mainly upon RdDM. One possibility for this difference in behavior is that the DE is proximal to transposon sequences which are subject to heterochromatic silencing whereas the b1TRs do not contain such sequences [15]. Future studies of P1-rr’ with additional mutants would further elucidate the complexity of chromatin regulation needed for various aspects of paramutation in maize.
Conclusions
Alleles of the maize p1 gene display unique and tissue-specific expression patterns and contain regions important for transcriptional regulation. This study identifies the proximal enhancer as a candidate for Mop1-mediated somatic repression of P1-wr, while also showing Mop1 does not affect the known pericarp distal enhancer element. We also provided evidence for the basis of the differential requirement of Mop1 for somatic silencing of P1-rr’ as compared to B’. Namely, the sequences necessary for paramutation have overlapping silencing mechanisms, such that even in the presence of mop1-1, symmetric DNA methylation and H3K9me2 remain relatively high compared to the fully-expressed, paramutable allele. Similar mechanisms might be present at other paramutagenic loci which have different requirements for establishing paramutation and maintaining somatic repression. This also raises the question of why P1-rr’ does not fully transition into non-RdDM maintenance of silencing a question which may be addressed by further study of the non-paramutagenic epiallele P1-prtp as well as additional RdDM mutants.
Materials and methods
Genetic stocks, genetic crosses and genotyping
All of the populations discussed here were planted at the Penn State University Agronomy Farm, Rock Springs, PA.
In the P1-wr; mop1-1 study, the P1-wr allele has been exposed for three generations to mop1-1 mutant background and the detailed crossing scheme has been described in details previously (Sidorenko and Chandler, 2008). Briefly, the P1-wr stock carrying Mop1/mop1-1 was outcrossed to a mop1-1/mop1-1 plant. The subsequent generation was self-pollinated and the plants carrying P1-wr/P1-wr; mop1-1/mop1-1 were designated as mutants and selected using the similar method as explained below for P1-rr’ allele. Plants with genotype P1-wr/P1-wr; Mop1/mop1-1 genotype were used as wild types.
The paramutagenic P1-rr’ allele was derived from a transgene-induced silencing event from a cross between P1-rr and a transgenic line containing a P1.2b:: GUS transgene (Sidorenko and Peterson, 2001). The P1-rr’ stock used in this study was the progeny of a homozygous (P1-rr’/P1-rr’) plant that showed very strong silencing and had colorless pericarps and light pink to colorless cob glumes (Sidorenko and Peterson, 2001). The progenies were generated by intercrossing P1-rr’/P1-rr’; Mop1/mop1-1; B’ and P1-rr’/P1-rr’; mop1-1/mop1-1; B’ (S2 Fig). These progeny plants were further genotyped by multiplex PCR using specific primers to distinguish homozygous and heterozygous mop1-1 plants. The primers used for genotyping are listed in S1 Table and PCR conditions are as follows: 94°C for 5 min, 30 cycles of (94°C for 30 sec, 56°C for 45 sec, 72°C for 45 sec), and final extension step at 72°C for 10 min. Increased plant pigmentation (purple plant) conferred by up-regulation of the B’ was used to identify the mop1-1 mutant (mop1-1/mop1-1) plants. The progenies showing gain of pericarp pigmentation used for molecular analysis have been exposed to mop1-1 background for three generations. Plants carrying homozygous and heterozygous mop1-1 were further selected as mutants and wild types respectively, for molecular characterization including DNA methylation, chromatin modifications, and gene expression analysis. The control P1-rr allele used in this study was derived from the standard P1-rr-4B2 genetic stock (Grotewold et al., 1991).
Plant genomic DNA extraction and genomic bisulfite sequencing
Leaf (v6 stage) and pericarp (18 days after pollination; DAP) tissue samples were harvested, snap frozen in liquid nitrogen, and stored at -80°C. Leaf genomic DNA was used from P1-wr/P1-wr; Mop1/mop1-1 and P1-wr/P1-wr; mop1-1/mop1-1) plants. Pericarp tissues (18 DAP) from P1-rr’ carrying homozygous (red) and heterozygous (colorless) mop1-1 plants were used for bisulfite sequencing assays. Genomic DNA was extracted using a modified CTAB (cetyltrimethylammonium bromide) method [58]. DNA was precipitated with 7.5M ammonium acetate and the pellet was re-suspended in 1X TE (1 M Tris pH 8.0 and 0.5 M EDTA pH 8.0). The genomic DNA from two independent plants was used for the bisulfite sequencing analysis. Bisulfite treatment was performed with EZ DNA Methylation-Gold Kit (Zymo Research, Orange, CA) and PCR amplified using gene-specific primers (S1 Table). To get high-yielding PCR products from bisulfite treated DNA, nested PCR was performed using two pairs of primers. For the distal enhancer region (fragment 15), RBS8F and RBS1R were used as external primers, and then RBS9F and RBS3R were used as nested primers to amplify a 499-bp region. For P1-rr, the DE region is 503 bp. For the 3’ end of intron 2 (fragment 8C), RBS11F and RBS11R were used as external primers, and then RBS12F and RBS12R were used as nested primers to amplify a 487-bp region. All PCR products were subcloned into pSC-A-amp/kanamycin vector using StrataClone PCR Cloning Kit (Agilent Technologies, Santa Clara, CA). The plasmid DNA was isolated using a StrataPrep Plasmid Miniprep Kit (Agilent Technologies, Santa Clara, CA) and DNA sequencing was performed using ABI Hitachi 3730XL DNA Analyzer in the Penn State Nucleic Acid Facility. For each bisulfite sequencing experiment, two biological replicates were used and at least ten technical replicates (clones) were obtained to determine the average methylation per sample. The DNA sequence data was aligned using ClustalW (http://www.ebi.ac.uk/Tools/msa/clustalw2/). To analyze the DNA methylation patterns of the compiled clones for each plant from each genotype, CyMATE was used to illustrate each DNA methylation sequence context (CG, CHG, CHH) (Yu et al., 2008). Overall DNA methylation in each context was calculated by dividing the number of methylated cytosines in a given context by total number of cytosines in that context in all clones. Average methylation is calculated as the mean of biological replicates with error bars indicating SE of the mean. The percent methylation between genotypes was compared with Student’s t tests and were considered significant at a P ≤ 0.05.
Small RNA sequencing and analysis
Young cobs (4 cm) were collected from three wild type (Mop1/mop1-1) and from two mop1-1 plants from a BC1F2 population segregating for mop1-1. For small RNA libraries, total RNA from the materials described above was isolated using Tri Reagent™ (Molecular Research Center, Inc., Cincinnati, OH). Small RNA libraries were constructed using the Illumina TruSeq Small RNA Sample Preparation Kit (RS-200-0012), and sequenced on an Illumina HiSeq2000 instrument at University of Delaware. Raw sequencing data was first trimmed of adapter sequences and then the read counts were normalized based on the total abundance of genome-matched reads, excluding structural sRNAs originating from annotated tRNA, rRNA, small nuclear (sn) and small nucleolar (sno) RNAs. The genome sequences were from maize B73 RefGen_v2. The small RNA sequence data are available from NCBI's Gene Expression Omnibus (GEO) under GEO Series accession number GSE68510 https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE68510. Small RNA reads were aligned to the P1-wr gene (GenBank: EF165349) using bowtie. Only reads with perfect match to p1 sequences were kept. Read counts were normalized to 1M reads per library as well as to 22 nt abundances. The 22-nt abundances were used for normalization control as mop1-1 causes a drastic reduction of 24-nt siRNA that leads to an overrepresentation of other size classes [44]. A single copy of the P1-wr sequence was analyzed as a representative for the remaining copies. Read depth across the gene was calculated using Samtools. To assess regions targeted by 24-nt siRNAs, reads were counted in 50 bp bins across the gene and results shown in Table 1.
Reverse transcription and Quantitative Real-Time PCR
Total RNA was isolated from pericarp tissues using RNAzol following the standard protocol (Molecular research center, Cincinnati, OH). The first strand cDNA templates were reverse transcribed with Superscript III reverse transcriptase (Invitrogen, Grand Island, NY) using oligo dT as a primer. Quantitative real-time PCR (qRT-PCR) was performed with ABI7500 Fast real-time PCR system using SYBR Green I (Roche, Madison, WI) as the detection system and the default program: 10 minutes of pre-incubation at 95°C followed by 40 cycles of (95°C for 15 sec, 60°C) for 1 min for PCR amplification. The primers used for qRT-PCR are p1 primers (RT_PWREx_2F and RT_PWREx_2R) and actin primers (actin_exon2_Fw and actin_exon2_Rev) (S1 Table). Actin1 gene was used as an endogenous control. The relative expression level of each gene was calculated using the 2–ΔΔCt method (Livak and Schmittgen, 2001). Gene expression analysis was performed with three independent samples.
Chromatin immuno-precipitation assay and quantitative real-time PCR (ChIP-qPCR)
ChIP assays were performed using pericarp tissues following a modified protocol (Haring et al., 2007; Kimura et al., 2008). Briefly, 18-day-old pericarp tissues were cross-linked with 3% formaldehyde and the chromatin complex was extracted and sheared to a size range of 0.5 to 1 kb fragments using a Bioruptor (Diagenode, Denville, NJ). The antibody used for ChIP was an anti-H3K9me2 kindly provided by Dr. Hiroshi Kimura [59]. This antibody was then coupled with sheep anti-mouse IgG Dynabeads M-280 (Life technologies, Carlsbad, CA). In addition, normal mouse IgG was used as a no-antibody control (NoAb). The ChIPed DNA was further purified using QIAquick PCR Purification Kit (QIAGEN, Valencia, CA) and quantified with qPCR. The primers used here were specific to distal enhancer (DE) region (PW_RTF15-2_Fw and PW_RTF15-2_Rev) as listed in S1 Table. ChIP-qPCR data was normalized using percentage-of-input method (See the details of ChIP analysis online guide: https://www.thermofisher.com/us/en/home/life-science/epigenetics-noncoding-rna-research/chromatin-remodeling/chromatin-immunoprecipitation-chip/chip-analysis.html). The relative differences between ChIP assays and input samples were determined using the ΔCT method and presented as a percentage of the input (taken as 100%). Data shown in this study are representative results of three independent experiments.
Supporting information
Methylation profile at CG, CHG and CHH contexts were obtained by genomic bisulfite sequencing. The y-axis shows percentage of DNA methylation. Individual sites are shown as circles.
(TIF)
Crosses between P1-rr’/P1-rr’ carrying homozygous (mop1-1/mop1-1) and heterozygous (Mop1/ mop1-1) were made to test whether mop1-1-induced reactivation of P1-rr’ is associated with hypomethylation. Since the stocks also carried a silenced B’ allele, plants containing homozygous mop1-1 were identified by the purple color of plant body, whereas plants containing heterozygous Mop1/mop1-1 showed green plant without purple pigmentation.
(TIF)
(A) Size distribution of small RNAs in P1-wr;Mop1 and P1-wr;mop1-1 samples with sum of small RNA abundances normalized to 1 million reads. (B) Size distribution of small RNA abundance after normalization to the abundance of 22-nt class. Three independent Mop1 samples and two independent mop1-1 samples are shown.
(TIF)
(A) Line diagram of P1-wr showing gene structure and different regulatory elements (see Fig 1 legend for details). (B) Abundance of siRNA was shown on sense and antisense strands from P1-wr;Mop1/mop1-1 and P1-wr;mop1-1/mop1-1 samples as reads per million (RPM) and normalized to the 22-nt size class (see Methods). Green, grey, and red shaded areas indicate regulatory regions, TEs, and exons, respectively. Proximal enhancer is shown as region between black dashed lines. siRNA abundances from P1-wr;Mop1/mop1-1 samples (blue) and from P1-wr;mop1-1/mop1-1 (red) are shown.
(TIF)
(XLSX)
Acknowledgments
We are thankful to Scott Harkcom, the Penn State Agronomy farm manager, and the farm crew for their help with land preparation and for tending maize genetic nurseries. We are thankful to Dr. Lyudmila Sidorenko for providing the materials of P1-wr; mop1-1 and P1-rr’; mop1. We appreciate Dr. Hiroshi Kimura for his kindness in providing the antibody used for ChIP assays.
Data Availability
Data is available from the GEO database with accession numbers GSM2652417 and GSE68510 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE68510).
Funding Statement
This work was supported by National Science Foundation collaborative awards 0619330 and 1051654 to SC and 1051576 to BCM. Support from Hatch awards PEN04452 and PEN04613 to SC is acknowledged. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Fultz D, Choudury SG, Slotkin RK. Silencing of active transposable elements in plants. Curr Opin Plant Biol. Elsevier Ltd; 2015;27: 67–76. doi: 10.1016/j.pbi.2015.05.027 [DOI] [PubMed] [Google Scholar]
- 2.Gehring M. Genomic Imprinting: Insights From Plants. Annu Rev Genet. 2013;47: 187–208. doi: 10.1146/annurev-genet-110711-155527 [DOI] [PubMed] [Google Scholar]
- 3.Chandler VL. Paramutation’s properties and puzzles. Sci (New York, NY). 2010;330: 628–629. doi: 10.1126/science.1191044 [DOI] [PubMed] [Google Scholar]
- 4.Erhard KF, Hollick JB. Paramutation: A process for acquiring trans-generational regulatory states. Curr Opin Plant Biol. Elsevier Ltd; 2011;14: 210–216. doi: 10.1016/j.pbi.2011.02.005 [DOI] [PubMed] [Google Scholar]
- 5.Hollick JB. Paramutation: A trans-homolog interaction affecting heritable gene regulation. Curr Opin Plant Biol. Elsevier Ltd; 2012;15: 536–543. doi: 10.1016/j.pbi.2012.09.003 [DOI] [PubMed] [Google Scholar]
- 6.Giacopelli BJ, Hollick JB. Trans-Homolog Interactions Facilitating Paramutation in Maize. Plant Physiol. 2015;168: 1226–1236. doi: 10.1104/pp.15.00591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huang J, Lynn JS, Schulte L, Vendramin S, Mcginnis K. Epigenetic Control of Gene Expression in Maize [Internet]. International Review of Cell and Molecular Biology. Elsevier Inc.; 2016. doi: 10.1016/bs.ircmb.2016.08.002 [DOI] [PubMed] [Google Scholar]
- 8.Quadrana L, Colot V. Plant Transgenerational Epigenetics. Annu Rev Genet. 2016;50: 467–491. doi: 10.1146/annurev-genet-120215-035254 [DOI] [PubMed] [Google Scholar]
- 9.Hollick JB. Paramutation and related phenomena in diverse species. Nat Rev Genet. Nature Publishing Group; 2016;18: 5–23. doi: 10.1038/nrg.2016.115 [DOI] [PubMed] [Google Scholar]
- 10.Brink R a. A Genetic Change Associated with the R Locus in Maize Which Is Directed and Potentially Reversible. Genetics. 1956;41: 872–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coe EH. A Regular and Continuing Conversion-Type Phenomenon at the B Locus in Maize. Proc Natl Acad Sci USA. 1959;45: 828–832. doi: 10.1073/pnas.45.6.828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hollick JB, Patterson GI, Coe EH, Cone KC, Chandler VL. Allelic interactions heritably alter the activity of a metastable maize pl allele. Genetics. 1995;141: 709–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sidorenko L V, Peterson T. Transgene-induced silencing identifies sequences involved in the establishment of paramutation of the maize p1 gene. Plant Cell. 2001;13: 319–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pilu R, Panzeri D, Cassani E, Cerino Badone F, Landoni M, Nielsen E. A paramutation phenomenon is involved in the genetics of maize low phytic acid1-241 (lpa1-241) trait. Heredity (Edinb). 2009;102: 236–45. doi: 10.1038/hdy.2008.96 [DOI] [PubMed] [Google Scholar]
- 15.Stam M, Belele C, Dorweiler JE, Chandler VL. Differential chromatin structure within a tandem array 100 kb upstream of the maize b1 locus is associated with paramutation. Genes Dev. 2002;16: 1906–1918. doi: 10.1101/gad.1006702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Belele CL, Sidorenko L, Stam M, Bader R, Arteaga-Vazquez MA, Chandler VL. Specific Tandem Repeats Are Sufficient for Paramutation-Induced Trans-Generational Silencing. PLoS Genet. 2013;9 doi: 10.1371/journal.pgen.1003773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haring M, Bader R, Louwers M, Schwabe A, Van Driel R, Stam M. The role of DNA methylation, nucleosome occupancy and histone modifications in paramutation. Plant J. 2010;63: 366–378. doi: 10.1111/j.1365-313X.2010.04245.x [DOI] [PubMed] [Google Scholar]
- 18.Sidorenko L, Chandler V. RNA-dependent RNA polymerase is required for enhancer-mediated transcriptional silencing associated with paramutation at the maize p1 gene. Genetics. 2008;180: 1983–1993. doi: 10.1534/genetics.108.095281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hollick JB, Kermicle JL, Parkinson SE. Rmr6 maintains meiotic inheritance of paramutant states in Zea mays. Genetics. 2005;171: 725–740. doi: 10.1534/genetics.105.045260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sloan AE, Sidorenko L, McGinnis KM. Diverse gene-silencing mechanisms with distinct requirements for RNA polymerase subunits in Zea mays. Genetics. 2014;198: 1031–42. doi: 10.1534/genetics.114.168518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sidorenko L, Dorweiler JE, Cigan AM, Arteaga-Vazquez M, Vyas M, Kermicle J, et al. A dominant mutation in mediator of paramutation2, one of three second-largest subunits of a plant-specific RNA polymerase, disrupts multiple siRNA silencing processes. PLoS Genet. 2009;5 doi: 10.1371/journal.pgen.1000725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stonaker JL, Lim JP, Erhard KF, Hollick JB. Diversity of Pol IV function is defined by mutations at the maize rmr7 locus. PLoS Genet. 2009;5 doi: 10.1371/journal.pgen.1000706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hale CJ, Stonaker JL, Gross SM, Hollick JB. A novel Snf2 protein maintains trans-generational regulatory states established by paramutation in maize. PLoS Biol. 2007;5: 2156–2165. doi: 10.1371/journal.pbio.0050275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barbour J-ER, Liao IT, Stonaker JL, Lim JP, Lee CC, Parkinson SE, et al. Required to maintain repression2 is a novel protein that facilitates locus-specific paramutation in maize. Plant Cell. 2012;24: 1761–1775. doi: 10.1105/tpc.112.097618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arteaga-Vazquez M, Sidorenko L, Rabanal F a, Shrivistava R, Nobuta K, Green PJ, et al. RNA-mediated trans-communication can establish paramutation at the b1 locus in maize. Proc Natl Acad Sci U S A. 2010;107: 12986–12991. doi: 10.1073/pnas.1007972107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dorweiler JE, Carey CC, Kubo KM, Hollick JB, Kermicle JL, Chandler VL. Mediator of Paramutation1 Is Required for Establishment and Maintenance of Paramutation At Multiple Maize Loci. Plant Cell. 2000;12: 2101–2118. doi: 10.1105/tpc.12.11.2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lisch D, Carey CC, Dorweiler JE, Chandler VL. A mutation that prevents paramutation in maize also reverses Mutator transposon methylation and silencing. Proc Natl Acad Sci U S A. 2002;99: 6130–6135. doi: 10.1073/pnas.052152199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McGinnis KM, Springer C, Lin Y, Carey CC, Chandler V. Transcriptionally silenced transgenes in maize are activated by three mutations defective in paramutation. Genetics. 2006;173: 1637–1647. doi: 10.1534/genetics.106.058669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Woodhouse MR, Freeling M, Lisch D. Initiation, establishment, and maintenance of heritable MuDR transposon silencing in maize are mediated by distinct factors. PLoS Biol. 2006;4: 1678–1688. doi: 10.1371/journal.pbio.0040339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woodhouse MR, Freeling M, Lisch D. The mop1 (mediator of paramutation1) mutant progressively reactivates one of the two genes encoded by the MuDR transposon in maize. Genetics. 2006;172: 579–592. doi: 10.1534/genetics.105.051383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grotewold E, Athma P, Peterson T. Alternatively spliced products of the maize P gene encode proteins with homology to the DNA-binding domain of myb-like transcription factors. Proc Natl Acad Sci U S A. 1991;88: 4587–4591. doi: 10.1073/pnas.88.11.4587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grotewold E, Drummond BJ, Bowen B, Peterson T. The myb-homologous P gene controls phlobaphene pigmentation in maize floral organs by directly activating a flavonoid biosynthetic gene subset. Cell. 1994;76: 543–553. doi: 10.1016/0092-8674(94)90117-1 [DOI] [PubMed] [Google Scholar]
- 33.Brink R, Styles ED. A collection of pericarp factors. Maize Genet Coop News Lett. 1966;40: 149–160. [Google Scholar]
- 34.Das OP, Messing J. Variegated phenotype and developmental methylation changes of a maize allele originating from epimutation. Genetics. 1994;136: 1121–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goettel W, Messing J. Paramutagenicity of a p1 epiallele in maize. Theor Appl Genet. 2013;126: 159–177. doi: 10.1007/s00122-012-1970-z [DOI] [PubMed] [Google Scholar]
- 36.Sidorenko L V., Li X, Cocciolone SM, Chopra S, Tagliani L, Bowen B, et al. Complex structure of a maize Myb gene promoter: Functional analysis in transgenic plants. Plant J. 2000;22: 471–482. doi: 10.1046/j.1365-313X.2000.00750.x [DOI] [PubMed] [Google Scholar]
- 37.Chopra S, Athma P, Li XG, Peterson T. A maize Myb homolog is encoded by a multicopy gene complex. Mol Gen Genet. 1998;260: 372–380. doi: 10.1007/s004380050906 [DOI] [PubMed] [Google Scholar]
- 38.Goettel W, Messing J. Epiallele biogenesis in maize. Gene. Elsevier B.V.; 2013;516: 8–23. doi: 10.1016/j.gene.2012.12.034 [DOI] [PubMed] [Google Scholar]
- 39.Sekhon RS, Wang PH, Sidorenko L, Chandler VL, Chopra S. Maize Unstable factor for orange1 Is Required for Maintaining Silencing Associated with Paramutation at the pericarp color1 and booster1 Loci. PLoS Genet. 2012;8 doi: 10.1371/journal.pgen.1002980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rhee Y, Sekhon RS, Chopra S, Kaeppler S. Tissue culture-induced novel epialleles of a Myb transcription factor encoded by pericarp color1 in maize. Genetics. 2010;186: 843–855. doi: 10.1534/genetics.110.117929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sekhon RS, Peterson T, Chopra S. Epigenetic modifications of distinct sequences of the p1 regulatory gene specify tissue-specific expression patterns in maize. Genetics. 2007;175: 1059–1070. doi: 10.1534/genetics.106.066134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chopra S, Cocciolone SM, Bushman S, Sangar V, McMullen MD, Peterson T. The maize unstable factor for orange1 is a dominant epigenetic modifier of a tissue specifically silent allele of pericarp color1. Genetics. 2003;163: 1135–46. Available: http://www.ncbi.nlm.nih.gov/pubmed/1266355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sekhon RS, Chopra S. Progressive loss of DNA methylation releases epigenetic gene silencing from a tandemly repeated maize Myb gene. Genetics. 2009;181: 81–91. doi: 10.1534/genetics.108.097170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nobuta K, Lu C, Shrivastava R, Pillay M, De Paoli E, Accerbi M, et al. Distinct size distribution of endogeneous siRNAs in maize: Evidence from deep sequencing in the mop1-1 mutant. Proc Natl Acad Sci U S A. 2008;105: 14958–14963. doi: 10.1073/pnas.0808066105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fujimoto R, Kinoshita Y, Kawabe A, Kinoshita T, Takashima K, Nordborg M, et al. Evolution and control of imprinted FWA genes in the genus Arabidopsis. PLoS Genet. 2008;4 doi: 10.1371/journal.pgen.1000048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gent JI, Ellis NA, Guo L, Harkess AE, Yao Y, Zhang X, et al. CHH islands: De novo DNA methylation in near-gene chromatin regulation in maize. Genome Res. 2013;23: 628–637. doi: 10.1101/gr.146985.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li Q, Gent JI, Zynda G, Song J, Makarevitch I, Hirsch CD, et al. RNA-directed DNA methylation enforces boundaries between heterochromatin and euchromatin in the maize genome. Proc Natl Acad Sci U S A. 2015; 1514680112-. doi: 10.1073/pnas.1514680112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gent JI, Madzima TF, Bader R, Kent MR, Zhang X, Stam M, et al. Accessible DNA and relative depletion of H3K9me2 at maize loci undergoing RNA-directed DNA methylation. Plant Cell. 2014;26: 4903–17. doi: 10.1105/tpc.114.130427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu C, Chen J, Zhang Y, Hu Q, Su W, Kuang H. Miniature inverted-repeat transposable elements (MITEs) have been accumulated through amplification bursts and play important roles in gene expression and species diversity in oryza sativa. Mol Biol Evol. 2012;29: 1005–1017. doi: 10.1093/molbev/msr282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei L, Gu L, Song X, Cui X, Lu Z, Zhou M, et al. Dicer-like 3 produces transposable element-associated 24-nt siRNAs that control agricultural traits in rice. Proc Natl Acad Sci. 2014;111: 3877–3882. doi: 10.1073/pnas.1318131111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eichten SR, Briskine R, Song J, Li Q, Swanson-Wagner R, Hermanson PJ, et al. Epigenetic and genetic influences on DNA methylation variation in maize populations. Plant Cell. 2013;25: 2783–97. doi: 10.1105/tpc.113.114793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jackson JP, Lindroth AM, Cao X, Jacobsen SE. Control of CpNpG DNA methylation by the KRYPTONITE histone H3 methyltransferase. Nature. 2002;416: 556–60. doi: 10.1038/nature731 [DOI] [PubMed] [Google Scholar]
- 53.Bernatavichute Y V., Zhang X, Cokus S, Pellegrini M, Jacobsen SE. Genome-wide association of histone H3 lysine nine methylation with CHG DNA methylation in Arabidopsis thaliana. PLoS One. 2008;3 doi: 10.1371/journal.pone.0003156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoekenga O a, Muszynski MG, Cone KC. Developmental patterns of chromatin structure and DNA methylation responsible for epigenetic expression of a maize regulatory gene. Genetics. 2000;155: 1889–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Enke RA, Dong Z, Bender J. Small RNAs prevent transcription-coupled loss of histone H3 lysine 9 methylation in Arabidopsis thaliana. PLoS Genet. 2011;7: 1–10. doi: 10.1371/journal.pgen.1002350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jia Y, Lisch DR, Ohtsu K, Scanlon MJ, Nettleton D, Schnable PS. Loss of RNA-dependent RNA polymerase 2 (RDR2) function causes widespread and unexpected changes in the expression of transposons, genes, and 24-nt small RNAs. PLoS Genet. 2009;5 doi: 10.1371/journal.pgen.1000737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walker EL, Panavas T. Structural features and methylation patterns associated with paramutation at the r1 locus of Zea mays. Genetics. 2001;159: 1201–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Saghai-Maroof M a, Soliman KM, Jorgensen R a, Allard RW. Ribosomal DNA spacer-length polymorphisms in barley: mendelian inheritance, chromosomal location, and population dynamics. Proc Natl Acad Sci U S A. 1984;81: 8014–8018. doi: 10.1073/pnas.81.24.8014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kimura H, Hayashi-Takanaka Y, Goto Y, Takizawa N, Nozaki N. The organization of histone H3 modifications as revealed by a panel of specific monoclonal antibodies. Cell Struct Funct. 2008;33: 61–73. doi: 10.1247/csf.07035 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Methylation profile at CG, CHG and CHH contexts were obtained by genomic bisulfite sequencing. The y-axis shows percentage of DNA methylation. Individual sites are shown as circles.
(TIF)
Crosses between P1-rr’/P1-rr’ carrying homozygous (mop1-1/mop1-1) and heterozygous (Mop1/ mop1-1) were made to test whether mop1-1-induced reactivation of P1-rr’ is associated with hypomethylation. Since the stocks also carried a silenced B’ allele, plants containing homozygous mop1-1 were identified by the purple color of plant body, whereas plants containing heterozygous Mop1/mop1-1 showed green plant without purple pigmentation.
(TIF)
(A) Size distribution of small RNAs in P1-wr;Mop1 and P1-wr;mop1-1 samples with sum of small RNA abundances normalized to 1 million reads. (B) Size distribution of small RNA abundance after normalization to the abundance of 22-nt class. Three independent Mop1 samples and two independent mop1-1 samples are shown.
(TIF)
(A) Line diagram of P1-wr showing gene structure and different regulatory elements (see Fig 1 legend for details). (B) Abundance of siRNA was shown on sense and antisense strands from P1-wr;Mop1/mop1-1 and P1-wr;mop1-1/mop1-1 samples as reads per million (RPM) and normalized to the 22-nt size class (see Methods). Green, grey, and red shaded areas indicate regulatory regions, TEs, and exons, respectively. Proximal enhancer is shown as region between black dashed lines. siRNA abundances from P1-wr;Mop1/mop1-1 samples (blue) and from P1-wr;mop1-1/mop1-1 (red) are shown.
(TIF)
(XLSX)
Data Availability Statement
Data is available from the GEO database with accession numbers GSM2652417 and GSE68510 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE68510).