

Abstract
Nickel-substituted rubredoxin (NiRd) is a functional enzyme mimic of hydrogenase, highly active for electrocatalytic and solution-phase hydrogen generation. Spectroscopic methods can provide valuable insight into the catalytic mechanism, provided the appropriate technique is used. In this study, we have employed multi-wavelength resonance Raman spectroscopy coupled with DFT calculations on an extended active-site model of NiRd to probe the electronic and geometric structures of the resting state of this system. Excellent agreement between experiment and theory is observed, allowing normal mode assignments to be made on the basis of frequency and intensity analyses. Both metal-ligand and ligand-centered vibrational modes are enhanced in the resonance Raman spectra. The latter provide information about the hydrogen bonding network and structural distortions due to perturbations in the secondary coordination sphere. To reproduce the resonance enhancement patterns seen for high-frequency vibrational modes, the secondary coordination sphere must be included in the computational model. The structure and reduction potential of the NiIIIRd state have also been investigated both experimentally and computationally. This work begins to establish a foundation for computational resonance Raman spectroscopy to serve in a predictive fashion for investigating catalytic intermediates of NiRd.
TOC image
The electronic and geometric structures of nickel-substituted rubredoxin, a highly active enzyme mimic of hydrogenase, were investigated using multi-wavelength resonance Raman spectroscopy. DFT calculations on an extended active site model showed excellent agreement with experiment only when the full secondary coordination environment was considered, emphasizing the importance of outer sphere interactions. Validation of this model across multiple oxidation states establishes a foundation for using calculations synergistically with experiment to examine enzymatic intermediates during hydrogen evolution.
Introduction
Hydrogen is an energy-dense, clean-burning fuel that has long been touted as an ideal energy storage medium for either direct use or for the generation of secondary transport fuels.1,2 Because of its industrial value, great effort has been aimed at developing sustainable methods for hydrogen generation and processing, with significant advances made on diverse fronts, from engineering hydrogen storage materials to developing efficient catalysts. In nature, hydrogen production and oxidation is carried out through the use of specialized metalloenzymes called hydrogenases (H2ases).3 These large, multicofactor systems contain either di-iron ([FeFe]) or heterobimetallic nickel-iron ([NiFe]) active sites and feature earth-abundant metals, high turnover frequencies, activity under mild reaction conditions in aqueous solutions, and, notably, very high thermodynamic efficiencies for H2 conversion reactions.3,4 These favorable characteristics have motivated diverse spectroscopic investigations to arrive at a general understanding of the enzymatic mechanism (Figure 1).3–10 The negligible catalytic overpotential renders the enzymes bidirectional, with important consequences both in vivo and in vitro.11–13 However, H2ases are relatively fragile, costly proteins, and as such, are generally poorly suited for large-scale application.14–16 Alternative methods for sustainable hydrogen generation are needed.
Figure 1.
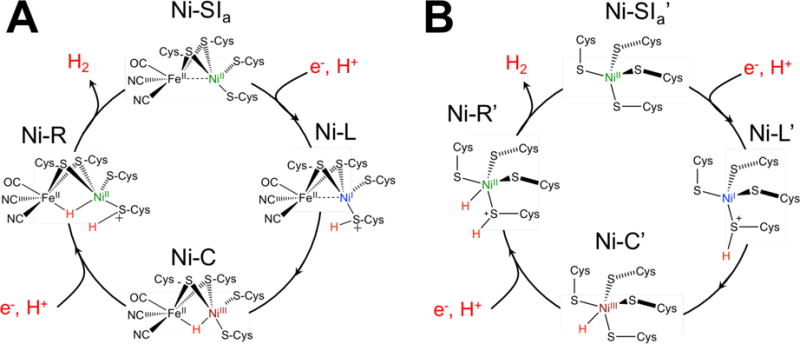
Proposed catalytic mechanisms for hydrogen generation by (A) [NiFe] hydrogenases and (B) NiRd.
Extensive research in the synthetic inorganic community aims to develop molecular catalysts that reproduce the advantages of the protein systems. A number of important advances have been made in recent years, including the use of first-row transition metals, low catalytic overpotentials that lead to bidirectionality, activity in aqueous solutions under mild conditions, electrocatalytic turnover frequencies that exceed solution-phase enzymatic rates, and incorporation of protein-like elements in the secondary coordination sphere.17–20 Many excellent reviews describing these compounds and highlighting progress towards increased activity have been written in recent years.21–26 However, no single catalyst has been able to incorporate all of the unique features of the native H2ase enzymes.
We have elected to address this challenge by developing protein-based models of the [NiFe] hydrogenases. Using rubredoxin (Rd), a small, iron-containing electron transfer protein, as a scaffold, we have replaced the native metal center with nickel to generate a monomeric, tetrathiolate-coordinated nickel(II) active site (NiRd). This system serves as a basic structural model of the native enzyme as it directly mimics the primary atomic coordination sphere of the redox-active metal in the [NiFe] H2ases. Further illustrating the relevance of this model protein system, we have recently been able to show high levels of solution-phase and electrochemical hydrogen evolution by NiRd, with electrocatalytic rates approaching that of the native enzyme and a modest overpotential of 540 mV.27 Nickel-substituted rubredoxin (NiRd) shows great promise as an artificial hydrogenase; unlike the native H2ases, it is easily generated, chemically and thermally robust, and highly tunable to enable catalyst optimization. The simplicity of the system also provides an opportunity to better understand the native [NiFe] hydrogenases, including investigation into the role that the FeII(CN)2CO fragment plays during catalysis and identifying the impact of having two bridging cysteines rather than four terminal ones, as found in NiRd. Towards this end, we are very interested in understanding the mechanism of catalytic hydrogen evolution by NiRd.
A preliminary mechanism for NiRd based on the current model for the [NiFe] hydrogenases is proposed in Figure 1. Like the [NiFe] hydrogenases, at least one nickel-hydride species is likely involved in catalysis.9,28,29 Additionally, while this is a controversial point in the native [NiFe] enzymes,6,28 the thiolate ligands in NiRd likely serve as the basic site for protonation (Ni-L’ and Ni-R’), showcasing the chemical competence of nickel-bound thiolates for intramolecular proton transfer. The mechanisms of the protein-based systems differ from the well-studied DuBois-type nickel-phosphine systems, which rely on intramolecular proton transfer from an amine group in the outer coordination sphere and proceed through distinct reduction and protonation steps, with Ni0 intermediates identified during the catalytic process.30,31 As with the model compounds and the native systems, detailed spectroscopic studies will be of great utility to characterize NiRd intermediates and identify mechanistic details, particularly when considering short-lived or reactive species.
Vibrational techniques are ideally suited to address questions of structure and bonding. However, conventional infrared (IR) spectroscopy is limited by the large absorption of water, and both IR and Raman spectroscopies suffer from a lack of selectivity, which is detrimental in the case of proteins. By contrast, resonance Raman (RR) spectroscopy is a highly selective and sensitive method that has been extensively applied to the study of metalloprotein active sites and a number of model compounds, including those related to hydrogenases.32–37 This scattering technique uses laser excitation wavelengths that are resonant with an electronic transition of a chromophore, resulting in significant intensity increases exclusively for vibrations coupled to that electronic transition.38,39 Because the enhancement mechanism relies on coupling between the electronic and vibrational excited states, RR spectroscopy provides information on both electronic and geometric structure.39 Use of multiple excitation wavelengths can access different transitions, which gives distinct RR intensity patterns that report on the nature of the excited states.40,41
In conjunction with experimental methods, theoretical calculations can be an invaluable aid for the interpretation of spectroscopic results. Computational methods are also increasingly employed to guide experimental design, which can preserve resources and accelerate progress.42 Prior to being used in a predictive fashion, however, a structural model must be developed and validated with respect to the computational methodology and available experimental data.43,44 Because the costs of computing an entire protein at the density functional theory (DFT) or ab initio levels would be prohibitive, this step is particularly important in theoretical studies on enzymes to ensure an appropriate level of truncation was applied.45,46
In this work, we have used multiple excitation wavelengths to obtain resonance Raman spectra of NiRd across different excited states to fully characterize the electronic and vibrational structure of the resting state of the enzyme. Isotope experiments were used to distinguish modes that are sensitive to hydrogen bonding within the active site, which will report on perturbations to this network during catalysis. In parallel, DFT studies were performed on models of the NiRd active site that varied in size to probe the importance of the outer coordination sphere. Structural, spectroscopic, and redox properties of these active-site models were calculated and compared to experimental data. Excellent agreement was obtained between the observed RR spectra and predicted vibrational frequencies and intensity patterns for the extended model, indicating that the calculations accurately reflect the electronic structure of the NiRd active site. Together, these results provide a foundation upon which to investigate the mechanism of hydrogen evolution by NiRd using resonance Raman spectroscopy synergistically with DFT calculations.
Materials and Methods
Sample Preparation
Expression, purification, and metal substitution of rubredoxin was carried out as previously described with minor modifications.27 Briefly, an additional purification step was included. The protein was run on a size-exclusion column (5 mL Superdex-75, prep-grade, Sigma-Aldrich) in 50 mM Tris buffer, pH 8.0, following the trichloroacetic acid precipitation and metal exchange to ensure complete purity of all protein samples. Isotopic substitution experiments were prepared by exchanging protein samples into 15 mM phosphate-buffered D2O (99.9% isotopic purity, Sigma-Aldrich) at pH 8.0 by centrifugal filtration (Millipore Centricons, 3.5 kDa MWCO). Solvent equilibration was allowed to proceed at 4 ˚C for at least 12 hrs prior to data collection.
Electrochemistry
All electrochemical experiments were carried out on a WaveNow potentiostat (Pine Research Instrumentation, Durham, NC) inside a nitrogen atmosphere glovebox, O2 <10 ppm (Vigor Technologies). Solution-phase electrochemical studies of NiRd were performed using 150 μL of sample contained within a 10 mm ID borosilicate tube. A glassy carbon working electrode (CH Instruments) was used in a conventional three-electrode set-up, with a miniature Ag/AgCl reference electrode (Pine Research Instrumentation) and a platinum wire as the counter electrode. The reported potentials against NHE were calculated by the addition of +198 mV to the experimentally measured potentials.47
Spectrophotometric Titrations
Potentiometric titrations were performed using a Shimadzu UV-2600 Spectrophotometer to monitor changes in the UV-visible absorbance spectrum of NiRd. Aliquots of potassium ferricyanide (Sigma-Aldrich) were added to a solution of NiIIRd in the indicated buffer and allowed to equilibrate for 15 minutes prior to recording the solution potential and optical spectrum. Potential measurements were made using a digital multimeter with a platinum working electrode and a Ag/AgCl reference electrode. As the optical absorption of K3[FeIII(CN)6] overlaps with the NiRd charge transfer transitions, the absorbance values at 740 nm reflecting the NiII d-d transitions were monitored to obtain the concentration of NiIIRd as a function of solution potential for the Nernstian curves shown in Figures 7B and S21.
Figure 7.
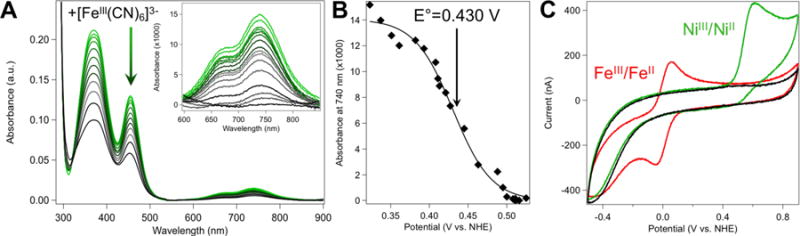
(A) Representative absorbance spectra showing the potentiometric titration of NiIIRd with potassium ferricyanide (40 μM NiRd, 50 mM Tris-buffer, pH 8.0; spectra shown reflect up to 1 equiv. ferricyanide added). (B) Absorbance of NiRd at 740 nm as increasing amounts of ferricyanide are added (50 mM Tris, pH 8.0). The line shown represents a least-squares fit of the data to the Nernst equation (E° = 0.430 V vs. NHE). (C) Cyclic voltammogram of NiRd (green), FeRd (red) and the blank glassy carbon electrode (black) in 100 mM KCl, 50 mM acetate buffer, pH 4.5 (ν = 10 mV/s).
Resonance Raman Spectroscopy
Resonance Raman (RR) spectroscopy was performed at room temperature with samples contained within a flame-sealed sodium borosilicate capillary tube (~1 mm ID). A mixed gas Kr-Ar laser (Coherent Innova Spectrum 70-C, Laser Innovations) provided excitation at 458 nm and 488 nm. The spectra obtained with 364 nm and 407 nm excitation employed a tunable titanium: sapphire laser (Spectra-Physics Tsunami) pumped by a 25 W DPSS laser (Spectra-Physics Millennia eV) and configured with a 10-ps Gires-Tournais interferometer, similar to the system described by Doig and coworkers.48 The ~4.0 W fundamental beam at 728 nm or 814 nm was focused into a β-barium borate (BBO) crystal (Eksma Optics) for second-harmonic generation, and the ~20 mW of 364 nm or ~10 mW of 407 nm light was separated from the fundamental with a Pellin-Broca prism and a dichroic mirror. The excitation beam of ~20 mW, ~10 mW, 25–30 mW, and 25–30 mW at 364 nm, 407 nm, 458 nm, and 407 nm, respectively was focused onto the sample using a 100 mm RFL, 90° off-axis parabolic mirror (Thorlabs). Scattered light was collected at a 135° backscattering geometry by a 55 mm FD f/1.2 mounted camera lens (Canon, 458 nm and 488 nm excitation) or a 50 mm FD f/0.8 UV fused silica aspheric lens (Edmund Optics, 364 nm and 407 nm excitation) and focused onto the 100 μm entrance slit of a spectrograph using a 50 mm, f/4 achromatic lens (Newport Corporation, 458 nm and 488 nm excitation) or a 50 mm, f/4.6 uncoated UV fused silica lens (CVI Optics, 364 nm and 407 nm excitation). Rayleigh scattering was rejected by the appropriate long-pass edge filter (Semrock RazorEdge). Raman scattered light was dispersed in an f/4.6, 0.32 m imaging spectrograph (Princeton Instruments, IsoPlane SCT 320) equipped with an 1800 gr/mm (407 nm, 458 nm, and 488 nm excitation) or 3600 gr/mm (364 nm excitation) holographic grating and imaged onto a Peltier-cooled CCD detector (Princeton Instruments, Pixis 100B).
Raman signal intensity was optimized and calibrated using known bands from a 1:1 v/v mixture of toluene and acetonitrile.49 The band pass and accuracy were found to be 9 cm−1 and +/− 1 cm−1, respectively. One-minute signal integrations were summed up over two hours for each sample to obtain accumulated spectra of NiRd and ZnRd as well as their respective protiated and deuterated buffers. Controls were performed at varying powers (0.7 – 115 mW) to ensure experiments were carried out under conditions of linear response (Figure S1). Single-pixel spikes due to cosmic rays impinging upon the detector were removed manually. The raw data is shown in Figure S2. Features from the appropriate buffer were subtracted from the spectra of NiRd (typically in a 0.6 –1 ratio to account for self-absorption), and a spline was drawn along the baseline of the resulting spectrum and subtracted to remove a broad fluorescent background; this process is shown in Figure S2, inset.50,51 Intensity normalization was performed using the non-resonant Raman phenylalanine peak at 1004 cm−1.52 The summed raw ZnRd spectra were subtracted from the raw NiRd spectra to obtain better signal-to-noise ratios and clearly identify bands attributed to the nickel-thiolate chromophore; these spectra were then baseline-corrected and scaled to the appropriate, Phenormalized, buffer-subtracted spectra using the remaining peaks (Figure S3). Peak positions of overlapping bands were resolved by Gaussian decomposition of the corrected NiRd spectra (Figure S4).
Construction of the Active-Site Model
The nickel-substituted rubredoxin (NiRd) computational model was constructed using the UCSF Chimera visualization software53 starting from the X-ray crystal structure of oxidized iron rubredoxin from Desulfovibrio desulfuricans (Dd) ATCC 27774 (PDB Code: 6RXN).54 Amino acid numbering refers to the Dd Rd. For the full model ([NiII(CNVC)(CPVC)]2−), the complete side chains of the C6N7V8C9 and C32P33V34C35 loops were retained (where C6 refers to cysteine, the 6th residue in the polypeptide chain), and the metal center was changed to a nickel ion. To model the secondary coordination sphere and retain natural hydrogen bonding networks, G10, G36, and V37 were also included.55 The loops were terminated with N-acetylation and C-amidation for charge neutrality. All other atoms were removed. For the backbone-only model ([NiII(CGGC)(CPGC)]2−), all side chains other than the coordinating cysteines and the proline residue were replaced by glycine residues, with G10, G36, and “G”37 still included. These loops were also capped with N-terminal acetylation and C-terminal amidation. The [NiII(NACA)4]2− model included only the cysteine residues capped with N- and C-terminal acetylation for charge neutrality. The NiIIRd and NiIIIRd structures were optimized with charges of −2 and −1 and multiplicities of 3 and 2, respectively. The complete geometry-optimized structures of the three active-site models are shown in the Supporting Information (Figure S5).
Calculations
All calculations were carried out using the ORCA computational chemistry program56–58 at the Ohio Supercomputer Center.59 Geometry optimizations were performed at the density functional theory (DFT) level of computation employing the BP8660,61 functional with the RI approximation62 followed by the B3LYP63,64 functional employing the RIJCOSX approximation in the gas phase.65 The Cα atoms of each amino acid were constrained in space to retain the fold of the protein without restricting the flexibility of the active-site nickel and ligands.66–68 In the absence of these constraints, distortion of the protein backbone (Figure S6) and slight differences in the TD-DFT calculations were observed (Figure S7), and significantly higher computational costs were incurred. The def2-SVP basis set was used for all atoms except the cysteine sulfur atoms and the nickel center, for which the def2-TZVPP basis set was used.69 The basis sets were benchmarked and the active site geometries optimized using both the TZVP and def2-TZVPP basis sets on all non-coordinating atoms, with few changes seen (Figure S8 and Table S1). Given the size of these models and resource constraints, the use of different basis sets for the primary and outer coordination spheres is necessary and is often used when considering extended models of metalloprotein active sites.44,66,67,70 Scalar relativistic effects were corrected for using the all-electron zero-order-relativistic-approximation (ZORA) applied to all atoms, and dispersion interactions were treated with the DFT-D3 atom-pairwise correction as implemented in ORCA.71–74
The absorption spectra of the geometry-optimized NiIIRd structures were calculated using time-dependent density functional theory (TD-DFT) calculations. The Tamm-Dancoff approximation was performed for the first 80 roots using the B3LYP functional.75–77 Vertical excitation energies and molar extinction coefficients for TD-DFT transitions were calculated using the ORCA_MAPSPC module to give the transitions shown in Figure 3. The transitions were convolved with a Gaussian linewidth of 2500 cm−1 to generate the absorption spectra in Figures S7 and S9. Vibrational frequencies were calculated using two-sided numerical differentiation. Resonance Raman intensities for each relevant electronic transition were obtained within the independent mode displaced harmonic oscillator (IMDHO) model using the gradient of the excited state energy at the ground state equilibrium geometry to calculate the dimensionless normal coordinate displacements (Δs) for each of the normal modes.78 The program ORCA_ASA was used to simulate the RR spectra for the first-order vibrational transitions.41,78,79 Normal modes were assigned by comparison to experimental spectra on the basis of similar frequencies and intensities. We note that absolute RR cross-sections are not calculated with this method. Instead, the theoretical spectral intensities were kept constant relative to each other and scaled collectively for comparison to experiment.
Figure 3.
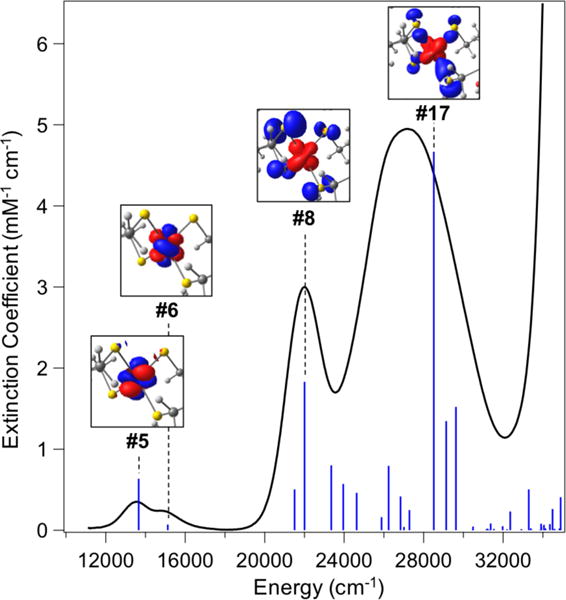
Optical absorption spectrum (black) of NiIIRd overlaid with TD-DFT-calculated optical transitions for the full model of NiIIRd (blue sticks). (Inset) Electron difference densities for the dominant transitions in the visible and near-UV region of the spectrum; blue lobes reflect loss of electron density, while red lobes reflect increased electron density in the transition. Difference density figures are labeled according to the energy-ordered number of that transition.
The reduction potential of the NiIIIRd/NiIIRd one-electron couple was calculated using a thermodynamic cycle as in Ref. 79 (for a detailed description, see the Supporting Information, Scheme S1).80 Solvation free energies were computed at the optimized gas phase geometries using the conductor-like screening model (COSMO) with the dielectric constant of ε = 80.4 for water.81 This dielectric constant was chosen because the full computational model represents a highly surface-exposed portion of the protein (Figure S10). Additional calculations showed only a weak dependence of reduction potential on dielectric constant above ε~10 (Figure S11). The relative reduction potential was calculated with respect to the calculated SHE value of 4.281 V.82 The reduction potential of ferrocene (D5d symmetry) was also calculated to estimate the accuracy of the computational method and compared to experimental data with respect to the SCE reference electrode (+0.241 V vs. SHE).83
Results
DFT-Optimized Model Geometries
The full computational model of NiIIRd includes the complete side chains of both CXXC loops as well as an extended backbone with N-terminal acetylation and C-terminal amidation to more accurately mimic the protein environment. For structural validation, the DFT-optimized geometry of the model was compared to the crystal structure of the NiII-substituted rubredoxin from Clostridium pasteurinum (Figure 2A and Table 1), which is the only nickel-substituted rubredoxin that has been crystallographically characterized.84 The protein backbone imposes a tetrahedral configuration at the metal center, which, for nickel(II), results in a high-spin, S=1 configuration that undergoes Jahn-Teller distortion to a tetragonally elongated pseudo-D2d symmetry; a semi-quantitative molecular orbital diagram for the nickel-based d-orbitals is shown in the Supporting Information (Figure S12). While these general trends are also observed crystallographically, the experimental structure shows a slightly higher level of distortion at the active site, with different cysteines participating in the longer nickel-sulfur bonds. Specifically, the Cp NiIIRd structure shows elongation by almost 0.2 Å of the Ni-S(Cys9) and Ni-S(Cys32) bond lengths, while the optimized DFT model shows only 0.05 Å elongation along the Ni-S(Cys6) and Ni-S(Cys32) bonds. However, the reported structure of the Cp NiIIRd is of relatively low resolution (2.0 Å) with a B-factor of 38.4 Å2 for the metal ion, which limits the extent to which the calculated and experimental active site geometries can be compared. In the secondary coordination sphere, the close hydrogen bonding network that has been noted in both the NMR and x-ray diffraction structures of iron- and nickel-substituted Rds is preserved, with distances of ~3.4 – 3.7 Å between the amide nitrogen and cysteine sulfur atoms (Figure 2A).55,84,85
Figure 2.
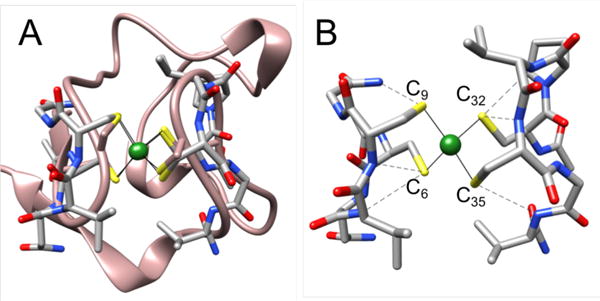
DFT-optimized geometry of full NiIIRd model (gray) (A) overlaid with the crystal structure of Cp NiIIRd (pink, PDB ID 1R0J) and (B) with the hydrogen-bonding network indicated with dashed lines. The full sidechains that are included in the model are shown; hydrogen atoms are omitted for clarity.
Table 1.
Selected bond distances of the geometry optimized NiRd models.
| X-Ray 1R0J.pdb | Full NiIIRd | Backbone NiIIRd | [NiII(NACA)4]2− | Full NiIII Rd | Backbone NiIIIRd | |
|---|---|---|---|---|---|---|
| Bond Lengths (Å) | ||||||
| Ni-S6a | 2.254 | 2.345 | 2.335 | 2.318 | 2.234 | 2.227 |
| Ni-S9 | 2.381 | 2.282 | 2.294 | 2.297 | 2.204 | 2.212 |
| Ni-S32 | 2.477 | 2.332 | 2.359 | 2.318 | 2.217 | 2.234 |
| Ni-S35 | 2.134 | 2.287 | 2.285 | 2.290 | 2.198 | 2.209 |
| Hydrogen Bonding Network Distancesb (Å) | ||||||
| Cys6-Val8 | 3.629 | 3.557 | 3.449 | — | 3.614 | 3.585 |
| Cys6-Cys9 | 3.672 | 3.676 | 3.609 | 3.399 | 3.521 | 3.595 |
| Cys9-Tyr11 | 3.543 | 3.314 | 3.437 | — | 3.393 | 3.367 |
| Cys32-Val34 | 3.513 | 3.436 | 3.345 | — | 3.471 | 3.361 |
| Cys32- Cys35 | 3.580 | 3.493 | 3.496 | 3.522 | 3.364 | 3.352 |
| Cys35-Val37 | 4.077 | 3.649 | 3.536 | — | 3.510 | 3.441 |
Sn refers to the sulfur atom on the nth cysteine.
Hydrogen bonding values shown as distances between heavy atoms, i.e., d(S-N).
Truncated models of the NiRd active site were constructed and compared to the full model to investigate the role of the outer coordination sphere (Table 1). The simplest “[NiII(NACA4)]2−” model includes only the coordinating cysteine residues terminated with N- and C-terminal acetylation. This model is highly symmetric yet retains the pattern of two long and two short Ni-S bonds (Figure S13). A larger “backbone” model was also developed that retains the full CXXC loops and extended backbone atoms but replaces the non-cysteine side chains with glycine residues. The effects of this truncation on the structure of the active site are minor, as shown in Table 1 and Supporting Information, Table S1 and Figure S14. However, as discussed below, these changes had notable effects on the predicted spectroscopic properties of the nickel center.
A computational model for the experimentally-accessible oxidized NiIIIRd was also constructed.86,87 The NiIIIRd structure was obtained by starting with the optimized NiIIRd geometry for the full model, removing an electron, and re-optimizing the coordinates of the oxidized species. As expected, the nickel-thiolate bonds contract upon oxidation, and the Jahn-Teller distortion is lifted to give a near-ideal tetrahedral geometry for the d7 configuration (Table 1 and Figure S15). The hydrogen-bonding network is only slightly perturbed. While no crystal structure exists of the NiIIIRd species, experimental support for the DFT-optimized structure was gained by comparison to spectroscopic properties and the thermodynamic reduction potential (vide infra).
Absorption spectroscopy
Nickel-substituted rubredoxin exhibits strong absorption bands in the visible region of the spectrum, owing to the tetrahedral geometry and orbital overlap between the metal center and sulfur thiolates.88–90 To validate the computational model of NiRd beyond structural markers, predicted spectroscopic properties were compared to experimental values. The optical absorption spectrum of NiIIRd is overlaid with the calculated absorption spectrum of the full NiIIRd model in Figure 3. Across the visible and near-UV wavelength range, there is good agreement for both the energies and intensities of the transitions. Notably, no energy offset or correction was needed for the direct comparison of the experimental and calculated absorption spectrum. The low-energy transitions at 13,654 cm−1 (ε = 636 M−1 cm−1) and 15,118 cm−1 (ε = 70 M−1 cm−1) had previously been assigned to d-d transitions on the basis of ligand field theory.88,90 The calculations agree with this assignment, with the majority of the electron difference density for States 5 and 6 arising from metal-based electron rearrangement from the dxy to the dx2-y2 orbital (Figure 3, inset). At 22,001 cm−1, State 8 can largely be ascribed to a ligand-to-metal charge-transfer transition in the sulfur p-π manifold; this is in good agreement, again, with ligand-field arguments and early MCD experiments on a related NiRd and nickel thiolate model compounds.88,91 Finally, in the near-UV region of the spectrum, charge transfer transitions from the sulfur p-σ orbitals to nickel are observed at 28,508 cm−1, as shown in the difference density plot for State 17. Similar energies and transition difference densities are obtained with TD-DFT calculations on the backbone NiIIRd model (Table S2 and Figure S16), though State 8 from the full model is split from one intense band into two similar, near-degenerate transitions in the backbone NiIIRd model. In the [NiII(NACA)4]2− model, however, much poorer agreement is seen between experiment and theory (Figure S9). Instead of two dominant charge-transfer transitions, many weak electronic excitations are seen, giving rise to a broad, featureless absorption spectrum. A single-point TD-DFT calculation using the x-ray diffraction structure of Cp NiRd also gave relatively poor agreement between experiment and theory (Figure S17).
Resonance Raman spectroscopy
The excellent agreement between the experimental and calculated optical absorption spectrum supports the use of this computational model to predict more sophisticated spectroscopic properties, such as resonance Raman (RR) intensities.39 While resonance enhancement from excitation into d-d transitions is known to be weak due to the low associated nuclear displacements,92 the multiple ligand-to-metal charge transfer bands in the visible and near-UV region of the spectrum are well-suited for RR investigations. Resonance Raman spectra of NiRd were obtained with excitation wavelengths of 488 nm, 458 nm, 407 nm, and 364 nm. Subtraction of ZnRd control samples was performed to ensure that all bands observed in Figure 4 can be attributed to the resonantly enhanced nickel-thiolate chromophore. Corresponding theoretical resonance Raman spectra were calculated for the full NiRd model considering excitation into State 8 at 20,000 cm−1 and 25,000 cm−1 and State 17 at 25,000 cm−1 and 30,000 cm−1 to simulate the experimental spectra obtained at 488 nm, 458 nm, 407 nm, and 364 nm, respectively. The direct comparison between experiment and theory is shown in Figure 4.
Figure 4.
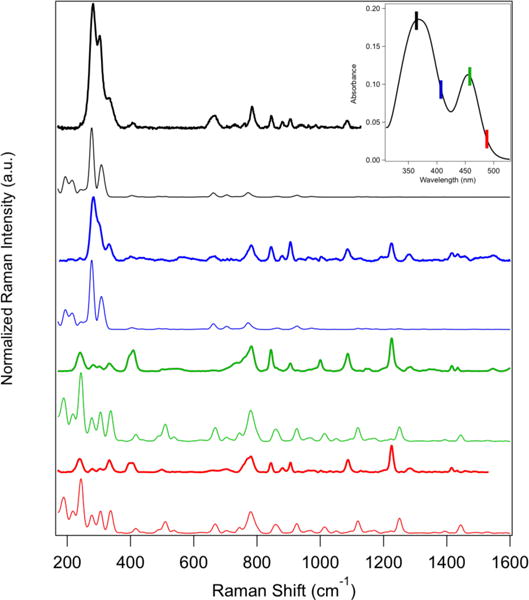
Experimental (thick lines, top) and calculated (thin lines, bottom) resonance Raman spectra of nickel-substituted rubredoxin at excitation wavelengths of 364 nm (black), 407 nm (blue), 458 nm (green), and 488 nm (red). (Inset) UV-vis absorption spectrum of NiRd with corresponding excitation wavelengths indicated.
The RR spectrum obtained with 364 nm excitation is dominated by three low-frequency modes at 282 cm−1, 303 cm−1, and 334 cm−1. Additional bands from ~650–900 cm−1 and ~1000–1100 cm−1, typical frequency ranges for sulfur-carbon and carbon-carbon stretching modes, respectively, are observed with approximately 10-fold lower intensities. The calculated RR spectrum of the full NiRd model using excitation directly into State 17 shows similar trends, with three large bands predicted in the low-frequency region of the spectrum and bands of significantly lower intensity in the high-frequency region. While the spectra are generally similar, a slightly different enhancement pattern is seen with excitation at 407 nm. Specifically, the low frequency modes are less enhanced relative to the high frequency modes. This change in intensity across the spectrum is also predicted in the RR calculations.
With excitation into the principal visible transition at 458 nm and 488 nm, the relative intensities of the low frequency peaks are approximately reversed. At these wavelengths, the most intense bands occur at 241 cm−1, 394 cm−1, and 409 cm−1. Additionally, the vibrational modes at higher frequencies are increasingly enhanced, with the most prominent high frequency peaks occurring at 783 cm−1, 844 cm−1, 1087 cm−1, and 1226 cm−1. These bands ultimately dominate the spectrum measured at 488 nm, though lower absolute intensities (when normalized to the Phe band) are seen. Analogous changes are seen in the calculated spectra.
Vibrational mode assignments
The vibrational frequencies of the RR bands seen at low energies are consistent with those expected for nickel-sulfur stretching and bending modes and can be qualitatively assigned on that basis. A more quantitative approach to vibrational mode assignments was carried out using the DFT-calculated vibrational frequencies in conjunction with predicted intensities (Table S3), which is necessary due to the high density of vibrational states found in such a large active-site model. To facilitate mode assignments, experimental and theoretical RR spectra were normalized to the intensity of the highest band at each wavelength for direct comparison (Figure 5).
Figure 5.
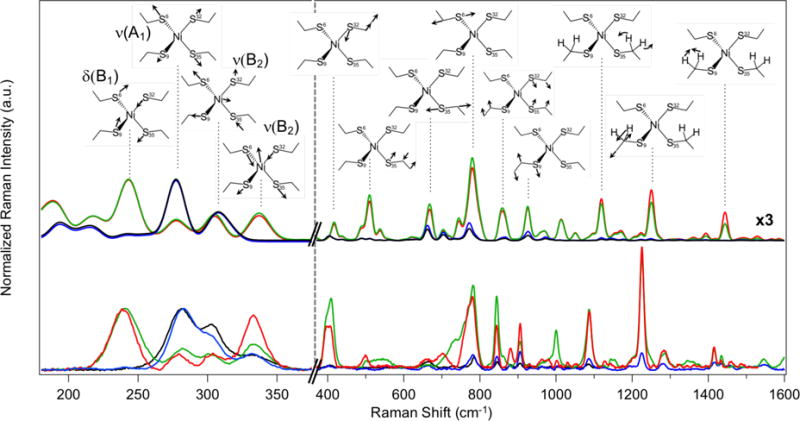
Intensity-normalized resonance Raman spectra of NiRd showing relative peak intensities across the four excitation wavelengths. Experimental (bottom) and calculated (top) spectra at 364 nm (black) and 407 nm (blue) were normalized to the 282 cm−1 peak, while the 458 nm (green) and 488 nm (red) spectra were normalized to the 241 cm−1 band to facilitate relative intensity comparisons. The high frequency region of the spectra is shown on a different abscissa, and the intensities of the calculated spectra in that region were scaled three-fold to better visualize the peaks. (Inset) Vector diagrams and approximate symmetry labels of the vibrational modes associated with the calculated spectra.
The low-frequency vibrations are dominated by metal-ligand normal modes. As discussed below, the vibrational modes were considered in a D2d framework, in which the tetrahedral (Td) T2-symmetric metal-ligand stretches and E and T2 bending modes are split into B2 + E stretching and A1 + B2 and B2 + E bending modes, respectively. The band calculated at 244 cm−1 corresponds to the δ(B1) S-Ni-S bend and has been assigned to the experimental band at 241 cm−1. The totally1 symmetric ν(A1) Ni-S stretch is calculated at 277 cm−1, in good agreement with the experimental band at 282 cm−1. The two medium-intensity bands at 308 cm−1 and 336 cm−1 correspond to the ν(B2) Ni-S anti-symmetric stretching modes, assigned to the experimental bands at 303 cm−1 and 334 cm−1. As mentioned above, the Jahn-Teller distortion at the active site results in longer Ni-S6 and Ni-S32 bonds than those of Ni-S9 and Ni-S35. As a result, the Ni-S anti-symmetric stretching modes occur at two different energies. The lower energy band at 308 cm−1 reflects the normal mode of the longer Ni-S bonds, while the higher energy band at 339 cm−1 reflects that of the shorter Ni-S bonds. The different nuclear displacement vectors for each of the modes are shown as insets in Figure 5. The bands calculated at 416 cm−1 and 418 cm−1 also show some ν(B2) Ni-S anti-symmetric stretching character; however, these modes are coupled into the Ni-S6-C bend and Ni-S32-C bend, respectively, resulting in increased frequencies relative to what would be anticipated for pure metal-ligand vibrational modes. These higher energy modes are assigned to the experimental bands at 394 cm−1 and 408 cm−1.
At higher frequencies, the vibrational bands are dominated by sulfur-carbon and carbon-carbon stretching modes, with some mixing into C-H stretching and bending motion as well. Again, owing to the asymmetry of the active site, two different vibrational frequencies are seen for the sulfur-carbon stretching modes. The bands at 661 cm−1 and 664 cm−1 represent the ν(S35-C) and ν(S9-C) modes, respectively. These cysteine residues exhibit shorter Ni-S bonds and, accordingly, S-C bonds that are longer by an average of 0.01 Å. The bands calculated at 743 cm−1 and 779 cm−1 are attributed to the ν(S6-C) and ν(S32-C) modes, respectively, which correspond to the two cysteine ligands featuring longer Ni-S bonds and thus shorter S-C bond lengths. These modes are closer in frequency to typical carbon-carbon bond vibrations and, as a result, some C-C wagging motion is mixed into the mode, causing increased frequencies relative to standard S-C stretching modes. The intense, narrow bands calculated at 926 cm−1 and 928 cm−1 represent the Cys9 C-C-C and Cys35 C-C-C bending modes, respectively, while the bands at 1011 cm−1 and 1019 cm−1 correspond to the Cys35 C-C and Cys9 C-C stretches. Above 1000 cm−1, significant proton motion is mixed into the heavy atom displacements, with the 1119 cm−1 and 1246 cm−1 bands assigned to the Cys35 Hβ-C-Hβ twist and Cys9 Hβ-C-Hβ twist coupled to ν(Cα-N) stretching motions, respectively. Finally, the calculated bands at 1443 cm−1 and 1447 cm−1 reflect the Cys9 δ(H-C9-H) and Cys35 δ(H-C35-H) bending modes. Calculated vibrational modes were assigned to experimental bands on the basis of visual correspondence in both intensity and frequency owing to the qualitative agreement between the intensity-normalized spectra, as is further described in Figure S18 and Table S3.
Isotope Shifts
Rubredoxin proteins are known to have a conserved set of hydrogen bonds between the backbone amide groups and the cysteine thiolate ligands that modulate structure and reduction potential.55,85 Short Namide - S distances of 3.4 – 3.7 Å were retained in the DFT-optimized geometry of NiIIRd (Table 1), with principal hydrogen-bonding interactions shown in Figure 2B. To probe this hydrogen-bonding network and identify vibrational modes that are coupled to the amide backbone, hydrogen/deuterium exchange experiments were performed. Resonance Raman spectra were obtained at all four excitation wavelengths for NiIIRd in protiated and deuterated buffer; representative spectra for both isotopes as well as the difference spectrum using 458 nm excitation are shown in Figure 6.
Figure 6.
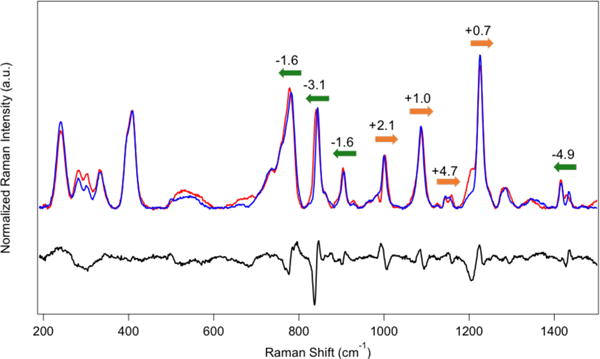
Resonance Raman spectra of NiRd (458 nm excitation) in buffered H2O (blue), D2O (red), and the corresponding difference spectrum (H – D, black). Frequency shifts indicated with colored arrows.
A number of modes are seen to shift experimentally upon isotopic exchange. In the low frequency region, where metal-ligand stretching and bending vibrations are prevalent, only minor changes in intensity are seen for the deuterated sample; vibrational frequencies of those modes generally remain constant. At higher frequencies, more pronounced changes are observed. Many bands shift to lower frequencies, including the S-C stretching mode at 780 cm−1 and C-C-C backbone wag at 840 cm−1; those modes are also calculated to shift in that direction (Table S3). However, the bands at 1000 cm−1, 1087 cm−1, 1153 cm−1, and 1225 cm−1, assigned to the backbone C-C stretching, Cys9/35 Hβ-C-Hβ twisting motion, and Cys6/32 Hβ-C-Hβ twisting motions, respectively, shift to higher frequencies in the presence of the heavier isotope. The calculated isotope shifts for these modes are also predicted to be positive, in support of the normal mode assignments. The increase in frequency can be attributed to coupling of N-H bending motion into the C-H torsional displacement. These amide protons are involved in strong hydrogen bonds to the coordinating cysteine residues. Owing to differences in mass, the vibrational coupling between the hydrogen-bonded amide backbone and the rest of the peptide will change upon isotopic substitution. This normal mode redistribution results in apparent frequency increases when deuterium is used. The same experimental frequency shifts are seen for analogous bands at different excitation wavelengths, albeit with different relative intensities (Figure S19). Because energy redistribution among normal modes upon isotopic substitution affects the calculated intensities significantly, some calculated shifts are overemphasized in the theoretical spectrum (Table S3 and Figure S20).
NiIIIRd
Spectrophotometric titrations using potassium ferricyanide (K3[Fe(CN)6], E˚’ = +420 mV vs. NHE at pH 7)93 were carried out by monitoring the NiIIRd charge transfer and d-d absorption bands (Figure 7A) as a function of solution potential. These data were fit to a standard Nernst equation to obtain a solution-phase reduction potential for the NiIII/II couple of +430 mV vs. NHE at pH 8 (Figure 7B). This potential was found to be pH-dependent, with an approximate −30 mV/pH unit shift from pH 4.5 to pH 10 (Figure S21). Protein film and solution-phase cyclic voltammetry experiments were also performed to directly measure the reduction potential. While the protein film experiments showed no electrochemical transition on the timescale of an experimentally accessible scan, possibly indicating slow electron transfer or film desorption, a quasi-reversible couple was observed using solution-phase electrochemistry, with an anodic peak potential of +610 mV at pH 4.5 (Figure 7C). This value is similar to that seen in the potentiometric titrations at pH 4.5, while the irreversibility of the transition is consistent with the lack of any signal in protein film voltammetry.
Using a thermodynamic square scheme, the reduction potential was calculated using DFT methods. To benchmark the level of theory and the accuracy of the computational methods, the reduction potential of the [FeCp2]0/+ couple was first examined in acetonitrile. The absolute reduction potential obtained by considering the change in the Gibbs free energy for the reduced and oxidized states was calculated to be +5.00 V. To convert this potential into a relevant reference frame, accepted values for the absolute potential of the standard hydrogen electrode (SHE) of +4.281 V and the standard calomel electrode (SCE) of +0.241 V vs. SHE were used.47 With this, the reduction potential for the [FeCp2]0/+ couple was calculated to be +0.44 V vs. SCE, in good agreement with experimental values of approximately +0.38 V vs. SCE.47 Analogous methods were employed for NiRd. The calculated reduction potential of the full NiIII/IIRd model was found to be +0.106 V vs. SHE, in modest agreement with the experimental values observed in the potentiometric and electrochemical measurements. The absolute reduction potentials and electronic energies of the [FeCp2]0/+ and NiIII/IIRd couples are presented in the Supporting Information (Tables S4–S5).
Because of the instability of the NiIIIRd species, no distinctive absorption peaks could be identified for the oxidized species. However, at low temperatures, EPR spectra of NiIIIRd were obtained (Figure S22). The spectral features are similar to those previously seen in the oxidized NiIIIRd from Pyrococcus furiosus,86 with g-values of gz = 2.27, gy = 2.09, and gx = 2.06 (giso = 2.14; Δgxy = 0.03). Given the known propensity of DFT to underestimate experimental g-tensor shifts,94 the calculated g-values are in satisfactory agreement with experiment at gz = 2.19, gy = 2.13, and gy = 2.10 (giso = 2.14; Δgxy = 0.03). The singly occupied molecular orbital (SOMO) represents a combination of the nickel dyz orbital and significant π* involvement from the sulfur px and py orbitals, with the spin density almost entirely localized on the metal center (Figure S22, inset).
Discussion
Comparison between experiment and theory
Developing accurate theoretical models for enzyme active sites presents a significant challenge across fields. Even with the rapidly increasing advances in computational power, it remains generally intractable to treat all of the atoms in an entire protein quantum mechanically. Conversely, considering only the active-site atoms can be a faulty approach, as many key markers for energetics and structure are incorrectly predicted.44,45 From an experimental standpoint, a similar argument can be made: Recreating an entire enzyme poses great biochemical difficulties, but generating a synthetic model that includes only the metal center and the primary coordination sphere typically fails to reproduce the reactivity of the enzyme.26,95 It is clear that, on both fronts, an intermediate approach is necessary.
The NiIIRd theoretical model developed in this work incorporates the dominant secondary sphere interactions within the CXXC loops and preserves the general structure imposed by the protein backbone.55,84 The model is moderately sized, containing 77 heavy atoms and 149 atoms total, and is readily processed with modest use of supercomputer resources.59 Importantly, with this level of the outer coordination environment included, we are able to accurately reproduce the geometric and electronic structures of NiIIRd. This is evident when considering the TD-DFT-predicted absorption spectrum, which shows excellent agreement with experimental results when considering both energetics and intensities of the transitions. As the electronic absorption spectrum of a system is highly sensitive to coordination environment and symmetry around the metal center,96–98 this high level of correspondence suggests the distortions predicted by the DFT optimization reflect the solution-phase geometry.
Further validation of the computational model is found in the predicted resonance Raman spectra. Resonance Raman spectroscopy relies on the coupling between electronic and vibrational states, with intensities governed by the displacement of the electronic excited state with respect to the ground state for each mode. The ground state vibrational frequencies that are measured in the experiment reflect bond length (bond order) and interatomic angles. Thus, RR spectroscopy is a direct probe of the electronic and geometric structures of a system.39,99
It is straightforward to calculate the vibrational frequencies for a given geometry; these parameters are directly related to each normal coordinate force constant, which is the second derivative of the potential energy surface.42 To predict the resonance Raman spectrum, the absorption cross-section must be coupled into the dimensionless displacements of each normal mode, which requires more sophisticated methods of computation.39 Traditionally, excited-state geometry optimizations must be performed and the excited state normal mode displacement is obtained by projecting the change in the mass-weighted Cartesian coordinates for each mode onto the excited-state normal coordinates.100–102 This process is quite cumbersome and virtually impossible to perform for large models of protein active sites. However, assuming harmonic potentials, the excited-state gradient can be used to predict the displacement along each normal mode. This method has been efficiently implemented in the ORCA_ASA program,75,78,103 developed by Petrenko and Neese, and integrated within the ORCA quantum chemistry software package.
To calculate the resonance Raman spectra of NiIIRd, the ORCA_ASA program was used within the IMDHO model considering only Albrecht A-term enhancement.103 Because the effects of overlapping excited states can introduce significant complications,104 a simplistic approach was taken in which RR intensities were calculated for only one excited state at a time, using a range of theoretical excitation energies, and the lower intensity transitions were ignored. Even with this primitive approach, notably good correspondence is seen between the experimental and predicted RR spectra, suggesting this approximation is valid. Considering that the relative intensity patterns of vibrational modes reflect the nature of the electronic transition probed, the spectra effectively serve as a “fingerprint” for the excited state.
Better agreement between experiment and theory is seen for the low frequency, metal-ligand-based modes relative to the higher energy, ligand-coupled modes, which has been seen previously in RR spectra of model inorganic compounds.37 The discrepancy at higher energies is ascribed to the fact that RR intensities are proportional to ν2Δ2, which causes increased enhancement for high frequency modes even in the case of smaller dimensionless displacements. For example, the calculated maximum Δs for the metal-ligand modes at 244 cm−1 and 277 cm−1 are approximately 1, while the corresponding Δs for the ligand-centered NiRd modes at 1000, 1200, and 1400 cm−1 are approximately only 0.05–0.1 (Table S3), approaching the accuracy limit of the methodology. As a result, the intensities of the higher energy vibrational transitions are often underestimated. Nevertheless, when this region of the spectrum is scaled appropriately and compared to experiment (Figure 5, right hand side), similar trends are observed between the calculated and measured spectral intensities.
Effects of the outer coordination sphere on the calculated spectroscopic properties
To assess the impact and necessity of including the CXXC side chains in the computational model, two smaller models were generated and analogous calculations performed. In the simplest [NiII(NACA)4]2− model, only the four cysteine amino acids were included, giving a symmetric active site without any contribution from the protein backbone or the outer coordination sphere. The optimized structure retained the tetragonal elongation, with two long nickel-sulfur bonds and two short ones, that would be expected for a high-spin Ni(II) system, illustrating the electronic rather than steric origin of this distortion. However, the [NiII(NACA)4]2− model poorly reproduced both the experimental absorption and resonance Raman spectra of NiRd, as shown in the Supporting Information (Figures S9 and S23).
As a next step, a minimal “backbone” model was also developed and analyzed. In this system, the CXXC binding loops with the peptide backbone were included, but all of the non-cysteine side chains were replaced by glycine. This model is significantly smaller, with only 64 heavy atoms and 115 total atoms. As such, it requires fewer resources for computational analysis and therefore could be a desirable replacement for the full model in future studies. Examining structural properties, only minor differences are observed between the optimized active-site geometries of the two models for both the NiIIRd and NiIIIRd redox states. In addition, the predicted energies, intensities, and nature of the electronic transitions are also very similar. Slight frequency shifts (<10 cm−1) were observed for the calculated vibrational bands, though the nuclear displacements of equivalent normal modes remained comparable. The same calculated g-tensor was obtained upon oxidation of the backbone model, further confirming the similarity in active-site electronic structure. On this basis, it would be reasonable to assume that the truncated backbone model accurately represents the NiRd active site.
However, important distinctions between the models become evident in the calculated resonance Raman intensities (Figure S23). While the calculated RR spectra of both models are dominated to some extent by the low frequency metal-ligand vibrations, greater intensity is transferred to the higher frequency, ligand-centered modes when the full side chains are included. The higher mode intensities can be attributed to increased coupling between the peptide backbone and side chains and the active site. The increased intensities thus reflect experimentally relevant contributions to the electronic excited state from the outer coordination sphere. This is reminiscent of the extended vibrational couplings seen between the heme group and the axial methionine ligand in cytochrome c and illustrates the potential relevance of these motions for modulating reactivity.105 The backbone model underestimates the intensity of those bands by approximately an order of magnitude when compared to experiment (Figure S23). In the full model, these bands are still slightly underestimated relative to the low frequency modes (Figure 5); however, it is important to note that the intensities scale as ν2Δ2, so smaller errors in calculated dimensionless displacements (Δs) result in larger deviations. Because a primary objective of this study is to develop and validate an accurate model of the NiRd active site for use in predicting RR features, reproducing the intensities and overall “fingerprint” across the entire vibrational spectrum is an important criterion. From this analysis, it seems necessary to use the full model of NiRd in future computational studies.
Using calculated RR spectra for mode assignments
In addition to providing a direct probe of the electronic transitions, the calculated resonance Raman spectra offer a convenient method for performing vibrational mode assignments. The traditional method for normal mode assignments typically requires systematic isotopic substitution and comparison to both calculated frequencies and frequency shifts. However, within a protein system, selective isotopic replacement can be prohibitively expensive and is often problematic. Further complicating analysis, substitution of heavy metals and sulfur-derived ligands only results in minor shifts (often <1 cm−1) that may fall within the resolution of the experiment and produce ambiguous assignments (Figure S24).106 By combining frequency and intensity analyses, as we have done in this study, robust vibrational mode assignments can be made.37
A visual depiction of how vibrational modes were assigned is shown in Figure S18, with the assignments and normal mode character given in Table S3 and expanded active-site displacements shown in Figure S25. Under an idealized tetrahedral geometry, the nickel thiolate core should have ν(A1) and ν(T2) stretching modes and δ(T2) and δ(E) sets of bending modes.106 Distortion to a D2d geometry lowers the symmetry and splits the T2 sets into a B2 + E set and the δ(E) modes into A1 + B1 modes (Figure S26). Briefly, considering the different enhancement patterns across the multiple excitation wavelengths employed, these fundamental normal modes were assigned in a pseudo-D2d framework (using the D2d point group symmetry labels). The totally symmetric ν(A1) stretching mode is calculated at 277 cm−1 and assigned to the band at 282 cm−1. This mode is particularly intense at the high-energy excitation wavelengths, as would be expected for enhancement with State 17, a sulfur-to-nickel σ* charge transfer transition (Figure 3, inset). At lower frequencies, the δ(E) set is split into two bands, with a B1 component observed at 244 cm−1, and the bands at 307 cm−1 and 335 cm−1 can be assigned to components of the ν(T2) set. This assignment is consistent with the ordering expected for a tetragonally elongated distortion, as seen in the DFT-optimized structure. Owing to vibrational coupling between the electronic and vibrational states, these typically weak normal modes are strongly enhanced with excitation into the lower energy, sulfur-to-nickel π* charge transfer transition. The bands at 394 cm−1 and 408 cm−1, previously attributed by Johnson and coworkers to cysteine deformation modes,106 have also been assigned here to Ni-S-C bending modes, with different frequencies attributed to the different cysteine thiolates.
The enhancement of high frequency bands (400 – 1500 cm−1) in the RR spectra arises because of mixing between metal-ligand modes and ligand-based vibrations and reflects coupling between the normal modes of the metal-ligand core and the protein backbone. Because of this vibrational mixing, RR spectroscopy can be used to monitor geometry and distortions beyond the metal-thiolate center. These modes provide information on local changes that arise from minor perturbations to the active site, for example, as a result of secondary sphere mutations. Additionally, we would expect significant frequency changes as a result of thiolate ligand protonation, which is involved in the proposed mechanism for hydrogen evolution. Assignment of these bands in the resting state will facilitate identification of changes in the ligand structure during catalysis.
Validation of the model across different oxidation states
In addition to obtaining a complete electronic and geometric description of the resting-state of the NiIIRd enzyme, we are interested in ultimately probing structures of catalytic intermediates. While NiIIIRd is not necessarily a proposed intermediate in the NiRd-catalyzed hydrogen evolution pathway, prior work by our lab and others have shown that this species can be generated near quantitatively and characterized,86,90 which renders it an ideal system for initial study and validation. The calculated EPR g-tensor, while of similar magnitude, is somewhat underestimated when compared to experiment. These systematic deviations have been reported previously for nickel-containing proteins.42,94,107,108 The NiIII/II reduction potential is also slightly underestimated by the calculations, though within the level of error seen for other calculated reduction potentials of metalloproteins;109 further, the pH effects noted experimentally are neglected in the computational model, which could account for the modest differences seen. These results provide proof-of-concept evidence that, with the moderately sized computational model developed here, other oxidation states of the system can be accurately described by density functional theory. Ultimately, these calculations may be able to serve in a predictive as well as interpretive role.
Conclusions
Resonance Raman spectroscopy is a powerful technique for selective investigation of the electronic and geometric structure of a molecule, which, ultimately, reflects the reactivity of the system. We have obtained a detailed description of the electronic structure of nickel-substituted rubredoxin, which is a highly active hydrogenase mimic, using resonance Raman spectroscopy across multiple excitation wavelengths coupled to DFT calculations on an extended active site model. Inclusion of the complete outer coordination sphere in the computational model is necessary to reproduce experimentally observed spectroscopic properties, such as optical absorption and resonance Raman intensities. In this way, vibrational mode assignments and a quantitative description of the electronic excited states have been obtained. Additionally, spectroscopic markers indicating active site distortion and ligand-based vibrational modes were identified. The excellent agreement between experiment and theory for the resting state of NiIIRd and the one-electron-oxidized NiIIIRd highlights the potential utility of this method to characterize the structures of catalytic intermediates and obtain insight into the mechanism of NiRd-catalyzed hydrogen evolution.
Supplementary Material
Acknowledgments
The authors would like to thank Haleigh Monaco for assistance with sample preparation and the OSC staff for technical support.
Funding Sources
This research was supported by the National Science Foundation (CHE-1454289); this work was also supported in part by The Ohio State University Materials Research Seed Grant Program, funded by the Center for Emergent Materials, an NSF-MRSEC, grant DMR-1420451, the Center for Exploration of Novel Complex Materials, and the Institute for Materials Research. Computational resources were provided through a grant from the Ohio Supercomputer Center (PAS1001).
ABBREVIATIONS
- RR
resonance Raman
- NiRd
nickel-substituted rubredoxin
- NACA
N- and C-acetylated cysteinate
- EPR
electron paramagnetic resonance; standard single-letter abbreviations used for all amino acids
Footnotes
Supporting Information. Power dependence, raw data and work-up procedure for resonance Raman spectra; detailed description of reduction potential calculations; semi-quantitative molecular orbital and d-orbital splitting diagrams; DFT-optimized structures, TD-DFT, calculated energies, and vibrational analyses of backbone, full, and (NACA)4 NiIIRd models in different oxidation states; EPR spectrum and SOMO of NiIIIRd; calculated IR spectra for NiIIRd; sample input files for ORCA calculations; and Cartesian coordinates for all optimized models. The following file is available free of charge.
References
- 1.Rifkin J. The Hydrogen Economy. Tarcher/Putnam; New York: 2002. [Google Scholar]
- 2.Turner JA. Sustainable Hydrogen Production. Science. 2004;305:972–974. doi: 10.1126/science.1103197. [DOI] [PubMed] [Google Scholar]
- 3.Lubitz W, Ogata H, Rüdiger O, Reijerse E. Hydrogenases. Chem Rev. 2014;114:4081–4148. doi: 10.1021/cr4005814. [DOI] [PubMed] [Google Scholar]
- 4.Shafaat HS, Rüdiger O, Ogata H, Lubitz W. [NiFe] hydrogenases: A common active site for hydrogen metabolism under diverse conditions. Biochim Biophys Acta BBA - Bioenerg. 2013;1827:986–1002. doi: 10.1016/j.bbabio.2013.01.015. [DOI] [PubMed] [Google Scholar]
- 5.Hidalgo R, Ash PA, Healy AJ, Vincent KA. Infrared Spectroscopy During Electrocatalytic Turnover Reveals the Ni-L Active Site State During H 2 Oxidation by a NiFe Hydrogenase. Angew Chem Int Ed. 2015;54:7110–7113. doi: 10.1002/anie.201502338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Evans RM, Brooke EJ, Wehlin SAM, Nomerotskaia E, Sargent F, Carr SB, Phillips SEV, Armstrong FA. Mechanism of hydrogen activation by [NiFe] hydrogenases. Nat Chem Biol. 2016;12:46–50. doi: 10.1038/nchembio.1976. [DOI] [PubMed] [Google Scholar]
- 7.Pandelia ME, Ogata H, Lubitz W. Intermediates in the Catalytic Cycle of [NiFe] Hydrogenase: Functional Spectroscopy of the Active Site. ChemPhysChem. 2010;11:1127. doi: 10.1002/cphc.200900950. [DOI] [PubMed] [Google Scholar]
- 8.Lubitz W, Reijerse E, van Gastel M. [NiFe] and [FeFe] hydrogenases studied by advanced magnetic resonance techniques. Chem Rev. 2007;107:4331–4365. doi: 10.1021/cr050186q. [DOI] [PubMed] [Google Scholar]
- 9.Ogata H, Krämer T, Wang H, Schilter D, Pelmenschikov V, van Gastel M, Neese F, Rauchfuss TB, Gee LB, Scott AD, et al. Hydride bridge in [NiFe]-hydrogenase observed by nuclear resonance vibrational spectroscopy. Nat Commun. 2015;6 doi: 10.1038/ncomms8890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horch M, Schoknecht J, Mroginski MA, Lenz O, Hildebrandt P, Zebger I. Resonance Raman Spectroscopy on [NiFe] Hydrogenase Provides Structural Insights into Catalytic Intermediates and Reactions. J Am Chem Soc. 2014;136:9870–9873. doi: 10.1021/ja505119q. [DOI] [PubMed] [Google Scholar]
- 11.Kwan P, McIntosh CL, Jennings DP, Hopkins RC, Chandrayan SK, Wu C-H, Adams MWW, Jones AK. The [NiFe]-Hydrogenase of Pyrococcus furiosus Exhibits a New Type of Oxygen Tolerance. J Am Chem Soc. 2015;137:13556–13565. doi: 10.1021/jacs.5b07680. [DOI] [PubMed] [Google Scholar]
- 12.McIntosh CL, Germer F, Schulz R, Appel J, Jones AK. The [NiFe]-Hydrogenase of the Cyanobacterium Synechocystis sp PCC 6803 Works Bidirectionally with a Bias to H-2 Production. J Am Chem Soc. 2011;133:11308–11319. doi: 10.1021/ja203376y. [DOI] [PubMed] [Google Scholar]
- 13.Parkin A, Sargent F. The hows and whys of aerobic H-2 metabolism. Curr Opin Chem Biol. 2012;16:26–34. doi: 10.1016/j.cbpa.2012.01.012. [DOI] [PubMed] [Google Scholar]
- 14.Fritsch J, Lenz O, Friedrich B. Structure, function and biosynthesis of O2-tolerant hydrogenases. Nat Rev Micro. 2013;11:106–114. doi: 10.1038/nrmicro2940. [DOI] [PubMed] [Google Scholar]
- 15.Leach MR, Zamble DB. Metallocenter assembly of the hydrogenase enzymes. Curr Opin Chem Biol. 2007;11:159–165. doi: 10.1016/j.cbpa.2007.01.011. [DOI] [PubMed] [Google Scholar]
- 16.Friedrich B, Fritsch J, Lenz O. Oxygen-tolerant hydrogenases in hydrogen-based technologies. Curr Opin Biotechnol. 2011;22:358–364. doi: 10.1016/j.copbio.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 17.Dutta A, DuBois DL, Roberts JAS, Shaw WJ. Amino acid modified Ni catalyst exhibits reversible H2 oxidation/production over a broad pH range at elevated temperatures. Proc Natl Acad Sci. 2014;111:16286–16291. doi: 10.1073/pnas.1416381111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dempsey JL, Brunschwig BS, Winkler JR, Gray HB. Hydrogen Evolution Catalyzed by Cobaloximes. Acc Chem Res. 2009;42:1995–2004. doi: 10.1021/ar900253e. [DOI] [PubMed] [Google Scholar]
- 19.Helm ML, Stewart MP, Bullock RM, DuBois MR, DuBois DL. A Synthetic Nickel Electrocatalyst with a Turnover Frequency Above 100,000 s−1 for H2 Production. Science. 2011;333:863–866. doi: 10.1126/science.1205864. [DOI] [PubMed] [Google Scholar]
- 20.Rodriguez-Maciá P, Dutta A, Lubitz W, Shaw WJ, Rüdiger O. Direct Comparison of the Performance of a Bio-inspired Synthetic Nickel Catalyst and a [NiFe]-Hydrogenase, Both Covalently Attached to Electrodes. Angew Chem Int Ed. 2015;54:12303–12307. doi: 10.1002/anie.201502364. [DOI] [PubMed] [Google Scholar]
- 21.Onoda A, Hayashi T. Artificial hydrogenase: biomimetic approaches controlling active molecular catalysts. Curr Opin Chem Biol. 2015;25:133–140. doi: 10.1016/j.cbpa.2014.12.041. [DOI] [PubMed] [Google Scholar]
- 22.Simmons TR, Berggren G, Bacchi M, Fontecave M, Artero V. Mimicking hydrogenases: From biomimetics to artificial enzymes. Coord Chem Rev. 2014;270–271:127–150. doi: 10.1016/j.ccr.2013.12.018. [DOI] [Google Scholar]
- 23.Schilter D, Camara JM, Huynh MT, Hammes-Schiffer S, Rauchfuss TB. Hydrogenase Enzymes and Their Synthetic Models: The Role of Metal Hydrides. Chem Rev. 2016;116:8693–8749. doi: 10.1021/acs.chemrev.6b00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lansing JC, Manor BC, Rauchfuss TB. Encyclopedia of Inorganic and Bioinorganic Chemistry. John Wiley & Sons, Ltd; 2011. Hydrogenase Models. [Google Scholar]
- 25.Losse S, Vos JG, Rau S. Catalytic hydrogen production at cobalt centres. Coord Chem Rev. 2010;254:2492–2504. doi: 10.1016/j.ccr.2010.06.004. [DOI] [Google Scholar]
- 26.Behnke SL, Shafaat HS. Heterobimetallic Models of the [NiFe] Hydrogenases: A Structural and Spectroscopic Comparison. Comments Inorg Chem. 2016;36:123–140. doi: 10.1080/02603594.2015.1108914. [DOI] [Google Scholar]
- 27.Slater JW, Shafaat HS. Nickel-Substituted Rubredoxin as a Minimal Enzyme Model for Hydrogenase. J Phys Chem Lett. 2015;6:3731–3736. doi: 10.1021/acs.jpclett.5b01750. [DOI] [PubMed] [Google Scholar]
- 28.Ogata H, Nishikawa K, Lubitz W. Hydrogens detected by subatomic resolution protein crystallography in a [NiFe] hydrogenase. Nature. 2015;520:571–574. doi: 10.1038/nature14110. [DOI] [PubMed] [Google Scholar]
- 29.Brecht M, van Gastel M, Buhrke T, Friedrich B, Lubitz W. Direct detection of a hydrogen ligand in the [NiFe] center of the regulatory H-2-sensing hydrogenase from Ralstonia eutropha in its reduced state by HYSCORE and ENDOR spectroscopy. J Am Chem Soc. 2003;125:13075–13083. doi: 10.1021/ja036624x. [DOI] [PubMed] [Google Scholar]
- 30.Wiedner ES, Yang JY, Chen S, Raugei S, Dougherty WG, Kassel WS, Helm ML, Bullock RM, Rakowski DuBois M, DuBois DL. Stabilization of Nickel Complexes with Ni0⋯H–N Bonding Interactions Using Sterically Demanding Cyclic Diphosphine Ligands. Organometallics. 2012;31:144–156. doi: 10.1021/om200709z. [DOI] [Google Scholar]
- 31.Dupuis M, Chen S, Raugei S, DuBois DL, Bullock RM. Comment on “New Insights in the Electrocatalytic Proton Reduction and Hydrogen Oxidation by Bioinspired Catalysts: A DFT Investigation”. J Phys Chem A. 2011;115:4861–4865. doi: 10.1021/jp111479z. [DOI] [PubMed] [Google Scholar]
- 32.Spiro TG, Czernuszewicz RS. Resonance Raman-Spectroscopy Of Metalloproteins. Methods Enzymol. 1995;246:416–460. doi: 10.1016/0076-6879(95)46020-9. [DOI] [PubMed] [Google Scholar]
- 33.Fiedler AT, Brunold TC. Combined spectroscopic/computational study of binuclear Fe(I)-Fe(I) complexes: Implications for the fully-reduced active-site cluster of Fe-only hydrogenases. Inorg Chem. 2005;44:1794–1809. doi: 10.1021/ic048739n. [DOI] [PubMed] [Google Scholar]
- 34.Galinato MGI, Whaley CM, Lehnert N. Vibrational Analysis of the Model Complex (mu-edt[Fe(CO)3]2 and Comparison to Iron-Only Hydrogenase: The Activation Scale of Hydrogenase Model Systems. Inorg Chem. 2010;49:3201–3215. doi: 10.1021/ic9022135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Siebert E, Horch M, Rippers Y, Fritsch J, Frielingsdorf S, Lenz O, Velazquez-Escobar F, Siebert F, Paasche L, Kuhlmann U, et al. Resonance Raman Spectroscopy as a Tool to Monitor the Active Site of Hydrogenases. Angew Chem Int Ed. 2013;52:5162–5165. doi: 10.1002/anie.201209732. [DOI] [PubMed] [Google Scholar]
- 36.Katz S, Noth J, Horch M, Shafaat HS, Happe T, Hildebrandt P, Zebger I. Vibrational spectroscopy reveals the initial steps of biological hydrogen evolution. Chem Sci. 2016;7:6746–6752. doi: 10.1039/C6SC01098A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shafaat HS, Weber K, Petrenko T, Neese F, Lubitz W. Key Hydride Vibrational Modes in [NiFe] Hydrogenase Model Compounds Studied by Resonance Raman Spectroscopy and Density Functional Calculations. Inorg Chem. 2012;51:11787–11797. doi: 10.1021/ic3017276. [DOI] [PubMed] [Google Scholar]
- 38.Albrecht AC, Hutley MC. Dependence Of Vibrational Raman Intensity On Wavelength Of Incident Light. J Chem Phys. 1971;55:4438–4443. doi: 10.1063/1.1676771. [DOI] [Google Scholar]
- 39.Myers AB, Mathies RA. Resonance Raman Intensities: A Probe of Excited-State Structure and Dynamics. In: Spiro TG, editor. Biological Applications of Raman Spectroscopy, Volume 2: Resonance Raman Spectra of Polyenes and Aromatics. Vol. 2 John Wiley and Sons; New York: 1987. [Google Scholar]
- 40.Han J, Loehr TM, Lu Y, Valentine JS, Averill BA, Sandersloehr J. Resonance Raman Excitation Profiles Indicate Multiple Cys-> Cu Charge-Transfer Transitions In Type-1 Copper Proteins. J Am Chem Soc. 1993;115:4256–4263. doi: 10.1021/ja00063a048. [DOI] [Google Scholar]
- 41.Petrenko T, Ray K, Wieghardt KE, Neese F. Vibrational markers for the open-shell character of transition metal bis-dithiolenes: An infrared, resonance Raman, and quantum chemical study. J Am Chem Soc. 2006;128:4422–4436. doi: 10.1021/ja0578451. [DOI] [PubMed] [Google Scholar]
- 42.Neese F. Prediction of molecular properties and molecular spectroscopy with density functional theory: From fundamental theory to exchange-coupling. Coord Chem Rev. 2009;253:526–563. doi: 10.1016/j.ccr.2008.05.014. [DOI] [Google Scholar]
- 43.Kampa M, Lubitz W, Gastel M, Neese F. Computational study of the electronic structure and magnetic properties of the Ni–C state in [NiFe] hydrogenases including the second coordination sphere. JBIC J Biol Inorg Chem. 2012;17:1269–1281. doi: 10.1007/s00775-012-0941-9. [DOI] [PubMed] [Google Scholar]
- 44.Krämer T, Kampa M, Lubitz W, van Gastel M, Neese F. Theoretical Spectroscopy of the NiII Intermediate States in the Catalytic Cycle and the Activation of [NiFe] Hydrogenases. ChemBioChem. 2013;14:1898–1905. doi: 10.1002/cbic.201300104. [DOI] [PubMed] [Google Scholar]
- 45.Siegbahn PEM. Modeling aspects of mechanisms for reactions catalyzed by metalloenzymes. J Comput Chem. 2001;22:1634–1645. doi: 10.1002/jcc.1119. [DOI] [Google Scholar]
- 46.Hu L, Söderhjelm P, Ryde U. Accurate Reaction Energies in Proteins Obtained by Combining QM/MM and Large QM Calculations. J Chem Theory Comput. 2013;9:640–649. doi: 10.1021/ct3005003. [DOI] [PubMed] [Google Scholar]
- 47.Pavlishchuk VV, Addison AW. Conversion constants for redox potentials measured versus different reference electrodes in acetonitrile solutions at 25°C. Inorganica Chim Acta. 2000;298:97–102. doi: 10.1016/S0020-1693(99)00407-7. [DOI] [Google Scholar]
- 48.Doig SJ, Prendergast FG. Continuously Tuneable, Quasi-Continuous-Wave Source for Ultraviolet Resonance Raman Spectroscopy. Appl Spectrosc. 1995;49:247–252. doi: 10.1366/0003702953963814. [DOI] [Google Scholar]
- 49.McCreery RL. Raman spectroscopy for chemical analysis. John Wiley & Sons; New York: 2000. [Google Scholar]
- 50.Sanchez KM, Neary TJ, Kim JE. Ultraviolet Resonance Raman Spectroscopy of Folded and Unfolded States of an Integral Membrane Protein. J Phys Chem B. 2008;112:9507–9511. doi: 10.1021/jp800772j. [DOI] [PubMed] [Google Scholar]
- 51.Shafaat HS, Leigh BS, Tauber MJ, Kim JE. Spectroscopic Comparison of Photogenerated Tryptophan Radicals in Azurin: Effects of Local Environment and Structure. J Am Chem Soc. 2010;132:9030–9039. doi: 10.1021/ja101322g. [DOI] [PubMed] [Google Scholar]
- 52.Harada I, Takeuchi H. Raman and Ultraviolet Resonance Raman Spectra of Proteins and Related Compounds. In: Clark RJH, Hester RE, editors. Spectroscopy of Biological Systems. John Wiley and Sons Ltd; Chichester, U.K.: 1986. [Google Scholar]
- 53.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera-A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 54.Stenkamp RE, Sieker LC, Jensen LH. The structure of rubredoxin from Desulfovibrio desulfuricans strain 27774 at 1.5 Å resolution. Proteins. 1990;8:352–364. doi: 10.1002/prot.340080409. [DOI] [PubMed] [Google Scholar]
- 55.Goodfellow BJ, Duarte ICN, Macedo AL, Volkman BF, Nunes SG, Moura I, Markley JL, Moura JJG. An NMR structural study of nickel-substituted rubredoxin. JBIC J Biol Inorg Chem. 2009;15:409–420. doi: 10.1007/s00775-009-0613-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Neese F. The ORCA program system. Wiley Interdiscip Rev Comput Mol Sci. 2012;2:73–78. doi: 10.1002/wcms.81. [DOI] [Google Scholar]
- 57.Pantazis DA, Neese F. All-Electron Scalar Relativistic Basis Sets for the Lanthanides. J Chem Theory Comput. 2009;5:2229–2238. doi: 10.1021/ct900090f. [DOI] [PubMed] [Google Scholar]
- 58.Pantazis DA, Chen X-Y, Landis CR, Neese F. All-Electron Scalar Relativistic Basis Sets for Third-Row Transition Metal Atoms. J Chem Theory Comput. 2008;4:908–919. doi: 10.1021/ct800047t. [DOI] [PubMed] [Google Scholar]
- 59.Ohio Supercomputer Center. 1987 [Google Scholar]
- 60.Becke AD. Density-Functional Exchange-Energy Approximation With Correct Asymptotic Behavior. Phys Rev A. 1988;38:3098–3100. doi: 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- 61.Perdew JP. Density-Functional Approximation for the Correlation-Energy of the Inhomogeneous Electron-Gas. Phys Rev B. 1986;33:8822–8824. doi: 10.1103/PhysRevB.33.8822. [DOI] [PubMed] [Google Scholar]
- 62.Eichkorn K, Weigend F, Treutler O, Ahlrichs R. Auxiliary basis sets for main row atoms and transition metals and their use to approximate Coulomb potentials. Theor Chem Acc. 1997;97:119–124. doi: 10.1007/s002140050244. [DOI] [Google Scholar]
- 63.Becke AD. Density-Functional Thermochemistry 3. The Role of Exact Exchange. J Chem Phys. 1993;98:5648–5652. doi: 10.1063/1.464913. [DOI] [Google Scholar]
- 64.Lee CT, Yang WT, Parr RG. Development of the Colle-Salvetti Correlation Energy Formula into a Functional of the Electron Density. Phys Rev B. 1988;37:785–789. doi: 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- 65.Kossmann S, Neese F. Efficient Structure Optimization with Second-Order Many-Body Perturbation Theory: The RIJCOSX-MP2 Method. J Chem Theory Comput. 2010;6:2325–2338. doi: 10.1021/ct100199k. [DOI] [PubMed] [Google Scholar]
- 66.Jackson TA, Karapetian A, Miller A-F, Brunold TC. Probing the Geometric and Electronic Structures of the Low-Temperature Azide Adduct and the Product-Inhibited Form of Oxidized Manganese Superoxide Dismutase. Biochemistry. 2005;44:1504–1520. doi: 10.1021/bi048639t. [DOI] [PubMed] [Google Scholar]
- 67.Wu H, Hall MB. Density functional theory on the larger active site models for [NiFe] hydrogenases: Two-state reactivity? Comptes Rendus Chim. 2008;11:790–804. doi: 10.1016/j.crci.2008.04.005. [DOI] [Google Scholar]
- 68.Liao R-Z, Himo F. Theoretical Study of the Chemoselectivity of Tungsten-Dependent Acetylene Hydratase. ACS Catal. 2011;1:937–944. doi: 10.1021/cs200242m. [DOI] [Google Scholar]
- 69.Weigend F, Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys Chem Chem Phys. 2005;7:3297. doi: 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- 70.Liptak MD, Fleischhacker AS, Matthews RG, Telser J, Brunold TC. Spectroscopic and Computational Characterization of the Base-off Forms of Cob(II)alamin. J Phys Chem B. 2009;113:5245–5254. doi: 10.1021/jp810136d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van Lenthe E, Baerends EJ, Snijders JG. Relativistic Regular 2-Component Hamiltonians. J Chem Phys. 1993;99:4597–4610. [Google Scholar]
- 72.van Lenthe E, Baerends EJ, Snijders JG. Relativistic Total-Energy Using Regular Approximations. J Chem Phys. 1994;101:9783–9792. doi: 10.1063/1.467943. [DOI] [Google Scholar]
- 73.van Lenthe E, van Leeuwen R, Baerends EJ, Snijders JG. Relativistic regular two-component Hamiltonians. Int J Quantum Chem. 1996;57:281–293. doi: 10.1002/(sici)1097-461x(1996)57:3<281∷aid-qua2>3.0.co;2-u. [DOI] [Google Scholar]
- 74.Grimme S, Antony J, Ehrlich S, Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J Chem Phys. 2010;132 doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- 75.Petrenko T, Kossmann S, Neese F. Efficient time-dependent density functional theory approximations for hybrid density functionals: Analytical gradients and parallelization. J Chem Phys. 2011;134:54116. doi: 10.1063/1.3533441. [DOI] [PubMed] [Google Scholar]
- 76.Furche F. On the density matrix based approach to time-dependent density functional response theory. J Chem Phys. 2001;114:5982. doi: 10.1063/1.1353585. [DOI] [Google Scholar]
- 77.Neese F, Olbrich G. Efficient use of the resolution of the identity approximation in time-dependent density functional calculations with hybrid density functionals. Chem Phys Lett. 2002;362:170–178. doi: 10.1016/S0009-2614(02)01053-9. [DOI] [Google Scholar]
- 78.Petrenko T, Neese F. Analysis and prediction of absorption band shapes, fluorescence band shapes, resonance Raman intensities, and excitation profiles using the time-dependent theory of electronic spectroscopy. J Chem Phys. 2007;127:164319. doi: 10.1063/1.2770706. [DOI] [PubMed] [Google Scholar]
- 79.Ray K, Petrenko T, Wieghardt K, Neese F. Joint spectroscopic and theoretical investigations of transition metal complexes involving non-innocent ligands. Dalton Trans. 2007;(16):1552. doi: 10.1039/b700096k. [DOI] [PubMed] [Google Scholar]
- 80.Konezny SJ, Doherty MD, Luca OR, Crabtree RH, Soloveichik GL, Batista VS. Reduction of Systematic Uncertainty in DFT Redox Potentials of Transition-Metal Complexes. J Phys Chem C. 2012;116:6349–6356. doi: 10.1021/jp300485t. [DOI] [Google Scholar]
- 81.Klamt A, Schuurmann G. COSMO – A New Approach To Dielectric Screening In Solvents With Explicit Expressions For The Screening Energy And Its Gradient. J Chem Soc Perkin Trans 2 Phys Org Chem. 1993;(5):799–805. doi: 10.1039/p29930000799. [DOI] [Google Scholar]
- 82.Isse AA, Gennaro A. Absolute Potential of the Standard Hydrogen Electrode and the Problem of Interconversion of Potentials in Different Solvents. J Phys Chem B. 2010;114:7894–7899. doi: 10.1021/jp100402x. [DOI] [PubMed] [Google Scholar]
- 83.Connelly NG, Geiger WE. Chemical Redox Agents for Organometallic Chemistry. Chem Rev. 1996;96:877–910. doi: 10.1021/cr940053x. [DOI] [PubMed] [Google Scholar]
- 84.Maher M, Cross M, Wilce MCJ, Guss JM, Wedd AG. Metal-substituted derivatives of the rubredoxin from Clostridium pasteurianum. Acta Crystallogr Sect D. 2004;60:298–303. doi: 10.1107/S090744490302794X. [DOI] [PubMed] [Google Scholar]
- 85.Lin I-J, Gebel EB, Machonkin TE, Westler WM, Markley JL. Changes in hydrogen-bond strengths explain reduction potentials in 10 rubredoxin variants. Proc Natl Acad Sci U S A. 2005;102:14581–14586. doi: 10.1073/pnas.0505521102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Huang YH, Park JB, Adams MW, Johnson MK. Oxidized nickel-substituted rubredoxin as a model for the Ni-C EPR signal of NiFe hydrogenases. Inorg Chem. 1993;32:375–376. doi: 10.1021/ic00056a004. [DOI] [Google Scholar]
- 87.Gee LB, Lin C-Y, Jenney FE, Adams MWW, Yoda Y, Masuda R, Saito M, Kobayashi Y, Tamasaku K, Lerche M, et al. Synchrotron-based Nickel Mössbauer Spectroscopy. Inorg Chem. 2016;55:6866–6872. doi: 10.1021/acs.inorgchem.5b03004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kowal AT, Zambrano IC, Moura I, Moura JJG, Legall J, Johnson MK. Electronic and Magnetic Properties of Nickel-Substituted Rubredoxin- A Variable-Temperature Magnetic Circular Dichroism Study. Inorg Chem. 1988;27:1162–1166. doi: 10.1021/ic00280a015. [DOI] [Google Scholar]
- 89.Moura I, Teixeira M, Xavier AV, Moura JJG, Lespinat PA, Legall J, Berlier M, Saintmartin P, Faugue G. Spectroscopic Studies of Cobalt and Nickel Substituted Rubredoxin. Recl Trav Chim Pays-Bas-J R Neth Chem Soc. 1987;106:418–418. doi: 10.1016/0162-0134(91)84025-5. [DOI] [Google Scholar]
- 90.Moura I, Teixeira M, Legall J, Moura JJG. Spectroscopic Studies of Cobalt and Nickel-Substituted Rubredoxin and Desulforedoxin. J Inorg Biochem. 1991;44:127–139. doi: 10.1016/0162-0134(91)84025-5. [DOI] [PubMed] [Google Scholar]
- 91.Van Heuvelen KM, Cho J, Dingee T, Riordan CG, Brunold TC. Spectroscopic and Computational Studies of a Series of High-Spin Ni(II) Thiolate Complexes. Inorg Chem. 2010;49:6535–6544. doi: 10.1021/ic100362q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sathyanarayana DN. Vibrational Spectroscopy: Theory and Applications. New Age International; 2015. [Google Scholar]
- 93.Goldberg M, Pecht I. Kinetics And Equilibria Of Electron-Transfer Between Azurin And Hexacyanoiron-(II/III) Couple. Biochemistry. 1976;15:4197–4208. doi: 10.1021/bi00664a011. [DOI] [PubMed] [Google Scholar]
- 94.Neese F. Prediction of electron paramagnetic resonance g values using coupled perturbed Hartree–Fock and Kohn–Sham theory. J Chem Phys. 2001;115:11080–11096. doi: 10.1063/1.1419058. [DOI] [Google Scholar]
- 95.Kaur-Ghumaan S, Stein M. [NiFe] hydrogenases: how close do structural and functional mimics approach the active site? Dalton Trans. 2014;43:9392. doi: 10.1039/c4dt00539b. [DOI] [PubMed] [Google Scholar]
- 96.Ballhausen CJ. Ligand field theory. Vol. 256 McGraw-Hill; New York: 1962. [Google Scholar]
- 97.Smith WE. The effect of distortion on the spin allowed ligand field bands of the spectrum of tetrahedral nickel(II) Spectrochim Acta Part Mol Spectrosc. 1982;38:1063–1068. doi: 10.1016/0584-8539(82)80035-4. [DOI] [Google Scholar]
- 98.Cotton FA, Faut OD, Goodgame DML. Preparation, Spectra and Electronic Structures of Tetrahedral Nickel(II) Complexes Containing Triphenylphosphine and Halide Ions as Ligands. J Am Chem Soc. 1961;83:344–351. doi: 10.1021/ja01463a021. [DOI] [Google Scholar]
- 99.Albrecht AC. Theory Of Raman Intensities. J Chem Phys. 1961;34:1476–1484. doi: 10.1063/1.1701032. [DOI] [Google Scholar]
- 100.Dierksen M, Grimme S. Density functional calculations of the vibronic structure of electronic absorption spectra. J Chem Phys. 2004;120:3544–3554. doi: 10.1063/1.1642595. [DOI] [PubMed] [Google Scholar]
- 101.Santoro F, Improta R, Lami A, Bloino J, Barone V. Effective method to compute Franck-Condon integrals for optical spectra of large molecules in solution. J Chem Phys. 2007;126:84509. doi: 10.1063/1.2437197. [DOI] [PubMed] [Google Scholar]
- 102.Berger R, Fischer C, Klessinger M. Calculation of the Vibronic Fine Structure in Electronic Spectra at Higher Temperatures. 1. Benzene and Pyrazine. J Phys Chem A. 1998;102:7157–7167. doi: 10.1021/jp981597w. [DOI] [Google Scholar]
- 103.Petrenko T, Neese F. Efficient and automatic calculation of optical band shapes and resonance Raman spectra for larger molecules within the independent mode displaced harmonic oscillator model. J Chem Phys. 2012;137:234107. doi: 10.1063/1.4771959. [DOI] [PubMed] [Google Scholar]
- 104.Shin KSK, Zink JI. Interference effects on resonance Raman excitation profiles caused by two electronic excited states. J Am Chem Soc. 1990;112:7148–7157. doi: 10.1021/ja00176a011. [DOI] [Google Scholar]
- 105.Galinato MGI, Kleingardner JG, Bowman SEJ, Alp EE, Zhao J, Bren KL, Lehnert N. Heme-protein vibrational couplings in cytochrome c provide a dynamic link that connects the heme-iron and the protein surface. Proc Natl Acad Sci. 2012;109:8896–8900. doi: 10.1073/pnas.1200345109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Huang YH, Moura I, Moura JJG, Legall J, Park JB, Adams MWW, Johnson MK. Resonance raman studies of nickel tetrathiolates and nickel-substituted rubredoxins and desulforedoxin. Inorg Chem. 1993;32:406–412. doi: 10.1021/ic00056a012. [DOI] [Google Scholar]
- 107.Shearer J, Neupane KP, Callan PE. Metallopeptide Based Mimics with Substituted Histidines Approximate a Key Hydrogen Bonding Network in the Metalloenzyme Nickel Superoxide Dismutase. Inorg Chem. 2009;48:10560–10571. doi: 10.1021/ic9010407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Manesis AC, Shafaat HS. Electrochemical, Spectroscopic, and Density Functional Theory Characterization of Redox Activity in Nickel-Substituted Azurin: A Model for Acetyl-CoA Synthase. Inorg Chem. 2015;54:7959–7967. doi: 10.1021/acs.inorgchem.5b01103. [DOI] [PubMed] [Google Scholar]
- 109.Niu S, Ichiye T. Density functional theory calculations of redox properties of iron-sulphur protein analogues. Mol Simul. 2011;37:572–590. doi: 10.1080/08927022.2011.582111. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.